
ABSTRACT
In the present study, we applied our ‘precise annotation’ to the mitochondrial (mt) genomes of human, chimpanzee, rhesus macaque and mouse using 5′ and 3′ end small RNAs. Our new annotations updated previous annotations. In particular, our new annotations led to two important novel findings: (1) the identification of five Conserved Sequence Blocks (CSB1, CSB2, CSB3, LSP and HSP) in the control regions; and (2) the annotation of Transcription Initiation and novel Transcription Termination Sites. Based on these annotations, we proposed a novel model of mt transcription which can account for the mt transcription and its regulation in mammals. According to our model, Transcription Termination Sites function as switches to regulate the production of short, long primary transcripts and uninterrupted transcription, rather than simply terminate the mt transcription. Moreover, the expression levels of mitochondrial transcription termination factors control the proportions of rRNAs, mRNAs and lncRNAs in total mt RNA. Our findings point to the existence of many other, as yet unidentified, Transcription Termination Sites in mammals.
Introduction
The annotation of animal mitochondrial (mt) genomes is pivotal to the study of the molecular phylogenetics and evolution of animals, and to the investigation of RNA processing, maturation, degradation and gene expression regulation [Citation1]. Although the annotation of mt mRNAs, tRNAs and rRNAs can be easily performed using web servers (e.g. MITOS [Citation2]), these methods (e.g. Blastx or structure-based covariance models) result in gaps and overlaps in the annotations of mt genomes and thus the resolution of the annotations may be severely limited. In 2017, we constructed the first quantitative transcription map of animal mt genomes by sequencing the full-length transcriptome of the insect Erthesina fullo Thunberg [Citation3] on the PacBio platform [Citation4] and established a straightforward and concise method to improve genome annotation. In 2019, we used 5′ and 3′ end small RNAs (5′ and 3′ sRNAs) [Citation1,Citation5] to improve genome annotation further, indeed to 1 bp resolution, which was later dubbed by us ‘precise annotation’ [Citation6]. In the present study, we aimed to show how to use our method to update the annotations of the human, chimpanzee, rhesus macaque and mouse (Homo sapiens, Pan troglodytes, Macaca mulatta and Mus musculus) mt genomes, and to discover novel features.
An animal mt genome contains at least one control region (CR) with a few exceptions (e.g. some ticks [Citation7]), except mt genomes of the known Cnidaria and other lower animals. The CR is also known as the Displacement-loop (D-loop), although CR and D-loop are not the same region in most species of animals. Although the existence of D-loop has been known for over fifty years, it is still not understood why this structure is synthesized and maintained [Citation8]. The prevailing hypothesis is that D-loop is involved, somehow, in mtDNA replication and/or transcription, and thus indirectly regulates these processes. In the present study, we performed precise annotation of human, chimpanzee, rhesus macaque and mouse mt genomes to cover both entire strands of them without leaving any gaps or overlaps. Further, we precisely annotated the H-strand promoters (HSPs) and the L-strand promoters (LSPs) of these four mt genomes. Five Conserved Sequence Blocks (CSB1, CSB2, CSB3, LSP and HSP) in the Control Region (CR) were annotated based on the analysis of their motifs in 52 mammalian mt genomes. We also precisely annotated mt non-coding genes MDL1, MDL1AS, MDL2 and MDL2AS [Citation9].
The two strands of an animal mt genome are differentiated by their nucleotide content: there is a guanine-rich (G-rich) strand referred to as the Heavy strand (H-strand) and a cytosine-rich strand referred to as the Light strand (L-strand). Early studies of human and mouse led to the idea that the H-strand involves two overlapping primary transcripts that are initiated at two different Transcription Initiation Termination Sites (TISs) in the CR, denoted as the first TIS and the second TIS (TISH1 and TISH2). The first primary transcript (the short H-strand transcript) was thought to span the CR, tRNAPhe, 12S rRNA, tRNAVal and 16S rRNA, and to terminate in the region of tRNALeu, whereas the second primary transcript (the long H-strand transcript) was thought to span almost the entire length of the H-strand and then terminate at the termination-associated sequence (TAS) [Citation10] in the CR (Results). Initiated at the TIS of the L-strand (TISL) in the CR, the L-strand is transcribed to produce the first primary transcript (the short L-strand transcript) which also terminates in the region of tRNALeu and/or the second primary transcript (the long L-strand transcript) which terminates in the region of CR (Results). The short H-strand transcript was predicted to explain why the steady-state levels of the 12S and 16S rRNA are 50 times higher than those of the mRNAs encoded on the same strand. There is no consensus, however, as of yet about the existence of TISH1 for the short H-strand transcript because attempts to reconstruct its activity in vitro have given conflicting results [Citation11]. A very recent study was most surprising in that MTERF1 [Citation12], a well-known mitochondrial transcription termination factor binding to TTSH1 and TTSL2 (Results), was reported to block the L-strand transcription, rather than regulate the H-strand transcription [Citation11]. In summary, the TISs and Transcription Termination Sites (TTSs) of the mt primary transcripts of human and mouse had not been determined before the present study and thus, the transcription process of these mammalian mt genomes was unclear.
In one of our previous studies of human mt genomes [Citation9], we reported for the first time the precise positions of TISH2 (for the long H-strand transcript) and TISL, and concluded that TISH1 (for the short H-strand transcript) does not exist. Furthermore, we reported the uninterrupted transcription of the H-strand. In the present study, we precisely annotated all the TISs and TTSs () of the mt primary transcripts of human, chimpanzee, rhesus macaque and mouse. This led to an important result: a novel ‘mt transcription’ model (below) of mammals which can account for the mt transcription and its regulation.
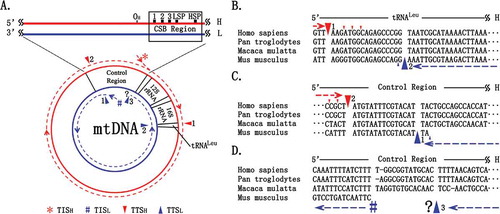
Figure 1. A novel mt transcription model of mammals.
TISH and TISL are indicated by # and * respectively. A. Transcripts from the H-strand (dashed lines) are indicated in red colour, whereas transcripts from the L-strand (in dashed lines) are indicated in blue colour. The mammalian reference genomes follow the direction of the H-strand (in red solid line) clockwise. B. Mammalian mt genomes terminate the transcription of the H-strand and the L-strand at TTSH1 or TTSL2, respectively. C. Mammalian mt genomes terminate the transcription of the H-strand and the L-strand at TTSH2 or TTSL1. D. The annotations of TTS H2 and TTSL3 in human, chimpanzee and rhesus macaque were weakly supported, whereas TTSL3 in mouse was not determined. All TTSs have several positions (indicated by triangles) as the last nucleotides for adenylation. Our findings point to the existence of other, as yet undiscovered, TTSs.
Results
A novel mt transcription model of mammals
In the present study, we precisely annotated two TISs (TISH and TISL) and five TTSs (TTSH1, TTSH2, TTSL1, TTSL2 and TTSL3) in human, chimpanzee, rhesus macaque and mouse mt genomes (Supplementary file 1), using 5′ and 3′ sRNAs. Our findings point to the existence of other, as yet undiscovered, TTSs. Of particular note is that our new annotation of a unique mouse TISH at the positions 16,285 corrected the previous annotations of two TISHs at the positions 16,295 and 16,287 (corrected using NC_005089.1) or even more TISHs proposed in the previous study [Citation13]. Our precise annotation of mouse TISH and TISL at the positions 16,285 and 16,188 corrected the previous annotations at the positions 16,287 and 16,187 (corrected using NC_005089.1) [Citation13], respectively.
The precise annotations of these four mt genomes improved the ‘mt transcription’ model proposed in our previous study [Citation6]. According to this model: (1) one CR initiates the transcription of the H-strand and the L-strand of the mammalian mt genome at the TISH and TISL, respectively (); (2) multiple TTSs function as switches by binding mitochondrial transcription termination factors, to thus regulate the production of short, long primary transcripts and uninterrupted transcription, rather than simply terminate the mt transcription; (3) the expression levels of mitochondrial transcription termination factors control the proportions of rRNAs, mRNAs and lncRNAs in total mt RNA; (4) RNA precursors of mt genes are seamlessly cleaved from primary transcripts with only a few intergenic regions between tRNAs; and (5) although all antisense transcripts are processed from primary transcripts, long antisense transcripts (e.g. tRNAMetAS/ND2AS/tRNATrpAS) degrade quickly as transient RNAs, making them unlikely to perform specific functions.
The first important evidence to support our ‘mt transcription’ model was the presence of only one TISH and one TISL, rather than multiple TISs, in each of the many species we studied [Citation1,Citation6]. Second, we reported that RNA polymerase has the ability to ‘read through’ the TISH after the long H-strand transcript has been synthesized at TTSH2, which indicated uninterrupted transcription [Citation9]. The third important evidence is the existence of multiple TTSs: TTSH1 and TTSL2 locate in the region of tRNALeu (); TTSH2 locates at the 5′ end of TAS and TTSL1 is close to TTSH2 (), and the discovery of TTSL1 explained why the transcription level of the L-strand has a substantial decrease in the region of TAS. However, we did not find that RNA polymerase has the ability to ‘read through’ the TISL but rather found TTSL3 close to TISL (). TTSH2 and TTSL3 are not well supported by sRNA-seq data due to the low expression levels of antisense genes.
Correction of previous annotations
Using 5′ and 3′ end small RNAs (5′ and 3′ sRNAs), the human, chimpanzee, rhesus macaque and mouse mt genomes were annotated precisely (Supplementary file 1) to update the previous version of annotations (NCBI RefSeq: NC_012920.1, NC_001644.1, NC_005943.1 and NC_005089.1), respectively. Our new annotations of the human and mouse mt genomes were confirmed by the PacBio full-length transcriptome data (Materials and Methods). The precise annotations generated in the present study covered the entire H-strand and L-strand of the human, chimpanzee, rhesus macaque and mouse mt genomes without leaving any gaps or overlaps (). Particularly, it was confirmed that two polycistronic transcripts, COI/tRNASerAS and ND5/ND6AS/tRNAGluAS, are not further cleaved but rather are used as templates for protein synthesis. The Coding Sequences (CDSs) of COI/tRNASerAS in human, chimpanzee, rhesus macaque and mouse have AGA, AGA, TAA and TAA as stop codons, respectively ().
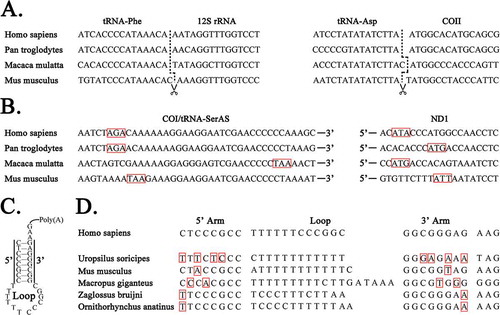
Figure 2. Correction of previous annotations.
A. RNA precursors of mt genes are seamlessly cleaved from primary transcripts. B. The Coding Sequences (CDSs) of COI/tRNASerAS in human, chimpanzee, rhesus macaque and mouse use AGA, AGA, TAA and TAA as stop codons (in red box), respectively. AC, ACACACCC, CC and GTGTTCTTT are before start codons (in red box) of ND1 in human, chimpanzee, rhesus macaque and mouse mt genomes respectively. C. The sRNA ncMT1 has polyA at its 3ʹ end and forms a typical stem-loop structure with 8-bp perfect matched nucleotides in the stem D. Among 52 mammalian mt genomes, five orthologs have co-varied mutations C-G/A-T (in red box), while others have the identical 8-bp perfect matched nucleotides in the stem (not showed in this figure).
Long antisense genes (usually above 1000 bp) in the four mt genomes (human, chimpanzee, rhesus macaque and mouse) had to be predicted and then annotated using the ‘mt transcription’ model (above), since they are transcribed as transient RNAs and thus are usually not well-covered by aligned reads from sRNA-seq or RNA-seq data owing to their rapid degradation. In the present study, long antisense genes were named as H-strand Antisense Segments (HASn) or L-strand Antisense Segments (LASn), where n indicates the number of HASs or LASs away from the D-loop region downstream. Two long non-coding genes (lncRNAs) MDL2 and MDL1AS and one small RNA (sRNA) non-coding mt RNA 1 (ncMT1) [Citation1] were identified as functional ncRNAs in mammalian mt genomes, while all other reported mt ncRNAs (e.g. lncND5 and lncND6 [Citation14]) were degraded fragments of transient RNAs or random breaks that occurred during experimental processing. Among three previously reported lncRNAs (lncND5, lncND6 and lncCytb [Citation14]), lncCytb was annotated as LAS3 (CytbAS/tRNAThrAS) in the present study, whereas lncND6 and lncND5 were degraded fragments of two other annotated RNAs (ND5/ND6AS/tRNAGluAS and LAS2) respectively (Supplementary file 1).
Our precise annotations corrected the previous annotations of mt coding genes (mRNA genes) by the analysis of their Open Reading Frames (ORFs). The mistakes in the previous annotations of mt coding genes were usually attributed to the irregularity of the start codons (e.g. TTG in Erthesina fullo [Citation3]) and stop codons (e.g. TA and T) in their CDSs, which usually begin at the 5ʹ ends by start codons ATG, ATA or ATT and end at the 3ʹ ends by stop codons TAA, TA and T. However, TA and T are completed by polyadenylation to form TAA after RNA cleavage and are thus often ignored. If one A/G or AA/AG is downstream of TA (e.g. ATP6) or T (e.g. ND1 and ND2), TAA or TAG is often incorrectly annotated as the stop codon. Using the precise annotation method, a coding gene is annotated by its mature RNA rather than its CDS, which may result in several nucleotides being annotated before the start codon in the CDS. A typical example is the three nucleotides before ATG of COI in mammals. These three nucleotides CTT, CTG, CTT, CTC, CTA and CAT appear 32, 5, 5, 4, 3, 2 and 1 times in 52 mammalian mt genomes, respectively (Supplementary file 1). These three nucleotides are not likely to belong to start codons, since one, two or more than three nucleotides appear frequently before start codons of other mt coding genes in mammals. For example, ACATA (start codons underlined), ACACACCCATG, CCATG and GTGTTCTTTATT were discovered at the beginning of ND1 in human, chimpanzee, rhesus macaque and mouse mt genomes respectively (). ATC is often annotated as a start codon for mt coding genes [Citation15], but the frequency of ATC is substantially lower than the other start codons, ATG, ATA and ATT. As it often appears before ATG, ATA or ATT (e.g. ATCAATATT in mouse ND5), we wonder if in fact ATC is a start codon.
In the present study, we improved the annotations of consecutive tRNAs (tRNATyr/tRNACys/ncMT1/tRNAAsn/tRNAAla) in chimpanzee, rhesus macaque and mouse mt genomes as we did in human [Citation1]. There are 1, 1, 2 and 3 intergenic regions () between tRNAs in the mt genomes of human, chimpanzee, rhesus macaque and mouse. Although these intergenic regions are also cleaved from the primary transcripts to form 1–2 bp RNAs, they are not likely to have biological functions, in our view. In particular, the novel 31-nt ncRNA named non-coding mt RNA 1 (ncMT1) is present in human [Citation1], chimpanzee, rhesus macaque and mouse mt genomes. In addition, we obtained the full-length sequences of ncMT1s in human and mouse using sRNA-seq data. Further investigation showed that the mature RNA of ncMT1 formed a typical stem-loop structure with 8-bp perfect matched nucleotides in the stem and whats more had polyA at its 3ʹ end (). Among the potential orthologs of ncMT1 genes in the 52 mammalian mt genomes we studied (Supplementary file 1), 47 orthologs had the identical 8-bp perfect matched nucleotides and five orthologs () had co-varied mutations (C-G/A-T) to maintain a conserved stem-loop structure. This high degree of evolutionary conservation implied that ncMT1 may have specific functions in mammals. The precise annotations of mt tRNAs revealed interesting differences in overhangs of the amino acid acceptor stems () between mouse and the other three primates (human, chimpanzee and rhesus macaque). Lastly, the present study confirmed that the acceptor stems of the tRNA precursors (e.g. tRNATyr in human) are adenylated to contain a 1-bp overhang at the 3ʹ ends of their mature RNAs.
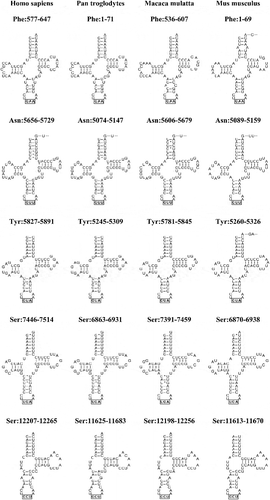
Figure 3. Precise annotation of mitochondrial tRNAs.
In the precise annotations, mt tRNAs are annotated as their precursors rather than their mature RNAs. The mouse tRNATyr, tRNASer(6870−6938) and tRNASer(11,613−11,670) have A, A and none as the overhang in the amino acid acceptor stems, while three primates (human, chimpanzee and rhesus macaque) have different overhangs. The intergenic nucleotides are appended at the 3ʹ ends of their upstream tRNAs. In the mouse mt genome, C, UU and GA are appended at the 3ʹ ends of their upstream tRNAPhe, tRNAAsn and tRNATyr, respectively.
MDL1, MDL2, MDL1AS and MDL2AS
The long H-strand transcript spans MDL1, tRNAPhe, 12S rRNA, tRNAVal, 16S rRNA, tRNALeu, ND1, tRNAIle, tRNAGlnAS, tRNAMet, ND2, tRNATrp, HAS1(tRNAAlaAS/tRNAAsnAS/ncMT1AS/tRNACysAS/tRNATyrAS), COI/tRNASerAS, tRNAAsp, COII, tRNALys, ATP8/6, COIII, tRNAGly, ND3, tRNAArg, ND4L/4, tRNAHis, tRNASer, tRNALeu, ND5/ND6AS/tRNAGluAS, Cytb, tRNAThr and MDL2, whereas the long L-strand transcript spans MDL1AS, tRNAPro, CytbAS/tRNAThrAS, tRNAGlu, LAS2(tRNAAspAS/COIIAS/tRNALysAS/(ATP8/6)AS/COIIIAS/tRNAGlyAS/ND3AS/tRNAArgAS/(ND4L/4)AS/tRNAHisAS/tRNASerAS/tRNALeuAS/ND5AS/ND6), tRNASer, COIAS, tRNATyr, tRNACys, ncMT1, tRNAAsn, tRNAAla, LAS1(tRNAMetAS/ND2AS/tRNATrpAS), tRNAGln and MDL2AS (). MDL2 is annotated to include tRNAProAS and the CR, whereas MDL2AS is annotated to include tRNAIleAS, ND1AS, tRNALeuAS, 16S rRNAAS, tRNAValAS, 12S rRNAAS, tRNAPheAS and the CR.
The CR is involved in the synthesis of four RNAs (Supplementary file 1). On the H-strand, the shorter RNA is defined as Mt D-loop 1 (MDL1), whereas the longer RNA is defined as MDL2. On the L-strand, the shorter RNA is defined as Mt D-loop 1 antisense gene (MDL1AS), whereas the longer RNA is defined as MDL2AS [Citation6]. MDL1 and MDL1AS start at the TISH and TISL, and end at the downstream cleavage sites on the H-strand and the L-strand, respectively. MDL2 is between the two nearest cleavage sites on the H-strand, respectively. In our previous study, the longer RNA on the H-strand was defined as the human MDL1 (hsa-MDL1), because the shorter RNA (NC_012920: 561–576) is only 16 bp long, which was unlikely to have specific functions. The discovery of 5′ and 3′ sRNAs, however, suggested that this 16-bp RNA may well have regulatory functions. Therefore, MDL1 was renamed as MDL2, thus MDL2 and MDL1AS were annotated as lncRNAs in the new annotations of mammalian mt genomes. The full-length transcripts of MDL1, MDL2 and MDL1AS [Citation9] have been detected in many animals using PacBio full-length transcriptome data, whereas, the transcripts of MDL2AS in the mt genomes of human, chimpanzee, rhesus macaque and mouse have been assembled from sRNA-seq data but have not yet been detected in their full-lengths.
Precise annotation of five conserved sequence blocks
Comparisons of D-loops in human and mouse mtDNAs revealed the presence of three Conserved Sequence Blocks (CSBs), referred to as CSB1, −2, and −3 [Citation16,Citation17] (). Compared with CSB2 that is responsible for the transition from RNA to DNA in the H-strand replication (Conclusion and Discussion), the biological functions of CSB1 and CSB3 are still unclear. Based on precise annotations of CSB1s, CSB2s, CSB3s, LSPs and HSPs in human, chimpanzee, rhesus macaque and mouse mt genomes, we predicted CSBs in 48 of 52 mammalian mt genomes (Supplementary file 1). Then, we used all the precisely annotated CSBs in human, chimpanzee, rhesus macaque and mouse, and predicted CSBs in 48 (totally 52) mammals to identify the sequence motifs. On the H-strand, the D-loop region has many ployA (An) and ployC (Cn) patterns allowing single nucleotide polymorphisms (SNPs) or Insertions/Deletions (InDels), which may provide signals for specific functions (e.g. recognition by enzymes for the initiation of DNA replication or transcription). In these An and Cn patterns, the most frequent SNPs are A/G or C/T. As the most evolutionarily conserved one of three CSBs, LSPs and HSPs, CSB3 has a motif A3C4A5 allowing SNPs or InDels, where A5 is the core of this motif. The previous study reported two closely related 15-nt sequence motifs ATGN9CAT on each side of D-loop [Citation18]. One motif forms part of CSB1, whereas the other motif is located the region of TAS (above). CSB1s in 52 mammalian mt genomes were identified to share this motif with three exceptions, which are ATAN9CAT in Pteropus vampyrus and ATGN8CAT in Pongo abelii and Pongo pygmaeus. In 17 primates (), HSP has a motif A3C4AAAGA, where AAAGA is the core of this motif. The reverse complimentary sequence of LSP has the same core AAAGA as HSP and a similar motif but allows more SNPs or InDels than HSP. CSB2 has a motif A3C6 allowing more SNPs or InDels than HSP, where C6 is the core of this motif. All of the motifs in CSB2, CSB3 and HSP contain A3C4, which is strong evidence that the An and Cn patterns provide signals for specific but as yet unknown functions.
Table 1. The arrangement of five CSBs.
Since our results (above) showed that HSP and LSP are CSBs, we define the CSB region to span CSB1, CSB2, CSB3, LSP and HSP (). One noteworthy finding is that the arrangement of five CSBs is evolutionarily conserved among 17 primates (). HSP is always close to the 3′ end of the D-loop region, 10 to 50 bp from the 3′ end, although the HSP motif has much sequence diversity among order and families of mammals. The core of HSP motif in primates is AAAGA, whereas the core of the HSP motif in rodents, although also A-rich, allows Ts (e.g. AATAA in mouse), yet the core of HSP motif in carnivores allows more Ts (e.g. AATTT in Canis lupus familiaris or AAATT in Puma concolor, Catopuma temminckii and Lynx rufus). From 17 mt genomes, we estimated the distance between CSB1 and CSB2, CSB2 and CSB3, CSB3 and LSP, and LSP and HSP to be about 100, 50, 50, and 100–150 bp, respectively. The distance between LSP and HSP varies greatly among the 17 primates.
Based on the findings above, we propose a simple way to identify the five CSBs. First, TISHs and TISLs can be identified using 5′ and 3′ sRNAs (Materials and Methods). Then, CSB2 can be identified by the polyC motif at upstream of LSP in mt genomes. The arrangement of five CSBs and the estimated distances between them can be used to determine CSB1 and CSB3 (ATGN9CAT). As five CSBs are evolutionarily conserved, information of closely related species may be used to predict the five CSBs within families and orders. In this way, we predicted five CSBs in 48 of 52 mammalian mt genomes (Supplementary file 1). Tandem repeats in the D-loop region are obstacles to estimating distances between the five CSBs. However, tandem repeats are present in at least 19 of 52 mammalian mt genomes, notably in all 12 carnivores. Although the biological functions of the tandem repeats in the D-loop region are still unclear, the tandem repeats in the E. fullo mt genome containing the repeated HSPs were discovered in one of our previous studies [Citation6]. In the E. fullo mt genome, HSP is also close to 3′ end of the D-loop region as it is in mammals. This suggests that the conserved arrangement of five CSBs or at least three CSBs (CSB2, LSP and HSP) may be present in a wide range of animal mt genomes.
5′ sRNAs of MDL1 and MDL1AS
Although a series of proteins (e.g. TFAM, TFB2M and MTERF1 [Citation19]) encoded by nuclear genes were reported to regulate mtDNA transcription [Citation15], it had not been considered that mtDNAs may regulate their transcription themselves until the discovery of MDL1, MDL1AS and their 5′ sRNAs in a previous study [Citation9]. In that study, our hypothesis was that 5′ sRNAs of MDL1 and MDL1AS may regulate the expression levels of mt genes by a negative feedback mechanism. In particular, the excessive 5′ sRNAs of MDL1 may inhibit H-strand transcription, whereas those of MDL1AS may inhibit L-strand transcription and thus H-strand replication, since the H-strand replication is intimately linked with the L-strand transcription (Conclusion and Discussion).
Since the H-strand and the L-strand are transcribed as primary transcripts, the 5′ sRNAs of MDL1 and MDL1AS may be used to quantify the expression levels of the total H-strand and L-strand transcripts, respectively (). Using the human931 sRNA-seq dataset, we found that expression of the 5′ sRNAs of MDL1 increased in cancer samples in comparison to normal samples, whereas expression of the 5′ sRNAs of MDL1AS decreased in the cancer samples (Supplementary file 2). The cancer samples were hepatocellular carcinoma (SRA: SRP002272), colorectal cancer (SRA: SRP022054), adrenocortical carcinoma (SRA: SRP028291) and head and neck squamous cell carcinoma (ENA: ERP001908).
Table 2. Quantification of 5′ small RNAs of MDL1 and MDL1AS.
Our ‘mt transcription’ model leads us to propose that the increase of 5′ sRNAs of MDL1 in cancer cells may be due to more frequent transcription initiation of the H-strand that produces more 5′ sRNAs of MDL1. More frequent transcription initiation in cancer cells may be induced to compensate for the lower amount of total mt RNAs caused by the lower frequency of uninterrupted transcription. The lower frequency of uninterrupted transcription thus may result from more binding of TTSs on the H-strand with mitochondrial transcription termination factors, since these TTSs may function as switches to regulate the production of short, long primary transcripts and uninterrupted transcription of the H-strand. One well-known mitochondrial transcription termination factor is MTERF1 [Citation12], a DNA-binding protein of 39 kDa that interacts with TTSH1 and TTSL2 in the tRNALeu region (). The increase of 5′ sRNAs of MDL1 suggests to us that MTERF1 may be up-regulated in cancer cells.
Conclusion and discussion
In the present study, we performed the precise annotation of human, chimpanzee, rhesus macaque and mouse mt genomes. Most of our findings confirmed findings from the previous studies and two potentially important findings were reported for the first time. These two findings include the identification of the motifs of five Conserved Sequence Blocks (CSB1, CSB2, CSB3, LSP and HSP) in the D-loop regions and the proposal of a novel ‘mt transcription’ model.
A long-standing hypothesis is that the H-strand replication is intimately linked with the L-strand transcription [Citation20]. Further studies of D-loops in human and mouse mtDNAs have suggested that short mt transcripts, originating at TISL, serve as primers for the initiation of synthesis of nascent H-strands. Based on these findings, CSB2 needs to be downstream of LSP for the RNA-DNA transition. This hypothesis was supported by the conserved arrangement of the five CSBs in most of the 52 mammals we surveyed. Unexpectively, we found a few order rearrangements of the three CSBs (). In the Pongo abelii mt genome, CSB2 is not downstream of LSP on the L-strand transcript. Future studies need be conducted to investigate the conserved arrangement of the five CSBs in a wider range of animal species. This will help to reveal mechanisms in the RNA-DNA transition and even other functions of D-loop.
In the present study, we proposed a novel mt transcription model of, but not limited to mammals. Binding with mitochondrial transcription termination factors, TTSs function as switches to determine the production of short, long primary transcripts and uninterrupted transcription of the H-strand. As a result, MTERF1 may control the ratio of rRNA to mRNA expressed. The ratio of rRNA to mRNA expressed should be regulated in a range of different cell cycles under different living conditions. The over-expression of MTERF1 in cancer cells could result in a higher rRNA/mRNA ratio and the loss of the mt transcription regulation. Moreover, the ratios of 16S rRNA/COI and 16S rRNAAS/ND1AS might be used to indicate the function of MTERF1 in the future studies.
Materials and methods
The reference sequences of human, chimpanzee, rhesus macaque and mouse mt genomes were downloaded from the NCBI RefSeq database under the accession numbers NC_012920.1, NC_001644.1, NC_005943.1 and NC_005089.1, respectively. The sRNA-seq data were downloaded from the NCBI SRA database under the accession numbers SRP017691, SRP012018 and SRP059657. The human mt genome was annotated using 5′ and 3′ sRNAs from the human931 sRNA-seq dataset, which had been constructed in one of our previous study [Citation21]. The mouse mt genome was annotated in the present study using 5′ and 3′ sRNAs from the datasets SRP017691 and SRP012018. The chimpanzee and rhesus macaque mt genomes were also annotated in the present study using 5′ and 3′ sRNAs from the dataset SRP059657. The mouse full-length transcriptome data was downloaded from the NCBI SRA database under the accession numbers SRP067402 and SRP101446.
Our precise annotation using 5′ and 3′ sRNAs followed the protocol published in one of our previous study [Citation1]. Our precise annotation of human mt genome was confirmed using pan RNA-seq analysis [Citation1], whereas our precise annotation of mouse mt genome was confirmed using the mouse full-length transcriptome data from datasets SRP067402 and SRP101446. Data cleaning and quality control were performed using Fastq_clean [Citation22]. Fastq_clean is a Perl based pipeline to clean DNA-seq [Citation23], RNA-seq [Citation24] and sRNA-seq data [Citation21] with quality control and had included some tools to process Pacbio data in the version 2.0. Statistics and plotting were conducted using the software R v2.15.3 with the package ggplot2 [Citation25]. The 5ʹ and 3ʹ ends of mature transcripts, polycistronic transcripts, antisense transcripts and the positions of 5ʹ m7G caps were observed and curated using the software Tablet v1.15.09.01 [Citation26].
Supplemental Material
Download Zip (232.7 KB)Acknowledgments
We appreciate the help equally from the people listed below. They are Professor Jishou Ruan from School of Mathematical Sciences, Yanqiang Liu, Guoqing Liu and Dawei Huang, Associate Professor Bingjun He and Qiang Zhao from College of Life Sciences, Nankai University.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplemental material
Supplemental data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Xu X, Ji H, Jin X, et al. Using pan RNA-seq analysis to reveal the ubiquitous existence of 5ʹ and 3ʹ end small RNAs. Front Genet. 2019;10:1–11.
- Bernt M, Donath A, Jühling F, et al. MITOS: improved de novo metazoan mitochondrial genome annotation. Mol Phylogen Evol. 2013;69(2):313–319.
- Gao S, Ren Y, Sun Y, et al. PacBio Full-length transcriptome profiling of insect mitochondrial gene expression. RNA Biol. 2016;13(9):820–825.
- Ren Y, Jiaqing Z, Sun Y, et al. Full-length transcriptome sequencing on PacBio platform (in Chinese). Chinese Sci Bull. 2016;61(11):1250–1254.
- Chen Z, Sun Y, Yang X, et al. Two featured series of rRNA-derived RNA fragments (rRFs) constitute a novel class of small RNAs. Plos One. 2017;12(4):e0176458.
- Ji H, Xu X, Jin X, et al. Using high-resolution annotation of insect mitochondrial DNA to decipher tandem repeats in the control region. RNA Biol. 2019;16(6):830–837.
- Shao R, Barker S. Mitochondrial genomes of parasitic arthropods: implications for studies of population genetics and evolution. Parasitology. 2007;134(2):153–167.
- Nicholls TJ, Minczuk M. In D-loop: 40 years of mitochondrial 7S DNA. Exp Gerontol. 2014;56(Complete):175–181.
- Gao S, Tian X, Chang H, et al. Two novel lncRNAs discovered in human mitochondrial DNA using PacBio full-length transcriptome data. Mitochondrion. 2017;38:41–47.
- Freyer C, Park CB, Ekstrand MI, et al. Maintenance of respiratory chain function in mouse hearts with severely impaired mtDNA transcription. Nucleic Acids Res. 2010;38(19):6577–6588.
- Terzioglu M, Ruzzenente B, Harmel J, et al. MTERF1 binds mtDNA to prevent transcriptional interference at the light-strand promoter but is dispensable for rRNA gene transcription regulation. Cell Metab. 2013;17(4):618–626.
- Fernandez-Silva P, Martinez-Azorin F, Micol V, et al. The human mitochondrial transcription termination factor (mTERF) is a multizipper protein but binds to DNA as a monomer, with evidence pointing to intramolecular leucine zipper interactions. Embo J. 1997;16(5):1066–1079.
- Chang DD, Clayton DA. Precise assignment of the heavy-strand promoter of mouse mitochondrial DNA: cognate start sites are not required for transcriptional initiation. Mol Cell Biol. 1986;6(9):3262–3267.
- Rackham O, Shearwood AM, Mercer TR, et al. Long noncoding RNAs are generated from the mitochondrial genome and regulated by nuclear-encoded proteins. RNA. 2011;17(12):2085.
- Fernandez SP, Enriquez JJ. Replication and transcription of mammalian mitochondrial DNA. Exp Physiol. 2010;88(1):41–56.
- Walberg MW, Clayton DA. Sequence and properties of the human KB cell and mouse L cell D-loop regions of mitochondrial DNA. Nucleic Acids Res. 1981;9(20):5411–5421.
- Taanman JW. The mitochondrial genome: structure, transcription, translation and replication. Biochim Biophys Acta. 1999;1410(2):103.
- Gustafsson CM, Falkenberg M, Larsson N-G. Maintenance and expression of mammalian mitochondrial DNA. Annu Rev Biochem. 2016;85(1):133–160.
- Wanrooij PH, Uhler JP, Shi Y, et al. A hybrid G-quadruplex structure formed between RNA and DNA explains the extraordinary stability of the mitochondrial R-loop. Nucleic Acids Res. 2012;40(20):10334–10344.
- Xu, B, Clayton DA. A persistent RNA-DNA hybrid is formed during transcription at a phylogenetically conserved mitochondrial DNA sequence. Mol Cell Biol. 1995;15(1):580–589.
- Wang F, Sun Y, Ruan J, et al. Using small RNA deep sequencing data to detect human viruses. Biomed Res Int. 2016;2016(2016):2596782.
- Zhang M, Zhan F, Sun H, et al. Fastq_clean: an optimized pipeline to clean the Illumina sequencing data with quality control. Bioinformatics and Biomedicine (BIBM), 2014 IEEE International Conference on. IEEE; Belfast, UK; 2014.
- Wang Y, Wang Z, Chen X, et al. The complete genome of brucella suis 019 provides insights on cross-species infection. Genes (Basel). 2016;7(2):7.
- Cao Q, Li A, Chen J, et al. Transcriptome sequencing of the sweet potato progenitor (Ipomoea Trifida (HBK) G. Don.) and discovery of drought tolerance genes. Tropical Plant Biology. 2016;9(2):63–72.
- Wickham H. ggplot2: elegant graphics for data analysis. Springer Science & Business Media; Dordrecht; 2009.
- Milne I, Stephen G, Bayer M, et al. Using Tablet for visual exploration of second-generation sequencing data. Brief Bioinform. 2012;14(2):193–202.