
ABSTRACT
The duck represents an important reservoir of influenza viruses for transmission to other avian and mammalian hosts, including humans. The increased pathogenicity of the recently emerging clades of highly pathogenic avian influenza (HPAI) viruses of the H5N1 subtype in ducks features systemic viral spread and organ-to-organ variation in viral transcription and tissue damage. We previously reported that experimental infection of Sudani ducks (Cairina moschata) with an Egyptian HPAI (H5N1) virus (clade 2.2.1.2) features high viral replication and severe tissue damage in lung, but lower viral replication and only mild histological changes in brain. Little is known about the involvement of miRNA in organ-specific responses to H5N1 viruses in ducks, and involvement of the other classes of small noncoding RNA (sncRNA) has not been investigated so far. Following RNA sequencing, we have annotated the duck sncRNome and compared global expression changes of the four major sncRNA classes (miRNAs, piRNAs, snoRNAs, snRNAs) between duck lung and brain during a 120 h time course of infection with this HPAI strain. We find major organ-specific differences in miRNA, piRNA and snoRNA populations even before infection and substantial reprogramming of all sncRNA classes throughout infection, which was less pronounced in brain. Pathway prediction analysis of miRNA targets revealed enrichment of inflammation-, infection- and apoptosis-related pathways in lung, but enrichment of metabolism-related pathways (including tryptophan metabolism) in brain. Thus, organ-specific differences in sncRNA responses may contribute to differences in viral replication and organ damage in ducks infected with isolates from this emerging HPAI clade, and likely other strains.
Introduction
Infection with the highly pathogenic avian influenza (HPAI) viruses of the subtype H5N1 is still enzootic in poultry in many parts of the world, notably in Egypt [Citation1], and due to its potential to be transmitted to humans, poses risks to public health. Despite years of efforts to control this infection in Egyptian poultry, the virus has established an enzootic situation in various bird species. The risk resulting from the genetic changes that have occurred in HPAI viruses was highlighted when some of these viruses exhibited increased pathogenicity in avian hosts that were previously believed to be asymptomatic carriers, such as ducks [Citation2]. In ducks, HPAI H5N1 viruses can cause systemic spread and infect many organs with characteristic organ-to-organ variation in virus replication and end-organ damage [Citation3]. Indeed, the preferential replication of the virus in certain organs could determine the sequelae of the infection. Previous studies have reported that newly evolved HPAI H5N1 strains have switched their organ tropism in ducks to be more neurotropic in nature, which has been linked to demise of infected ducks [Citation2]. The mechanisms behind the increased pathogenicity of HPAI (H5N1) viruses in ducks are still unclear.
Most of the studies on infection dynamics of HPAI (H5N1) viruses in ducks and the consequent cellular and/or organismal host responses have focused on infection-driven changes in protein expression [Citation4,Citation5]. Investigations at the RNA level (coding and non-coding), on the other hand, have been underutilized. About 80% of the eukaryotic genome are transcribed, but only 1–2% of the transcripts encode proteins [Citation6] and the remaining 78% of expressed RNA correspond to non-coding RNAs (ncRNA), the functions of many of which remain to be elucidated. ncRNA <200 nucleotide in length are referred to as small noncoding RNAs (sncRNAs) and comprise four major classes: microRNA (miRNAs), small nucleolar RNAs (snoRNAs), small nuclear RNAs (snRNAs), and Piwi-interacting RNAs (piRNA) [Citation7]. miRNAs are commonly considered the most abundant, or at least the best studied, sncRNA population. Although they were first discovered in the nematode Caenorhabditis elegans [Citation8], the spectrum of their regulatory activities and functions have greatly expanded and span nearly all known biological processes [Citation9]. In ducks, miRNAs have been shown to mediate gene regulation in a multitude of processes, including lipid metabolism [Citation10], feather formation [Citation11] and responses to viral infections [Citation12]. However, only a single study this far has addressed the role of miRNA in the host response of duck (Anas platyrhynchos) to HPAI (H5N1) virus infection [Citation13], and none of the other sncRNA classes have been investigated. This is all the more surprising as miRNA expression constitutes an integral part of the host response to influenza viruses in non-avian (e.g. [Citation14,Citation15]) and avian hosts (e.g. [Citation16,Citation17]). Our previous analysis of published data sets [Citation14] revealed a group of miRNAs that are commonly expressed in various hosts following infection with influenza A virus (IAV). Li et al [Citation13] argued that reprogramming of miRNA populations in H5N1-infected thymus, spleen and bursa of Fabricius of chicken and ducks accounts for susceptibility of these bird species to H5N1 virus infection. Along the same line, experimental infection of leghorn chicken with IAV (H5N3) led to differential miRNA responses in lung and trachea [Citation16] and different chicken breeds (e.g. layers and broilers) might have characteristic miRNA expression patterns following infection with the same virus [Citation17], suggesting that miRNA could play roles in differences in immunity between the two breeds.
In contrast to the extensive work on miRNA in host responses to IAV infection, essentially nothing is known about roles of the other classes of sncRNA in cellular responses to IAV infection in general, or in the duck response to HPAI (H5N1) infection in particular. This is surprising as accumulating evidence suggests that at least global piRNA and snoRNA expression can change markedly during physiologic and pathologic adaptation of eukaryotic cells [Citation18–Citation20], that snoRNA can be processed to miRNA (reviewed in [Citation21]), and that snRNAs can regulate the IAV-induced blocking of cell pre-mRNA splicing [Citation22]. The lack of studies of sncRNA other than miRNA in the duck is likely due to the incomplete annotation of its sncRNAome, as only its miRNome has been annotated this far (e.g. [Citation11,Citation23]).
We have recently shown that experimental infection of Sudani ducks (Cairina moschata) with an Egyptian HPAI (H5N1) strain (clade 2.2.1.2) led to disseminated infection and major pathology in several organs including lung [Citation24]. Brain seemed to represent a unique ecological niche for the virus in that, even though the animals developed substantial neurological deficits, the extent of viral RNA replication and virus-induced tissue lesions were much lower than in the other organs. Considering the potential of all four major sncRNA classes to reflect cellular responses, we reasoned that global changes in the expression of each of the four major sncRNA classes might reflect the differences in viral spread and tissue destruction observed between lung and brain in this avian host. To address this question, we have annotated the duck sncRNAome, then compared the extent of differential expression (DE) of the four major sncRNA classes in lung and brain after infection with this HPAI (H5N1) strain, and finally predicted pathways regulated by the miRNAs DE in the two organs.
Results
Differences between lung and brain in virus transcription, and tissue inflammation and degeneration throughout the infection time course
The virus transcription kinetics and growth rate differed between lung and brain in that in lung there was a steep exponential rise in HA mRNA that peaked at 40–48 hpi, decreased markedly by 96 hpi and was followed by a rebound towards 120 hpi, whereas in brain exponential growth in HA mRNA transcription was milder (peaking at about 10-fold less than in lung) and the decline after 48 hpi was slower (). To test whether there were concomitant differences in inflammation and histological changes between lung and brain across the entire time course, we assessed the extent of histopathological alterations (inflammation and degenerative lesions) in the two organs across the time course. Indeed, inflammatory infiltration was detected in lung at the earliest time point and continued to increase throughout most time points, whereas practically no inflammatory cells were detected in brain (). Likewise, degenerative changes (necrosis of several cell types including airway epithelium) were detected in lung early on and persisted throughout most of the time course, whereas only spurious changes (a mild neuronal necrosis at 16 hpi) were detected in brain (). Thus, the observed higher viral replication in lung was paralleled by a much more intense inflammatory response and cell/organ damage in this organ compared to brain.
Figure 1. Viral transcription and tissue inflammation and degeneration in lung and brain of HPAI (H5N1)-experimentally infected ducks.
(A) Relative quantification of HA mRNA in lung and brain (three animals/time point), respectively. The FC in expression was calculated using the 2−∆∆CT method with duck β-actin mRNA as internal reference; the HA mRNA background signal detected in mock infected ducks was arbitrarily assigned the reference value of 1. This figure was adapted with permission from the publisher using data originally published in [Citation24]. (B, C) Semi-quantitative scoring of inflammation (b) and degeneration (c) in lung and brain throughout the time course, based on H&E-stained sections from three animals per time point. Each score consists of the average of subscores evaluating intensity and area involved, with a maximal possible score of 3.5. Data are plotted as the mean (±95% CI of SD) of three animals. The statistical significance of the differences between the two organs in terms of virus transcription at each time point was calculated using unpaired, two-tailed t-test. *P ≤ 0.05; **P ≤ 0.01;***P ≤ 0.001.
![Figure 1. Viral transcription and tissue inflammation and degeneration in lung and brain of HPAI (H5N1)-experimentally infected ducks.(A) Relative quantification of HA mRNA in lung and brain (three animals/time point), respectively. The FC in expression was calculated using the 2−∆∆CT method with duck β-actin mRNA as internal reference; the HA mRNA background signal detected in mock infected ducks was arbitrarily assigned the reference value of 1. This figure was adapted with permission from the publisher using data originally published in [Citation24]. (B, C) Semi-quantitative scoring of inflammation (b) and degeneration (c) in lung and brain throughout the time course, based on H&E-stained sections from three animals per time point. Each score consists of the average of subscores evaluating intensity and area involved, with a maximal possible score of 3.5. Data are plotted as the mean (±95% CI of SD) of three animals. The statistical significance of the differences between the two organs in terms of virus transcription at each time point was calculated using unpaired, two-tailed t-test. *P ≤ 0.05; **P ≤ 0.01;***P ≤ 0.001.](/cms/asset/423cf10a-c119-4d61-9f70-186d1dfba978/krnb_a_1669879_f0001_oc.jpg)
The repertoire of sncRNAs in duck lung and brain
As shown in Fig. S1, and Table S1, the average number of generated reads was somewhat higher in lung than in brain. More than half of these (68.2 and 61.4% in lung and brain, respectively) passed the length filter (default thresholds for removal were <32 and >15 nts). Of the unique mapped reads, 52.4% (lung) and 41.8% (brain) mapped uniquely to one of the small RNA classes (miRNA, piRNA, snoRNAs, and snRNAs) or to miscellaneous RNAs (rRNAs [ribosomal RNAs] and other RNA that mapped to the duck genome) (Fig. S1). A reads-vs.-sequence length plot revealed highest expression around 22 bases, which agreed with the expected size distribution of small RNAs (Fig. S2).
Table 1. Overview of the reads generated by small RNA-seq. of infected and mock-treated duck lung and brain. Values correspond to the average of the 30 libraries per organ representing all time points and replicates.
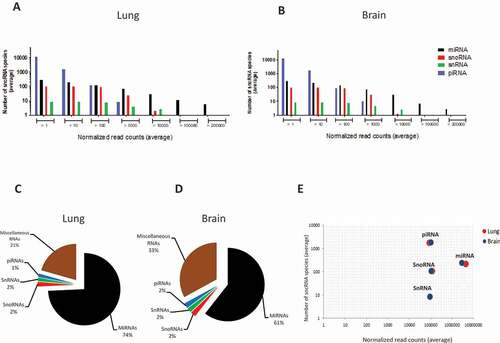
A total of 93,598 piRNAs, 518 miRNAs, 110 snoRNAs and 10 snRNAs were expressed in various abundance in lung and brain across all samples. As shown in , both the number of detected sncRNA species and their expression levels varied by sncRNA class, following similar patterns in lung and brain. Even though piRNA constituted the highest number of expressed sncRNA, about 90% were expressed at extremely low levels (mean <10 CPM per library). On the average, less than 1% of all expressed piRNAs contained >100 CPM and none were expressed at >10,000 CPM. miRNA expression, on the other hand, followed a different pattern in that there were much fewer lowly expressed species and that most sncRNA >10,000 and all >100,000 CPM were miRNA. Indeed, when considering only sncRNA with an average of >10 CPM, miRNA constituted the sncRNA class with the greatest number of reads in both organs (), followed by miscellaneous RNAs. miRNA were somewhat more abundant in lung than in brain, but there were no differences between lung and brain in terms of the other sncRNA classes. Also, lung and brain did not differ in the total number of species in each sncRNA class (). The mirdeep2 algorithm predicted seven novel miRNAs, three of which were highly abundant (mean >1000 CPM/sample) in both organs. The structures of two of these highly abundant predicted miRNAs are shown in Fig. S3.
Figure 2. Overview of the distribution of the four major sncRNA classes in lung and brain of mock- and HPAI H5N1-infected ducks (n = 30 in each organ). (A, B) Distribution of the average number of sncRNA species grouped according to expression level (average normalized read count, CPM). X-axes show ranges of mean expression level (reads), y-axes the number of species of each of the four sncRNA classes within the respective range. (C, D) Global abundance of each sncRNA class based on their average normalized read counts, based on all sncRNA species of >10 CPM/sample. (E) Comparison by sncRNA class of the average number of identified sncRNA species (y-axis) vs average expression level (x-axis), based on all sncRNA species with expression of >10 reads/sample.
Reprogramming of sncRNA expression following H5N1 infection in duck lung and brain
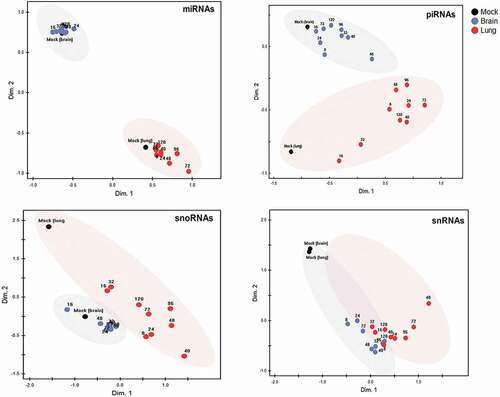
nMDS plots () and the underlying Euclidean distances (Fig. S4) were then used to compare the two organs in terms of extent and kinetics of reprogramming of the four sncRNA classes throughout infection. miRNA, piRNA and, to a somewhat lesser extent, snoRNA expression patterns clearly differentiated between the two organs even in the uninfected state (mock). In general, expression changes in all four sncRNA classes were greater in lung than in brain, but were evident as early as 8 hpi in both organs. In lung, reprogramming of miRNA and piRNA peaked by 72 h, of snRNA between 72 and 96 h, and that of snoRNA somewhat sooner (40 hpi) (). Changes in expression were less clear in brain. Visualization of Euclidean distances (Fig. S4) supported the impression from the nMDS that the differences in organ-specific expression patterns (i.e. lung vs. brain at most time points) of miRNA were greater than the changes induced within each organ by the evolving infection. In contrast, even though piRNA populations differed clearly between uninfected lung and brain, there were additional substantial infection-induced changes in piRNA expression, particularly in lung. Of note, expression of snRNA in lung and brain was nearly identical, but was affected markedly by infection in both organs as early as 8 hpi, i.e. approximately the duration of one cycle of IAV infection.
Figure 3. Global reprogramming of sncRNA classes in lung and brain of HPAI H5N1 virus-infected ducks.
Non-metric multidimensional scaling (nMDS) analysis, based on Euclidian distance, was applied to normalized expression values (CPM) of 518 miRNAs, 470 piRNAs (with a mean expression of >10 CPM), 110 snoRNAs, and 10 snRNAs. Each dot is based on the average read count (three biological replicates) of each sncRNA species at the respective time point in lung and in brain. Both organ-specific and infection-driven changes are seen in miRNA, piRNA and snoRNA, but only infection-driven changes in snRNA.
Differential expression of miRNAs in lung and brain
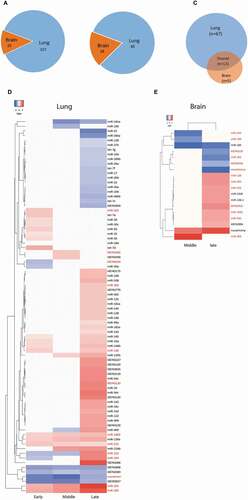
To obtain an overview of the differences of miRNA reprogramming between lung and brain at different phases of infection, the number of miRNA DE events across the time course (i.e. if one miRNA was DE at two time points it was counted as two; n = 157 in lung and 23 in brain) was compared between lung and brain ( and Fig. S5). In both organs, there were more DE events featuring up-regulation than down-regulation (Fig. S5) except during the middle infection phase in brain. There were 80 and 18 DE miRNAs in lung and brain, respectively (). Sixty-seven miRNAs were exclusively DE in lung, but only 5 exclusively in brain. Additionally, 13 miRNAs were DE in both organs at least at one time point (). Although these 13 miRNAs were DE in both organs, extent and timing of their DE differed substantially between the two organs throughout the course of the infection ().
Figure 4. Differential expression patterns of miRNAs in lung and brain of H5N1-infected ducks across the three major phases of infection.
(A) Number of miRNA DE events (i.e. if one miRNA was DE at two time points it was counted as two) in lung and brain at all time points. (B) Number of miRNAs that are DE (regardless of the number of time points) in lung and/or brain. (C, D) Heat maps showing the log2 FC values of the miRNAs that were DE in lung (n = 80) and in brain (n = 18). The red coloured miRNAs refer to those that are DE in both organs (n = 13). The remaining ones in each organs (black coloured miRNAs) are DE uniquely in each organ (lung = 67, brain = 5). Early = 8–24 hpi, middle = 32–48 hpi and late = 72–120 hpi. miRNAs were considered DE if they had log2 FC values > 0 (up-regulated, red coloured cells in the heat maps) or <0 (down-regulated, blue coloured cell in the heat maps) and adjusted P-values <0.05. MiRNAs were named after comparison with miRBase. Several miRNAs are either identical in sequence, but located on different genomic regions, or differ in 1–2 nucleotides and are located in the same genomic region; these were considered the same miRNA and were assigned the same name. MiRNAs that start with ‘KB’ could not be matched with any miRBase entry but were present in Ensemble. Novelmirna are predicted miRNAs that are not present in any of the databases.
Kinetics of differential miRNA expression in lung and brain
miRNA reprogramming started sooner and reached a higher extent in lung than in brain. In lung, there was a burst of DE during the first 24 hpi (early), followed by a period of relative quiescence (32–48 hpi, middle) and then a major peak of DE at late time points (72–120 hpi, late) (). DE in brain was not observed during the early phase, to a mild degree during the middle phase, and, again, was maximal during late infection (although it was less intense than in lung) (). These results were corroborated when considering the total number of DE events, i.e. counting two events if a miRNA was DE at two time points, etc. (Fig. S5). In an attempt to determine the potentially most robust miRNAs related to H5N1 virus infection in each organ, we tested whether any of the organ-specific miRNAs were DE in all infection phases. As shown in Fig. S6, 62.6%, 13.4% and 2.9% of the 67 lung-unique miRNAs were DE in late, middle and early infection phase, respectively, and five of these were DE in all infection phases. Of the five brain-unique DE miRNAs, three were DE only during the late infection phase and two (one known and one predicted) miRNAs were DE in both middle and late phases. Taken together, the above results agree with the findings from our previous study [Citation24], where we found that weight loss commenced at 16 hpi and continued at a relatively stable rate, whereas mortality was by far greatest in the late phase starting at 72 hpi; this phase also coincided with the greatest degree of miRNA DE in both organs.
Genomic loci of selected differentially expressed miRNAs
Considering the strong organ specificity of the miRNA responses, we tested whether there was an association between the organ-specific expression pattern of certain miRNAs and their co-localization in the duck genome. In the most recently released draft genome of the mallard duck (A. platyrhynchos) [Citation25], the duck genome sequence has not been assigned chromosome locations. Instead, 78,487 sequences were arranged into scaffolds, i.e. contigs that have not been assembled into known chromosomes. Therefore, it was only possible to retrieve each scaffold position and the transcripts of the 85 miRNAs DE in lung and/or brain contained in it (). As shown in Table S2, the 85 miRNAs are predicted to originate from 65 transcripts (ranging in size from 64 to 159 base pairs) positioned in 63 scaffolds. Fourteen of these 63 scaffolds (22.2%) were found to host more than 1 miRNA (range = 2–5 miRNAs). The majority of these 14 scaffold (11, 78.5%) contained miRNAs that exhibit the same organ-specific DE pattern (i.e. scaffolds that contain miRNAs DE specifically in lung (n = 10) or those that contain miRNAs DE commonly in lung and brain (n = 1)), whereas three of them (21.4%) contained miRNAs that were DE in a non-organ-specific manner (i.e. the scaffolds contain miRNAs that are DE in lung and others that are DE commonly in lung and brain). Performing a χ2 analysis on these data revealed that the miRNAs that were DE specifically in lung had the propensity to also co-localize on the same scaffold (Fisher’s exact test, P = 0.009). Interestingly, all miRNA commonly DE in both organs localized on the same scaffold. We also found that miRNAs that were positioned on one scaffold might originate from different (n = 9) or the same (n = 5) transcripts. In contrast, all five brain-specific miRNAs localized on separate scaffolds. There were differences between our analysis and information available in Ensemble in that 12 miRNAs for which our analysis revealed scaffold positions did not have annotated sncRNA positions in Ensemble (duck). By searching their orthologous positions in chicken (Gallus gallus), four were found to constitute miRNAs, one a snoRNA (snoRNA58), but the remaining seven could not be assigned. Analysing the chicken orthologs of the 85 studied miRNAs also revealed that three duck miRNAs are predicted to originate from snoRNAs. Of these, only one was annotated as a snoRNA in Ensemble (duck), one was identified as miscellaneous miRNA, and one was not found.
Prediction of organ-specific and common targets of the differentially expressed miRNAs
The regulatory functions of miRNAs rely mainly on targeting genes which themselves form components of various regulatory networks. As shown in Fig. S7, there was considerable overlap among the predicted target genes of the organ-specific DE miRNAs. However, functional prediction revealed that miRNAs regulate different pathways in lung and brain ( and Table S3). Overall, the identified KEGG pathways tended to be more numerous and more highly enriched in lung (n = 9; mean 13.3%) than in brain (n = 6; mean 2.5%). In lung, seven of the nine enriched KEGG pathways (RNA polymerase, proteasome, apoptosis, toll-like receptor signalling pathway, focal adhesion, and extracellular matrix) as well as the endothelin receptor B component of the neuroactive ligand receptor interaction pathway could be directly related to IAV infection and/or inflammation, and the enrichment of apoptosis agreed well with our histological data of increased cell/tissue degeneration in lung (). In contrast, in brain, most of the identified pathways related to metabolism. As tabulated in Table S3, enriched lung GO terms related to immunological process (e.g. natural killer cell mediated cytotoxicity), molecular processes (e.g. TGF-β activated receptor and phosphatidylinositol 3-phosphate), and biological processes (e.g. quinone and ubiquinone biosynthesis). In brain, GO terms such as positive regulation of axon extension and protein catabolic processes were enriched.
Figure 5. KEGG pathway enrichment predicted to be regulated by miRNAs DE in lung or brain during HPAI (H5N1) infection.
1358 predicted mRNA targets for the 67 miRNA DE in lung and 87 predicted mRNA targets of the 5 miRNA DE only in brain were used as input to retrieve the KEGG pathways enriched in the respective organ (P-value ≤0.05). The degree of enrichment of each KEGG pathway was determined by the percentages of the associated genes (number of the predicted target genes/overall number of gene entities in the corresponding pathway).
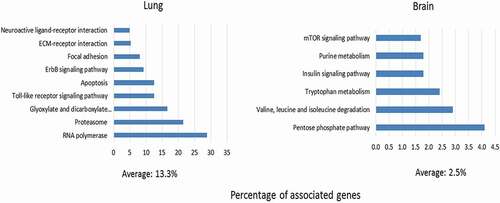
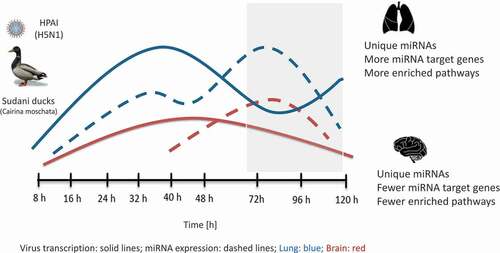
Figure 6. Schematic diagram summarizing the dynamics of HPAI (H5N1) virus and host-encoded miRNA expression in lung and brain of Sudani duck.
Virus transcription rate was higher in lung than in brain. This was followed by miRNA reprogramming, which was higher in lung than in brain and culminated during late infection phase (grey box). This was associated with higher enrichment of KEGG pathways targeted by the DE miRNAs in lung than in brain.
Discussion
Based on a de novo annotation of the duck sncRNome, we provide the first integrated analysis of expression changes in all major sncRNA classes following HPAI H5N1 virus (clade 2.2.2.1) infection in a natural host, the duck. In addition to the expected changes in miRNA expression, we identified major reprogramming also in piRNA and, less so, snoRNA and snRNA populations. Consistent with the previously described higher viral replication and stronger histopathologic changes in lung than in brain, expression changes of sncRNAs were greater in lung than in brain and predicted miRNA-regulated pathways in lung related more to anti-viral, inflammation and tissue damage responses, whereas in brain mostly to basic metabolic pathways. These findings are summarized schematically in Fig. 6.
Reprogramming of the major sncRNA classes
Studies on sncRNA dysregulation during IAV infection in avian hosts have mostly neglected the non-miRNA classes of sncRNA, likely due to the lack of bioinformatics tools and/or annotated sncRNomes. As expected, we found a strong reprogramming of miRNA expression in the majorly affected target organ, the lung, but also a small number of miRNA that are specifically DE in brain. Our results revealed that, in addition, populations of piRNA and, less so, snoRNA differed considerably between these two organs, even before infection, but that a substantial degree of expression change also took place after infection.
Although piRNAs were the sncRNA class with the highest species number, the majority of detected piRNAs were expressed at a low level. This suggests (1) that the activity and function of piRNAs might depend more on synergism between multiple species rather than on the activity of a single molecule, or (2) that a fair number of lowly expressed piRNA (<10 CPM reads/library) species may constitute biologically not very relevant RNA ‘noise’. On the other hand, the strong organ-specific and infection-driven expression changes in the more abundant piRNA (>10 CPM reads/library) ( and Fig. S4) clearly point to a strong association with organ-specific and infection-related processes. The latter finding agrees well with the previously documented roles of piRNAs in silencing the activity of transposable elements (TEs) [Citation26] and as anti-viral factors in insects [Citation27], and strongly suggests that piRNA play roles also in higher organisms (in this case, a vertebrate) in cellular responses to viral infections. In addition, the clear differences in piRNA populations between uninfected lung and brain add to the accumulating evidence that piRNAs may play important roles in eukaryotic gene regulation in general (reviewed in [Citation20,Citation28]). Clearly, further research is needed to empirically validate the expression and elucidate the function of this interesting class of sncRNA in host responses to influenza infection in the duck and other hosts. Our results showed that miRNAs (which were much lower in number than piRNAs) tended to be the most highly expressed sncRNA class. This was comparable to the results obtained in previous studies in lungs of mice infected with PR8 H1N1 viruses [Citation29,Citation30] and in thymus, bursa and spleen of H5N1-infected chicken and ducks [Citation13]. SnoRNA expression was intermediate. These molecules are known to regulate basic aspects of ribosome function and can give rise to other regulatory short RNAs [Citation21]. Considering that influenza virus infection impacts strongly on host cell translation, it will be of interest to study whether DE of snoRNAs participates in this cross-talk between virus and the host cell machinery.
Pathophysiologically important differences between lung and brain in predicted miRNA functions and regulated pathways
Due to the lack of bioinformatics tools to predict pathways regulated by piRNA or snoRNA, we restricted the predicted pathway analysis to miRNAs. Nevertheless, we found strong evidence of inter-organ differences in piRNA and snoRNA expression and also a significant degree of expression change throughout infection. It remains to be tested, once the corresponding pathway prediction tools for these sncRNA classes are available, whether the identified factors/pathways relate to specific pathophysiological differences as in the identified miRNA-regulates pathways, or whether the processes regulated by piRNA and snoRNA relate more to basic aspects of metabolism and homeostasis.
Our analysis revealed substantial differences between lung and brain in terms of the predicted miRNA-regulated pathways. The kinetic of histopathological analysis in as well as our previous histopathologic analysis of organs derived from the same duck infection experiment [Citation24] clearly showed marked cellular infiltration, histological alterations and cell damage in lung, but only mild changes (without any evidence of inflammatory cell infiltration into parenchyma) in brain. Consistent with this, the predicted KEGG pathways in lung related to antiviral responses, inflammation, and cell damage response/apoptosis. In addition, the GO terms revealed PI3-phosphate binding. These two latter findings agree well with our results from a mouse model of IAV infection, where an analysis of miRNA expression in infected lungs from DBA/2J mice (more susceptible to IAV infection) vs. lungs from C57BL/6J mice (less susceptible to IAV infection) revealed a strong induction of miRNA known to regulate apoptosis and the PI3K pathway [Citation30]. Furthermore, the rapid and early induction of apoptosis in duck lung has been previously reported in experimental infection of duck embryos and primary lung cells with H3N2, H1N1 and H5N1 [Citation31]. The proteasome pathway was among the highly enriched KEGG pathways in lung, with seven genes within it being targets of lung-specific DE miRNAs. Given the importance of the ubiquitin proteasome system in the entry of IAVs into host cells [Citation32], it is tempting to speculate that the invading H5N1 virus may use miRNAs to fine tune the host machinery for its own benefit. Glyoxylate and dicarboxylate metabolism was the third most highly enriched KEGG pathway in lung. This observation suggests that basic metabolic pathways may also be altered in H5N1 infected duck lung, but the exact implications of this intriguing observation remain to be elucidated.
In the absence of experimental verification, investigating the predicted miRNA–mRNA interactions might help to determine whether the observed miRNA reprogramming is part of the tissue-intrinsic anti-viral response or a virus-driven effect. For example, miR-145 and miR-125b, which were DE only in lung, target the zinc finger proteins ZFYVE19 and ZBTB8OS, which are known to mediate type-1 IFN induction during seasonal influenza virus infection [Citation33]. Thus, it is likely that the induction of these miRNAs in lung is driven by H5N1 virus to dampen host immunity. Conversely, other lung-specific miRNAs (e.g. miR-194, miR-20b, miR-106 and miR-146) were predicted to target protein tyrosine phosphatase (PTPN2), a suppressor of interferon responses [Citation34], suggesting that this may be part of the intrinsic host response geared towards initiating IFN-mediated anti-viral activities.
Interestingly, many of the identified lung-unique miRNAs have been reported previously to be DE in lung or trachea of IAV-infected chicken or ducks [Citation16,Citation17]. Two of the lung specific miRNAs, miR-130a and miR-125b are particularly important because they were DE with respect to mock infection in all phases of H5N1 infection. miR-130a expressed in chicken lung was previously predicted to target the polymerase basic 1 (PB1) segment of H5N1 virus [Citation35] and was significantly up-regulated in lungs of H5N1-infected mice irrespective of virus pathogenicity [Citation36]. miR-125b was de-regulated upon H5N3 virus infection in lung and trachea of chickens [Citation16] and exhibited a similar DE pattern in thymus, bursa and spleen of HPAI H5N1-infected chicken and ducks [Citation13]. This miRNA was found also to be DE after IAV infection in humans and pig [Citation15,Citation37].
In brain, only one of the six predicted pathways (mTOR) related to known anti-IAV responses. It is tempting to speculate that (in analogy to a recent publication using a cellular model of IAV infection), regulation of mTOR in H5N1-infected duck brain relates to autophagy, as a manifestation of cell stress and/or viral replication [Citation38]. The other five KEGG pathways related to cellular metabolism. This agrees well with the well-established finding that viral CNS infections can effect profound changes in CNS metabolism, for instance by altering energy substrate utilization or inducing the tryptophan-kynurenine-NAD+ pathway [Citation39]. Indeed, tryptophan metabolism was one of the predicted KEGG pathways in brain. In addition, these findings fully support the histological observation that we detected essentially no inflammatory or degenerative changes in brain in our study. They also suggest that the strongly manifested neurological signs of these ducks at the time of death [Citation24] are not due to destruction of brain parenchyma (as is often observed in HPAI H5N1 infection of susceptible avian hosts) but due to subcellular events of neurodegeneration or a metabolic encephalopathy.
This is the first study designed to identify brain-specific miRNA in influenza virus infection of any species. Only five miRNAs were DE only in brain, three of which have been studied in other models. miR-183, which was down-regulated in brain in both middle and late time points, was down-regulated in the brain of mice infected with rabies virus [Citation40], arguing that the down-regulation of this miRNAs may be a general feature of neurotropic viruses. miR-138 has previously been shown to regulate cyclin-dependent kinase 13 (CDK-13) in IAV-infected human respiratory epithelial A549 cells [Citation41]. miR-1416 is expressed in chicken lung and has been predicted to target the PB1 segment of H5N1 virus [Citation35]. Further studies are needed to explore the potential function of these miRNAs in the context of HPAI (H5N1) virus infection in ducks.
Genomic localization of the significant DE miRNAs
A large proportion of host-encoded miRNAs (36% of human, 46% of mouse miRNAs) reside in clusters on the genome [Citation42]. MiRNAs within a single cluster are usually transcribed as single polycistronic primary transcripts and thus might be cooperatively expressed during the same biological processes [Citation43]. Albeit our analysis was done on a small set of miRNAs, our results showed that the majority (11/14, 78.5%) of miRNA clusters harboured miRNAs that exhibited an organ-unique DE pattern, suggesting the possibility that the organ-specific DE pattern is linked to the co-localization of the respective miRNAs. Additional wide-scale bioinformatics analyses of other experimental systems featuring duck are needed to unravel the link between the co-localization of miRNAs and their biological functions in the context of IAV infection. The observation that three miRNAs were found to be homologs to snoRNAs in chicken support the notion that snoRNAs could generate miRNAs as a part of their regulatory function [Citation44].
miR-223 and miR-155
It is worth mentioning that while miR-223 and miR-155 have been implicated in various aspects of IAV pathogenesis [Citation30,Citation45,Citation46], in our study, miR-223 was not detected and miR-155 was detected but not DE (adjusted p = 0.1–0.8). This clearly substantiates the notion that the miRNA response to IAV infection differs depending on host species and/or viral strain.
Limitations of the presented study
The low number of biological replicates per time point, which was necessary in order to find a compromise between sampling a sufficient number of time points, achieving an acceptable statistical power per treatment, and animal welfare considerations, limits some of the interpretations. Future work should be directed at using higher numbers of biological replicates, but focusing on fewer time points. In addition, including one or more time points beyond 120 hpi would be important, as both viral transcription and miRNome reprogramming were clearly still ongoing at 120 hpi. This would help unravel long-term effects of the virus, particularly on the brain. Since the duck genome is still incomplete, the annotations done in this study are mostly based on similarity searches to closely related species (i.e. chicken) and the duck sncRNAome will certainly experience some revision once genome annotation and chromosome assignment have been completed.
Conclusions
The results of the current study provide first insights into tissue-specific responses of all major sncRNA classes, with most detailed focus on miRNA, during HPAI H5N1 virus infection in a natural avian host, the duck . Expression of specific sncRNA will differ depending on tissue tropism and virulence of the viral strain used, but the overall principle that expression in all four sncRNA classes is reprogrammed in response to organ- and virus-specific factors will likely to be a recurring theme. These data open new avenues for future studies on sncRNA classes in the responses of avian and other hosts (including humans) to influenza virus infection, particularly their functions in regulating end-organ damage and permissiveness of an organ to entry, replication and transmission of the virus.
Materials and methods
Virus propagation and titration
The A/chicken/Faquos/amn12/2011 (H5N1) strain (PubMed accession number: JQ627585.1) was kindly provided by Dr. Abdo Nagy (Virology Department, Faculty of Veterinary Medicine, Zagazig University, Zagazig, Egypt) and was used in this experiment. This virus was isolated from household broiler chickens following a fulminant outbreak in El-Sharkia province, Egypt. Viral propagation and titration were done as previously described [Citation47]. Briefly, 200 µl of the virus was inoculated into 11-day-old specific pathogen-free (SPF) embryonated chicken eggs (SPF-ECEs), and equal numbers of SPF-ECEs were used as control groups. All SPF-ECEs were incubated and monitored daily for viability. The propagated virus was identified in the harvested allantoic fluid by haemagglutination inhibition (HAI) test as described previously [Citation47]. Virus titres were calculated according to the Reed and Muench method [Citation48].
Experimental infection
Detailed infection protocols are described in [Citation24]. Briefly, prior to virus inoculation, previous H5N1 infection was excluded by testing blood samples using HAI test and enzyme-linked immunosorbant assay (ELISA). Two-week-old ducks (average weight 730 g) were separated into two groups: mock treatment (n = 30) and HPAI H5N1 infection (n = 60) groups. Ducks in both groups were inoculated intranasally with 100 µl of PBS or 100 µl of a viral suspension containing 106.7 embryonated egg infective dose 50 (EID50), respectively. At least three infected ducks that showed clinical signs were euthanized at 8, 16, 24, 32, 40, 48, 72, 96 and 120 h post-infection (hpi). For comparison, three randomly selected ducks from the mock-treated group were euthanized. Lung and brain tissues were kept in RNAlater (Qiagen) at −80°C until RNA extraction. All experimental work was done in compliance with the local rules on animal welfare and was approved ahead by the Ethics Committee for Animal Studies at the Faculty of Veterinary Medicine, Zagazig University, Egypt.
RNA extraction and mRNA expression analysis
The biosamples were collected in the context of the studies published in [Citation24]. Extraction of total RNA, reverse transcription (RT) and reverse transcriptase real-time PCR (RT-qPCR)-based detection of viral haemagglutinin (HA) mRNA was performed as described previously [Citation24].
Kinetics of histological alterations in lung and brain
4-µm-thick sections of formalin-fixed, paraffin-embedded tissues were stained with haematoxylin-eosin (H&E) and evaluated by a board-certified veterinary pathologist (co-author FS), who was blinded to the identity of the slides. Inflammation (inflammatory infiltrates) and cell/tissue degeneration (necrotic lesions) were assessed separately in samples from three ducks at each time point. For each of the two parameters, degree (intensity) (0 = not detected, 1 = low intensity, 2 = intermediate intensity, and 3 = high intensity) and tissue area involved (0 = not detected, 1 = 1–25%, 2 = 26–50%, 3 = 51–75%, and 4 = 76–100%) of total area were scored and the intensity and area involved subscores was taken to yield the final score. Representative H&E stains of lung (16 hpi) and brain (48 hpi) are shown in [Citation24].
Library preparation and small RNA sequencing (RNA-seq.)
Small RNA-seq. was carried out as described previously [Citation30] using the TruSeqTM Small RNA-seq. Kit and a HiSeq 2500 Illumina sequencer (both Illumina, Inc., San Diego, CA, USA). The TruSeqTM Small RNA-seq Kit is specific for miRNAs and other small RNAs (sRNAs) that have a 3ʹ-OH group resulting from enzymatic cleavage by RNA processing enzymes. RNA (approximately 1250 ng/sample) was used to synthesize complementary DNA (cDNA), which was then amplified through 15 PCR cycles. Gel electrophoresis was used to separate cDNA library fragments (nucleotide length = 150 nucleotides (nts)) and the cDNA was then isolated from the excised gel slice. Tissues from at least three animals per condition were analysed. Eight samples per lane were sequenced using Illumina molecular barcode. To reduce batch effects, samples from the same time point were never run on the same lane.
Identification, prediction, and annotation of Sudani duck sncRNAs
Small RNA prediction and identification were conducted using OASIS [Citation49]. To annotate known and to predict novel sncRNAs including miRNAs, the sRNA detection module of OASIS was used to align the sequenced reads to the duck (A. platyrhynchos) genome (BGI_duck_1.0) [Citation25] available in Ensemble [Citation50]. Primers and adaptor sequences were removed using the cutadapt software (version 1.8.1) [Citation51]. Quality control was done using fastqc version 0.11.2. Since only four duck miRNA species were annotated in the most recent version of miRBase (version 22.1) [Citation52], putative duck miRNAs were annotated using the Mirdeep2 software (version.2) [Citation53] by similarity with miRNAs annotated for other species in miRBase version 22.1 [Citation52]. snoRNAs, snRNAs and rRNAs were retrieved from the Ensemble database by matching the sequenced reads against human (version GRCh38.74), mouse (version GRCh38.74) and Drosophila melanogaster (version BDGP5.77) [Citation50]. Duck genome (version ap1) was mapped against all libraries using Spliced Transcripts Alignment to a Reference (STAR) software [Citation54]. We used the following restricting mapping parameters to minimize the number of mapped contamination and artefacts: out filter Mismatch NoverLmax 0.05 (allowing 0 mismatches for reads with length 15–19 and not more than 1 mismatch for reads with length 20–32), out-filter-Match-N-min15, out Filter Score Min Over Lread 0, out filter Match N min Over Lread 0, align Intron Max 1, reads mapping to more than five positions on the genome were not considered. The size limit of ≥15 bases was chosen because a particular sequence of this length would be expected to occur 1 in 4.2 Gb, whereas the duck genome is only 1.1 Gb in size. To annotate the piRNAs, the sequenced reads (fastq files) were mapped against known chicken piRNAs (n = 17,167,95) included in piRBase [Citation55]. In order to count the number of reads on each predicted and annotated miRNA, we used the featurecounts software (version 1.5.0) [Citation56] with default parameters.
Non-metric multidimensional scaling analysis
In order to describe global changes in the expression of sncRNAs in lung and brain and across the time course of the infection, mean expression (count per million reads, CPM) of each identified sncRNA species (miRNAs = 518, snoRNAs = 110, snRNAs = 10) at each time point was used as input into a non-metric multidimensional scaling (nMDS) analysis based on Euclidean distances. Because it was not feasible to use all the 93,598 predicted potential piRNAs, only piRNAs with mean >10 CPM across all samples (n = 470) were used for the nMDS. The analysis was done using PC-ORD (V. 5) software, Gleneden Beach, OR, USA.
Differential expression analysis of the identified miRNAs
The count files produced from the small RNA detection module of OASIS were used as inputs into the DE module of OASIS [Citation49], which includes the R package DeSeq2 (version 1.10.1) [Citation57]. MiRNAs with adjusted p-values <0.05 and a FC of ≥1 or ≤1 were considered significantly DE.
Genomic position of selected differentially expressed miRNAs
We used the draft assembly of the mallard duck (A. platyrhynchos) genome (BGI_duck_1.0) [Citation25] available in Ensemble [Citation50], which had been assembled to the scaffold level, to assign the genomic location of the DE miRNAs. We defined the location of each miRNA with respect to the genome scaffolds and the pri-miRNA transcript by using a restricted mapping alignment in order to avoid contaminants and artefacts.
In silico target prediction and functional analyses of differentially expressed miRNAs
The sequences of the 3ʹ-untranslated regions (UTRs) of the annotated duck mRNAs were retrieved from Ensemble selecting duck as background and using biomaRt queries (version 2.27.2) (http://www.biomart.org/). For RNA target prediction, RNAhybrid target predictor software [Citation58] was used. For calibration purposes, the frequencies of dinucleotides were calculated using all 3ʹ-UTR sequences. Using the dinucleotide frequencies, RNA calibrate (parameters ‘-d 3 UTR_duck.freq -k 5000 -l 500,300’) was executed to estimate position and shape parameters of the 3ʹ-UTRs. The resulting values were 2.20 and 0.17 for position and shape, respectively, and were used as input for the RNA hybrid. All targets with p-values <0.001 were selected. To predict functions of the DE miRNAs, the target mRNA genes for the significant DE miRNAs were used as inputs in ClueGo V2.1.7 [Citation59], a Cytoscape [Citation60] (V 3.2.0) plug-in, using the default parameter and duck (A. platyrhynchos) as background species. This allowed retrieving the Gene Ontology (GO) terms and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways. These analyses identified a total of 2121 gene targets, comprising 1358 predicted targets for the 67 miRNAs DE in lung, 87 predicted targets for the 5 miRNAs DE in brain, and 676 target genes for the 13 miRNAs DE in both organs. The enriched GO and KEGG terms were selected with a cut-off of P ≤ 0.05. The degree of enrichment of a certain GO term or KEGG pathway was determined by the percentage of associated genes (calculated as number of identified genes/total number of gene entities in the respective GO term or KEEG pathway).
Author contributions
M.S., F.A. and F.P. designed the experiments. M.S., F.A. and A.H. performed the experiments. R.G. performed the small RNA sequencing. M.S., V.C., R.O.V. and S.B. performed the bioinformatics and statistical analyses of the data. F.A. contributed to data analysis. F.S. performed the histopathological examination. M.S. prepared the figures and tables and wrote the initial draft of the manuscript. F.P. supervised the project and revised the final version of the manuscript. All authors reviewed the final version of the manuscript and agree with its content.
Supplemental Material
Download Zip (16.7 MB)Acknowledgments
We thank Michael Jarek and Sabin Bhuju for providing technical support with RNA seq., Alessandro Boianelli for his involvement in the early phase of the project. We also thank Leonardo S. de Araujo (TWINCORE) and Matthias Preusse (Helmholtz Centre for Infection Research) for helpful discussions. The TWINCORE is a joint venture of Hannover Medical School and the Helmholtz Centre for Infection Research, Braunschweig, Germany.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplemental material
Supplemental data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- ElMasry I, Elshiekh H, Abdlenabi A, et al. Avian influenza H5N1 surveillance and its dynamics in poultry in live bird markets, Egypt. Transbound Emerg Dis. 2017;64(3):805–814. PubMed PMID: 26608470.
- Sturm-Ramirez KM, Ellis T, Bousfield B, et al. Reemerging H5N1 influenza viruses in Hong Kong in 2002 are highly pathogenic to ducks. J Virol. 2004;78(9):4892–4901. Epub 2004/04/14.PubMed PMID: 15078970; PubMed Central PMCID: PMCPMC387679.
- Pantin-Jackwood MJ, Costa-Hurtado M, Shepherd E, et al. Pathogenicity and transmission of H5 and H7 highly pathogenic avian influenza viruses in mallards. J Virol. 2016;90(21):9967–9982. PubMed PMID: 27558429; PubMed Central PMCID: PMC5068544.
- Smith J, Smith N, Yu L, et al. A comparative analysis of host responses to avian influenza infection in ducks and chickens highlights a role for the interferon-induced transmembrane proteins in viral resistance. BMC Genomics. 2015;16:574. Epub 2015/08/05. PubMed PMID: 26238195; PubMed Central PMCID: PMCPmc4523026.
- Fleming-Canepa X, Aldridge JR Jr., Canniff L, et al. Duck innate immune responses to high and low pathogenicity H5 avian influenza viruses. Vet Microbiol. 2019;228:101–111. Epub 2018/12/30. PubMed PMID: 30593354; PubMed Central PMCID: PMCPmc6365012.
- Mudge JM, Harrow J. Creating reference gene annotation for the mouse C57BL6/J genome assembly. Mamm Genome. 2015;26(9–10):366–378. 10.1007/s00335-015-9583-x. PubMed PMID: 26187010; PubMed Central PMCID: PMC4602055.
- Ghildiyal M, Zamore PD. Small silencing RNAs: an expanding universe. Nat Rev Genet. 2009;10(2):94–108. Epub 2009/ 01/17. PubMed PMID: 19148191; PubMed Central PMCID: PMCPmc2724769.
- Lee RC, Feinbaum RL, Ambros V, et al. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993;75(5):843–854.
- Tomankova T, Petrek M, Kriegova E. Involvement of microRNAs in physiological and pathological processes in the lung. Respir Res. 2010;11:159. PubMed PMID: 21092244; PubMed Central PMCID: PMC3001429
- He J, Wang W, Lu L, et al. Analysis of miRNAs and their target genes associated with lipid metabolism in duck liver. Sci Rep. 2016;6:27418. Epub 2016/06/09. PubMed PMID: 27272010; PubMed Central PMCID: PMCPmc4897641.
- Chen X, Ge K, Wang M, et al. Integrative analysis of the pekin duck (Anas anas) microRNAome during feather follicle development. BMC Dev Biol. 2017;17(1):12. 10.1186/s12861-017-0153-1. PubMed PMID: 28728543; PubMed Central PMCID: PMC5520360.
- Wu X, Jia R, Zhou J, et al. Virulent duck enteritis virus infected DEF cells generate a unique pattern of viral microRNAs and a novel set of host microRNAs. BMC Vet Res. 2018;14(1):144. 10.1186/s12917-018-1468-2. PubMed PMID: 29704894; PubMed Central PMCID: PMC5923184.
- Li Z, Zhang J, Su J, et al. MicroRNAs in the immune organs of chickens and ducks indicate divergence of immunity against H5N1 avian influenza. FEBS Lett. 2015;589(4):419–425. Epub 2014/ 12/30. PubMed PMID: 25541489
- Samir M, Vaas LA, Pessler F. MicroRNAs in the host response to viral infections of veterinary importance. Front Vet Sci. 2016;3:86. PubMed PMID: 27800484; PubMed Central PMCID: PMC5065965.
- Jiang P, Zhou N, Chen X, et al. Integrative analysis of differentially expressed microRNAs of pulmonary alveolar macrophages from piglets during H1N1 swine influenza A virus infection. Sci Rep. 2015;5:8167. PubMed PMID: 25639204.
- Wang Y, Brahmakshatriya V, Zhu H, et al. Identification of differentially expressed miRNAs in chicken lung and trachea with avian influenza virus infection by a deep sequencing approach. BMC Genomics. 2009;10:512. Epub 2009/11/07. PubMed PMID: 19891781; PubMed Central PMCID: PMC2777939.
- Wang Y, Brahmakshatriya V, Lupiani B, et al. Integrated analysis of microRNA expression and mRNA transcriptome in lungs of avian influenza virus infected broilers. BMC Genomics. 2012;13:278. PubMed PMID: 22726614; PubMed Central PMCID: PMC3496578.
- Yamashiro H, Siomi MC. PIWI-interacting RNA in drosophila: biogenesis, transposon regulation, and beyond. Chem Rev. 2018;118(8):4404–4421. PubMed PMID: 29281264.
- Dupuis-Sandoval F, Poirier M, Scott MS. The emerging landscape of small nucleolar RNAs in cell biology. Wiley Interdiscip Rev RNA. 2015;6(4):381–397. Epub 2015/ 04/17. PubMed PMID: 25879954; PubMed Central PMCID: PMCPMC4696412.
- Yu Y, Xiao J, Hann SS. The emerging roles of PIWI-interacting RNA in human cancers. Cancer Manag Res. 2019;11:5895–5909. Epub 2019/07/16. PubMed PMID: 31303794; PubMed Central PMCID: PMCPMC6612017.
- Scott MS, Ono M. From snoRNA to miRNA: dual function regulatory non-coding RNAs. Biochimie. 2011;93(11):1987–1992. PubMed PMID: 21664409; PubMed Central PMCID: PMC3476530.
- Wang W, Krug RM. U6atac snRNA, the highly divergent counterpart of U6 snRNA, is the specific target that mediates inhibition of AT-AC splicing by the influenza virus NS1 protein. RNA. 1998;4(1):55–64. PubMed PMID: 9436908.
- Apopo S, Liu H, Jing L, et al. Identification and profiling of microRNAs associated with white and black plumage pigmentation in the white and black feather bulbs of ducks by RNA sequencing. Anim Genet. 2015;46(6):627–635. PubMed PMID: 26369256.
- Samir M, Hamed M, Abdallah F, et al. An Egyptian HPAI H5N1 isolate from clade 2.2.1.2 is highly pathogenic in an experimentally infected domestic duck breed (Sudani duck). Transbound Emerg Dis. 2018. Epub 2018/ 01/25. DOI:10.1111/tbed.12816. PubMed PMID: 29363279.
- Huang Y, Li Y, Burt DW, et al. The duck genome and transcriptome provide insight into an avian influenza virus reservoir species. Nat Genet. 2013;45(7):776–783. Epub 2013/ 06/12. PubMed PMID: 23749191; PubMed Central PMCID: PMCPMC4003391.
- Brennecke J, Aravin AA, Stark A, et al. Discrete small RNA-generating loci as master regulators of transposon activity in drosophila. Cell. 2007;128(6):1089–1103. Epub 2007/03/10. PubMed PMID: 17346786.
- Varjak M, Leggewie M, Schnettler E. The antiviral piRNA response in mosquitoes? J Gen Virol. 2018 December;99:1551–1562. PubMed PMID: 30372405.
- Peng JC, Lin H. Beyond transposons: the epigenetic and somatic functions of the Piwi-piRNA mechanism. Curr Opin Cell Biol. 2013;25(2):190–194. PubMed PMID: 23465540; PubMed Central PMCID: PMC3651849.
- Peng X, Gralinski L, Ferris MT, et al. Integrative deep sequencing of the mouse lung transcriptome reveals differential expression of diverse classes of small RNAs in response to respiratory virus infection. MBio. 2011;2(6). DOI:10.1128/mBio.00198-11 PubMed PMID: 22086488; PubMed Central PMCID: PMC3221602.
- Preusse M, Schughart K, Pessler F. Host genetic background strongly affects pulmonary microRNA expression before and during influenza A virus infection. Front Immunol. 2017;8:246. Epub 2017/04/06. PubMed PMID: 28377766; PubMed Central PMCID: PMCPMC5359533.
- Kuchipudi SV, Dunham SP, Nelli R, et al. Rapid death of duck cells infected with influenza: a potential mechanism for host resistance to H5N1. Immunol Cell Biol. 2012;90(1):116–123. PubMed PMID: 21423263; PubMed Central PMCID: PMC3257048.
- Khor R, McElroy LJ, Whittaker GR. The ubiquitin-vacuolar protein sorting system is selectively required during entry of influenza virus into host cells. Traffic. 2003;4(12):857–868. Epub 2003/11/18.PubMed PMID: 14617349.
- Lee SM, Chan RW, Gardy JL, et al. Systems-level comparison of host responses induced by pandemic and seasonal influenza A H1N1 viruses in primary human type I-like alveolar epithelial cells in vitro. Respir Res. 2010;11:147. PubMed PMID: 21029402; PubMed Central PMCID: PMC2988725.
- Diaz-Guerra MJ, Castrillo A, Martin-Sanz P, et al. Negative regulation by protein tyrosine phosphatase of IFN-gamma-dependent expression of inducible nitric oxide synthase. J Immunol. 1999;162(11):6776–6783. PubMed PMID: 10352298.
- Kumar A, Vn MA, Raut AA, et al. Identification of chicken pulmonary miRNAs targeting PB1, PB1-F2, and N40 genes of highly pathogenic avian influenza virus H5N1 in silico. Bioinform Biol Insights. 2014;8:135–145. PubMed PMID: 25002813; PubMed Central PMCID: PMC4069037.
- Choi EJ, Kim HB, Baek YH, et al. Differential microRNA expression following infection with a mouse-adapted, highly virulent avian H5N2 virus. BMC Microbiol. 2014;14:252. Epub 2014/10/01. PubMed PMID: 25266911; PubMed Central PMCID: PMCPMC4189662.
- Song H, Wang Q, Guo Y, et al. Microarray analysis of microRNA expression in peripheral blood mononuclear cells of critically ill patients with influenza A (H1N1). BMC Infect Dis. 2013;13:257. Epub 2013/ 06/05. PubMed PMID: 23731466; PubMed Central PMCID: PMCPMC3679792.
- Wang R, Zhu Y, Zhao J, et al. Autophagy promotes replication of influenza A virus in vitro. J Virol. 2019;93(4). Epub 2018/ 12/14. PubMed PMID: 30541828; PubMed Central PMCID: PMCPMC6363991. DOI:10.1128/JVI.01984-18
- Suhs KW, Novoselova N, Kuhn M, et al. Kynurenine is a cerebrospinal fluid biomarker for bacterial and viral CNS infections. J Infect Dis. 2019. Epub 2019/02/06. PubMed PMID: 30721966. DOI:10.1093/infdis/jiz048
- Zhao P, Zhao L, Zhang T, et al. Changes in microRNA expression induced by rabies virus infection in mouse brains. Microb Pathog. 2012;52(1):47–54. Epub 2011/ 10/22. PubMed PMID: 22015383.
- Bakre A, Andersen LE, Meliopoulos V, et al. Identification of host kinase genes required for influenza virus replication and the regulatory role of microRNAs. PLoS One. 2013;8(6):e66796. PubMed PMID: 23805279; PubMed Central PMCID: PMC3689682.
- Olena AF, Patton JG. Genomic organization of microRNAs. J Cell Physiol. 2010;222(3):540–545. PubMed PMID: 20020507; PubMed Central PMCID: PMCPMC4028663.
- Zhang Y, Zhang R, Su B. Diversity and evolution of microRNA gene clusters. Sci China C Life Sci. 2009;52(3):261–266. PubMed PMID: 19294351.
- Scott MS, Avolio F, Ono M, et al. Human miRNA precursors with box H/ACA snoRNA features. PLoS Comput Biol. 2009;5(9):e1000507. Epub 2009/09/19. PubMed PMID: 19763159; PubMed Central PMCID: PMCPMC2730528.
- Li Y, Chan EY, Li J, et al. MicroRNA expression and virulence in pandemic influenza virus-infected mice. J Virol. 2010;84(6):3023–3032. Epub 2010/01/15. PubMed PMID: 20071585; PubMed Central PMCID: PMCPMC2826040.
- Gracias DT, Stelekati E, Hope JL, et al. The microRNA miR-155 controls CD8(+) T cell responses by regulating interferon signaling. Nat Immunol. 2013;14(6):593–602. PubMed PMID: 23603793; PubMed Central PMCID: PMC3664306.
- Salem MHI. Pathogenesis of highly pathogenic avian influenza virus in native ducks experimentally infected [Master thesis]. Faculty of Veterinary Medicine, Zagazig University; 2014.
- LJ REED, MUENCH H. A simple method of estimating fifty percent endpoints. Am J Epidemiol. 1938;27(3):493–497.
- Capece V, Garcia Vizcaino JC, Vidal R, et al. Oasis: online analysis of small RNA deep sequencing data. Bioinformatics. 2015;31(13):2205–2207. PubMed PMID: 25701573; PubMed Central PMCID: PMC4481843.
- Zerbino DR, Achuthan P, Akanni W, et al. Ensembl 2018. Nucleic Acids Res. 2018;46(D1):D754–D61.
- Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnetJournal. 2011;17(1):3. Epub 2011-08-02.
- Kozomara A, Birgaoanu M, Griffiths-Jones S. miRBase: from microRNA sequences to function. Nucleic Acids Res. 2019;47(D1):D155–d62. Epub 2018/ 11/14PubMed PMID: 30423142; PubMed Central PMCID: PMCPMC6323917.
- Friedlander MR, Mackowiak SD, Li N, et al. miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res. 2012;40(1):37–52. PubMed PMID: 21911355; PubMed Central PMCID: PMC3245920.
- Dobin A, Davis CA, Schlesinger F, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29(1):15–21. PubMed PMID: PMC3530905.
- Wang J, Zhang P, Chen R, et al. piRBase: a comprehensive database of piRNA sequences. Nucleic Acids Res. 2018;47(D1):D175–D80.
- Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30(7):923–930. PubMed PMID: 24227677.
- Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):1–21.
- Kruger J, Rehmsmeier M. RNAhybrid: microRNA target prediction easy, fast and flexible. Nucleic Acids Res. 2006;34(Web Server issue):W451–4. Epub 2006/ 07/18. PubMed PMID: 16845047; PubMed Central PMCID: PMCPMC1538877.
- Bindea G, Mlecnik B, Hackl H, et al. ClueGO: a cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics. 2009;25(8):1091–1093. PubMed PMID: 19237447; PubMed Central PMCID: PMC2666812.
- Shannon P, Markiel A, Ozier O, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–2504. PubMed PMID: 14597658; PubMed Central PMCID: PMC403769.