
ABSTRACT
How have the branchpoint motifs evolved in organisms of different complexity? Here we identified and examined the consensus motifs (R1C2T3R4A5Y6, R: A or G, Y: C or T) of 898 fungal genomes. In Ascomycota unicellular yeasts, the G4/A4 ratio is mostly (98%) below 0.125 but increases sharply in multicellular species by about 40 times on average, and in the more complex Basidiomycota, it increases further by about 7 times. The global G4 increase is consistent with A4 to G4 transitions in evolution. Of the G4/A4-interacting amino acids of the branchpoint binding protein MSL5 (SF1) and the HSH155 (SF3B1), as well as the 5ʹ splice sites (SS) and U2 snRNA genes, the 5ʹ SS G3/A3 co-vary with the G4 to some extent. However, corresponding increase of the G4-complementary GCAGTA-U2 gene is rare, suggesting wobble-base pairing between the G4-containing branchpoint motif and GTAGTA-U2 in most of these species. Interestingly, the G4/A4 ratio correlates well with the abundance of alternative splicing in the two phyla, and G4 enriched significantly at the alternative 3' SS of genes in RNA metabolism, kinases and membrane proteins. Similar wobble nucleotides also enriched at the 3' SS of multicellular fungi with only thousands of protein-coding genes. Thus, branchpoint motifs have evolved U2-complementarity in unicellular Ascomycota yeasts, but have gradually gained more wobble base-pairing nucleotides in fungi of higher complexity, likely to destabilize branchpoint motif-U2 interaction and/or branchpoint A protrusion for alternative splicing. This implies an important role of relaxing the branchpoint signals in the multicellularity and further complexity of fungi.
Introduction
The complexity of different life forms has evolved with many changes such as the increased number of genes and diverse regulations including their usage of alternatively spliced exons from yeast to human beings [Citation1–Citation3]. Among the different elements/factors involved in alternative splicing, the canonical splice signals have not been well explored in genome-wide large-scale analysis for their evolutionary changes in relation to species complexities.
Alternative splicing of pre-mRNA contributes greatly to the transcriptomic/proteomic complexity of eukaryotes, particularly metazoans [Citation2]. Aberrant splicing causes many diseases [Citation4,Citation5], attesting to the diverse impact on different aspects of human and other living organisms by this important step of gene regulation. In fungi, however, the reported percentages of genes with alternative splicing have been relatively low compared to that in mammals [Citation6,Citation7]. Yet, as more and more genomes/transcriptomes of fungi are available, this appears to be changing at least for some of the species, for example 18% of genes with alternative splicing in C. neofromans [Citation8], a common pathogen of lung infection. Even higher abundance of alternative splicing has been reported in several other species [Citation9]. The splice variants are from genes that affect virulence and multicellular complexity [Citation8]. Studies of specific variants have demonstrated non-complementary functions [Citation10]. Together, these observations support that alternative splicing in fungi is perhaps more prevalent and important than what was thought previously.
Alternative splicing is controlled by cis-acting RNA elements that are often bound by trans-acting factors [Citation11–Citation13]. Others and we have shown previously that some splicing regulatory RNA elements have evolved genome-wide, for instance G tracts [Citation14–Citation16], contributing to the increased abundance of alternative splicing in mammals [Citation16,Citation17]. The resulting variants control cell cycle or differentiation in mammalian cells in the case of PRMT5 [Citation17,Citation18], and also expected to widely impact many other biological processes among hundreds of species [Citation16]. Moreover, different regulatory elements have also evolved differentially and respond to regulation by upstream cell signalling through their bound splicing factors [Citation13,Citation19–Citation21].
Besides the regulatory elements, the canonical splice sites 5ʹGT/3ʹAG and adjacent nucleotides are also important for alternative splicing with their variable strengths among different species [Citation7,Citation16], particularly in humans [Citation22]. The branchpoint (BP) motif, one of the 3ʹ splice site motifs (with polypyrimidine tract (Py) and the 3ʹ AG), is also a target of regulation for alternative or aberrant splicing [Citation23–Citation25]. However, due to its more variable sequence and distance from the 3ʹ AG, it has not been included in the consensus sequence and strength analyses of the 3ʹ splice sites in our previous genome-wide studies of hundreds of Ensembl species [Citation7,Citation16,Citation22], though a number of other studies have done so using single or a smaller number of genomes [Citation26–Citation28]. Of the different divisions of eukaryotes, fungi often show more conserved branchpoint motifs yet with significant variations [Citation29,Citation30]. These diverse species also show distinct differences in uni-/multi-cellularity as well as reproductive bodies such as the sacs of Ascomycota and the more complex basidia of Basidiomycota [Citation31,Citation32]. Here we analysed the most enriched branchpoint motifs of all 3ʹ splice sites of the hundreds of Ensembl-annotated fungal genomes and report a surprisingly distinct feature of the branchpoint motifs of uni- and multi-cellular fungi and phyla Ascomycota and Basidiomycota.
Results
Identification of the consensus branchpoint motifs of 898 fungal species/strains
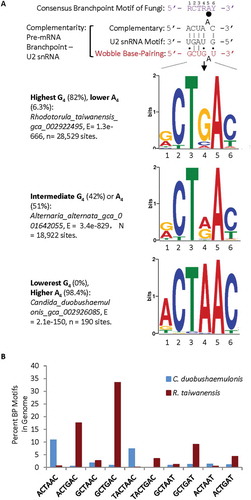
To identify the genome-wide consensus branchpoint motifs of different fungal species, we run the scripts of Multiple Em for Motif Elicitation (MEME) [Citation33], with the regions from −30 to +2 positions of all annotated 3ʹ splice sites of the fungal species/strains of the Ensembl genome release 42/95 [Citation16,Citation34]. The search identified significantly enriched motifs of 725 species/strains that have the branchpoint-like consensus R1C2T3R4A5Y6 (R: A or G, Y: C or T, branchpoint A in bold) based on at least one hundred 3ʹ splice sites per genome, representing 83.1% of the genome-wide 3ʹ splice sites on average (± 0.5%, SEM, n = 725 genomes). Ninety-seven per cent of them (Supplementary Table I) have a CTAAC (~60.4% of the species/strains) or CTGAC (37.0%) as the most enriched motif and varying G4/A4 ratios in different genomes (); the rest of species have CTGAT or CTAAT. At the extremes of the G4/A4 ratios are the distinct enrichment of the GCTGAC and ACTAAC, for instance in the genomes of R. taiwanensis and C. duobushaemulonis, respectively (). Further search between the −50 and −3 and other regions identified branchpoint motifs in a total of 898 out of 1,014 genomes (~90%, Supplementary Table Ia), including the S. cerevisiae that contain most of its motifs upstream the −30nt [Citation35]. The rest of the 116 genomes have either less than 100 of 3ʹ splice sites in the MEME motif (52 genomes) or no consensus BP motif identified in these searches.
Figure 1. Representative branch point motifs identified among the Ensembl-annotated fungal species using MEME. A. Consensus of the fungal branchpoint motifs in the genomes and complementarity of the corresponding pre-mRNA motifs with U2 snRNA without or with wobble base-pairing nucleotides (Top), and three representative sequence logos of the information content (bits) of the enriched branchpoint motifs between the −30 and +2 positions of annotated 3ʹ splice sites in the genome of each species, from MEME analysis. The big black dot indicates the branchpoint nucleotide and the smaller ones the wobble pairings. B. Per cent distribution of the branch point motifs of extreme examples of G4/A4 levels in the consensus sequences of two fungal species. Percentages of G4: of all 3ʹ splice sites containing the MEME branchpoint motif in a species. The nucleotide positions are numbered according to the consensus sequence in A.
Distinct distribution of the most enriched branchpoint motifs in uni- versus multi-cellular species of ascomycota, and in ascomycota versus basidiomycota species
With the increasing percentages of G4, the corresponding A4 of each species/strain decreases proportionally overall (). Closer examination indicated that 97% of the CTAAC as the most enriched motifs are present in 80% of the Ascomycota (where A4% > G4%) but rarely (2%) in the Basidiomycota species. In contrast, the CTGAC was the most enriched motifs (where G4% > A4%) in 98% of the Basidiomycota species but only in 20% of the Ascomycota species.
Figure 2. Distinct distribution of G4 and A4 among species/strains of different complexity. A. G4 and A4 percentages of 683 Ascomycota and 197 Basidiomycota species/strains, ranked by increasing G4%. Pezizomycotina comprises 99% of the subphyla of 475 multicellular Ascomycota species within the G4 range between 0.125 and 5.0. Note that the sudden decrease of A4 and increase of G4 separates multicellular from unicellular Ascomycota. B. Percentages of uni- or multi-cellular Ascomycota species with different thresholds of G4/A4 ratios (n = 212 unicellular, and 467 multicellular species). The ovals represent uni- or multi-cellular Ascomycota species. The Basidiomycota group also contains both uni- and multi-cellular species, but they are not clearly separated by their G4/A4 ratios. *: a group of C. neoformans strains with similar percentages of G4 or A4.
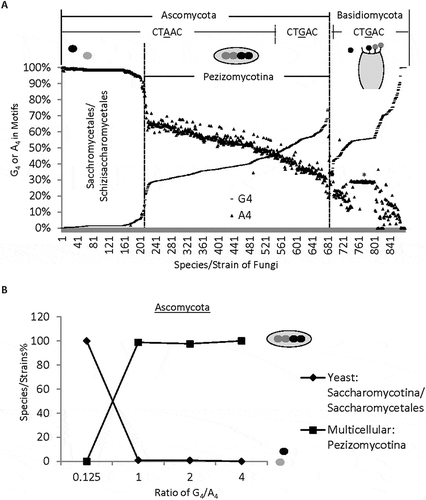
Of the Ascomycota, interestingly 208 genomes with extremely low G4 but high A4 and thus a G4/A4 ratio of less than 0.125 (0.02 ± 0.001, average ± SEM, or A4 > 85%) are all (100%) of unicellular yeasts saccharomycetales or schizosaccharomycetales (), with the latter at the higher end. Two hundred and twenty three Ascomycota genomes have ratios between 0.125 and 1 but include only 3 yeast genomes (0.4%), including the saccharomycetale Lipomyces starkeyi [Citation36]. This lipid-producing yeast has relatively more 3ʹ splice sites (14,340 in total) than the other Ascomycota and a G4/A4 ratio of 0.49 representing 99.7% of its 3ʹ splice sites in the genome. One hundred and forty genomes have ratios between 1 and 4 but with only one yeast species (0.7%). In contrast to the decreasing percentages of yeasts with the increasing G4/A4 ratios, the percentages of the multicellular subphyla (majority as Pezizomycotina, ) increased dramatically from 0% to about 98%, 98% and then 100%. The corresponding G4/A4 ratio threshold increased from 0.125 to 1, 2 and then 4, respectively (), by about 40 times on average (from 0.022 to 0.89).
Of the Basidiomycota genomes, most of them have a G4/A4 ratio between 1 and 34 with an average of 6.5 (± 2.2, SEM, n = 197 genomes), significantly higher than that of the Ascomycota (p = 5.2E-07). However, unlike the Ascomycota yeasts, the unicellular species of Basidiomycota have G4/A4 ratios ranging from 1.8 to 13.
In contrast to the above BP-G4 or -A4 changes in the different fungal groups, the level of the first nucleotide G1 of 3' exons stayed relatively constant without corresponding changes in a separate analysis of 516 fungal genomes (Supplemental Table Ib). Thus, the G4 and A4 nucleotides have distinct distribution patterns between Ascomycota and Basidiomycota and in uni- versus multi-cellular Ascomycota genomes.
We also observed G4/A4 ratios higher than 0.1 in a small number (17 in total) of genomes that belong to the phylum Blastocladiomyceta, Chytridiomycota, Glomeromycota, Mortierellomycota, Mucoromycota or Zoopagomyceta (Supplementary Table I and Ia); their prevalence remains to be determined with data from a larger number of species/strains. However, these ratios suggest that the BP-G4 has evolved convergently though to different extents in different phyla of fungi.
The G4/A4 ratio increases with the number of genome-wide 3ʹ splice sites, likely through A to G transition
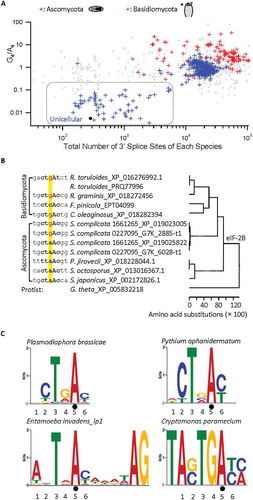
Besides its enrichment/increase in many of the Basidiomycota or multicellular Ascomycota species, the G4 nucleotide and G4/A4 ratio also increased with the growing number of 3ʹ splice sites overall (). The genome-wide total numbers of annotated 3ʹ splice sites are small in the unicellular yeasts of Ascomycota (on average 2,765 ± 1,332, n = 68 species), but more than tripled in the multicellular Ascomycota (20,383 ± 322, n = 326) and further doubled in the Basidiomycota (42,173 ± 2502, n = 109) species. The G4/A4 ratio increased from 0 to 34 with the increasing 3ʹ splice site counts. In contrast, the ratios of C−3/T−3 or G−4/A−4 of the 3ʹ splice site (N−4Y−3A−2G−1) of more than 470 fungal genomes in a similar MEME search did not have such a correlation. Thus, the G4/A4 ratio is positively correlated with the total number of 3ʹ splice sites in the fungal species.
Figure 3. Relationship between the G4/A4 ratio and the total number of 3ʹ splice sites of different genomes and the G4 – A4 evolvement among different species. A. G4/A4 ratio versus the number of 3ʹ splice sites in each of 503 fungal species (395 Ascomycota, 108 Basidiomycota), in logarithmic scales. Blue markers boxed in grey-dotted line: unicellular Ascomycota species. Grey markers: C−3/T−3 (spades) or G−4/A−4 (dots) ratios of the 3ʹ splice site of 470 or 488 fungal genomes as controls for comparison. B. The G4 or A4 within the potential branchpoint motifs of a 3ʹ splice site of the conserved eIF-2B beta gene in different species/strains. The homology tree is according to the eIF-2B beta proteins (with protein IDs) aligned by ClustalW. Note that both S. Complicata strains contain duplicated eIF-2B beta genes. C. The branchpoint MEME motifs of four protist species also containing G4 and/or A4. Note that the E. invadens branchpoint A has a fixed position (−8) relative to the 3ʹ AG, and the C. paramecium consensus has extra nucleotides beyond the 6 positions focused in this study. Black dot: the branchpoint A. The nucleotide positions are numbered according to the consensus sequence in .
The extremely low G4/A4 ratio (< 0.15, or A4 > 85%) in the unicellular Ascomycota species with less 3ʹ splice sites is consistent with the conservation of CTAAC branchpoint motifs in intron-poor species [Citation29]. What is more interesting is that the conserved A gradually decreased with G4 increased, and thus G4/A4 increased as well, as the complexity of the groups of the species and number of 3ʹ splice sites or introns increase in general ( and ).
The distinctively enriched G4/A4 nucleotides of the two closely related phyla or their subphyla with increasing numbers of 3ʹ splice sites suggested that nucleotide transition likely occurred during expansion of the genomes in evolution. We thus examined a highly conserved 3ʹ splice site of the eukaryotic initiation factor eIF-2B beta gene, which we found duplicated in one species (). Three of four Basidiomycota species examined contain a G4 and one contains a C4 in the branchpoint motif. Based on the accompanying decrease of A4 and increase of G4 among different species/strains as the complexity of fungi increases (), it is likely that the G4 or C4 was transited or transversed from A4. Interestingly, we also identified a transition from A to G in the duplicated genes of both strains of the Ascomycota S. complicata (, strains nrrl_y_17804_gca_001661265 and nrrl_y_17804_gca_000227095) while the other three species of Ascomycota contain a nucleotide A at the same positions. The two strains have G4/A4 ratios of 0.96 and 1.02 with 8,268 and 13,775 annotated 3ʹ splice sites, respectively, well beyond the threshold ratio of 0.125 for most unicellular Ascomycota with highly conserved CTAAC. The branchpoint nucleotide changes during the gene duplication of S. complicata eIF-2B thus demonstrates an example of the A to G transition that has likely contributed to the increase of G4/A4 ratios with the increased number of 3ʹ splice sites and/or duplicated genes of a species.
We also examined the homologous protist gene eIF-2B but did not identify an intron in the corresponding region. However, either G or A can still be found as the most enriched nucleotide at this position of the consensus branchpoint motifs of three protist species with G4/A4 ratios ranging from about 0.55 to 2.91 (, representing 50-100% of their >100 splice sites). In a fourth species, C. paramecium, only two introns were identified in the genome of the remnant nucleus nucleomorph [Citation37], both of which have G4 instead of A4, with BP motifs of CTGAT and GTGAC. Therefore, the G4 and A4 exist in both fungi and protists to different extents, suggesting that both nucleotides have evolved to different extents among different species/divisions. Taken together with the gradual changes of the G4/A4 ratio () and enriched G4 in other phyla of fungi (S_Table I), the CTGAC branchpoint motifs likely evolved and enriched in fungi of higher complexity since the divergence of Ascomycota and Basidiomycota at about 400 or of fungi and protists at about 1500 million years ago [Citation38–Citation40]. In at least some cases, this was achieved through A4 to G4 transition.
Corresponding changes of the A4/G4-interacting factors/elements
The branchpoint interacts with the proteins MSL5 (SF1) or HSH155 (SF3B1) [Citation41–Citation44], as well as the 5ʹ splice site and U2 snRNA at different steps of the spliceosomal assembly [Citation45–Citation47]. We thus examined their potential covariations with the G4 for possible changes of their interaction.
For MSL5, its amino acids N163, V165 and K196 interact directly with the adenine base of A4 [Citation41]. N163 contacts the base via a water-bridged hydrogen bond, and V165 and K196 sandwich the base ring without distinguishing adenine from guanine [Citation41]. By comparing these amino acids of the protein from different fungal genomes, we found that either N163 or K196 was highly conserved among 453 Ascomycota (99.6% or 97.6%) and 159 Basidiomycota genomes (94.3% or 100%). The V165 had some more changes, from 14.1% in Ascomycota to 100% in Basidiomycota genomes but not in a distinct unicellular versus multicellular distribution. For HSH155, it has mutations (E291D, R294L and D450G) corresponding to those of SF3b1 in myelodysplastic syndromes showing genetic interaction with the A4, again without distinguishing adenine from guanine [Citation42]. We only identified 6 changes (5 E291D and 1 D450N), all in Ascomycota genomes (of 58 Ascomycota and 72 Basidiomycota genomes). The five E291Ds are all in unicellular yeasts but absent in another five of such organisms. Thus, there are distinct changes in the branchpoint motif-interacting proteins in yeast or Ascomycota but they do not co-vary consistently with or distinguish G4 from A4 according to the current evidence.
For the 5ʹ splice site, we searched with the MEME between the −5 and +10 positions based on the previous splice site matrices [Citation7]. We found that as the G4 increased, the 5ʹ SS nucleotides G−1, A3, T4, G5, T6 and T7 decreased in the multicellular Ascomycota compared to those in the unicellular yeast groups, and G3, A4, C4 and to some extent A5 increased (, and S_Table II). These appear to be overall changes mostly in a group-specific way instead of co-varying proportionally with the BP-G4 increase in each genome. Their changes in Basidiomycota are not as dramatic as in Ascomycota. Interestingly the 5' SS A3/G3 decreases/increases slightly as the BP-G4 increases among the yeast, multicellular Ascomycota and some Basidiomycota with BP-G4 less than 60% (). The 5ʹ SS G3 increase is consistent with more wobble base pairing of the 5ʹ splice site with the U1 snRNA. Accompanying this change is a much more dramatic overall increase of 5' SS A4 in the multicellular Ascomycota over that in yeasts, in favour of A:U pairing with the ACUUACC-motif-containing U1 snRNA [Citation48]. Thus, there are distinct changes of multiple nucleotides at the 5ʹ splice site between the unicellular yeasts and multicellular Ascomycota groups in favour of either wobble or canonical base pairing with the U1 snRNA. More interestingly, the 5' SS A3/G3 co-vary with the BP-G4 in a genome-by-genome manner in Ascomycota and some Basidiomycota.
Figure 4. Distribution of the nucleotides of the 5ʹ splice site and the U2 snRNA gene entries among different fungal genomes. A. Representative distribution of the nucleotides of the 5ʹ splice site among the different fungal genomes as the branchpoint motif G4 increases or A4 decreases. Shown are A3, G3 or A4 within the MEME motif of the 5ʹ splice site. B. Bar graph showing the distribution of 1,336 unique U2 gene entries in the Rfam database with different motifs among 361 of the Ascomycota or Basidiomycota species with branchpoint G4/A4 ratios in .
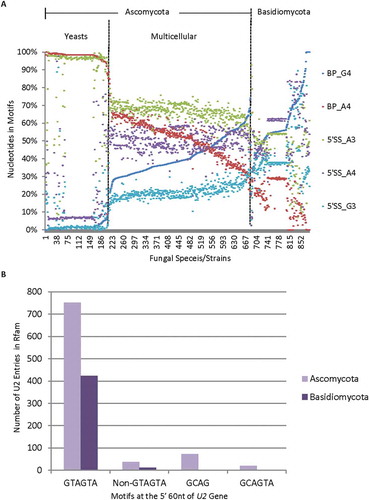
Co-evolution of U2 snRNA with a G4 -complementary Cytidine is rare
Could the G4-containing branchpoint motifs have co-evolved with their trans-acting U2 to keep their complementarity unchanged? To address this question, we searched the Rfam database [Citation49], and identified 1,336 U2 gene entries of 361 species of Ascomycota or Basidiomycota in that have at least one entry per species ( and S_Table IIa). Most (1,176, 88%) of them contain GTAGTA (TACTAAC-complementary) within their 5ʹ 60nt in 358 species (99%), suggesting that the database has well covered the orthologous U2. In contrast, we only identified 160 non-GTAGTA U2 entries in 49 species, 48 of which have higher G4/A4 ratios (>0.3) or large numbers of introns (5,000–80,000). Although this is consistent with the idea for enriched non-GTAGTA U2 in fungi of higher complexity rather than in unicellular Ascomycota yeasts, they are about 3 times in the Ascomycota over that in Basidiomycota species (), unlike the CTGAC distribution pattern between the two phyla (). More importantly, of the 1,336 genes, we identified only 75 GCAG-containing U2, mostly (73) in the Ascomycota but only 2 in the Basidiomycota species, and only 19 of them (1.4%) also contain the GCAGTA that is complementary to the branchpoint motif TACTGAC. The numbers of GCAGTA-U2 entries are also less than one-third of the GTAGTA-U2 genes in the same species (ratio = 0.27 ± 0.1, n = 3 species). In contrast, a separate search of the database indicates that the GCAGTA-U2 genes are present more often (by 2–20 folds) in metazoa and plants than in fungi or protists. Therefore, even if any of these other non-GTAGTA U2 snRNAs, particularly the GCAG- or GCAGTA-containing U2, were functional, their rarity in fungi in the high coverage database is incomparable with the increased CTGAC branchpoint motifs in most Ascomycota and all of Basidiomycota species. We thus conclude that co-evolution of U2 complementary to the G4 is rare and the branchpoint motif G4-U2 interaction should be mostly in wobble G•U pairing as the G4 evolved, particularly in Basidiomycota.
The G4/A4 ratio is correlated with the overall abundance of alternative splicing of fungal species/strains
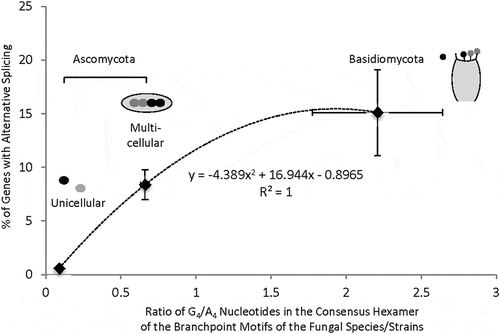
With the 5ʹ splice site or trans-acting factor changes considered, the most obvious effect of the evolved CUGAC distinct from CUAAC is likely to wobble (G•U) pair with the GUAG of U2 snRNA. The G•U pair is not well tolerated causing a dominant phenotype of slow growth in the yeast S. cerevisiae [Citation50]. In humans, where CTGAC is also one of the most abundant branchpoint motifs [Citation16], weakened strengths of branchpoint motifs in binding to U2 snRNA are associated with alternative splicing [Citation23]. Alternative splicing after the ATP-dependent step involves the control of U2 snRNP in other organism systems [Citation51–Citation53]. Together these suggest that the CTGAC branchpoint motif is likely associated with alternative splicing in fungi as well. We thus plotted the reported percentages of genes with at least one alternative splicing event in different species against the species G4/A4 ratio. The different species of Ascomycota and Basidiomycota have various abundance of alternative splicing [Citation54–Citation65]. Their percentages of alternative splicing increase overall as the G4/A4 ratio goes up (). Specifically, the abundance of alternative splicing in Basidiomycota is about 2 folds (p = 0.03) as the G4/A4 ratio goes up by 3.5 folds (p = 1.9E-7) over those in multicellular Ascomycota on average. Therefore, the evolvement of the G4/A4 ratio among the species/phyla of different complexity is correlated with their overall abundance of alternative splicing.
Figure 5. Relationship of the percentages of genes with at least one alternative splicing event to the G4/A4 ratios of different fungal species/phyla. Here the unicellular species of Ascomycota is S. pombe. The other (multicellular) Ascomycota species are: A. oryzae, A. flavus, F. graminearum, T. melanosporum, P. brasiliensis Pb01, C. immitis, P. brasiliensis Pb18, P. brasiliensis Pb03, A. niger, N. crassa, A. nidulans, P. anserina, P. nodorum (n = 13 species/strains). The three Basidiomycota species are: C. neoformans, S. commune, C. cinerea (n = 3). The abundance of alternative splicing in each species can be found in the references in the text. The points with error bars represent the mean (± SEM) values of each axis. The equation of the dotted trendline with the Pearson correlation coefficient is based on the mean values of the points.
Wobble base-pairing nucleotides are enriched in the branchpoint motifs of alternative 3ʹ splice sites destabilizing their interaction with the GUAGUA motif of U2 snRNA
We next searched with the MEME for enriched branchpoint motifs within the alternative 3ʹ splice sites (from the −50 to −3 positions) in the genome of the Basidiomycota C. neoformans, where the constitutive exons are annotated in the Ensembl database and high abundance of alternative splicing has been reported [Citation8]. This search identified a highly enriched consensus of GCTGA(C/T) at 348 alternative 3ʹ splice sites (out of 454) compared to that of the 35,048 3ʹ splice sites in the species (, E = 4.0e-205). G1 and T6 wobble bases are also enriched in the alternative 3ʹ splice sites of multicellular Ascomycota N. crassa in a separate analysis (data not shown).
Figure 6. Enrichment of G4 and other wobble nucleotides in the MEME branchpoint motifs of the alternative 3ʹ splice sites (A), its potential effect on U2 snRNA binding (B) and functional impact (C-D), in Basidiamycota C. neoformans. The nucleotide positions are numbered according to the consensus sequence in as in previous figures. a: p= 1.3E-13, b: p= 2.6E-07, compared to the genome-wide 3ʹ splice sites, in hypergeometric test. The binding strength to the GUAGUA motif of U2 snRNA was measured by the changes in free energy dG (kcal/mol) upon transition from A4 to G4 and other nucleotides (G1 or T6) within the different branchpoint motifs of the alternative 3ʹ splice sites. The functional clusters were obtained using DAVID. Shown in D are the alternative 3ʹ splice sites of the prmt gene between exons 10 and 11 (boxes) with details of the sequence features including the branchpoint motif, 3ʹ AGs and the splice variants’ last codons (underlined, with the coded amino acids under them), as well as the variant protein domains and terminal amino acids from the two splicing pathways (a or b).
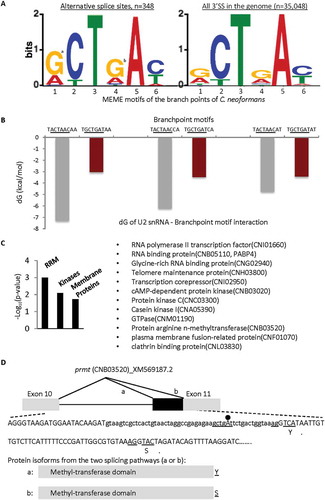
To determine if the enriched wobble bases correlate with the increased occurrences of motifs with reduced affinity to U2, we searched the constitutive and alternative 3ʹ splice sites separately for U2-high or -lower affinity BP motifs. For C. neoformans, four U2 snRNA entries were found in the Rfam database RF00004 [Citation49], all containing the GTGTAGTA but not GTGCAGTA motif. The abundance of the high-affinity TACTAAC in the upstream 3ʹ splice sites (−50 – −3) is not different between the constitutive and alternative exons (0.85% versus 0.88%, p = 0.2). In contrast, the lower affinity TGCTGAT motif is significantly less abundant in the upstream 3ʹ splice sites of constitutive exons (4.0% versus 6.6%, p = 0.003, n = 35,644 sites). Each splice site has only a single copy of one of the two motifs. Therefore, the low-affinity branchpoint motif is enriched in the alternative splice sites.
We then measured the binding of different branchpoint motifs of the alternative splice sites to the GUGUAGUA motif of U2 in RNAcofold free energy analysis [Citation66]. The result indicates that enrichment of the wobble bases G1, G4 and T6 consistently causes much less net reduction of free energy upon pairing between the branchpoint and U2 snRNA motifs, compared to those motifs containing A1, A4 and C6 (). Even considering that the G4 wobble-pairs with the pseudouridine (ψ) of U2 snRNA, where the ψ induces the branchpoint A to protrude into the minor groove of the helix [Citation67], the largest effect on helix stability upon replacing U with ψ is observed for A:ψ followed by G:ψ pairing [Citation68]. Further considering the context effect of flanking nucleotides when G:ψ showed more reduced free energy than A:ψ pairs, the difference is no more than 0.7kcal/mol [Citation68], far less than the effect by replacing the A4 with G4 (as much as 4.2kcal/mol, ). Therefore, the enriched G1, G4 and T6 likely destabilize the branchpoint motif-U2 interaction and/or protrusion of the branchpoint A.
The functions of the alternatively spliced genes cluster significantly for RNA processing and signalling as well as a large group of membrane proteins in DAVID functional clustering analysis (). For example, alternative usage of the 3ʹ splice sites of the prmt gene results in either a tyrosine or serine of the terminal amino acid in the protein isoforms (), likely to impact fruiting body development, an essential role of prmt gene for basidiomycetes [Citation69]. Thus, the G4 and other wobble nucleotides-associated alternative splicing likely modulates the transcriptome, signalling, cell communication and development.
Wobble base-pairing nucleotides are also enriched in multicellular fungi that have relatively small numbers of protein-coding genes
Are the wobble nucleotides also enriched in the fungi that have relatively small numbers of protein-coding genes but high levels of complexity particularly multicellularity? Most multicellular species have more than 10,000 genes [Citation31]. However, three species, the Ascomycota Neolecta irregularis, Basidiomycota Tremella mesenterica and Ustilago Maydis, have only 5,546, 6,902 and 8,313 protein-coding genes, respectively [Citation31,Citation70–Citation72]. The Neolecta irregularis and Tremella mesenterica have G4/A4 ratios of 1.21 and 4.72 (-B), corresponding to an estimated alternative splicing level of 10% and 20% of the genes, respectively, according to the result in . They also have high levels of T6 or G1 wobble nucleotides, respectively.
Figure 7. Enrichment of G4 in the MEME branchpoint motifs of the genome-wide 3ʹ splice sites of gene-poor multicellular species and alternative 3ʹ splice sites of U. maydis. The nucleotide positions are numbered according to the consensus sequence in as in the previous figures.
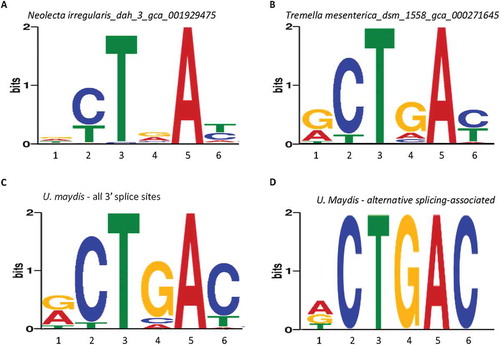
For the Ustilago maydis, it did not give a branchpoint motif in the MEME (−30 – +2) search of the fungal genomes (–). Further manual examination of its genome-wide 3ʹ splice site sequences indicated that this was due to its more distant, further upstream branchpoints, with about 5 times of CTAAC/CTGAC between −50 and −3 over the −30 – +2 region. Search with MEME between the −50 and −3 positions of its genome-wide 3ʹ splice sites identified a motif GCTGAC with an even higher G4/A4 ratio of 10.4 due to the rarity of A4 within its consensus (, E = 3.6E-385, n = 1,594, and S_Table Ia). An unusual C4, instead of A4, showed up second to G4, which is expected to cause mismatch with U2. Therefore, these species have high G4 or G4/A4 ratios and other wobble or even mismatch nucleotides within their branchpoint motifs.
To determine if the high G4/A4 ratios are indeed associated with alternative splicing in U. maydis, we used DEXSeq with our stringent filter [Citation73,Citation74], to analyse the RNA-Seq data from teliopsore, haploid, filamentous or sporidial Ustilago maydis [Citation75]. The result indicates that 361 of (16%) of the 2,247 U. maydis intron-containing genes are alternatively spliced (S_Table III). Analysis of these 3ʹ splice sites with MEME resulted in a highly enriched ACTGAC motif (, E = 4.5e-5, n = 69 sites). Of the significant clusters of genes involved, 40 genes encode ATP-binding proteins in signalling or other cellular processes, and 26 genes have functions in the nucleus. Therefore, G4 enrichment and high G4/A4 ratio are significantly associated with alternative splicing, likely to compensate at least partly the reduced gene product diversity due to the relatively smaller numbers of genes, for higher complexity/multicellularity.
Discussion
Beyond the regulatory elements and canonical splice site motifs analysed with the Ensembl genomes [Citation7,Citation16], we have also tried to examine the branchpoint motifs for their evolutionary changes in hundreds of species of different complexity. In our initial attempt, we applied the MEME to the 3ʹ splice sites of all annotated Ensembl genomes. The search in the other three divisions metazoa, plants and protists identified 3ʹ Py or AG but not branchpoint motifs (except several protist species, ), likely due to their more degenerate sequences. In contrast, the search in fungal genomes successfully obtained significantly enriched branchpoint motifs in 898 genomes. The rest of the fungal genomes may have degenerate motifs or locations that would need further refined searches; however, the current search has given an unique opportunity for us to examine the genome-wide branchpoint motifs among hundreds of related species of different complexities.
Evolvement of wobble nucleotides of the fungal branchpoint motifs in most of the fungal genomes
Co-evolution of trans-acting factors and RNA splice signals appears to be a common phenomenon [Citation19,Citation74,Citation76–Citation79]. For instance, evolved SR proteins likely facilitate the usage of weakened branchpoints for alternative splicing [Citation77,Citation80]. U2AF2 has evolved with polypyrimidine tracts in both fungal and nematode species [Citation7,Citation81,Citation82], with strong preference to the changed nucleotides in the pre-mRNA [Citation83], though the opposite effect on RNA binding was proposed for fungi [Citation81]. The co-evolution effects likely contribute to the different constraints on the relaxation of splice signals suitable for the different abundance of alternative splicing in the different species.
Based on the MEME consensus of the fungal branchpoint motifs, we have focused on the most enriched 6 nucleotides RCTRAY. Of the different motifs, GCUG(A)U wobble base pairs (underlined) with the GUAGU of the orthologous U2 snRNA at three positions (). The wobble bases are enriched to different extents among the 898 fungal genomes. Alternative to this scenario would be compensatory U2 changes in the same species/genomes to restore complementarity and thus stability of branchpoint–U2 interaction and constitutive splicing. However, our current data and others seem to support wobble base-pairing in most of the fungal genomes examined here.
Unlike the protein factors whose effects on branchpoint usage cannot be assessed readily by examining their sequences, the changes of some U snRNA sequences can be measured quantitatively by complementarity or wobble base-pairing using the free energy analysis (). Compensatory changes of U1 snRNA with the corresponding 5ʹ splice sites in some fungal or protist species have been observed [Citation7]. For U2, such changes particularly to the GCAGTA motif seem to be rare in fungi (). These observations are similar to a previous study identifying limited U1- and no U2-correlated changes with the splice signals in 20 eukaryotes [Citation81].
The U snRNA genes also include many pseudogenes [Citation84], but some U snRNA variants or identical copies are indeed expressed and biologically functional in cells or animals [Citation85,Citation86]. Thus, complementary binding by the small group of non-GTAGTA U2 to the wobble nucleotide-containing branchpoint motifs and their potential function would need careful experimental examination before a conclusion can be drawn. Even compensatory U2 changes in the small number of genomes indeed occur (), their copy numbers are often much less than the GTAGTA-U2 and thus less dosage effect as observed in other cases [Citation50,Citation86]. Therefore, most of the fungal species here likely did not co-evolve U2 to complement the G4 wobble nucleotide of the branchpoint motif and even they do in rare cases, the U2 effect would need careful assessment [Citation81].
In contrast to U2, several A4/G4-interacting amino acids of MSL5 and HSH155 changed in some yeast or multicellular Ascomycota genomes but they do not distinguish G4 from A4 [Citation41,Citation42]. The 5ʹ SS G3/A3 covariation with the BP-G4 increase would need further investigation with compensatory mutations to substantiate a possible interaction.
Genome-wide relaxation of the canonical splice sites, alternative splicing and organismal complexity
Global relaxation of cis-acting splice signals at alternative splice sites had been observed before [Citation22,Citation87], but rarely in a large number of genomes for measurable, gradual changes among related species of different complexity. Our previous study of the canonical splice sites around the GT/AG motifs identified an optimal range of constraints that allow for maximal species levels of alternative splicing [Citation7]: either too randomized or constrained splice sites out of the window will sharply decrease the levels. The current study shows that the branchpoint is highly constrained with CTAAC in Ascomycota yeasts () but gradually relaxed with the wobble nucleotide G4 and others to various extents in multicellular Ascomycota and in Basidiomycota species. Their degeneracy, but not up to the level of metazoan species, has allowed us to obtain their genome-wide branchpoint motifs with the MEME. Their wobble or even mismatch nucleotides of the degenerate branchpoint motifs, together with the 5ʹ SS G3, likely destabilize U snRNA binding and/or the branchpoint A protrusion to contribute to alternative splicing (–).
The G4/A4 ratio of 0.125, representing the relative abundance of the CTGAC and CTAAC motifs of a species ( and ), well separates multicellular from unicellular species of Ascomycota except several yeast species including Lipomyces starkeyi that has a specialized function to produce lipids [Citation36]. Distinct from the unicellular yeasts of Ascomycota, most (95%) of the Basidiomycota species including unicellular ones have G4/A4 ratios higher than 1.0 (), consistent with the higher complexity of Basidiomycota such as their reproduction bodies [Citation31,Citation32]. In particular, prmt, one of the alternatively spliced genes, contains three wobble nucleotides within its branchpoint motif GCTGAT and the gene is essential for the fruiting body development [Citation69]. Other functions such as stress response or virulence are also parts of the complexity involving alternative splicing in C. neoformans [Citation8]. Moreover, our splicing analysis of three species of gene-poor multicellular fungi is also consistent with an earlier study using EST for the U maydis, which has an abundance of alternative splicing close to that of the C. neoformans, and about 40% of the events are not simply intron retentions causing mRNA to degrade [Citation88]. These splice sites are highly enriched of the G4 within their branchpoint motifs as well (, see also ). It is thus possible that the evolved wobble nucleotides contribute to the complexity of fungi by promoting alternative splicing.
The global changes of branchpoint motifs in fungi lend further support to that not only the regulatory elements but also the canonical splice sites have evolved to relax the splice sites with increasing organismal complexity, likely by promoting alternative splicing. The splice sites could set the tone of the abundance of alternative splicing to a certain level in a species [7]; the regulatory elements could allow further variation to fine-tune the transcriptome and proteome. Together with trans-acting factors and many other steps of gene regulation such as novel genes, regulatory networks and transcriptome remodelling [Citation9,Citation70,Citation89], the evolved wobble nucleotides of the splice signals likely help generate a fine repertoire of gene products of optimal complexity for refined cellular and physiological functions.
In summary, our analysis demonstrates that the CTAAC and CTGAC branchpoint motifs distribute distinctively among fungal species of different multicellularity or complexity. Wobble nucleotides, particularly of the CTGAC branchpoint motif, have evolved to replace the CTAAC likely through A to G transition to destabilize U2 snRNA binding and/or branchpoint A protrusion for alternative splicing. This implies an important role of the relaxed U2–branchpoint interaction in the multicellularity and/or complexity of fungi.
Materials and methods
Genome data
The 3ʹ splice sites between the −30 and +2 positions (or between −15 and −4 or −50 and −3) of annotated species were extracted from the Ensembl genome releases 42/95 [Citation34]. Only AG-type 3ʹ splice sites were included in the analysis with MEME [Citation33,Citation90]. The MEME scripts were downloaded from http://meme-suite.org/doc/download.html and run with these criteria: ‘-nmotifs 1 -mod zoops -dna’ for the branchpoint motifs. Whichever region contained the highest number of 3ʹ splice sites counted for the MEME motifs are listed in the final 898 genomes in S_Table Ia: 570 from search between the −50 and −3, 322 from the −30 and +2 and 6 from the −15 and −4 regions. For the C−3/T−3 and A−4/G−4 ratios within MEME motifs of the 3ʹ splice site, the value of nmotifs was set to 3. The nucleotide matrices within the most significant MEME motifs and the total numbers of input 3ʹ splice sites of each genome were extracted from the MEME results. The search with −12 and +2 regions also identified the branchpoint motif T(−8)NA(−6)Y that can account for the previously unexplained conserved R-6 and Y-8 of the Babesia and related protist species [Citation7,Citation16].
Phylogeny of fungi
We used the phylogeny of fungi from UNITE as by Tedersoo et al. [Citation91].
Analysis of the species/genome differences of the MSL5 or HSH155 proteins
We used the yeast S. cerevisiae amino acid sequences of these proteins to search the Ascomycota or Basidiomycota phyla in the NCBI database using BLASTP. We counted the respective amino acids from each genome based on the resulting alignment of identified hits of these proteins.
Free energy analysis of the motif binding to U2 snRNA
We used the RNAcofold at the ViennaRNA Web Services [Citation66], http://rna.tbi.univie.ac.at/, with the branchpoint motif and the corresponding U2 snRNA motif GUAGUA by forcing out the branchpoint A in the helix as in the U2-BP structure [Citation67], and unchecking the option ‘avoid isolated base pairs’.
Analysis of the alternative 3ʹ splice sites
From the Biomart website for fungi (http://fungi.ensembl.org/biomart/martview/), we downloaded and examined some representative species for their annotated non-‘Constitutive’ exons, which we considered as alternative exons. We analysed with MEME the 3ʹ splice sites (between the −50 and -3 positions) of C. neoformans, which has the most number of annotated alternative exons and highest percentage (18.8%) of genes alternatively spliced, for their enriched branchpoint motifs. Functional cluster analysis of the host genes was carried out using the DAVID with high stringency [Citation92].
For the 3ʹ splice sites involved in the alternative splicing of U. maydis genes, we used DEXSeq [Citation73], to analyse the raw reads of the RNA-Seq data of filamentous versus sporidial, teliospore versus haploid sequences deposited at the SRA database by Olgeiser [Citation75], or Donaldson [Citation93], with accession numbers SRP131368 and SRP099003, respectively. The resulting exon lists were further filtered through our stringent criteria, as reported [Citation74], to obtain 441 highly confident alternative exons. This list was combined with the BioMART list of non-constitutive exons to obtain a unique list of alternative exons of 361 genes in total for the DAVID functional cluster analysis.
Statistical analysis
Two-tailed Student’s t-test was used in all analyses except in nucleotide distribution analysis (hypergeometric density test) or unless otherwise specified.
Supplemental Material
Download MS Excel (871.1 KB)Disclosure statement
No potential conflict of interest was reported by the authors.
Supplemental material
Supplemental data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Barbosa-Morais NL, Irimia M, Pan Q, et al. The evolutionary landscape of alternative splicing in vertebrate species. Science. 2012;338:1587–1593.
- Nilsen TW, Graveley BR. Expansion of the eukaryotic proteome by alternative splicing. Nature. 2010;463:457–463.
- Levine M, Tjian R. Transcription regulation and animal diversity. Nature. 2003;424:147–151.
- Feng D, Xie J. Aberrant splicing in neurological diseases. Wiley Interdiscip Rev RNA. 2013;4:631–649.
- Chabot B, Shkreta L. Defective control of pre-messenger RNA splicing in human disease. J Cell Biol. 2016;212:13–27.
- Sieber P, Voigt K, Kämmer P, et al. Comparative study on alternative splicing in human fungal pathogens suggests its involvement during host invasion. Front Microbiol. 2018;9:2313.
- Nguyen H, Das U, Wang B, et al. The matrices and constraints of GT/AG splice sites of more than 1000 species/lineages. Gene. 2018;660:92–101.
- Grützmann K, Szafranski K, Pohl M, et al. Fungal alternative splicing is associated with multicellular complexity and virulence: a genome-wide multi-species study. DNA Res. 2014;21:27–39.
- Krizsán K, Almási É, Merényi Z, et al. Transcriptomic atlas of mushroom development reveals conserved genes behind complex multicellularity in fungi. Proc Natl Acad Sci U S A. 2019;116:7409–7418.
- Marshall AN, Montealegre MC, Jimenez-Lopez C, et al. Alternative splicing and subfunctionalization generates functional diversity in fungal proteomes. PLoS Genet. 2013;9:e1003376.
- Black DL. Mechanisms of alternative pre-messenger RNA splicing. Annu Rev Biochem. 2003;72:291–336.
- Lee Y, Rio DC. Mechanisms and Regulation of Alternative Pre-mRNA Splicing. Annu Rev Biochem. 2015;84:291–323.
- Sohail M, Xie J. Diverse regulation of 3ʹ splice site usage. Cell Mol Life Sci. 2015;72:4771–4793.
- Yeo G, Hoon S, Venkatesh B, et al. Variation in sequence and organization of splicing regulatory elements in vertebrate genes. Proc Natl Acad Sci U S A. 2004;101:15700–15705.
- Sohail M, Cao W, Mahmood N, et al. Evolutionarily emerged G tracts between the polypyrimidine tract and 3′ AG are splicing silencers enriched in genes involved in cancer. BMC Genomics. 2014;15:1143.
- Nguyen H, Xie J. Widespread separation of the polypyrimidine tract from 3ʹ ag by g tracts in association with alternative exons in metazoa and plants. Front Genet. 2018;9:741.
- Sohail M, Xie J. Evolutionary emergence of a novel splice variant with an opposite effect on the cell cycle. Mol Cell Biol. 2015;35:2203–2214.
- Sohail M, Zhang M, Litchfield D, et al. Differential expression, distinct localization and opposite effect on Golgi structure and cell differentiation by a novel splice variant of human PRMT5. Biochim Biophys Acta. 2015;1853:2444–2452.
- Xie J. Differential evolution of signal-responsive RNA elements and upstream factors that control alternative splicing. Cell Mol Life Sci. 2014;71:4347–4360.
- Liu G, Razanau A, Hai Y, et al. A conserved serine of heterogeneous nuclear ribonucleoprotein L (hnRNP L) mediates depolarization-regulated alternative splicing of potassium channels. J Biol Chem. 2012;287:22709–22716.
- Razanau A, Xie J. Emerging mechanisms and consequences of calcium regulation of alternative splicing in neurons and endocrine cells. Cell Mol Life Sci. 2013;70:4527–4536.
- Stamm S, Zhu J, Nakai K, et al. An alternative-exon database and its statistical analysis. DNA Cell Biol. 2000;19:739–756.
- Corvelo A, Hallegger M, Smith CW, et al. Genome-wide association between branch point properties and alternative splicing. PLoS Comput Biol. 2010;6:e1001016.
- Alsafadi S, Houy A, Battistella A, et al. Cancer-associated SF3B1 mutations affect alternative splicing by promoting alternative branchpoint usage. Nat Commun. 2016;7:10615.
- Darman RB, Seiler M, Agrawal A, et al. Cancer-Associated SF3B1 Hotspot Mutations Induce Cryptic 3′ Splice Site Selection through Use of a Different Branch Point. Cell Rep. 2015;13:1033–1045.
- Mercer TR, Clark MB, Andersen SB, et al. Genome-wide discovery of human splicing branchpoints. Genome Res. 2015;25:290–303.
- Taggart AJ, Lin C-L, Shrestha B, et al. Large-scale analysis of branchpoint usage across species and cell lines. Genome Res. 2017;27:639–649.
- Pineda JMB, Bradley RK. Most human introns are recognized via multiple and tissue-specific branchpoints. Genes Dev. 2018;32:577–591.
- Irimia M, Roy SW. Evolutionary convergence on highly-conserved 3ʹ intron structures in intron-poor eukaryotes and insights into the ancestral eukaryotic genome. PLoS Genet. 2008;4:e1000148.
- Kupfer DM, Drabenstot SD, Buchanan KL, et al. Introns and splicing elements of five diverse fungi. Eukaryot Cell. 2004;3:1088–1100.
- Nagy LG. Evolution: complex Multicellular Life with 5,500 Genes. Curr Biol. 2017;27:R609–R612.
- Choi J, Kim SH. A genome Tree of Life for the Fungi kingdom. Proc Natl Acad Sci U S A. 2017;114:9391–9396.
- Bailey TL, Boden M, Buske FA, et al. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 2009;37:W202–208.
- Kersey PJ, Allen JE, Allot A, et al. Ensembl Genomes 2018: an integrated omics infrastructure for non-vertebrate species. Nucleic Acids Res. 2018;46:D802–D808.
- Spingola M, Grate L, Haussler D, et al. Genome-wide bioinformatic and molecular analysis of introns in Saccharomyces cerevisiae. RNA. 1999;5:221–234.
- Liu H, Zhao X, Wang F, et al. The proteome analysis of oleaginous yeast Lipomyces starkeyi. FEMS Yeast Res. 2011;11:42–51.
- Tanifuji G, Onodera NT, Wheeler TJ, et al. Complete nucleomorph genome sequence of the nonphotosynthetic alga Cryptomonas paramecium reveals a core nucleomorph gene set. Genome Biol Evol. 2011;3:44–54.
- Feng DF, Cho G, Doolittle RF. Determining divergence times with a protein clock: update and reevaluation. Proc Natl Acad Sci U S A. 1997;94:13028–13033.
- Doolittle RF, Feng DF, Tsang S, et al. Determining divergence times of the major kingdoms of living organisms with a protein clock. Science. 1996;271:470–477.
- Taylor JW, Berbee ML. Dating divergences in the Fungal Tree of Life: review and new analyses. Mycologia. 2006;98:838–849.
- Jacewicz A, Chico L, Smith P, et al. Structural basis for recognition of intron branchpoint RNA by yeast Msl5 and selective effects of interfacial mutations on splicing of yeast pre-mRNAs. RNA. 2015;21:401–414.
- Carrocci TJ, Zoerner DM, Paulson JC, et al. SF3b1 mutations associated with myelodysplastic syndromes alter the fidelity of branchsite selection in yeast. Nucleic Acids Res. 2017;45:4837–4852.
- Finci LI, Zhang X, Huang X, et al. The cryo-EM structure of the SF3b spliceosome complex bound to a splicing modulator reveals a pre-mRNA substrate competitive mechanism of action. Genes Dev. 2018;32:309–320.
- Liu Z, Luyten I, Bottomley MJ, et al. Structural basis for recognition of the intron branch site RNA by splicing factor 1. Science. 2001;294:1098–1102.
- Wilkinson ME, Fica SM, Galej WP, et al. Postcatalytic spliceosome structure reveals mechanism of 3′–splice site selection. Science. 2017;358:1283–1288.
- Fica SM, Oubridge C, Wilkinson ME, et al. A human postcatalytic spliceosome structure reveals essential roles of metazoan factors for exon ligation. Science. 2019;363:710–714.
- Parker R, Siliciano PG, Guthrie C. Recognition of the TACTAAC box during mRNA splicing in yeast involves base pairing to the U2-like snRNA. Cell. 1987;49:229–239.
- Seraphin B, Kretzner L, Rosbash M. A U1 snRNA: pre-mRNAbase pairing interaction is required early in yeast spliceosome assembly but does not uniquely define the 5ʹ cleavage site. Embo J. 1988;7:2533–2538.
- Kalvari I, Argasinska J, Quinones-Olvera N, et al. Rfam 13.0: shifting to a genome-centric resource for non-coding RNA families. Nucleic Acids Res. 2018;46:D335–D342.
- Miraglia L, Seiwert S, Igel AH, et al. Limited functional equivalence of phylogenetic variation in small nuclear RNA: yeast U2 RNA with altered branchpoint complementarity inhibits splicing and produces a dominant lethal phenotype. Proc Natl Acad Sci U S A. 1991;88:7061–7065.
- House AE, Lynch KW. Regulation of alternative splicing: more than just the ABCs. J Biol Chem. 2008;283:1217–1221.
- Lallena MJ, Chalmers KJ, Llamazares S, et al. Splicing regulation at the second catalytic step by Sex-lethal involves 3ʹ splice site recognition by SPF45. Cell. 2002;109:285–296.
- House AE, Lynch KW. An exonic splicing silencer represses spliceosome assembly after ATP-dependent exon recognition. Nat Struct Mol Biol. 2006;13:937–944.
- Kim E, Magen A, Ast G. Different levels of alternative splicing among eukaryotes. Nucleic Acids Res. 2007;35:125–131.
- Aanes H, Winata CL, Lin CH, et al. Zebrafish mRNA sequencing deciphers novelties in transcriptome dynamics during maternal to zygotic transition. Genome Res. 2011;21:1328–1338.
- Daines B, Wang H, Wang L, et al. The Drosophila melanogaster transcriptome by paired-end RNA sequencing. Genome Res. 2011;21:315–324.
- Ramani AK, Calarco JA, Pan Q, et al. Genome-wide analysis of alternative splicing in Caenorhabditis elegans. Genome Res. 2011;21:342–348.
- Xiong J, Lu X, Zhou Z, et al. Transcriptome analysis of the model protozoan, Tetrahymena thermophila, using Deep RNA sequencing. PloS One. 2012;7:e30630.
- Sorber K, Dimon MT, DeRisi JL. RNA-Seq analysis of splicing in Plasmodium falciparum uncovers new splice junctions, alternative splicing and splicing of antisense transcripts. Nucleic Acids Res. 2011;39:3820–3835.
- Loftus, B.J., Fung, E., Roncaglia, P. et al. The genome of the basidiomycetous yeast and human pathogen Cryptococcus neoformans. Science. 2005;307:1321–1324.
- McGuire AM, Pearson MD, Neafsey DE, et al. Cross-kingdom patterns of alternative splicing and splice recognition. Genome Biol. 2008;9:R50.
- Zhao C, Waalwijk C, de Wit PJ, et al. RNA-Seq analysis reveals new gene models and alternative splicing in the fungal pathogen Fusarium graminearum. BMC Genomics. 2013;14:21.
- Gehrmann T, Pelkmans JF, Lugones LG, et al. Schizophyllum commune has an extensive and functional alternative splicing repertoire. Sci Rep. 2016;6:33640.
- Tisserant E, Da Silva C, Kohler A, et al. Deep RNA sequencing improved the structural annotation of the Tuber melanosporum transcriptome. New Phytol. 2011;189:883–891.
- Wang B, Guo G, Wang C, et al. Survey of the transcriptome of Aspergillus oryzae via massively parallel mRNA sequencing. Nucleic Acids Res. 2010;38:5075–5087.
- Gruber AR, Bernhart SH, Lorenz R. The ViennaRNA web services. Methods Mol Biol. 2015;1269:307–326.
- Newby MI, Greenbaum NL. Sculpting of the spliceosomal branch site recognition motif by a conserved pseudouridine. Nat Struct Biol. 2002;9:958–965.
- Kierzek E, Malgowska M, Lisowiec J, et al. The contribution of pseudouridine to stabilities and structure of RNAs. Nucleic Acids Res. 2014;42:3492–3501.
- Nakazawa T, Tatsuta Y, Fujita T, et al. Mutations in the Cc.rmt1 gene encoding a putative protein arginine methyltransferase alter developmental programs in the basidiomycete Coprinopsis cinerea. Curr Genet. 2010;56:361–367.
- Nguyen TA, Cissé OH, Yun Wong J, et al. Innovation and constraint leading to complex multicellularity in the Ascomycota. Nat Commun. 2017;8:14444.
- Kamper J, Kahmann R, Bölker M, et al. Insights from the genome of the biotrophic fungal plant pathogen Ustilago maydis. Nature. 2006;444:97–101.
- Floudas D, Binder M, Riley R, et al. The Paleozoic origin of enzymatic lignin decomposition reconstructed from 31 fungal genomes. Science. 2012;336:1715–1719.
- Anders S, Reyes A, Huber W. Detecting differential usage of exons from RNA-seq data. Genome Res. 2012;22:2008–2017.
- Lei L, Cao W, Liu L, et al. Multilevel Differential Control of Hormone Gene Expression Programs by hnRNP L and LL in Pituitary Cells. Mol Cell Biol. 2018;38(12):e00651–17.
- Olgeiser L, Haag C, Boerner S, et al. The key protein of endosomal mRNP transport Rrm4 binds translational landmark sites of cargo mRNAs. EMBO Rep. 2019;20(1):e46588.
- Busch A, Hertel KJ. Evolution of SR protein and hnRNP splicing regulatory factors. Wiley Interdiscip Rev RNA. 2012;3:1–12.
- Shen H, Green MR. RS domains contact splicing signals and promote splicing by a common mechanism in yeast through humans. Genes Dev. 2006;20:1755–1765.
- Das U, Nguyen H, Xie J. Transcriptome protection by the expanded family of hnRNPs. RNA Biol. 2019;16:155–159.
- Attig J, de los Mozos IR, Haberman N, et al.. Splicing repression allows the gradual emergence of new Alu-exons in primate evolution. eLife. 2016;5:e19545.
- Izquierdo JM, Valcarcel J. A simple principle to explain the evolution of pre-mRNA splicing. Genes Dev. 2006;20:1679–1684.
- Schwartz SH, Silva J, Burstein D, et al. Large-scale comparative analysis of splicing signals and their corresponding splicing factors in eukaryotes. Genome Res. 2007;18:88–103.
- Sickmier EA, Frato KE, Shen H, et al. Structural basis for polypyrimidine tract recognition by the essential pre-mRNA splicing factor U2AF65. Mol Cell. 2006;23:49–59.
- Hollins C, Zorio DA, MacMorris M, et al. U2AF binding selects for the high conservation of the C. elegans 3ʹ splice site. RNA. 2005;11:248–253.
- Denison RA, Van Arsdell SW, Bernstein LB, et al. Abundant pseudogenes for small nuclear RNAs are dispersed in the human genome. Proc Natl Acad Sci U S A. 1981;78:810–814.
- O’Reilly D, Dienstbier M, Cowley SA, et al. Differentially expressed, variant U1 snRNAs regulate gene expression in human cells. Genome Res. 2013;23:281–291.
- Jia Y, Mu JC, Ackerman SL. Mutation of a U2 snRNA gene causes global disruption of alternative splicing and neurodegeneration. Cell. 2012;148:296–308.
- Burset M, Seledtsov IA, Solovyev VV. SpliceDB: database of canonical and non-canonical mammalian splice sites. Nucleic Acids Res. 2001;29:255–259.
- Ho EC, Cahill MJ, Saville BJ. Gene discovery and transcript analyses in the corn smut pathogen Ustilago maydis: expressed sequence tag and genome sequence comparison. BMC Genomics. 2007;8:334.
- Prud’homme B, Gompel N, Carroll SB. Emerging principles of regulatory evolution. Proc Natl Acad Sci U S A. 2007;104(Suppl 1):8605–8612.
- Bailey TL, Elkan C. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc Int Confer Intellig Sys Mol Bio. 1994;2:28–36.
- Tedersoo L, Sánchez-Ramírez S, Kõljalg U, et al. High-level classification of the Fungi and a tool for evolutionary ecological analyses. Fungal Divers. 2018;90:135–159.
- Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57.
- Donaldson ME, Ostrowski LA, Goulet KM, et al. Transcriptome analysis of smut fungi reveals widespread intergenic transcription and conserved antisense transcript expression. BMC Genomics. 2017;18:340.