
ABSTRACT
We previously showed that miR-122 was frequently downregulated in hepatocellular carcinoma (HCC) and C/EBPα transactivated miR-122 expression. In this study, we found that Sp1 bound to the miR-122 promoter at two different sites. Interestingly, either inhibition or overexpression of Sp1 could decrease the miR-122 promoter activity and the cellular miR-122 level in hepatoma cells. Further investigations disclosed that Sp1 cooperated with C/EBPα to induce miR-122 transcription by binding to the positive regulatory site D in the miR-122 promoter, whereas eEF1A1 interacted with Sp1 to bind to the negative regulatory site E and inhibit miR-122 transcription. Significantly, both Sp1 and eEF1A1 levels were enhanced, but C/EBPα and miR-122 expression were reduced in HCC tissues. Knockdown of eEF1A1 enhanced miR-122 level and inhibited cell growth, and these effects were abrogated when Sp1 was silenced. Consistently, the promoter activity enhanced by site E deletion was attenuated by silencing Sp1. Moreover, reduction of miR-122 resulted from Sp1 overexpression was rescued by coexpressing C/EBPα. These data suggest that C/EBPα and eEF1A1 may play opposing roles in Sp1-regulating miR-122 transcription, and the eEF1A1 upregulation accompanied by C/EBPα downregulation in HCC may switch the regulatory functions of Sp1 and led to reduced miR-122 transcription. These findings highlight the complex regulatory network of miR-122 expression and its significance in hepatocarcinogenesis.
Abbreviations: MiRNA: microRNA; HCC, hepatocellular carcinoma; eEF1A1: eukaryote translation elongation factor 1A1; siRNA: small interfering RNA; qPCR: real-time quantitative RT-PCR; EMSA: electrophoretic mobility shift assay; ChIP: chromatin immunoprecipitation; TSS: transcription start site.
Introduction
MicroRNA-122 (miR-122) is the most abundant miRNA in the liver that can repress cell proliferation, migration and promotes cell apoptosis [Citation1]. Deregulation of miR-122 is implicated in various physiological and pathological processes, including hepatitis C virus replication [Citation2], lipid metabolism [Citation3], systemic iron metabolism [Citation4], hepatic insulin resistance [Citation5], hepatic fibrogenesis [Citation6,Citation7], and hepatocellular carcinoma (HCC) development [Citation8–Citation12]. Downregulation of miR-122 is a frequent event in HCC and is associated with HCC growth and metastasis [Citation1]. We have previously identified a GSK-3β-C/EBPα-miR-122-IGF-1R regulatory circuitry: miR-122 suppresses IGF-1R expression and in turn attenuates IGF-1R/Akt signalling and sustains GSK-3β activity. The activated GSK-3β not only represses cyclin D1 expression and cell proliferation, but also activated C/EBPα, which transactivates miR-122 expression and thereby enforces IGF-1R suppression. We also show that decreased C/EBPα expression or reduced C/EBPα activity results in downregulation of miR-122 in HCC [Citation10]. C/EBPα belongs to the family of basic region leucine zipper transcription factor and is widely expressed in numerous tissues, including liver, adipose tissue, small intestine, etc [Citation13]. C/EBPα knockdown promotes cell growth of a non-transformed liver cell line [Citation14], whereas re-expression of C/EBPα in HCC cell lines inhibits tumour development in vivo [Citation15].
Consistent with its enrichment in the liver, miR-122 is transactivated by a cohort of liver-enriched transcription factors, including C/EBPα, HNF-4 and HNF-6 [Citation10,Citation16,Citation17]. In the case of hepatic insulin resistance, HNF-4 is phosphorylated and inhibited by JNK1, thereby leads to decrease of miR-122 and consequent elevation of its target gene PTP1B, which inhibits hepatic insulin signalling [Citation5]. PPARγ/RXRα complex transactivates miR-122 expression, which is inhibited by the binding of corepressors N-CoR and SMRT or hepatitis B virus X protein in HCC cells [Citation18]. Moreover, farnesoid X receptor transactivates the expression of miR-122 and subsequently represses the growth of HCC cells [Citation19]. Considering its importance in liver function, more extensive explorations on the regulation of miR-122 expression and the underlying mechanisms of miR-122 dysregulation are required.
The transcription factor Sp1 is considered as a basal transcription factor that recruits the general transcription machinery to the promoter without a TATA box and activates or suppresses gene expression depending on the cellular context [Citation20]. Sp1 is overexpressed in numerous cancers and its high level is associated with poor prognosis of patients [Citation20]. The role of Sp1 in miR-122 expression has not been reported yet. The eukaryotic translation elongation factors 1 alpha 1 (eEF1A1) is known as a translation factor, which carries the amino acyl-tRNA into the ribosome during protein translation process. It is also a pleiotropic protein that is highly expressed in human tumours and controls cell proliferation, death and mobility [Citation21]. Although eEF1A is preferentially located in the cytoplasm, increased amount of nuclear eEF1A is found in HCC [Citation22]. However, whether eEF1A1 is involved in transcription regulation and HCC development remains largely unknown.
In this study, we predicted multiple Sp1 binding sites in the miR-122 promoter and showed that both knockdown and overexpression of Sp1 decreased the miR-122 level. Further in vitro and in vivo studies disclosed that Sp1 bound to two distinct sites in the miR-122 promoter. Binding of Sp1 to the positive regulatory site cooperated with C/EBPα to transactivate miR-122 expression, whereas Sp1 binding to the negative regulatory site mediated the inhibitory effect of eEF1A1 on miR-122 transcription. The increase of eEF1A1 accompanied by the decrease of C/EBPα in HCC switched the regulatory functions of Sp1 and led to downregulation of miR-122. These findings highlight the complex regulatory network of miR-122 transcription and may provide new target for anti-HCC therapy.
Result
Sp1 binds to the miR-122 promoter and displays both stimulatory and inhibitory roles on mir-122 transcription
We previously showed that a 450-bp region upstream of the transcription start site (TSS) of pri-miR-122 displayed phylogenetic conservation and strong promoter activity [Citation10]. Six putative Sp1 binding sites were predicted in the miR-122 promoter region (referred to site A-F, ). Significantly, Sp1 silencing (Supplementary Fig. S1A) reduced the levels of cellular pri-miR-122 () and mature miR-122 (). Intriguingly, ectopic Sp1 expression (Supplementary Fig. S1B) also decreased miR-122 level (). To confirm the role of Sp1 in miR-122 transcription, a luciferase reporter construct p122-WT that contained the −450 to +150 bp region of miR-122 promoter (TSS was assigned as +1) in the pGL3-basic vector was generated (). Consistent with the above observations, both Sp1 knockdown and overexpression significantly reduced the promoter activity of p122-WT (,).
Figure 1. Sp1 regulates miR-122 transcription. (A) Annotation of the miR-122 promoter sequence. The predicted consensus binding sites of Sp1 (site A to F) and C/EBPα are shadowed. TSS, transcription start site. (B) Sp1 silencing reduced cellular pri-miR-122 level. (C) Sp1 knockdown reduced cellular mature miR-122 level. Huh-7 cells were transfected with control RNA duplex (NC) or siRNA targeting Sp1 (siSp1-1 and siSp1-2) for 48 h before qPCR analysis. GAPDH and U6 were used as internal control for (B) and (C), respectively. (D) Overexpression of Sp1 reduced cellular miR-122 level. Huh-7 cells were infected with control lentivirus pCDH-CMV-MCS-EF1-copGFP (Ctrl), Sp1-expressing lentivirus pCDH-Sp1 (Sp1) for 72 h before qPCR analysis. U6 was used as an internal control. (E) Schematic diagram for firefly luciferase reporter constructs containing the indicated genomic sequences of miR-122 promoter. The predicted Sp1 binding sites are depicted as short vertical line. The C/EBPα binding site is depicted as diamond. Deletion of binding site is depicted as triangle. (F) Sp1 silencing reduced the miR-122 promoter activity. Huh-7 cells were cotransfected with pRL-PGK, p122-WT and NC or siSp1-1 for 48 h before luciferase assay. (G) Overexpression of Sp1 impaired the miR-122 promoter activity. Huh-7 cells were cotransfected with pRL-PGK, p122-WT and Ctrl or pCDH-Sp1 for 72 h before luciferase assay. ** P < .01; *** P < .001.
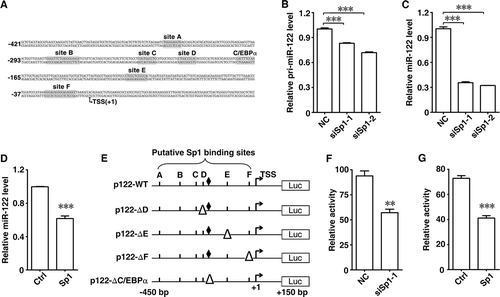
To further dissect the role of the predicted Sp1 binding sites, individual site deletion analysis was performed. Compared with p122-WT, removal of either Sp1-binding site D (p122-ΔD) or F (p122-ΔF) attenuated the miR-122 promoter activity, whereas deletion of site E (p122-ΔE) enhanced the promoter activity ( and ). However, deletion of either site A, B or C did not affect the miR-122 reporter activity (). These results suggest that sites D, E and F may be the cis-elements that regulate miR-122. Next, EMSA was used to verify the interaction between Sp1 and these putative Sp1 binding sites. As shown, the probes corresponding to sites D and E could form specific complexes with nuclear proteins, as manifested by the appearance of specific bands (, lane 2 and 6), and the band intensity of the probe-protein complexes decreased when excess (100x) unlabelled oligonucleotide with Sp1 consensus binding sequence was added (, lane 3 and 7), but remained unchanged in the presence of a non-specific scrambled oligonucleotide (, lane 4 and 8). However, the probe corresponding to site F did not likely form specific complex with nuclear proteins (Supplementary Fig. S2). Moreover, antibody supershift assay revealed that pre-incubation with anti-Sp1 antibody led to a decrease in the band intensity of the site D or E probe-protein complexes (), suggesting the binding of Sp1 with these two sites. Subsequent ChIP assay was performed to validate the in vivo interaction between Sp1 and the miR-122 promoter. The chromatin complexes were precipitated by anti-Sp1 antibody or control IgG and then amplified by PCR reaction using primers covering site D or E. As shown, the fragments of sites D and E were detected in the input DNA and the anti-Sp1 antibody-precipitated DNA, whereas the control IgG-precipitated DNA yielded no band (). These data suggest that Sp1 binds to the miR-122 promoter and exerts stimulatory or inhibitory function on miR-122 expression; Sp1-binding sites D and E may represent positive and negative regulatory site, respectively.
Figure 2. Sp1 binds to the different sites in the miR-122 promoter. (A) Deletion of Sp1 binding sites affected the miR-122 promoter activity. Huh-7 cells were cotransfected with pRL-PGK and the indicated luciferase reporters. (B) EMSA assay verified the interaction of nuclear proteins with Sp1 binding sites D and E sequences. The analysis was performed in the presence (+) or absence (-) of Huh-7 nuclear extracts. Each binding reaction included γ-32P-labelled probe that comprised site D or E sequences, with (+) or without (-) addition of excess (100x) unlabelled Sp1 consensus binding sequence or non-specific scrambled oligo. (C) Antibody supershift assay identified Sp1 as a potential nuclear protein interacting with sites D and E sequences. The nuclear extracts were preincubated with anti-Sp1 antibody (α-Sp1) or control IgG before incubation with γ-32P-labelled probe. (D) ChIP assay validated the in vivo interaction between Sp1 and the miR-122 promoter. Schematic diagram of amplicons is shown (top). The predicted Sp1 binding sites A-F are depicted as thick vertical lines and two primer sets as arrows. The chromatin complexes precipitated by anti-Sp1 antibody (α-Sp1) or control IgG were amplified using two primer sets that encompassed sites D and E, respectively. ** P < .01; ns, not significant.
C/EBPα and eEF1A1 distinctly affect the regulatory roles of Sp1 in miR-122 transcription
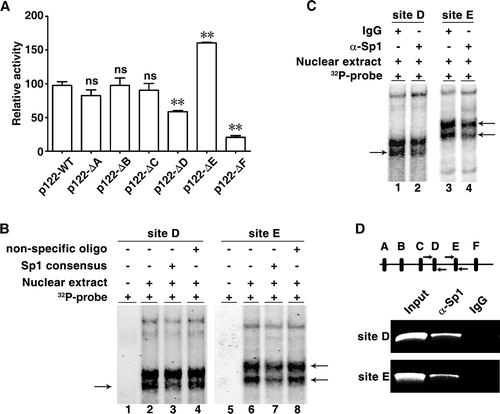
Sp1 is a basal transcription factor that is highly regulated by multiple cellular factors to positively or negatively modulate gene expression [Citation20]. It has been reported that Sp1 cooperates with C/EBPα to induce gene transcription [Citation23,Citation24]. In the miR-122 promoter region, the site D is adjacent to the C/EBPα binding site () that we have identified to be critical for miR-122 transcription [Citation10]. As shown, removal of either site D or C/EBPα binding site significantly impaired the miR-122 promoter activity (), and Sp1 silencing reduced the activity of wildtype miR-122 promoter, but could not affect the activity of mutant promoter with deletion of site D or C/EBPα binding site (). Interestingly, the promoter activity enhanced by site E deletion was attenuated by silencing Sp1 (). Notably, reduction of cellular miR-122 level resulted from Sp1 overexpression was rescued by coexpressing C/EBPα ( and Supplementary Fig. S3). These results suggest that Sp1 may bind to site D and cooperate with C/EBPα to induce miR-122 transcription.
Figure 3. C/EBPα and eEF1A1 distinctly affect the regulatory roles of Sp1 in miR-122 transcription. (A) Sp1 silencing could not affect the activity of mutant miR-122 promoter with deletion of site D or C/EBPα binding site. Huh-7 cells were cotransfected with pRL-PGK, the indicated luciferase reporters and NC or siSp1-1. (B) Decrease of miR-122 level caused by Sp1 overexpression was rescued by coexpression of C/EBPα. Huh-7 cells were coinfected with the indicated lentiviruses for 72 h before qPCR analysis. Ctrl, pCDH-CMV-MCS-EF1-copGFP; Sp1, pCDH-Sp1; C/EBPα, pCDH-C/EBPα. U6 was used as an internal control. (C) Identification of nuclear proteins that interacted with Sp1 binding site E in HCC cells. Nuclear extracts from HCC (T) or its paired non-tumour tissue (N) were incubated with DNA probe containing Sp1 binding site E sequence. The captured proteins were separated by SDS-PAGE and the bands with differential intensities (indicated by arrows) were isolated and subjected to MALDI-TOF mass spectrometry analysis. Three bands were identified as single protein, respectively, and other two bands were identified as mixtures, including mixture 1 (DHB4 and NCPR) and mixture 2 (RS4X and DECR2). M, marker. (D) Knockdown of Sp1 abrogated the elevation of miR-122 level induced by eEF1A1 silencing. Huh-7 cells were cotransfected with the indicated siRNA duplex for 48 h before qPCR analysis. U6 was used as an internal control. (E) Immunoprecipitation of cellular eEF1A1 with Sp1. Nuclear extracts from Huh-7 cells were immunoprecipitated using anti-eEF1A1 antibody (α-eEF1A1), followed by immunoblotting for Sp1. Isotype-matched control IgG was used as negative control. (F) Antibody supershift assays identified eEF1A1 as a potential component of the protein complex interacting with site E. Nuclear extracts of Huh-7 cells were incubated with γ-32P-labelled probe that comprised site E sequence. The radiolabelled intensity of the DNA-protein complexes (indicated by arrows) reduced significantly when α-eEF1A1 was added, compared with addition of control IgG. (G) eEF1A1 was associated with the miR-122 promoter in vivo. The chromatin complexes precipitated by α-eEF1A1 or control IgG were amplified using primer set that encompassed site E. * P < .05; ** P < .01; ns, not significant.
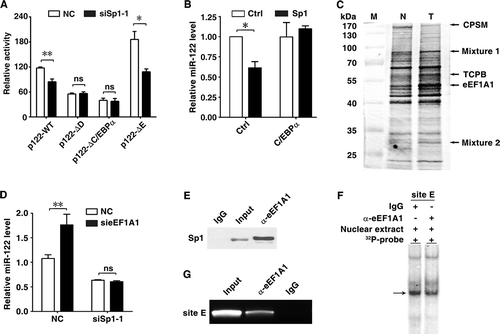
We further investigated whether some inhibitory factors interacted with Sp1 via site E to inhibit miR-122 expression in HCC cells. A DNA pull-down assay was performed using double-stranded DNA probe comprised of site E sequence. Among the pulled-down proteins, eEF1A1 displayed significantly higher levels in HCC than that in non-tumour liver tissues (). It has been shown that eEF1A1 promotes oncogenic transformation, cell proliferation and survival [Citation21]. We therefore explored whether eEF1A1 regulated miR-122 expression. As shown, eEF1A1 silencing (Supplementary Fig. S4) dramatically increased miR-122 level (), and this effect was abrogated when Sp1 was silenced (). Since eEF1A protein lacked the DNA binding domain [Citation21], we therefore examined whether it modulated miR-122 expression by interacting with Sp1. Immunoprecipitation with anti-eEF1A1 antibody showed an in vivo interaction between eEF1A1 and Sp1 (). Antibody supershift assays revealed that pre-incubation with anti-eEF1A1 antibody decreased the band intensity of the site E probe-protein complexes (), indicating the presence of eEF1A1 in the protein complex that bound to the sequence of site E. ChIP analysis further showed that the miR-122 promoter region containing site E sequence was precipitated by anti-eEF1A1 antibody (). These data suggest that eEF1A1 may bind to site E via Sp1 and inhibit miR-122 transcription.
Upregulation of eEF1A1 is associated with miR-122 downregulation and increased growth of HCC cells
The biological significance of the above findings was then examined. We have shown that miR-122 inhibits proliferation of HCC cells [Citation10]. As shown, both Sp1 and eEF1A1 were upregulated, whereas C/EBPα and miR-122 were downregulated in HCC tissues (,). Moreover, elevation of Sp1 or eEF1A1 and decrease of C/EBPα were associated with downregulation of miR-122 (). Subsequent investigations revealed that knockdown of Sp1 impaired miR-122 expression () and promoted growth of HCC cells (), whereas eEF1A1 silencing enhanced miR-122 level () and inhibited cell growth (), and these effects were abrogated when Sp1 was silenced ( and ).
Figure 4. Association of Sp1, C/EBPα and eEF1A1 levels with miR-122 expression and cell proliferation. (A) Sp1 and eEF1A1 were upregulated, whereas C/EBPα and miR-122 were downregulated in HCC tissues. Sp1, eEF1A1 and C/EBPα expression in paired HCC (T) and adjacent non-tumour tissues (N) were assessed by immunoblotting. GAPDH was used as an internal control. The intensity of each band, which represents the expression level, was densitometrically quantified. The value under each sample indicated the expression level of target protein that was normalized by GAPDH. The miR-122 level was detected by qPCR analysis. U6 was used as an internal control. (B) Comparison of Sp1, eEF1A1, C/EBPα or miR-122 level between HCC and adjacent non-tumour tissues. (C) The relationship among the Sp1, eEF1A1, C/EBPα and miR-122 levels. Three-dimensional scatter plot showed the relative expression of eEF1A1 (x-axis), Sp1 (y-axis) and C/EBPα (z-axis). The colour of dot indicated the relative level of miR-122. Sp1, eEF1A1 and C/EBPα protein was detected by immunoblotting. The levels of Sp1, eEF1A1, C/EBPα and miR-122 used for analysis in (B) and (C) were derived from (A). (D) Knockdown of Sp1 or eEF1A1 affected cell proliferation. Huh-7 cells were cotransfected with the indicated siRNA duplex for 48 h before cell counting. (E) A model of the regulatory mechanisms of miR-122 expression. *P < .05; **P < .01; ***P < .001; ns, not significant.
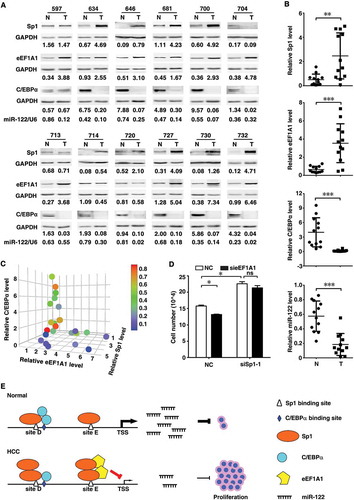
Collectively, these results suggest that in normal cells with high C/EBPα and low eEF1A1, binding of Sp1 in site D may cooperate with C/EBPα to induce miR-122 transcription; while in HCC cells, downregulation of C/EBPα accompanied by upregulation of eEF1A1 may lead to increased association of eEF1A1 with Sp1 on site E, which consequently results in decreased miR-122 expression and in turn uncontrolled cell proliferation ().
Discussion
In the past years, much of the effort has been dedicated to exploring the biological function of miR-122, but the regulatory mechanisms of miR-122 expression remain largely unknown. In this study, we identified Sp1 as a transcription factor that regulated miR-122 expression. In a previous study, we showed that the liver-enriched transcription factor C/EBPα transactivated miR-122 transcription [Citation10]. Sp1 is a basal transcription factor, which activates or suppresses the expression of genes depending on its interaction with coactivators or corepressors [Citation20]. Intriguingly, we found that in contrast to downregulation of miR-122, the level of Sp1 elevated in HCC tissues, and ectopic Sp1 expression significantly suppressed miR-122 expression in HCC cells, whereas this effect was abrogated by coexpression of C/EBPα. Furthermore, deletion of Sp1 binding site D abrogated the miR-122 promoter activity, and silencing Sp1 could not affect the activity of the miR-122 promoter with deletion of site D or C/EBPα binding site. These findings identified site D as the positive regulatory site and inferred that Sp1 might activate miR-122 transcription by cooperating with C/EBPα, and downregulation of C/EBPα in HCC cells may disable Sp1 from inducing miR-122 transcription.
eEF1A is a core component of the protein synthesis machinery. Emerging evidences suggest that eEF1A may contribute to tumorigenesis independent of its translational activity. eEF1A cooperates with c-Raf to antagonize IFN-α-induced apoptosis in lung cancer cells [Citation25]. eEF1A interacts with and stabilizes phospho-Akt, in turn promotes proliferation, survival and motility in breast cancer cells [Citation26]. Herein, we revealed that eEF1A1 interacted with Sp1 and associated with the Sp1-binding site E. Deletion of site E elevated the miR-122 promoter activity and silencing eEF1A1 increased the endogenous miR-122 level, which were attenuated by silencing Sp1. These data suggest that eEF1A1 may inhibit miR-122 transcription via interacting with Sp1 and enhanced eEF1A1 expression may represent a novel mechanism for miR-122 downregulation in HCC. Consistent with our result, it was reported that cervical cancer suppressor 3, an isoform of eEF1A, could cooperate with ZBTB7A to recruit the corepressors SMRT and BCoR, thereby repressed the transcription of p21CIP1 gene [Citation27].
Notably, removal of the predicted Sp1-binding site F, which was located only a few nucleotides upstream of the TSS, led to decrease of the miR-122 promoter activity, but EMSA assay revealed no binding of Sp1 to site F. This potential discrepancy may attribute to the damage of the miR-122 proximal promoter upon site F deletion. It has been reported that the integrality of the proximal sequence is required for promoter activity [Citation28].
Interestingly, we found that Sp1 and eEF1A1 were upregulated, but C/EBPα was downregulated in HCC tissues. Furthermore, expression of C/EBPα rescued the reduced miR-122 expression induced by Sp1 overexpression, whereas knockdown of Sp1 attenuated the increased miR-122 expression induced by eEF1A1 inhibition. These data suggest that in normal cells, Sp1 may cooperate with C/EBPα to induce miR-122 transcription, but in HCC cells, downregulation of C/EBPα may lead to increased amount of Sp1 to be associated with eEF1A1, which inhibits miR-122 transcription.
Collectively, we identified the opposing roles of C/EBPα and eEF1A1 in Sp1-regulated miR-122 transcription and the underlying mechanisms, which extends our understanding on the regulation of miR-122 transcription and the mechanisms responsible for miR-122 downregulation in HCC.
Materials and methods
Reagents
Reagents were purchased as follows: TRIzol, Lipofectamine RNAiMAX, Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA); Moloney murine leukaemia virus (MMLV) reverse transcriptase (Promega, Madison, WI, USA); restriction enzymes (MBI Fermentas, Hanover, MD, USA); Dulbecco’s modified Eagle’s medium (DMEM, Hyclone, Logan, UT, USA); foetal bovine serum (FBS, GIBCO, Grand Island, NY, USA).
Human tissues and cell lines
Paired HCC and adjacent non-tumour liver tissues were collected from patients undergoing HCC resection in Cancer Centre, Sun Yat-sen University. Both tumour and non-tumour tissues were histologically confirmed. Informed consent was obtained from each patient and the study was approved by the Institute Research Ethics Committee at Cancer Centre, Sun Yat-sen University.
The cells used in this study included HCC cell line (Huh-7) and HEK293T. All cells were cultured in DMEM with 10% FBS.
Oligonucleotides and plasmids
The following small interfering RNA (siRNA) oligonucleotides (Genepharma, Shanghai, China) were used: siSp1-1, siSp1-2 and sieEF1A1 that, respectively, targeted human Sp1 and eEF1A1 transcripts; the negative control RNA duplex (NC) for siRNA was non-homologous to any human genome sequence.
To analyse the promoter activity of miR-122, various genomic fragments upstream of pri-miR-122 were cloned into the KpnI and XhoI sites upstream of the firefly luciferase in pGL3-basic vector (Promega). The lentivirus expression vectors pCDH-Sp1 and pCDH-C/EBPα were generated by, respectively, inserting the coding sequence of Sp1 and C/EBPα genes into the XbaI and BamHI or EcoRI sites of pCDH-CMV-MCS-EF1-copGFP (System Biosciences, Mountain View, CA, USA).
All constructs were confirmed by direct sequencing. All oligonucleotide sequences are listed in Supplementary Table 1.
Cell transfection
A final concentration of 50 nM RNA oligonucleotides was transfected with Lipofectamine RNAiMAX. Lipofectamine 2000 was used to transfect plasmid or a mixture of plasmid/RNA oligonucleotides.
Luciferase reporter assay
To detect the promoter activity, Huh-7 cells were cotransfected with 100 ng firefly luciferase reporter, 5 ng pRL-PGK (Promega) and 50 nM RNA duplex in a 48-well plate. pRL-PGK, which expresses Renilla luciferase, was used to correct the differences in both transfection and harvest efficiencies. Forty-eight hours post-transfection, cells were harvested and subjected to the luciferase assay following the manufactory’s instruction.
Virus production and infection
For lentivirus production, HEK293T cells were cotransfected with the lentivirus expression vector and the Lenti-X™ HTX Packaging System (Clontech, Takara Bio Inc., Dalian, China), followed by replacement with fresh medium 16 h post-transfection and the supernatant was harvested 48 h later. The lentiviral supernatant was centrifuged at 1000 g for 10 min to remove cellular debris and the aliquots were stored at – 80°C. For in vitro infection, target cells were cultured in fresh medium supplemented with 20% lentiviral supernatant and 4 μg/mL polybrene (Millipore, Billerica, MA, USA).
Analysis of gene expression
Real-time quantitative RT-PCR (qPCR) was used to detect RNA levels. Total RNAs were extracted using the TRIzol reagent, subjected to DNase I digestion (1 U/μL) at 37°C for 30 min and then to heat inactivation of DNase I at 65°C for 10 min.
The expression levels of miR-122 and the reference gene U6 were quantified using the TaqMan MicroRNA Assay kit (Applied Biosystems, Foster City, CA, USA). qPCR analyses for the expression of Sp1, eEF1A1 and the reference gene GAPDH were performed using Power SYBR® Green PCR Master Mix (Applied Biosystems). The temperature cycle profile for the qPCR reactions was 95°C for 1 min and then 40 cycles of 95°C for 15 s and 60°C for 1 min. Melting curve analysis was performed to verify the specificity of the PCR product immediately after amplification, as follows: heating to 95°C for 20 s, cooling to 60°C for 20 s, followed by a temperature increase to 95°C with a transition rate of 0.11°C/s and the continuous detection of fluorescence.
All qPCR reactions were performed on a LightCycler 480 (Roche Diagnostics GmbH, Mannheim, Germany) and run in duplicate. The cycle threshold (Ct) values did not differ by more than 0.5 among the duplicate runs. The level of target genes was normalized to the level of the internal control gene to yield a 2−ΔΔCt value. Sequences for primers are listed in Supplementary Table 1.
Immunobloting was performed to evaluate protein levels. Protein samples were separated by SDS-PAGE, transferred to PVDF membranes, and probed with primary antibody against Sp1 (sc-59X), C/EBPα (sc-61X, Santa Cruz Biotechnology, Santa Cruz, CA, USA), eEF1A1 (11,402-1-AP, Proteintech, Chicago, IL, USA), GAPDH (BM1623, Boster, Wuhan, China), and then incubated with ImmunoPure Peroxidase Conjugated Goat Anti-Mouse IgG (H + L) (cat. 31,430) or Anti-Rabbit IgG (H + L) (cat. 31,460, Thermo Scientific, Waltham, MA, USA). Signals were detected with an enhanced chemiluminescence system (GE Healthcare, Chicago, IL, USA).
Electrophoretic mobility shift assay (EMSA) and antibody supershift assay
Nuclear extracts were prepared using NE-PER™ Nuclear and Cytoplasmic Extraction Reagents (Thermo Scientific). The probes, corresponding to the predicted Sp1 binding sequences on miR-122 promoter, were chemically synthesized (Supplementary Table 1), annealed and end-labelled with γ-32P-ATP. Binding reaction was carried out with 5 μg nuclear extract and 0.1 pmol γ-32P-labelled probe using a Gel Shift Assay System (Promega). For competition assay, nuclear extract was preincubated with 100-fold molar excess of unlabelled oligo for 30 min prior to adding labelled probe. For antibody supershift assay, nuclear extract was preincubated with 2 μg anti-Sp1 (sc-59X), anti-eEF1A1 (11,402-1-AP) antibody or isotype-matched control IgG (Sigma, St. Louis, MO, USA) at 4°C for 16 h before adding to the binding reaction. DNA-protein complexes were separated on a pre-electrophoresed polyacrylamide gel (5%) at 4°C. The gel was vacuum-dried at 80°C, and then exposed to a Storage Phosphor Screen and read with a Typhoon Phosphor Imager (Amersham Bioscience, Sunnyvale, CA, USA).
Chromatin immunoprecipitation (ChIP) assay
Huh-7 cells were cross-linked with 1% formaldehyde at room temperature for 10 min. Cells were washed with ice-cold 1× PBS, scraped in 1× PBS plus protease inhibitors and collected by centrifugation. Cell pellets were suspended in lysis buffer [50 mM Tris-HCl (pH 8.0), 10 mM EDTA and 1% SDS] with protease inhibitor cocktail (Roche Diagnostics GmbH). Chromatin was then sheared by sonicating on ice. Chromatin complexes were immunoprecipitated with 4 μg anti-Sp1, anti-eEF1A1 antibody or isotype-matched control IgG for 16 h at 4°C with rotation. The precipitated complexes were collected with Dynabeads® Protein G (Dynal, Oslo, Norway). The DNA-protein crosslink was reversed by heating the samples at 65°C overnight. The DNA was purified and then subjected to PCR amplification. Sequences for primers are listed in Supplementary Table 1.
Identification of DNA-binding proteins
The identification of DNA-binding proteins was performed as described previously [Citation29]. Briefly, 0.2 pmol of double-stranded DNA was immobilized onto 1 mg Dynabeads M-280 Streptavidin (Dynal) following the protocol supplied by the manufacturer. Approximately 100 μg nuclear extract was diluted to a final volume of 550 μl binding buffer [50 mM KCl, 20 mM HEPES (pH 7.9), 1 mM MgCl2, 0.5 mM DTT, 0.1 μg/μl poly-(dI-dC) and 10% glycerol] with protease inhibitors. The reaction mixtures were preincubated at room temperature for 10 min, followed by addition of 400 μg Dynabeads with immobilized DNA probe and further incubation for another 30 min. After magnetic separation, the beads were washed three times with 400 μl washing buffer [50 mM KCl, 20 mM HEPES (pH 7.9), 1 mM MgCl2, 0.5 mM DTT and protease inhibitors]. The captured proteins were separated by SDS-PAGE and visualized by silver staining. The differential protein bands were subjected to matrix-assisted laser desorption/ionization time-of-flight mass spectrometry analysis.
Coimmunoprecipitation assay
Nuclear extract of Huh-7 cells was diluted in RIPA buffer [150 mM NaCl, 50 mM Tris-HCl (pH 7.4), 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS and 1 mM EDTA] with protease inhibitor cocktail. The lysates were precleared with Dynabeads® Protein G for 1 h. The supernatants were equally divided and incubated with anti-eEF1A1 antibody or isotype-matched control IgG at 4°C for 3 h. The precipitated complexes were collected by incubation with Dynabeads® Protein G for 1 h. The beads were washed 4 times with washing buffer [150 mM NaCl, 50 mM Tris-HCl (pH 7.4) and 0.1% Triton X-100], and the proteins were eluted with SDS sample buffer at 95°C for 5 min.
Bioinformatics tool and statistical analysis
Alibaba2.1 (http://www.gene-regulation.com/pub/programs/alibaba2/index.html) was used for the prediction of transcription factor binding sites.
Data are presented as the mean±SEM of at least three independent experiments. Analyses on the differences between groups were conducted using GraphPad Prism version 4.0 (GraphPad Software, Inc., San Diego, CA). The differences between groups were analysed using a Student t test when only two groups were compared or assessed using one-way analysis of variance when more than two groups were compared. Two-factor analysis was performed using two-way analysis of variance with a posttest for subsequent comparisons of individual factors. All statistical tests were two-sided. P values <0.05 were considered statistically significant.
Author contributions
SMZ supervised the project. CZ and SMZ designed the experiments, analysed the data, and wrote the manuscript. CZ, YS and FYW conducted the experiments. All authors discussed the results, read and approved the manuscript.
Supplemental Material
Download Zip (2.2 MB)Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplementary data can be accessed here.
Additional information
Funding
Related Research Data
References
- Bandiera S, Pfeffer S, Baumert TF, et al. miR-122 – a key factor and therapeutic target in liver disease. J Hepatol. 2015;62:448–457.
- Jopling CL, Yi M, Lancaster AM, et al. Modulation of hepatitis C virus RNA abundance by a liver-specific MicroRNA. Science. 2005;309:1577–1581.
- Esau C, Davis S, Murray SF, et al. miR-122 regulation of lipid metabolism revealed by in vivo antisense targeting. Cell Metab. 2006;3:87–98.
- Castoldi M, Vujic Spasic M, Altamura S, et al. The liver-specific microRNA miR-122 controls systemic iron homeostasis in mice. J Clin Invest. 2011;121:1386–1396.
- Yang YM, Seo SY, Kim TH, et al. Decrease of microRNA-122 causes hepatic insulin resistance by inducing protein tyrosine phosphatase 1B, which is reversed by licorice flavonoid. Hepatology. 2012;56:2209–2220.
- Li J, Ghazwani M, Zhang Y, et al. miR-122 regulates collagen production via targeting hepatic stellate cells and suppressing P4HA1 expression. J Hepatol. 2013;58:522–528.
- Zeng C, Wang YL, Xie C, et al. Identification of a novel TGF-beta-miR-122-fibronectin 1/serum response factor signaling cascade and its implication in hepatic fibrogenesis. Oncotarget. 2015;6:12224–12233.
- Bai S, Nasser MW, Wang B, et al. MicroRNA-122 inhibits tumorigenic properties of hepatocellular carcinoma cells and sensitizes these cells to sorafenib. J Biol Chem. 2009;284:32015–32027.
- Tsai WC, Hsu PW, Lai TC, et al. MicroRNA-122, a tumor suppressor microRNA that regulates intrahepatic metastasis of hepatocellular carcinoma. Hepatology. 2009;49:1571–1582.
- Zeng C, Wang R, Li D, et al. A novel GSK-3 beta-C/EBP alpha-miR-122-insulin-like growth factor 1 receptor regulatory circuitry in human hepatocellular carcinoma. Hepatology. 2010;52:1702–1712.
- Tsai WC, Hsu SD, Hsu CS, et al. MicroRNA-122 plays a critical role in liver homeostasis and hepatocarcinogenesis. J Clin Invest. 2012;122:2884–2897.
- Hsu SH, Wang B, Kota J, et al. Essential metabolic, anti-inflammatory, and anti-tumorigenic functions of miR-122 in liver. J Clin Invest. 2012;122:2871–2883.
- Lourenço AR, Coffer PJ. A tumor suppressor role for C/EBPα in solid tumors: more than fat and blood. Oncogene. 2017;36:5221–5230.
- Zhang S, Jiang T, Feng L, et al. Yin Yang-1 suppresses differentiation of hepatocellular carcinoma cells through the downregulation of CCAAT/enhancer-binding protein alpha. J Mol Med. 2012;90:1069–1077.
- Reebye V, Sætrom P, Mintz PJ, et al. Novel RNA oligonucleotide improves liver function and inhibits liver carcinogenesis in vivo. Hepatology. 2014;59:216–227.
- Xu H, He JH, Xiao ZD, et al. Liver-enriched transcription factors regulate microRNA-122 that targets CUTL1 during liver development. Hepatology. 2010;52:1431–1442.
- Laudadio I, Manfroid I, Achouri Y, et al. A feedback loop between the liver-enriched transcription factor network and miR-122 controls hepatocyte differentiation. Gastroenterology. 2012;142:119–129.
- Song K, Han C, Zhang J, et al. Epigenetic regulation of MicroRNA-122 by peroxisome proliferator activated receptor-gamma and hepatitis b virus X protein in hepatocellular carcinoma cells. Hepatology. 2013;58:1681–1692.
- He J, Zhao K, Zheng L, et al. Upregulation of microRNA-122 by farnesoid X receptor suppresses the growth of hepatocellular carcinoma cells. Mol Cancer. 2015;14:163.
- Beishline K, Azizkhan-Clifford J. Sp1 and the ‘hallmarks of cancer’. Febs J. 2015;282:224–258.
- Abbas W, Kumar A, Herbein G. The eEF1A proteins: at the crossroads of oncogenesis, apoptosis, and viral infections. Front Oncol. 2015;5:75.
- Grassi G, Scaggiante B, Farra R, et al. The expression levels of the translational factors eEF1A 1/2 correlate with cell growth but not apoptosis in hepatocellular carcinoma cell lines with different differentiation grade. Biochimie. 2007;89:1544–1552.
- Dong J, Tsai-Morris CH, Dufau ML. A novel estradiol/estrogen receptor alpha-dependent transcriptional mechanism controls expression of the human prolactin receptor. J Biol Chem. 2006;281:18825–18836.
- Hein N, Jiang K, Cornelissen C, et al. TGFbeta1 enhances MAD1 expression and stimulates promoter-bound Pol II phosphorylation: basic functions of C/EBP, SP and SMAD3 transcription factors. BMC Mol Biol. 2011;12:9.
- Lamberti A, Longo O, Marra M, et al. C-Raf antagonizes apoptosis induced by IFN-alpha in human lung cancer cells by phosphorylation and increase of the intracellular content of elongation factor 1A. Cell Death Differ. 2007;14:952–962.
- Pecorari L, Marin O, Silvestri C, et al. Elongation factor 1 alpha interacts with phospho-Akt in breast cancer cells and regulates their proliferation, survival and motility. Mol Cancer. 2009;8:58.
- Choi WI, Kim Y, Yu MY, et al. Eukaryotic translation initiator protein 1A isoform, CCS-3, enhances the transcriptional repression of p21CIP1 by proto-oncogene FBI-1 (Pokemon/ZBTB7A). Cell Physiol Biochem. 2009;23:359–370.
- Sandelin A, Carninci P, Lenhard B, et al. Mammalian RNA polymerase II core promoters: insights from genome-wide studies. Nat Rev Genet. 2007;8:424–436.
- Nordhoff E, Krogsdam AM, Jorgensen HF, et al. Rapid identification of DNA-binding proteins by mass spectrometry. Nat Biotechnol. 1999;17:884–888.