
ABSTRACT
The tumour suppressor p53 and its paralogues, p63 and p73, are essential to maintain cellular homoeostasis and the integrity of the cell’s genetic material, thus meriting the title of ‘guardians of the genome’. The p53 family members are transcription factors and fulfill their activities by controlling the expression of protein-coding and non-coding genes. Here, we review how the latter group transcended from the ‘dark matter’ of the transcriptome, providing unexpected and intriguing anti-cancer therapeutic strategies.
Introduction
Almost twenty years ago, the sequencing of the human genome was officially completed [Citation1]. This Herculean effort lasted for almost two decades and had already produced its preliminary results in 2001 [Citation2,Citation3], when it became irrefutable that the protein coding portion of the genome – i.e. that behaving accordingly to the central dogma of molecular biology – was not as large as expected. Indeed, despite the fact that more than 85% of the human genome is actively transcribed [Citation4], only ~2% of this transcriptome corresponds to messenger RNAs (mRNAs). Most of the remaining 98% comprises a large amount of RNA species with poorly characterized or completely unknown functions, which were initially dubbed as the ‘dark matter’ of the genome [Citation5] and have since then represented an intensive field of research. This research has contributed to the expansion of the ever-growing classification of RNA species. In addition to the long-known yet still surprising groups of ribosomal RNAs (rRNAs), transfer RNAs (tRNAs), and small nuclear and nucleolar RNAs (snRNAs and snoRNAs), there are 4 other main classes of RNA molecules [Citation6]. The largest of these classes comprises the PIWI-interacting RNAs (piRNAs), whose expression is limited to specific developmental stages of germ cells [Citation7]. The remaining categories are more widely expressed and include the ~22 nucleotides long microRNAs (miRNAs) [Citation8], the longer than 200 nucleotides long non-coding RNAs (lncRNAs) [Citation9], and the back-splicing generated circular RNAs (circRNAs) [Citation10]. Notably, many members of these RNA species are dysregulated in human cancers [Citation11–Citation15] underscoring the relevance of these RNA molecules for tumour initiation and progression. This dysregulation is intertwined with the genetic alterations affecting one of the most important tumour suppressive pathways, the p53 pathway. Indeed, the tumour suppressor p53 is the main cellular hub responsible for maintaining cellular homoeostasis and genome integrity, and it fulfils its roles by controlling the expression of protein-coding and non-coding genes alike [Citation16–Citation18]. Given its centrality in counteracting stimuli that can perturb the physiological conditions of cells [Citation19,Citation20], the loss of p53 functions is a common feature of human cancers [Citation21], and numerous animal models have shown that either lack [Citation22,Citation23] of or mutations [Citation24,Citation25] in the TP53 gene determines the onset of a variety of tumours and can recapitulate the human tumour-predisposing syndrome, known as Li-Fraumeni.
Despite being considered unique to the point of deserving the title of the ‘guardian of the genome’[Citation26], it is now clear that p53 is supported in its activities by the other members of its family, p63 and p73 [Citation27–Citation29]. These three transcription factors share a similar DNA binding domain, that allows them to regulate the expression of a common pool of genes crucial to prevent tumorigenesis, including genes involved in cell-cycle arrest [Citation30], DNA repair [Citation31], apoptosis [Citation32], autophagy [Citation19], and cellular metabolism [Citation20]. Beyond these commonalities, however, different biological functions are associated with each of the p53 family members, as highlighted by the phenotypes of the respective knockout mouse models. Indeed, p63 emerged as crucial for the proper formation and differentiation of pluri-stratified epithelia [Citation33,Citation34], while p73 is essential for the development of the central nervous system [Citation35]. Further diversification in their biological roles is provided by the complex structure of the p53 family genes, all of which encode numerous isoforms due both to the usage of alternative promoters and to the splicing events involving the respective 3ʹ UTRs [Citation36–Citation38]. The various isoforms are grouped in two sets based either on the presence of a transcriptional activation domain resembling that present in p53 (TA isoforms) or on the absence thereof (ΔN isoforms). The TA isoforms have tumour suppressive properties and can be bound and inhibited by the ΔN isoforms, which can conversely exert oncogenic activities [Citation39–Citation41]. In line with this, human cancers generally show an imbalance among these isoforms in favour of the ΔN isoforms [Citation42]. The distinct roles of the various isoforms are corroborated by the unique phenotypes of the isoform-specific knockout mouse models. Indeed, even though both the TAp63−/− and the TAp73−/− mice are tumour prone [Citation43,Citation44], the former is characterized by premature ageing[Citation45], stem cell defects [Citation46] and tendency to diabetes and obesity [Citation47], while the latter cannot properly differentiate multiciliated cells, thereby having impaired functionality of several organs, such as ear, ependyma, fallopian tube, and trachea [Citation48,Citation49]. In the case of the ΔN isoforms, instead, loss of ΔNp63 prevents the terminal differentiation of the epidermis and causes craniofacial abnormalities and limb defects [Citation50–Citation52], while lack of ΔNp73 is associated with severe neurodegeneration [Citation53,Citation54].
The specific roles of the different isoforms of the p53 family members are ultimately achieved via the regulation of unique transcriptional programmes, which comprise both protein-coding and non-coding target genes. Here, we focus on the numerous connections between the p53 family members and the different classes of non-coding RNAs in physiological conditions as well as in cancers, and we discuss the possible therapeutic approaches targeting such connections.
Ribosomal biogenesis, nucleolar stress, and the p53 family
Ribosomal RNAs (rRNAs) represent ~85% of the RNA mass in eukaryotic cells [Citation6] and their biogenesis is finely regulated to guarantee a constant balance between their expression and that of the 80 ribosomal proteins (RPs) that ultimately constitute the ribosomes [Citation55]. To facilitate such regulation, ribosomal biogenesis is temporally and spatially confined in the nucleolus, where one the two rRNA precursors, 47S, is transcribed by the RNA polymerase I and processed into the mature 18S of the 40S ribosomal subunit, and the 28S and 5.8S rRNAs of the 60S subunit [Citation56]. The fourth rRNA, 5S, is instead transcribed by the RNA polymerase III in the nucleus, but it is then actively transported in the nucleolus, where it undergoes through the coordinated assembly with the remaining rRNAs and RPs [Citation56]. This multistep process, which concludes with the export of the 40S and 60S into the cytoplasm, requires numerous accessory factors comprising both non-ribosomal proteins and small nucleolar RNAs (snoRNAs) [Citation57] and is coordinated with cell growth and division so that ribosomal biogenesis may occur only during the interphase [Citation58].
Dysregulated ribosomal biosynthesis is a trait of both solid [Citation59,Citation60] and liquid [Citation61] tumours and entails the disruption of the nucleolar organization, an event known as ‘nucleolar stress’[Citation62]. An ever-growing body of evidence unequivocally demonstrates the link between nucleolar stress and p53 activation (). The presence of free RPs, which is indicative of a disequilibrium in the ratio between rRNAs and RPs, can be detected by MDM2, an E3 ubiquitin ligase acting as negative regulator of p53 [Citation63]. RPL5 and RPL11 were the first two RPs shown to interact with MDM2 and to block its function, thus leading to p53 accumulation in the disrupted nucleoli [Citation64–Citation66]. There, p53 interacts with the promyelocytic leukaemia (PML) tumour suppressor, which enhances p53 acetylation by p300 thus further preventing the MDM2-dependent ubiquitination of p53 [Citation67]. An additional stabilization of p53 is provided by another RP, RPL26, which interacts with both the 5ʹ and 3ʹ UTRs of the p53 mRNA and promotes its translation [Citation68]. In unstressed conditions, this is prevented by MDM2, which blocks the interaction between RPL26 and the p53 mRNA, thereby realizing an additional feedback loop linking the ribosomal biogenesis with the p53 pathway [Citation69]. The ultimate goal of this activation of p53 following nucleolar stress is to halt ribosomal biosynthesis. This is achieved by p53 at multiple levels including: i) the regulation of the transcriptional activity of RNA polymerase I and III, either directly [Citation70] or by counteracting the cMyc-dependent induction of the rRNA genes [Citation71]; ii) preventing the nuclear import of the RPs that are translated in the cytoplasm [Citation72]; and iii) blocking the nuclear export of the 40S and 60S ribosomal subunits [Citation72]. All these events contribute to suppress ribosomal biogenesis until the unbalance between rRNAs and RPs that led to the nucleolar stress is resolved [Citation73].
The pivotal role of p53 in controlling the homoeostasis of the ribosomal biogenesis is corroborated by the investigation of several RP-deficient mice. Although these mice display different phenotypes, such as embryonic (Rps6−/−) [Citation74] or perinatal (Rpl24−/−) [Citation75] lethality, impaired T cell development (Rpl22−/−) [Citation76], and low body weight and anaemia (Rpl27a−/−) [Citation77], all these defects are due to the hyperactivation of p53 and can be rescued by the concomitant deletion of the Trp53 gene [Citation74–Citation77].
During the past decade, a few reports have shed light on the connections between the ribosomal biogenesis and other members of the p53 family. For example, two papers have recently unveiled a role for TAp73 in controlling the translation of mRNAs encoding nucleolar proteins, thus in turn affecting rRNA processing and global protein synthesis [Citation78,Citation79] (). Acute downregulation or chemical inhibition of TAp73 impairs the translation of nucleolar protein, which reduces the rRNA processing and the polysomal/subpolysomal ratio, ultimately leading to an impaired global protein synthesis [Citation78]. In TAp73−/− mice, a compensatory mechanism occurs that allows the maintenance of global protein synthesis by bypassing the checkpoint set up by the translational elongation factor eEF2K [Citation79].
The other p53 family member reported to affect ribosomal biogenesis is ΔNp63, which directly induces the levels of Basonuclin1 (BCN1) [Citation80], a transcription factor controlling the expression of a subset of rRNA genes via RNA polymerase I and III [Citation81,Citation82] (see ). The ΔNp63-BCN1-rRNAs axis is particularly relevant in basal cell carcinomas and in head and neck squamous cell carcinomas, where the levels of both ΔNp63 [Citation42] and BCN1 [Citation80,Citation83] are upregulated in comparison with normal matched tissue and could increase the ribosomal biogenesis to sustain the higher demand for protein production by proliferating cells.
Given that upregulated ribosomal biogenesis is a feature of numerous human cancers, extensive efforts have been made to design inhibitors of this process and some of these small molecules are in advanced pre-clinical investigations or in early clinical trial phases [Citation84]. A promising compound, called CX-5461, is a non-genotoxic drug inhibiting the recruitment of the ribosomal DNA transcription factor Selectivity factor 1 (SL1) on the promoters of the rRNA genes [Citation85]. Notably, when tested in a Eµ-MYC mouse model of Burkitt lymphoma, CX-5461 triggered the p53-dependent cell death of malignant B cells without affecting normal cells and extended the survival of these mice [Citation86]. P53-dependent apoptosis is also induced by another inhibitor of rDNA transcription, hernandonine, which binds to RPA194, the large catalytic protein of the RNA polymerase I, and promotes its degradation via the proteasome [Citation87]. It will be interesting to evaluate in the future whether CX-5461 and hernandonine can stimulate the activities of the other pro-apoptotic members of the p53 family, namely TAp63 and TAp73, thus providing an alternative therapeutic strategy to treat tumours characterized by the functional loss of p53.
The p53 family and tRNA deregulation in cancer
In 1958, ‘soluble ribonucleic acid intermediates in protein synthesis’ were discovered [Citation88] making these molecules, later named transfer RNAs (tRNAs), the first class of non-coding RNAs to be identified. Given their essential role in translation together with rRNAs, upregulation of tRNAs in tumours has for too long been considered just a consequence of the high metabolic rate and increased demand in protein synthesis typical of cancer cells [Citation89]. However, the regulation of tRNA levels is more complex than previously envisioned. Indeed, not all of the 506 human tRNA genes are simultaneously overexpressed in a given tumour [Citation90]. Instead, the expression of tRNA isoacceptors (i.e. different tRNA isoforms loaded with the same amino acid) is tissue-specific and finely tuned to the expression of the mRNAs needed for the tissue’s optimal functions [Citation91]. Accordingly, unbalance in the coordinated transcription of tRNAs and mRNAs may lead to tissue degeneration and even death [Citation92].
The major element determining tRNA expression is the activity of the RNA polymerase III, which is synchronized to the cell cycle and kept under control by the tumour suppressor RB and p53 [Citation70]. On the contrary, numerous oncogenes have been proven to promote RNA polymerase III transcriptional activity, including cMyc [Citation71], Ras/ERK [Citation93], PI3K/AKT/mTOR [Citation94], and TERT [Citation95]. Notably, these oncogenes induce the expression of selective tRNAs in a tissue-specific manner, thus underlining the fact that the conditions in which specific tRNAs are upregulated are crucial for them to exert their pro-oncogenic effects. The best example of this crosstalk between oncogenes and tRNAs is that of TERT upregulating tRNALeu and tRNATyr in highly aggressive triple-negative breast cancers but inducing different tRNAs in other organs [Citation95].
Additional complexity in the regulation of tRNA levels has recently been provided by a report demonstrating that the p53 induced miR-34a post-transcriptionally regulates the initiator tRNAMet [Citation96] (). Although this is the only case of miRNA-tRNA interaction known so far, these two RNAs create a feedback loop that is highly relevant for human tumours. Indeed, if on one hand miR-34a decreases the levels of tRNAMet, on the other hand the overexpression of this tRNA bypasses the S/G2 cell cycle transition controlled by p53 and miR-34a thus promoting tumour initiation [Citation96]. Furthermore, tRNAMet can sustain tumour progression by increasing the migratory and invasive potential of melanoma cells [Citation97], a property shared with tRNAGlu and tRNAArg in metastatic breast cancer cells [Citation89].
Not only are tRNAs now established as key elements in tumour and metastasis formation, but they are also emerging as potential cancer biomarkers. In renal clear cell carcinomas, the overexpression of tRNAArg and the downregulation of tRNAPro and tRNAThr correlate with poor overall survival [Citation98], and in lung adenocarcinoma the overexpression of tRNAGlu and tRNATyr and the downregulation of tRNAAsn and tRNAThr are associated with increased recurrence risk [Citation99]. It is very likely that the constantly-growing availability of next generation sequencing data will aid in unveiling prognostic markers for tRNAs in other tumour types as well. It has been hypothesised that tRNAs might also be predictive of responsiveness to cancer treatment. This is based on the fact that the elevated protein synthesis rate present in fast proliferating tumours causes a reduction in translational accuracy [Citation100]. This augmented protein synthesis error (PSE) is due to tRNA misreading (i.e. the incorporation of a wrong amino acid in a protein), which leads to an in vivo tumour growth similar to what is achieved by a potent oncogene, such as K-rasG12V [Citation101], and can endow cancer cells with drug resistance and adaptation to tumour suppressive signals, a phenomenon referred to as adaptive mistranslation [Citation102]. The biological consequence of PSE is the induction of the unfolded protein response (UPR) also known as the ER stress response [Citation103]. Although UPR helps cancer cells to cope with translational inaccuracy by upregulating molecular chaperones and increasing the proteasome-dependent degradation of misfolded proteins [Citation103], it can also be an Achilles’ heel for the tumour. Indeed, if PSE is not cleared, USP triggers the pro-apoptotic members of the p53 family thus leading to tumour cell death [Citation104–Citation106]. This could be pharmacologically exploited, since tumours relying on tRNA misreading may be more responsive to UPR and/or proteasome inhibitors [Citation107].
The bidirectional crosstalk between the p53 family and miRNAs in cancer
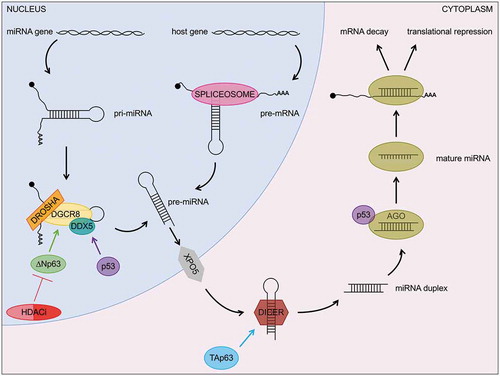
MicroRNAs (miRNAs) are a large group of small RNAs that function by regulating mRNA stability and translation in a sequence-specific manner [Citation108]. Although they are ~22 nucleotides long, they are produced as longer precursors either via splicing of host gene pre-mRNA transcripts (so called mirtrons) [Citation109] or via RNA polymerase II transcription of dedicated genes (primary miRNAs or pri-miRNAs) [Citation110]. In the latter case, the precursors undergo 5ʹ capping and 3ʹ polyadenylation similar to mRNAs [Citation111] but are characterized by a unique feature, the presence of a ~ 70 nucleotides long RNA stem-loop. This structure is recognized by the microprocessor, a multiprotein complex including DGCR8, which mediates the association with the stem-loop [Citation112], and Drosha, which processes pri-miRNAs into precursor miRNAs or pre-miRNAs [Citation113]. The resulting pre-miRNAs and the mirtrons, whose synthesis instead does not require the microprocessor, are then exported from the nucleus in an exporting-5 dependent manner [Citation114]. Once in the cytoplasm, they are further processed by the RNAse III enzyme Dicer into mature miRNAs [Citation115], which subsequently interact with the RNA-induced silencing complex (RISC), where the miRNA-mRNA interaction and the Argonaute-dependent cleavage of the mRNA occur [Citation116].
The miRNA biogenesis pathway is regulated by the p53 family members at various levels (). The first of such regulatory events to be unveiled was the interaction between p53 and the microprocessor component DDX5 to promote the maturation of tumour suppressive miRNAs in response to DNA damage [Citation117]. In similar conditions, p53 also affects the RISC complex through its binding to Ago2, hence globally perturbing the miRNA-mRNA interactions [Citation118]. Mutant p53 proteins were demonstrated to retain the capability to associate with both DDX5 and Ago2 and to highjack them as part of the so-called mutant p53 gain of function [Citation117,Citation118].
In addition to p53, other members of the family are involved in miRNA processing. For example, TAp63 was shown to induce Dicer expression [Citation43]. This is particularly relevant for tumorigenesis, since the tumour and metastatic suppressive functions of TAp63 rely on this mechanism [Citation43] and deletion of either TAp63 [Citation43] or Dicer [Citation119] predisposes mice to the onset of metastatic tumours. This crucial link between TAp63 and Dicer is further supported by the finding that the loss of both factors is a common feature of different types of aggressive human tumours, including breast cancers, head and neck squamous cell carcinomas, and lung adenocarcinomas [Citation43]. Importantly, in addition to having a global effect on miRNA biogenesis through Dicer, TAp63 achieves its tumour suppressive functions by directly inducing the expression of specific miRNAs, including miR-130b that abolishes the migratory and invasive potential of cancer cells [Citation43].
The other isoform of p63, ΔNp63, transcriptionally activates DGCR8 [Citation50], affecting in this way the cleavage of pri-miRNAs into pre-miRNAs. Notably, the ΔNp63/DGCR8 axis is necessary for the maturation of a group of miRNAs required for the proper terminal differentiation of the epidermis, thereby explaining the skin defects observed in the ΔNp63−/− mice [Citation50]. Because of the pleiotropic roles of ΔNp63 in the initiation and progression of multiple human cancer types [Citation120], the ΔNp63/DGCR8 axis is crucial in human tumours as well. Indeed, tumours overexpressing ΔNp63 are generally addicted to this oncogene and can be treated by targeting ΔNp63 either genetically [Citation40] or pharmacologically via histone deacetylase inhibitors (HDACi) based therapies [Citation41]. Resistance to HDACi treatment due to low levels of ΔNp63’s E3 ubiquitin ligase, Fbxw7, can be bypassed by directly targeting DGCR8 or oncogenic miRNAs processed in a ΔNp63/DGCR8-dependent manner, such as let-7d and miR-128 [Citation41].
In addition to control miRNA biogenesis, the p53 family can directly affect the expression of specific miRNAs (). One of the best characterized examples is the induction of miR-34 by p53 in response to DNA damaging agents, such as ionizing radiations [Citation121] and chemotherapeutic drugs [Citation117]. This induction is essential for the p53-dependent cell cycle arrest and senescence [Citation121], during which p53 also represses the expression of several miRNAs, including miR-17-5p, miR-106b, and miR-155 [Citation122]. P53-induced senescence is counteracted by ΔNp63 both through: i) the repression of senescence-specific miRNAs, such as miR-181 and miR-130b [Citation123], the latter being instead induced by TAp63 [Citation43]; and ii) the induction of pro-proliferative miRNAs, like miR-630 [Citation124]. Furthermore, ΔNp63 regulates the expression of the miR-200 family and miR-205, through which ΔNp63 promotes the epithelial-mesenchymal transition (EMT) [Citation125]. This is crucial for the development of epithelial tissues such as the mammary gland [Citation126] and for the pro-metastatic activity of ΔNp63 in bladder [Citation127] and prostate [Citation128] cancers.
Table 1. Connections between miRNAs and the p53 family.
One intriguing miRNA induced by TAp73 is miR-193b, which in turn binds to the 3ʹUTR of TP73, thus inducing a negative feedback loop that keeps TAp73 activity under check [Citation129]. This is not the only miRNA directly regulating the stability of the p53 family mRNAs. Additional examples are the oncogenic miR-125b, that inhibits p53 activation by interacting with its 3ʹ UTR [Citation130], and the tumour suppressive miR-203, which inhibits the ΔNp63-dependent proliferation of cancer cells by binding to its 3ʹ UTR [Citation131]. These findings demonstrate that the intricate crosstalk between the p53 family and miRNAs acts in both directions and has crucial repercussions for human cancers.
LncRNAs and the activity of the p53 family members in human tumours
The vast majority of the ‘dark matter’ comprises long non-coding RNAs (lncRNAs), RNA species arbitrarily defined as RNA polymerase II transcripts that are longer than 200 nucleotides and devoid of open reading frames [Citation132]. Due to the ever-growing identification of the lncRNAs thanks to the advances in RNA sequencing techniques, lncRNAs have been believed to represent transcriptional noise [Citation133]. However, a small but steadily growing list of lncRNAs has been confirmed to have biological roles, including modulation of gene expression (both in cis [Citation134] and in trans [Citation135]), mRNA stability control [Citation136], sequestration of miRNAs [Citation137], regulation of protein localization [Citation138], and organization of scaffolds both for RNA binding proteins [Citation139] and for subnuclear domains [Citation140].
Several lncRNAs have recently been identified as key components of the p53 pathway (), including known oncogenic lncRNAs that act as upstream modulators of p53, such as ANRIL, MALAT1, PURPL, and PVT1. The former is also known as CDKN2B-AS1, because it is transcribed from the same promoter but on the opposite strand compared to CDKN2B (also known as ARF) [Citation141]. During the last stage of the DNA damage response, i.e. when the DNA repair is completed and p53 levels return to normal, ANRIL directly binds to the nascent ARF transcript and recruits polycomb repressor complex (PRC) 1 and 2 to silence ARF expression [Citation142]. As a consequence of the reduced ARF levels, MDM2 is free to interact with p53 and to drive its degradation [Citation143]. In several human tumours, including breast [Citation142], lung [Citation144], and ovarian [Citation145] cancers, ANRIL is overexpressed thereby leading to the hypoactivation of p53. A similar result is obtained in tumours where cMYC is coamplified with the lncRNA PVT1 [Citation146]. This RNA promotes the loading of EZH2 on the promoter of the large tumour suppressor kinase 2 (LATS2) [Citation147], the kinase at the core of the Hippo pathway responsible for the phosphorylation and subsequent inactivation of YAP/TAZ [Citation148]. Similar to ARF, LATS2 also counteracts MDM2 binding to p53 [Citation149]. Therefore, PVT1 overexpression results in reduced LATS2 levels and increased MDM2-dependent degradation of p53 [Citation147]. In addition to regulating p53 ubiquitination levels via MDM2, there are lncRNAs affecting other p53 posttranslational modifications (PTMs). This is the case for MALAT1, an oncogenic lncRNA enhancing the deacetylation activity of SIRT1 on p53, thus reducing the ability of p53 to be recruited to promoters of its target genes [Citation150]. Besides these effects on p53 PTMs, another lncRNA, named WRAP53, can instead stabilize p53 post-transcriptionally. Indeed, this lncRNA is an antisense transcript of TP53, which binds to the 5ʹ UTR of the p53 mRNA via a perfectly complementary sequence and promotes p53 mRNA translation and the subsequent accumulation of the p53 protein in response to DNA damage [Citation151]. Finally, a special case of upstream modulator of p53 is that of PURPL. Indeed, not only does this lncRNA prevent the interaction between p53 and MYBBP1A thus counteracting p53 stabilization, but PURPL itself is a direct target of p53 [Citation152]. This creates a negative feedback loop between PURPL and p53, which is an important mechanism to avoid the hyperactivation of p53 in unstressed conditions [Citation152]. p53 has also been shown to induce the expression of other lncRNAs that act as downstream tuners of different p53’s biological responses, such as apoptosis (PINCR) [Citation153], cell cycle arrest (DINO) [Citation154], DNA repair (GUARDIN) [Citation155], and senescence (PANDA) [Citation156].
Table 2. Connections between lncRNAs and the p53 family.
In contrast to the large body of evidence linking p53 and lncRNAs, only a handful of reports have clearly demonstrated an interplay between these RNA species and the other members of the p53 family. Notable examples are SNHG1 [Citation157], which inhibits TAp63 via an uncharacterized mechanism leading to metastatic lung squamous cell carcinomas, and BLNCR [Citation158] and XIAP-AS1 [Citation159], two direct targets of ΔNp63 mediating its proliferative and invasive effects, respectively. Given the ever-growing interest in the lncRNA field, it is very likely that additional and surprising connections between lncRNAs and the p53 family will be unveiled in the near future.
Circular RNAs and their effects on the p53 pathway
In 1976, curious ‘single-stranded and covalently closed circular RNA molecules’ were described in pathogenic viroids [Citation160]. Since then, circular RNAs (circRNAs) have been discovered in most organisms, including Archaea, plants, and metazoans [Citation10]. Although initially disregarded as artefacts of splicing errors [Citation161], RNA-sequencing methods including the depletion of polyadenylated RNAs and the degradation of linear RNA molecules via RNAse R, revealed that ~10% of all the expressed human genes produce circRNA splice variants [Citation162]. Notably, in several instances, the levels of the circRNA isoforms exceed those of the respective linear transcripts [Citation163], hence confuting the linear RNA-centric view that circRNAs are just ‘scrambled exons’[Citation161]. The first functional circRNA to be reported was CDR1as [Citation164], also known as ciRS-7 [Citation165], which is the archetype for miRNA-sponging circRNAs. It indeed contains more than 70 binding sites for miR-7 and suppresses the activity of this miRNA. In addition to the inactivation of miRNAs, circRNAs can exert a variety of mechanisms of function, such as affecting protein localization [Citation166], regulating the transcription of their parental gene [Citation167], and acting as scaffolds for the formation of protein complexes [Citation168].
Intriguingly, a few circRNAs were demonstrated to encode small peptides, thus indicting that not every circRNA is an actual non-coding RNA. One of the peptide-encoding circRNAs is the circular form of the p53-induced transcript PINT, whose peptide acts as a tumour suppressor in glioblastomas by binding to PAF1c, an RNA polymerase II associated factor, and inhibiting the transcriptional elongation of multiple oncogenic transcripts [Citation169]. Another interesting circRNA is the circular form of the lncRNA ANRIL. While, as described above, linear ANRIL affects p53 stability via ARF [Citation141], circANRIL expression impairs rRNA processing and maturation, ultimately leading to nucleolar stress and p53 activation [Citation170]. The activation of p53 is also affected by other circRNAs, including: i) circ_0000263, which sponges miR-150-5p, in turn causing the up-regulation of the p53 inhibitor MDM4 [Citation171]; ii) circ_0055538, whose loss in oral squamous cell carcinoma attenuates p53’s pro-apoptotic response [Citation172]; and iii) circAMOTL1L, whose downregulation promotes prostate cancer progression by impairing the p53-dependent regulation of EMT [Citation173].
Despite the numerous reports demonstrating the functional interactions between circRNAs and the p53 pathway (), further efforts are still needed to show whether circRNAs can also affect the stability or the activity of the remaining members of the p53 family and what consequences this interplay may have in human cancers and other biological processes.
Table 3. Connections between circRNAs and the p53 family.
Conclusions and future perspectives
The world of ncRNAs is ever changing and leading to new venues of research and holds the promise of novel therapies for multiple diseases including cancer. Though initially shadowed by the interest captured by the protein-coding RNAs, the ‘dark matter’ has undeniably proven to comprise molecules with biological functions crucial for both cell physiology and several human diseases. These findings have provided fuel for the investigation in the ncRNA field to the point that it is now clear how intertwined the ncRNAs are with the components of one of the most important pathways in human cancers, the p53 pathway. Indeed, not only are ncRNAs at the centre of the phenotypes shown by the mouse models lacking specific isoforms of the p53 family members, as in the case of the skin defects of the ΔNp63−/− mice [Citation50] or the tumour predisposition of the TAp63−/− mice [Citation43], but small molecules targeting ncRNA biogenesis (as CX-5461 [Citation85] and hernandonine [Citation87]) and UPR and/or proteasome inhibitors [Citation107] exert their anti-tumour effects via the activation of the pro-apoptotic members of the p53 family.
In addition to these pharmacological approaches affecting global ncRNA biogenesis, several strategies have been designed to target specific ncRNAs via complementary base-pairing recognition, as provided by antisense oligonucleotides (ASOs) [Citation174], miRNA inhibitors [Citation175], and siRNAs [Citation176]. To guarantee their stability and delivery, these molecules are chemically modified (e.g. cholesterol [Citation177] and N-acetylgalactosamine [Citation178]) and/or loaded on either liposomal, polymeric, or inorganic nanoparticles [Citation179]. Many of these ncRNA-based therapeutics showed significant anti-cancer effects in in vivo models and are currently being tested in early clinical trial phases for the treatment of both solid tumours and haematological malignancies [Citation180].
Despite the constant progress made in the ncRNA field, there are still some uncharted territory and unanswered questions worthy of further investigation. For example, are there any lncRNAs or circRNAs whose levels or functions are affected by either TAp73 or ΔNp73? How successful will therapeutic strategies aiming to stabilize p53 by blocking lncRNAs, like ANRIL [Citation142], MALAT1 [Citation150], and PVT1 [Citation147] be? Furthermore, given that the inactivation of p53 and its downstream pathway as well as the overexpression of the oncogenic p53 family members, ΔNp63 and ΔNp73, are among the most common alterations in human cancers [Citation21,Citation42], are these ncRNA inhibitors as well as those currently under investigation effective in treating these tumours? We are certain that the paths leading to the answers of these questions are filled with novel and exciting ncRNA functions and will cast an everlasting light shining upon the world of the ‘dark matter’.
Figure 1. The p53 family modulates the nucleolar stress response. Nucleolar structure and function are impaired by multiple stressors, including inhibitors of ribosomal DNA transcription such as CX-5461 and hernandonine. These compounds induce p53 stabilization, which relies on its interaction with PML and subsequent p300-mediated acetylation. Once activated, p53 counteracts the ΔNp63-induced transcription of ribosomal genes and prompts cell death in concert with the other pro-apoptotic members of the family, TAp63 and TAp73.
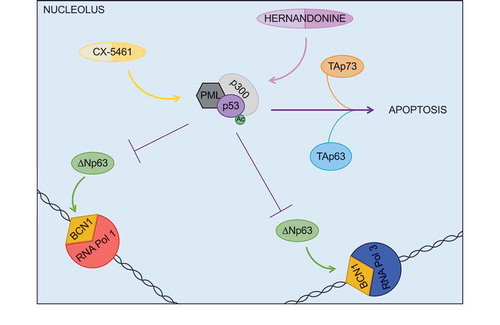
Figure 2. TAp73 controls global protein synthesis via the translation of ribosomal proteins. In physiological conditions, TAp73 promotes the translation of mRNAs encoding ribosomal proteins (RPs), which in turn are required to sustain global protein synthesis. When nucleolar stress occurs, instead, TAp73 forestalls the production of RPs thus ultimately halting global protein synthesis.
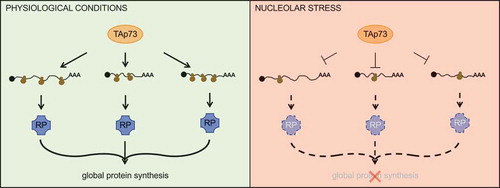
Figure 3. miR-34a inhibits the tRNA initiator tRNAMet. Following either endogenous (e.g. oncogene activation) or exogenous (e.g. DNA damaging agents) stimuli, p53 is activated and promotes the expression of miR-34a, which is the only miRNA known to target tRNAs. Specifically, miR-34a binds to tRNAMet and prevents it from promoting biological processes supporting tumour formation and progression.
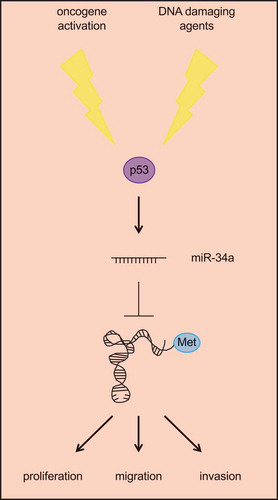
Figure 4. The p53 family regulates multiple layers of the miRNA biogenesis pathway. In addition to directly regulating the expression of miRNAs (see ), p53 and its family members, TAp63 and ΔNp63, affect miRNA processing as well as the activity of miRNAs on their mRNA targets.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- International Human Genome Sequencing C. Finishing the euchromatic sequence of the human genome. Nature. 2004;431:931–945.
- Lander ES, Linton LM, Birren B, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921.
- Venter JC, Adams MD, Myers EW, et al. The sequence of the human genome. Science. 2001;291:1304–1351.
- Hangauer MJ, Vaughn IW, McManus MT. Pervasive transcription of the human genome produces thousands of previously unidentified long intergenic noncoding RNAs. PLoS Genet. 2013;9:e1003569.
- Kapranov P, St Laurent G, Raz T, et al. The majority of total nuclear-encoded non-ribosomal RNA in a human cell is ‘dark matter’ un-annotated RNA. BMC Biol. 2010;8:149.
- Jankowsky E, Harris ME. Specificity and nonspecificity in RNA-protein interactions. Nat Rev Mol Cell Biol. 2015;16:533–544.
- Weick EM, Miska EA. piRNAs: from biogenesis to function. Development. 2014;141:3458–3471.
- Michlewski G, Caceres JF. Post-transcriptional control of miRNA biogenesis. RNA. 2019;25:1–16.
- Yao RW, Wang Y, Chen LL. Cellular functions of long noncoding RNAs. Nat Cell Biol. 2019;21:542–551.
- Patop IL, Wust S, Kadener S. Past, present, and future of circRNAs. Embo J. 2019;38(16):e100836.
- Chatterjee M, Sengupta S. Emerging roles of long non-coding RNAs in cancer. J Biosci. 2019;44:22.
- Dragomir M, Calin GA. Circular RNAs in cancer - lessons learned from microRNAs. Front Oncol. 2018;8:179.
- Hata A, Kashima R. Dysregulation of microRNA biogenesis machinery in cancer. Crit Rev Biochem Mol Biol. 2016;51:121–134.
- Huang SQ, Sun B, Xiong ZP, et al. The dysregulation of tRNAs and tRNA derivatives in cancer. J Exp Clin Cancer Res. 2018;37:101.
- Nguyen le XT, Raval A, Garcia JS, et al. Regulation of ribosomal gene expression in cancer. J Cell Physiol. 2015;230:1181–1188.
- Goeman F, Strano S, Blandino G. MicroRNAs as key effectors in the p53 network. Int Rev Cell Mol Biol. 2017;333:51–90.
- Kaiser AM, Attardi LD. Deconstructing networks of p53-mediated tumor suppression in vivo. Cell Death Differ. 2018;25:93–103.
- Lin T, Hou PF, Meng S, et al. Emerging roles of p53 related lncRNAs in cancer progression: a systematic review. Int J Biol Sci. 2019;15:1287–1298.
- Napoli M, Flores ER. The family that eats together stays together: new p53 family transcriptional targets in autophagy. Genes Dev. 2013;27:971–974.
- Napoli M, Flores ER. The p53 family orchestrates the regulation of metabolism: physiological regulation and implications for cancer therapy. Br J Cancer. 2017;116:149–155.
- Kandoth C, McLellan MD, Vandin F, et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502:333–339.
- Donehower LA, Harvey M, Slagle BL, et al. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature. 1992;356:215–221.
- Jacks T, Remington L, Williams BO, et al. Tumor spectrum analysis in p53-mutant mice. Curr Biol. 1994;4:1–7.
- Lang GA, Iwakuma T, Suh YA, et al. Gain of function of a p53 hot spot mutation in a mouse model of Li-Fraumeni syndrome. Cell. 2004;119:861–872.
- Olive KP, Tuveson DA, Ruhe ZC, et al. Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell. 2004;119:847–860.
- Lane DP. Cancer. p53, guardian of the genome. Nature. 1992;358:15–16.
- Flores ER, Sengupta S, Miller JB, et al. Tumor predisposition in mice mutant for p63 and p73: evidence for broader tumor suppressor functions for the p53 family. Cancer Cell. 2005;7:363–373.
- Flores ER, Tsai KY, Crowley D, et al. p63 and p73 are required for p53-dependent apoptosis in response to DNA damage. Nature. 2002;416:560–564.
- Pflaum J, Schlosser S, Muller M. p53 family and cellular stress responses in cancer. Front Oncol. 2014;4:285.
- Qian Y, Chen X. Senescence regulation by the p53 protein family. Methods Mol Biol. 2013;965:37–61.
- Lin YL, Sengupta S, Gurdziel K, et al. p63 and p73 transcriptionally regulate genes involved in DNA repair. PLoS Genet. 2009;5:e1000680.
- Prokhorova EA, Zamaraev AV, Kopeina GS, et al. Role of the nucleus in apoptosis: signaling and execution. Cell Mol Life Sci. 2015;72:4593–4612.
- Mills AA, Zheng B, Wang XJ, et al. p63 is a p53 homologue required for limb and epidermal morphogenesis. Nature. 1999;398:708–713.
- Yang A, Schweitzer R, Sun D, et al. p63 is essential for regenerative proliferation in limb, craniofacial and epithelial development. Nature. 1999;398:714–718.
- Yang A, Walker N, Bronson R, et al. p73-deficient mice have neurological, pheromonal and inflammatory defects but lack spontaneous tumours. Nature. 2000;404:99–103.
- Chen Y, Peng Y, Fan S, et al. A double dealing tale of p63: an oncogene or a tumor suppressor. Cell Mol Life Sci. 2018;75:965–973.
- Joruiz SM, Bourdon JC. p53 isoforms: key regulators of the cell fate decision. Cold Spring Harb Perspect Med. 2016;6:a026039.
- Vikhreva P, Melino G, Amelio I. p73 alternative splicing: exploring a biological role for the C-terminal isoforms. J Mol Biol. 2018;430:1829–1838.
- Candi E, Dinsdale D, Rufini A, et al. TAp63 and DeltaNp63 in cancer and epidermal development. Cell Cycle. 2007;6:274–285.
- Venkatanarayan A, Raulji P, Norton W, et al. IAPP-driven metabolic reprogramming induces regression of p53-deficient tumours in vivo. Nature. 2015;517:626–630.
- Napoli M, Venkatanarayan A, Raulji P, et al. DeltaNp63/DGCR8-dependent micrornas mediate therapeutic efficacy of HDAC inhibitors in cancer. Cancer Cell. 2016;29:874–888.
- Orzol P, Holcakova J, Nekulova M, et al. The diverse oncogenic and tumour suppressor roles of p63 and p73 in cancer: a review by cancer site. Histol Histopathol. 2015;30:503–521.
- Su X, Chakravarti D, Cho MS, et al. TAp63 suppresses metastasis through coordinate regulation of Dicer and miRNAs. Nature. 2010;467:986–990.
- Tomasini R, Tsuchihara K, Wilhelm M, et al. TAp73 knockout shows genomic instability with infertility and tumor suppressor functions. Genes Dev. 2008;22:2677–2691.
- Su X, Paris M, Gi YJ, et al. TAp63 prevents premature aging by promoting adult stem cell maintenance. Cell Stem Cell. 2009;5:64–75.
- Su X, Napoli M, Abbas HA, et al. TAp63 suppresses mammary tumorigenesis through regulation of the Hippo pathway. Oncogene. 2017;36:2377–2393.
- Su X, Gi YJ, Chakravarti D, et al. TAp63 is a master transcriptional regulator of lipid and glucose metabolism. Cell Metab. 2012;16:511–525.
- Napoli M, Flores ER. Unifying the p73 knockout phenotypes: tAp73 orchestrates multiciliogenesis. Genes Dev. 2016;30:1253–1254.
- Nemajerova A, Kramer D, Siller SS, et al. TAp73 is a central transcriptional regulator of airway multiciliogenesis. Genes Dev. 2016;30:1300–1312.
- Chakravarti D, Su X, Cho MS, et al. Induced multipotency in adult keratinocytes through down-regulation of DeltaNp63 or DGCR8. Proc Natl Acad Sci U S A. 2014;111:E572–81.
- Romano RA, Smalley K, Magraw C, et al. DeltaNp63 knockout mice reveal its indispensable role as a master regulator of epithelial development and differentiation. Development. 2012;139:772–782.
- Restelli M, Lopardo T, Lo Iacono N, et al. DLX5, FGF8 and the Pin1 isomerase control DeltaNp63alpha protein stability during limb development: a regulatory loop at the basis of the SHFM and EEC congenital malformations. Hum Mol Genet. 2014;23:3830–3842.
- Tissir F, Ravni A, Achouri Y, et al. DeltaNp73 regulates neuronal survival in vivo. Proc Natl Acad Sci U S A. 2009;106:16871–16876.
- Wilhelm MT, Rufini A, Wetzel MK, et al. Isoform-specific p73 knockout mice reveal a novel role for delta Np73 in the DNA damage response pathway. Genes Dev. 2010;24:549–560.
- Bohnsack KE, Bohnsack MT. Uncovering the assembly pathway of human ribosomes and its emerging links to disease. Embo J. 2019;38:e100278.
- Boisvert FM, van Koningsbruggen S, Navascues J, et al. The multifunctional nucleolus. Nat Rev Mol Cell Biol. 2007;8:574–585.
- Nerurkar P, Altvater M, Gerhardy S, et al. Eukaryotic ribosome assembly and nuclear export. Int Rev Cell Mol Biol. 2015;319:107–140.
- Mayer C, Grummt I. Ribosome biogenesis and cell growth: mTOR coordinates transcription by all three classes of nuclear RNA polymerases. Oncogene. 2006;25:6384–6391.
- Wang H, Zhao LN, Li KZ, et al. Overexpression of ribosomal protein L15 is associated with cell proliferation in gastric cancer. BMC Cancer. 2006;6:91.
- Wang M, Hu Y, Stearns ME. RPS2: a novel therapeutic target in prostate cancer. J Exp Clin Cancer Res. 2009;28:6.
- Derenzini E, Rossi A, Trere D. Treating hematological malignancies with drugs inhibiting ribosome biogenesis: when and why. J Hematol Oncol. 2018;11:75.
- Yang K, Yang J, Yi J. Nucleolar Stress: hallmarks, sensing mechanism and diseases. Cell Stress. 2018;2:125–140.
- Deisenroth C, Franklin DA, Zhang Y. The evolution of the ribosomal protein-MDM2-p53 pathway. Cold Spring Harb Perspect Med. 2016;6:a026138.
- Dai MS, Lu H. Inhibition of MDM2-mediated p53 ubiquitination and degradation by ribosomal protein L5. J Biol Chem. 2004;279:44475–44482.
- Lohrum MA, Ludwig RL, Kubbutat MH, et al. Regulation of HDM2 activity by the ribosomal protein L11. Cancer Cell. 2003;3:577–587.
- Zhang Y, Wolf GW, Bhat K, et al. Ribosomal protein L11 negatively regulates oncoprotein MDM2 and mediates a p53-dependent ribosomal-stress checkpoint pathway. Mol Cell Biol. 2003;23:8902–8912.
- Bernardi R, Scaglioni PP, Bergmann S, et al. PML regulates p53 stability by sequestering Mdm2 to the nucleolus. Nat Cell Biol. 2004;6:665–672.
- Takagi M, Absalon MJ, McLure KG, et al. Regulation of p53 translation and induction after DNA damage by ribosomal protein L26 and nucleolin. Cell. 2005;123:49–63.
- Ofir-Rosenfeld Y, Boggs K, Michael D, et al. Mdm2 regulates p53 mRNA translation through inhibitory interactions with ribosomal protein L26. Mol Cell. 2008;32:180–189.
- White RJ. RNA polymerases I and III, non-coding RNAs and cancer. Trends Genet. 2008;24:622–629.
- Oskarsson T, Trumpp A. The Myc trilogy: lord of RNA polymerases. Nat Cell Biol. 2005;7:215–217.
- Golomb L, Bublik DR, Wilder S, et al. Importin 7 and exportin 1 link c-Myc and p53 to regulation of ribosomal biogenesis. Mol Cell. 2012;45:222–232.
- Bursac S, Brdovcak MC, Donati G, et al. Activation of the tumor suppressor p53 upon impairment of ribosome biogenesis. Biochim Biophys Acta. 2014;1842:817–830.
- Panic L, Tamarut S, Sticker-Jantscheff M, et al. Ribosomal protein S6 gene haploinsufficiency is associated with activation of a p53-dependent checkpoint during gastrulation. Mol Cell Biol. 2006;26:8880–8891.
- Barkic M, Crnomarkovic S, Grabusic K, et al. The p53 tumor suppressor causes congenital malformations in Rpl24-deficient mice and promotes their survival. Mol Cell Biol. 2009;29:2489–2504.
- Anderson SJ, Lauritsen JP, Hartman MG, et al. Ablation of ribosomal protein L22 selectively impairs alphabeta T cell development by activation of a p53-dependent checkpoint. Immunity. 2007;26:759–772.
- Terzian T, Dumble M, Arbab F, et al. Rpl27a mutation in the sooty foot ataxia mouse phenocopies high p53 mouse models. J Pathol. 2011;224:540–552.
- Marini A, Rotblat B, Sbarrato T, et al. TAp73 contributes to the oxidative stress response by regulating protein synthesis. Proc Natl Acad Sci U S A. 2018;115:6219–6224.
- Rotblat B, Agostini M, Niklison-Chirou MV, et al. Sustained protein synthesis and reduced eEF2K levels in TAp73(-\-) mice brain: a possible compensatory mechanism. Cell Cycle. 2018;17:2637–2643.
- Boldrup L, Coates PJ, Laurell G, et al. p63 Transcriptionally regulates BNC1, a Pol I and Pol II transcription factor that regulates ribosomal biogenesis and epithelial differentiation. Eur J Cancer. 2012;48:1401–1406.
- Wang J, Zhang S, Schultz RM, et al. Search for basonuclin target genes. Biochem Biophys Res Commun. 2006;348:1261–1271.
- Zhang S, Wang J, Tseng H. Basonuclin regulates a subset of ribosomal RNA genes in HaCaT cells. PLoS One. 2007;2:e902.
- Cui C, Elsam T, Tian Q, et al. Gli proteins up-regulate the expression of basonuclin in basal cell carcinoma. Cancer Res. 2004;64:5651–5658.
- Brighenti E, Trere D, Derenzini M. Targeted cancer therapy with ribosome biogenesis inhibitors: a real possibility? Oncotarget. 2015;6:38617–38627.
- Drygin D, Lin A, Bliesath J, et al. Targeting RNA polymerase I with an oral small molecule CX-5461 inhibits ribosomal RNA synthesis and solid tumor growth. Cancer Res. 2011;71:1418–1430.
- Bywater MJ, Poortinga G, Sanij E, et al. Inhibition of RNA polymerase I as a therapeutic strategy to promote cancer-specific activation of p53. Cancer Cell. 2012;22:51–65.
- Chen YT, Chen JJ, Wang HT. Targeting RNA polymerase I with hernandonine inhibits ribosomal RNA synthesis and tumor cell growth. Mol Cancer Res. 2019;17:2294–2305.
- Hoagland MB, Stephenson ML, Scott JF, et al. A soluble ribonucleic acid intermediate in protein synthesis. J Biol Chem. 1958;231:241–257.
- Goodarzi H, HCB N, Zhang S, et al. Modulated expression of specific tRNAs drives gene expression and cancer progression. Cell. 2016;165:1416–1427.
- Santos M, Fidalgo A, Varanda AS, et al. tRNA deregulation and its consequences in cancer. Trends Mol Med. 2019;25(10):853–865.
- Dittmar KA, Goodenbour JM, Pan T. Tissue-specific differences in human transfer RNA expression. PLoS Genet. 2006;2:e221.
- Ishimura R, Nagy G, Dotu I, et al. RNA function. Ribosome stalling induced by mutation of a CNS-specific tRNA causes neurodegeneration. Science. 2014;345:455–459.
- Sriskanthadevan-Pirahas S, Deshpande R, Lee B, et al. Ras/ERK-signalling promotes tRNA synthesis and growth via the RNA polymerase III repressor Maf1 in Drosophila. PLoS Genet. 2018;14:e1007202.
- Kantidakis T, Ramsbottom BA, Birch JL, et al. mTOR associates with TFIIIC, is found at tRNA and 5S rRNA genes, and targets their repressor Maf1. Proc Natl Acad Sci U S A. 2010;107:11823–11828.
- Khattar E, Kumar P, Liu CY, et al. Telomerase reverse transcriptase promotes cancer cell proliferation by augmenting tRNA expression. J Clin Invest. 2016;126:4045–4060.
- Wang B, Li D, Kovalchuk I, et al. miR-34a directly targets tRNAi(Met) precursors and affects cellular proliferation, cell cycle, and apoptosis. Proc Natl Acad Sci U S A. 2018;115:7392–7397.
- Birch J, Clarke CJ, Campbell AD, et al. The initiator methionine tRNA drives cell migration and invasion leading to increased metastatic potential in melanoma. Biol Open. 2016;5:1371–1379.
- Zhang Z, Ye Y, Gong J, et al. Global analysis of tRNA and translation factor expression reveals a dynamic landscape of translational regulation in human cancers. Commun Biol. 2018;1:234.
- Kuang M, Zheng D, Tao X, et al. tRNA-based prognostic score in predicting survival outcomes of lung adenocarcinomas. Int J Cancer. 2019;145:1982–1990.
- Yang JR, Chen X, Zhang J. Codon-by-codon modulation of translational speed and accuracy via mRNA folding. PLoS Biol. 2014;12:e1001910.
- Santos M, Pereira PM, Varanda AS, et al. Codon misreading tRNAs promote tumor growth in mice. RNA Biol. 2018;15:773–786.
- Ribas de Pouplana L, Santos MA, Zhu JH, et al. Protein mistranslation: friend or foe? Trends Biochem Sci. 2014;39:355–362.
- Hetz C, Papa FR. The unfolded protein response and cell fate control. Mol Cell. 2018;69:169–181.
- Stavridi ES, Halazonetis TD. p53 and stress in the ER. Genes Dev. 2004;18:241–244.
- Pyati UJ, Gjini E, Carbonneau S, et al. p63 mediates an apoptotic response to pharmacological and disease-related ER stress in the developing epidermis. Dev Cell. 2011;21:492–505.
- Ramadan S, Terrinoni A, Catani MV, et al. p73 induces apoptosis by different mechanisms. Biochem Biophys Res Commun. 2005;331:713–717.
- Hetz C, Axten JM, Patterson JB. Pharmacological targeting of the unfolded protein response for disease intervention. Nat Chem Biol. 2019;15:764–775.
- Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–233.
- Ruby JG, Jan CH, Bartel DP. Intronic microRNA precursors that bypass drosha processing. Nature. 2007;448:83–86.
- Lee Y, Kim M, Han J, et al. MicroRNA genes are transcribed by RNA polymerase II. Embo J. 2004;23:4051–4060.
- Cai X, Hagedorn CH, Cullen BR. Human microRNAs are processed from capped, polyadenylated transcripts that can also function as mRNAs. RNA. 2004;10:1957–1966.
- Han J, Lee Y, Yeom KH, et al. Molecular basis for the recognition of primary microRNAs by the drosha-DGCR8 complex. Cell. 2006;125:887–901.
- Lee Y, Ahn C, Han J, et al. The nuclear RNase III drosha initiates microRNA processing. Nature. 2003;425:415–419.
- Yi R, Qin Y, Macara IG, et al. Exportin-5 mediates the nuclear export of pre-microRNAs and short hairpin RNAs. Genes Dev. 2003;17:3011–3016.
- Bernstein E, Caudy AA, Hammond SM, et al. Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature. 2001;409:363–366.
- Gregory RI, Chendrimada TP, Cooch N, et al. Human RISC couples microRNA biogenesis and posttranscriptional gene silencing. Cell. 2005;123:631–640.
- Suzuki HI, Yamagata K, Sugimoto K, et al. Modulation of microRNA processing by p53. Nature. 2009;460:529–533.
- Krell J, Stebbing J, Frampton AE, et al. The role of TP53 in miRNA loading onto AGO2 and in remodelling the miRNA-mRNA interaction network. Lancet. 2015;385(Suppl 1):S15.
- Kumar MS, Pester RE, Chen CY, et al. Dicer1 functions as a haploinsufficient tumor suppressor. Genes Dev. 2009;23:2700–2704.
- Abbas HA, Bui NHB, Rajapakshe K, et al. Distinct TP63 isoform-driven transcriptional signatures predict tumor progression and clinical outcomes. Cancer Res. 2018;78:451–462.
- He L, He X, Lim LP, et al. A microRNA component of the p53 tumour suppressor network. Nature. 2007;447:1130–1134.
- Brosh R, Shalgi R, Liran A, et al. p53-repressed miRNAs are involved with E2F in a feed-forward loop promoting proliferation. Mol Syst Biol. 2008;4:229.
- Rivetti Di Val Cervo P, Lena AM, Nicoloso, et al. p63-microRNA feedback in keratinocyte senescence. Proc Natl Acad Sci U S A. 2012;109:1133–1138.
- Huang Y, Kesselman D, Kizub D, et al. Phospho-DeltaNp63alpha/microRNA feedback regulation in squamous carcinoma cells upon cisplatin exposure. Cell Cycle. 2013;12:684–697.
- Gregory PA, Bert AG, Paterson EL, et al. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol. 2008;10:593–601.
- Lu Y, Cao J, Napoli M, et al. miR-205 regulates basal cell identity and stem cell regenerative potential during mammary reconstitution. Stem Cells. 2018;36:1875–1889.
- Tran MN, Choi W, Wszolek MF, et al. The p63 protein isoform DeltaNp63alpha inhibits epithelial-mesenchymal transition in human bladder cancer cells: role of MIR-205. J Biol Chem. 2013;288:3275–3288.
- Tucci P, Agostini M, Grespi F, et al. Loss of p63 and its microRNA-205 target results in enhanced cell migration and metastasis in prostate cancer. Proc Natl Acad Sci U S A. 2012;109:15312–15317.
- Ory B, Ramsey MR, Wilson C, et al. A microRNA-dependent program controls p53-independent survival and chemosensitivity in human and murine squamous cell carcinoma. J Clin Invest. 2011;121:809–820.
- Le MT, Teh C, Shyh-Chang N, et al. MicroRNA-125b is a novel negative regulator of p53. Genes Dev. 2009;23:862–876.
- Lena AM, Shalom-Feuerstein R, Rivetti Di Val Cervo P, et al. miR-203 represses ‘stemness’ by repressing DeltaNp63. Cell Death Differ. 2008;15:1187–1195.
- Mercer TR, Dinger ME, Mattick JS. Long non-coding RNAs: insights into functions. Nat Rev Genet. 2009;10:155–159.
- Ponting CP, Belgard TG. Transcribed dark matter: meaning or myth? Hum Mol Genet. 2010;19:R162–8.
- Wang X, Arai S, Song X, et al. Induced ncRNAs allosterically modify RNA-binding proteins in cis to inhibit transcription. Nature. 2008;454:126–130.
- Kornienko AE, Guenzl PM, Barlow DP, et al. Gene regulation by the act of long non-coding RNA transcription. BMC Biol. 2013;11:59.
- Faghihi MA, Modarresi F, Khalil AM, et al. Expression of a noncoding RNA is elevated in Alzheimer’s disease and drives rapid feed-forward regulation of beta-secretase. Nat Med. 2008;14:723–730.
- Cesana M, Cacchiarelli D, Legnini I, et al. A long noncoding RNA controls muscle differentiation by functioning as a competing endogenous RNA. Cell. 2011;147:358–369.
- Halic M, Becker T, Pool MR, et al. Structure of the signal recognition particle interacting with the elongation-arrested ribosome. Nature. 2004;427:808–814.
- Zappulla DC, Cech TR. Yeast telomerase RNA: a flexible scaffold for protein subunits. Proc Natl Acad Sci U S A. 2004;101:10024–10029.
- Mao YS, Sunwoo H, Zhang B, et al. Direct visualization of the co-transcriptional assembly of a nuclear body by noncoding RNAs. Nat Cell Biol. 2011;13:95–101.
- Yu W, Gius D, Onyango P, et al. Epigenetic silencing of tumour suppressor gene p15 by its antisense RNA. Nature. 2008;451:202–206.
- Meseure D, Vacher S, Alsibai KD, et al. Expression of ANRIL-polycomb complexes-CDKN2A/B/ARF genes in breast tumors: identification of a two-gene (EZH2/CBX7) signature with independent prognostic value. Mol Cancer Res. 2016;14:623–633.
- Li J, Poi MJ, Tsai MD. Regulatory mechanisms of tumor suppressor P16(INK4A) and their relevance to cancer. Biochemistry. 2011;50:5566–5582.
- Lin L, Gu ZT, Chen WH, et al. Increased expression of the long non-coding RNA ANRIL promotes lung cancer cell metastasis and correlates with poor prognosis. Diagn Pathol. 2015;10:14.
- Qiu JJ, Lin YY, Ding JX, et al. Long non-coding RNA ANRIL predicts poor prognosis and promotes invasion/metastasis in serous ovarian cancer. Int J Oncol. 2015;46:2497–2505.
- Tseng YY, Moriarity BS, Gong W, et al. PVT1 dependence in cancer with MYC copy-number increase. Nature. 2014;512:82–86.
- Wan L, Sun M, Liu GJ, et al. Long noncoding RNA PVT1 promotes non-small cell lung cancer cell proliferation through epigenetically regulating LATS2 expression. Mol Cancer Ther. 2016;15:1082–1094.
- Calses PC, Crawford JJ, Lill JR, et al. Hippo pathway in cancer: aberrant regulation and therapeutic opportunities. Trends Cancer. 2019;5:297–307.
- Aylon Y, Michael D, Shmueli A, et al. A positive feedback loop between the p53 and Lats2 tumor suppressors prevents tetraploidization. Genes Dev. 2006;20:2687–2700.
- Chen R, Liu Y, Zhuang H, et al. Quantitative proteomics reveals that long non-coding RNA MALAT1 interacts with DBC1 to regulate p53 acetylation. Nucleic Acids Res. 2017;45:9947–9959.
- Mahmoudi S, Henriksson S, Corcoran M, et al. Wrap53, a natural p53 antisense transcript required for p53 induction upon DNA damage. Mol Cell. 2009;33:462–471.
- Li XL, Subramanian M, Jones MF, et al. Long noncoding RNA PURPL suppresses basal p53 levels and promotes tumorigenicity in colorectal cancer. Cell Rep. 2017;20:2408–2423.
- Chaudhary R, Gryder B, Woods WS, et al. Prosurvival long noncoding RNA PINCR regulates a subset of p53 targets in human colorectal cancer cells by binding to matrin 3. Elife. 2017;6:pii:e23244.
- Schmitt AM, Garcia JT, Hung T, et al. An inducible long noncoding RNA amplifies DNA damage signaling. Nat Genet. 2016;48:1370–1376.
- Hu WL, Jin L, Xu A, et al. GUARDIN is a p53-responsive long non-coding RNA that is essential for genomic stability. Nat Cell Biol. 2018;20:492–502.
- Hung T, Wang Y, Lin MF, et al. Extensive and coordinated transcription of noncoding RNAs within cell-cycle promoters. Nat Genet. 2011;43:621–629.
- Zhang HY, Yang W, Zheng FS, et al. Long non-coding RNA SNHG1 regulates zinc finger E-box binding homeobox 1 expression by interacting with TAp63 and promotes cell metastasis and invasion in Lung squamous cell carcinoma. Biomed Pharmacother. 2017;90:650–658.
- Tanis SEJ, Koksal ES, van Buggenum J, et al. BLNCR is a long non-coding RNA adjacent to integrin beta-1 that is rapidly lost during epidermal progenitor cell differentiation. Sci Rep. 2019;9:31.
- Lu X, Yu Y, Tan S. Long non-coding XIAP-AS1 regulates cell proliferation, invasion and cell cycle in colon cancer. Artif Cells Nanomed Biotechnol. 2019;47:767–775.
- Sanger HL, Klotz G, Riesner D, et al. Viroids are single-stranded covalently closed circular RNA molecules existing as highly base-paired rod-like structures. Proc Natl Acad Sci U S A. 1976;73:3852–3856.
- Nigro JM, Cho KR, Fearon ER, et al. Scrambled exons. Cell. 1991;64:607–613.
- Jeck WR, Sorrentino JA, Wang K, et al. Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA. 2013;19:141–157.
- Salzman J, Gawad C, Wang PL, et al. Circular RNAs are the predominant transcript isoform from hundreds of human genes in diverse cell types. PLoS One. 2012;7:e30733.
- Memczak S, Jens M, Elefsinioti A, et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature. 2013;495:333–338.
- Hansen TB, Jensen TI, Clausen BH, et al. Natural RNA circles function as efficient microRNA sponges. Nature. 2013;495:384–388.
- Yang Q, Du WW, Wu N, et al. A circular RNA promotes tumorigenesis by inducing c-myc nuclear translocation. Cell Death Differ. 2017;24:1609–1620.
- Li Z, Huang C, Bao C, et al. Exon-intron circular RNAs regulate transcription in the nucleus. Nat Struct Mol Biol. 2015;22:256–264.
- Du WW, Yang W, Liu E, et al. Foxo3 circular RNA retards cell cycle progression via forming ternary complexes with p21 and CDK2. Nucleic Acids Res. 2016;44:2846–2858.
- Zhang M, Zhao K, Xu X, et al. A peptide encoded by circular form of LINC-PINT suppresses oncogenic transcriptional elongation in glioblastoma. Nat Commun. 2018;9:4475.
- Holdt LM, Stahringer A, Sass K, et al. Circular non-coding RNA ANRIL modulates ribosomal RNA maturation and atherosclerosis in humans. Nat Commun. 2016;7:12429.
- Cai H, Zhang P, Xu M, et al. Circular RNA hsa_circ_0000263 participates in cervical cancer development by regulating target gene of miR-150-5p. J Cell Physiol. 2019;234:11391–11400.
- Su W, Sun S, Wang F, et al. Circular RNA hsa_circ_0055538 regulates the malignant biological behavior of oral squamous cell carcinoma through the p53/Bcl-2/caspase signaling pathway. J Transl Med. 2019;17:76.
- Yang Z, Qu CB, Zhang Y, et al. Dysregulation of p53-RBM25-mediated circAMOTL1L biogenesis contributes to prostate cancer progression through the circAMOTL1L-miR-193a-5p-Pcdha pathway. Oncogene. 2019;38:2516–2532.
- Amodio N, Stamato MA, Juli G, et al. Drugging the lncRNA MALAT1 via LNA gapmeR ASO inhibits gene expression of proteasome subunits and triggers anti-multiple myeloma activity. Leukemia. 2018;32:1948–1957.
- Janssen HL, Reesink HW, Lawitz EJ, et al. Treatment of HCV infection by targeting microRNA. N Engl J Med. 2013;368:1685–1694.
- Mahmoodi Chalbatani G, Dana H, Gharagouzloo E, et al. Small interfering RNAs (siRNAs) in cancer therapy: a nano-based approach. Int J Nanomedicine. 2019;14:3111–3128.
- Krutzfeldt J, Rajewsky N, Braich R, et al. Silencing of microRNAs in vivo with ‘antagomirs’. Nature. 2005;438:685–689.
- Huang Y. Preclinical and clinical advances of GalNAc-decorated nucleic acid therapeutics. Mol Ther Nucleic Acids. 2017;6:116–132.
- Wang WT, Han C, Sun YM, et al. Noncoding RNAs in cancer therapy resistance and targeted drug development. J Hematol Oncol. 2019;12:55.
- Matsui M, Corey DR. Non-coding RNAs as drug targets. Nat Rev Drug Discov. 2017;16:167–179.