
ABSTRACT
Huntington’s Disease (HD) is a monogenetic neurodegenerative disorder mainly caused by the cytotoxicity of the mutant HTT protein (mHTT) encoded by the mutant HTT gene. Lowering HTT mRNA has been extensively studied as a potential therapeutic strategy, but how its level is regulated endogenously has been unclear. Here we report that the RNA-binding protein (RBP) HuR interacts with and stabilizes HTT mRNA in an mHTT-dependent manner. In HD cells but not wild-type cells, siRNA knockdown or CRISPR-induced heterozygous knockout of HuR decreased HTT mRNA stability. HuR interacted with HTT mRNA at a conserved site in exon 11 rather than the 3ʹ-UTR region of the mRNA. Interestingly, this interaction was dependent on the presence of mHTT, likely via the activation of MAPK11, which enhanced cytosolic localization of the HuR protein. Thus, mHTT, MAPK11 and HuR may form a positive feedback loop that stabilizes HTT mRNA and enhances mHTT accumulation, which may contribute to HD progression. Our data reveal a novel regulatory mechanism of HTT mRNA via non-canonical binding of HuR.
Introduction
Huntington’s disease (HD) is an important monogenetic neurodegenerative disorder mainly caused by the mutation of the HTT gene [Citation1], which encodes the mutant HTT protein (mHTT) with expanded polyglutamine tract (polyQ) [Citation2,Citation3]. mHTT cytotoxicity is the major cause of HD, while the underlying molecular mechanisms remain unclear and likely involve multiple pathways [Citation2]. Recently, gene therapy approaches targeting HTT mRNA have obtained tremendous success in developing potential treatment for HD: in several mammalian models, delivery of short hairpin RNAs (shRNAs) or small interference RNAs (siRNAs) or antisense oligonucleotides (ASOs) targeting HTT mRNA lowered HTT protein levels and attenuated neuropathology and disease-related phenotypes [Citation4–Citation7]; More importantly, an ASO targeting HTT mRNA, HTT-Rx developed by IONIS, has been investigated in clinical studies and obtained preliminary success in Phase I/IIa clinical trial [Citation8]. Meanwhile, how HTT mRNA is regulated endogenously has been less well understood. This mechanism may worth studying because it may provide novel insights into HTT biology and potential therapeutic targets for HD.
The stability of HTT mRNA could be influenced by non-coding RNAs including an anti-sense LncRNA expressed by the reverse strand of the HTT gene [Citation9]. On the other hand, RNA-binding proteins (RBPs) have been shown to modulate the stability of many mRNAs by interacting with them, playing key roles in many neurodegenerative disorders such as ALS/FTD [Citation10]. Thus, whether and which RBPs regulate HTT mRNA is of interest in the HD and the RNA stability research field and yet remains unknown.
We have previously demonstrated MAPK11 as a novel kinase modulator of HTT mRNA stability [Citation11], giving us an entry point to identify potential RBPs regulating HTT mRNAs. In this study, we identified HuR as the RBP that interacts with HTT mRNA and stabilizes it. We further elucidated relevant molecular mechanisms and the binding site, providing novel insights into HTT mRNA regulation and new pathobiology function of HuR.
Results
The RBP HuR is a potential modifier of HTT mRNA levels in HD cells in an mHTT-dependent manner
Our previous studies have shown that MAPK11 modulates the stability of HTT mRNA. To identify the potential downstream RBP(s) that interact with HTT mRNA, we investigated candidate RBPs that may interact with HTT mRNA based on the CLIP-seq data analysed by StarBase (http://starbase.sysu.edu.cn/) [Citation12] (). We then checked the brain expression of this panel of candidate RBPs based BioGPS (http://biogps.org) [Citation13] and selected those brain-expressing RBPs with more than 5 potential HTT mRNA targeting sites for further testing (). We then investigated their potential influences on mHTT levels in immortalized HD patient fibroblasts (Q45 and Q68) by the well-established HTRF (homogeneous time-resolved fluorescence) assay using the 2B7/MW1 antibody pair. Transfection of pooled siRNA targeting ELAVL1 (referred to as ‘HuR’ in the subsequent text to keep consistency with the literature), but not the other RBP expressing genes significantly lowered mHTT levels in both Q45 and Q68 patient fibroblasts (), revealing HuR as a candidate RBP that regulates HTT mRNA.
Figure 1. Miniscreen of potential mHTT mRNA-interacting RBPs that modulate HTT levels.
(A) The list of potential HTT mRNA interacting RBPs based on the CLIP-seq data analysed in StarBase (ref. [Citation12]). These candidates were then checked for brain tissue expression at BioGPS (ref. [Citation13]). The ‘# of target sites’ indicate the number of potential RBP-bound sequences detected in the HTT mRNA. The RBPs that are expressed in the brain with the ‘# of target sites’ >5 were prioritized for further testing (white rows). eIF4AIII was deprioritized since it is a translation initiation factor and may have non-specific effects.(B) mHTT levels in the HD patient fibroblasts were tested by HTRF using the 2B7/MW1 antibody pair. Two different lines (Q45 and Q68) were transfected with the pooled siRNA targeting the indicated RBPs. Bars indicate mean and SEM (4 biological replicates).The statistical analysis was performed by one-way ANOVA with the Dunnett’s post hoc tests. ****P < 0.0001, **P < 0.001.
![Figure 1. Miniscreen of potential mHTT mRNA-interacting RBPs that modulate HTT levels.(A) The list of potential HTT mRNA interacting RBPs based on the CLIP-seq data analysed in StarBase (ref. [Citation12]). These candidates were then checked for brain tissue expression at BioGPS (ref. [Citation13]). The ‘# of target sites’ indicate the number of potential RBP-bound sequences detected in the HTT mRNA. The RBPs that are expressed in the brain with the ‘# of target sites’ >5 were prioritized for further testing (white rows). eIF4AIII was deprioritized since it is a translation initiation factor and may have non-specific effects.(B) mHTT levels in the HD patient fibroblasts were tested by HTRF using the 2B7/MW1 antibody pair. Two different lines (Q45 and Q68) were transfected with the pooled siRNA targeting the indicated RBPs. Bars indicate mean and SEM (4 biological replicates).The statistical analysis was performed by one-way ANOVA with the Dunnett’s post hoc tests. ****P < 0.0001, **P < 0.001.](/cms/asset/c05cbdaa-50e2-4142-a950-79ff9e91ded6/krnb_a_1712894_f0001_oc.jpg)
To confirm HuR as an HTT mRNA regulator, we tested the effect of HuR knockdown in HD cells including the HD knockin mouse striatal cells STHdhQ7/Q111 [Citation14] and immortalized human HD patient fibroblasts (Q47), which express endogenous full-length mouse homolog of HTT (msHTT) and human HTT (hsHTT) mRNA, respectively. HuR knockdown by siRNAs significantly reduced HTT mRNA levels in both cell lines (, , note that the same set of siRNAs were able to knockdown both human and mouse HuR because of their sequence similarities, and thus we used the HuR symbol for both mouse and human cells; knock-down efficiency was also validated by Western-blots in Fig. S1). Consistent with this, HuR knockdown in other HD patient fibroblast lines (Q45 & Q68) reduced HTT mRNA levels as well (Fig. S2). Consistently, HuR knockdown lowered HTT protein levels as well (). On the other hand, over-expression of HuR by cDNA transfection in the STHdhQ7/Q111 significantly increased HTT mRNA levels (), confirming HuR as a positive regulator of HTT mRNA levels.
Figure 2. HuR modulates HTT mRNA levels in a mHTT dependent manner.
(A) RT-qPCR measurements of HuR and HTT mRNA level in wild-type (STHdhQ7/Q7) or HD (STHdhQ7/Q111) mouse striatal cells transfected with the siRNAs targeting HuR (HuR_siRNA 1 and HuR_siRNA 2) or with the non-targeting control siRNA (Neg_siRNA) for 48 hours (12 technical replicates from 3 biological replicates). All signals were normalized to the averaged signal of Neg_siRNA transfected controls.(B) Similar to (A), but in immortalized human wild-type (Q16) or HD (Q47) fibroblasts (12 technical replicates from 3 biological replicates).(C) HTRF measurements of HTT protein levels in the HuR siRNA or the non-targeting control siRNA (Neg_siRNA) transfected wild-type or HD cells (12 technical replicates from 3 biological replicates for STHdh cells, and 16 technical replicates from 4 biological replicates for immortalized human fibroblasts). The siRNAs were transfected for 72 hours. All signals were normalized to the Neg_siRNA transfected controls. Note that the HTRF antibody pair 2B7/2166 was used here to detect the total HTT (including mHTT and wild-type HTT proteins) levels, different from the mHTT-specific antibody pair used for .(D) RT-qPCR measurements of HuR and mouse HTT (msHTT) mRNA levels in the HD mouse striatal cells (STHdhQ7/Q111) transfected with cDNA plasmids expressing human HuR. All signals were normalized to the averaged signal of the empty vector transfected control group (mock) (12 technical replicates from 3 biological replicates). Note that the HuR mRNA measurement in the mock group utilized the mouse HuR qPCR primers, whereas the one in the HuR transfected group utilized human HuR qPCR primers. Since these two set of primers had similar efficiency (Fig. S5) and the same threshold was used for analysis, the comparison between these two groups is valid.(E) RT-qPCR measurements of HuR and mouse HTT (msHTT) mRNA level in the wild-type mouse striatal cells (STHdhQ7/Q7). The cells were transfected with the siRNAs targeting HuR or the non-targeting control siRNA (Neg_siRNA), and then after 24 hours with cDNA plasmids expressing human wild-type full-length HTT (hsHTT-Q23), mutant HTT exon1 (hsHTTexon1-Q72) or full-length (hsHTT-Q73) (12 technical replicates from 3 biological replicates). The mRNA levels were then measured 48 hours after cDNA transfection. All the signals were normalized to the averaged signals of the Neg_siRNA transfected control groups.For all plotted data, error bars represent mean and SEM. The statistical analysis was performed by one-way ANOVA and post-hoc Dunnett’s tests for A-C, and two-tailed unpaired t tests for D-E. **P < 0.01; ****P < 0.0001; n.s., P > 0.05.
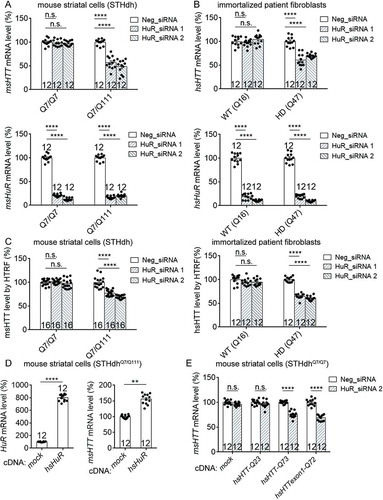
Interestingly, the HuR-mediated HTT mRNA regulation seemed to be missing in the wild-type cells (). This could be explained by two possibilities. First, HuR only regulates mutant HTT mRNA with expanded CAG repeats. Second, HuR regulates both mutant and wild-type HTT mRNA, but the regulation is dependent on the presence of the mutant HTT mRNA and/or protein.
To distinguish between these two possibilities, we exogenously expressed mutant hsHTT mRNA and proteins in the wild-type mouse cells (STHdhQ7/Q7), in which the endogenous wild-type msHTT mRNA level was insensitive to HuR knockdown. Expression of full-length mutant hsHTT (hsHTT-Q73) or the mutant hsHTT-exon1 fragment (hsHTTexon1-Q72) enabled the HuR to regulate the endogenous wild-type msHTT mRNA, whereas expressing wild-type hsHTT had no effects (), validating the second possibility. Thus, HuR may regulate both mutant and wild-type HTT mRNA, but in a mutant HTT dependent manner.
HuR positively regulates HTT mRNA stability
As a member of the ELAVL family RBPs, HuR may stabilize target mRNAs by its direct interaction with them [Citation15]. Thus, the most likely mechanism of HuR-mediated regulation of HTT mRNA levels is via RNA-stabilization. We thus examined HTT mRNA degradation in the HD cells by measuring the levels of mRNAs after treatment of actinomycin D, which stops new RNA synthesis by inhibiting RNA polymerase activity so that the degradation of pre-existing mRNAs could be measured by qPCR directly. In HD cells, including STHdhQ7/Q111 and immortalized human patient fibroblasts (Q47), transfection of HuR siRNAs significantly increased the degradation of HuR mRNA via RNA interference (, left panels). Importantly, both msHTT and hsHTT mRNAs were also degraded significantly faster in HuR knockdown cells than in the control cells, suggesting that HuR is required for HTT mRNA stabilization in these cells (, right panels).
Figure 3. Lowering HuR by siRNA knockdown or CRISPR heterozygous knockout decreased HTT mRNA stability in HD cells.
(A) HuR mRNA (left) and HTT mRNA (right) stability measurements in HD mouse striatal cells (STHdhQ7/Q111) (12 technical replicates from 3 biological replicates). Cells were transfected with indicated siRNAs for 36 hours, then treated with actinomycin D to stop new mRNA synthesis. HuR and HTT mRNA levels were then measured at different time points as indicated on the X-axis. The levels were then normalized to ms18S mRNA levels and plotted as the percentage of the time 0. Data values (mean ± S.E.M.) have been indicated for each point.(B) Similar to (A), but in immortalized HD patient fibroblasts (Q47) (12 technical replicates from 3 biological replicates). Data values (mean ± S.E.M.) have been indicated for each point.(C) Left panel: A representative Western-blot of HuR in HD mouse striatal cells (STHdhQ7/Q111) with or without HuR knocked out by the CRISPR system. α-Actin was blotted as a loading control. The sequencing chromatograms also verified the heterozygous knockout of HuR from the population (lower panel). Right panel: similar to A, but in HuR± or HuR+/+ STHdhQ7/Q111 cells without siRNA transfections (12 technical replicates from 3 biological replicates). Data values (mean ± S.E.M.) have been indicated for each point.For all plotted data, error bars represent mean and SEM. The statistical analysis was performed by two-way ANOVA, except the middle panel of C, which was analysed by two-tailed unpaired t tests. ***P < 0.001; ****P < 0.0001.
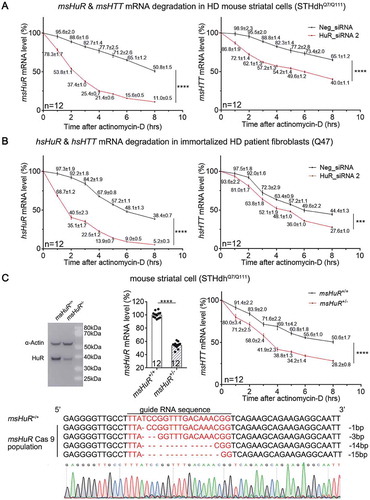
To further confirm this conclusion, we attempted to knock out the HuR gene in STHdhQ7/Q111 cells by the CRISPR/cas9 system, but only managed to obtain pooled colonies of heterozygous HuR knockout cells (HuR±, ), possibly because a complete loss of HuR is cytotoxic. The HTT mRNA degradation in the HuR± STHdhQ7/Q111 cells was also significantly faster than in the HuR+/+ STHdhQ7/Q111 cells (), consistent with the knockdown experiments. Taken together, HuR positively regulates HTT mRNA levels via increasing its stability.
The HuR protein interacts with HTT mRNA in its exon 11 in the coding region
As an RBP, HuR may directly interact with HTT mRNA to stabilize it. In fact, previous PAR-CLIP-Seq data analysed in Starbase [Citation12] suggested 25 potential HuR-binding sites on human HTT mRNA (). To confirm the binding between HuR and the HTT mRNA, we performed RNA-immunoprecipitation (RNA-IP) experiments in STHdhQ7/Q111 and immortalized Q47 fibroblasts. The HuR protein was pulled down by anti-HuR conjugated magnetic beads, and the amount of HTT mRNA that was pulled down with the HuR protein was remarkably higher than the control (pulled down by IgG-conjugated beads) (), suggesting that the HuR protein interacts with HTT mRNA in these HD cells.
Figure 4. HuR interacts with HTT mRNA in the HD cells, likely in the protein-coding region of HTT.
(A) A list of potential HuR’s binding sites in human HTT (hsHTT) mRNA based on StarBase (ref. [Citation12]). The highlighted lines indicate a candidate site (#105708) that has conserved sequences in human and mouse. ‘AU-rich’ indicates a site that is not listed in StarBase, but is still a possible interaction site predicted by HuR preferred binding sequences (ARE).(B) RT-qPCR quantifications of HuR-bound HTT mRNA levels in HD mouse striatal cells (STHdhQ7/Q111) or immortalized HD patient fibroblasts (Q47) as well as the wild-type controls (STHdhQ7/Q7 and Q16) by RNA-IP (12 technical replicates from 3 biological replicates). IgG was used as a negative control for the IP, and the 18S level was quantified as a baseline control to normalize the signals.(C) RT-qPCR quantifications of HuR-bound exogenously expressed human HTT (hsHTT) mRNA levels in WT mouse striatal cells (STHdhQ7/Q7) transfected with cDNA plasmids expressing full-length wild-type human HTT (hsHTT-Q23), full-length mutant HTT (hsHTT-Q73) or exon1 of mutant HTT (hsHTTexon1-Q72) by RNA-IP (12 technical replicates from 3 biological replicates). IgG was used as a negative control for the IP, and the 18S level was quantified as a baseline control to normalize the signals.(D) RT-qPCR quantifications of the exogenously expressed human HTT mRNA level in wild-type mouse striatal cells (STHdhQ7/Q7) transfected with the HuR siRNA or the non-targeting control siRNA (Neg_siRNA). The cDNA plasmids expressing exon1 (hsHTTexon1-Q72) or full-length (hsHTT-Q73) of human mutant HTT mRNA were transfected 24 hours after siRNA transfection, and their levels were measured after another 48 hours (12 technical replicates from 3 biological replicates).For all plotted data, error bars represent mean and SEM. The statistical analysis was performed by two tailed unpaired t tests, ****P < 0.0001; n.s.: P > 0.05.
![Figure 4. HuR interacts with HTT mRNA in the HD cells, likely in the protein-coding region of HTT.(A) A list of potential HuR’s binding sites in human HTT (hsHTT) mRNA based on StarBase (ref. [Citation12]). The highlighted lines indicate a candidate site (#105708) that has conserved sequences in human and mouse. ‘AU-rich’ indicates a site that is not listed in StarBase, but is still a possible interaction site predicted by HuR preferred binding sequences (ARE).(B) RT-qPCR quantifications of HuR-bound HTT mRNA levels in HD mouse striatal cells (STHdhQ7/Q111) or immortalized HD patient fibroblasts (Q47) as well as the wild-type controls (STHdhQ7/Q7 and Q16) by RNA-IP (12 technical replicates from 3 biological replicates). IgG was used as a negative control for the IP, and the 18S level was quantified as a baseline control to normalize the signals.(C) RT-qPCR quantifications of HuR-bound exogenously expressed human HTT (hsHTT) mRNA levels in WT mouse striatal cells (STHdhQ7/Q7) transfected with cDNA plasmids expressing full-length wild-type human HTT (hsHTT-Q23), full-length mutant HTT (hsHTT-Q73) or exon1 of mutant HTT (hsHTTexon1-Q72) by RNA-IP (12 technical replicates from 3 biological replicates). IgG was used as a negative control for the IP, and the 18S level was quantified as a baseline control to normalize the signals.(D) RT-qPCR quantifications of the exogenously expressed human HTT mRNA level in wild-type mouse striatal cells (STHdhQ7/Q7) transfected with the HuR siRNA or the non-targeting control siRNA (Neg_siRNA). The cDNA plasmids expressing exon1 (hsHTTexon1-Q72) or full-length (hsHTT-Q73) of human mutant HTT mRNA were transfected 24 hours after siRNA transfection, and their levels were measured after another 48 hours (12 technical replicates from 3 biological replicates).For all plotted data, error bars represent mean and SEM. The statistical analysis was performed by two tailed unpaired t tests, ****P < 0.0001; n.s.: P > 0.05.](/cms/asset/579872b1-be76-4257-97a2-d76f29ae23b4/krnb_a_1712894_f0004_oc.jpg)
We further explored the HuR-binding site(s) in the HTT mRNA. Based on the analysis of StarBase (), some of the potential binding sites are in the coding region, whereas some are in the 3ʹ-UTR. Thus, we exogenously expressed hsHTT cDNA with only the coding sequence but not the 3ʹUTR sequence to test if at least some of the binding site(s) are within the coding region. We transfected the wild-type mouse STHdhQ7/Q7 cells with the plasmids that exogenously express human mutant HTT with full-length coding sequences (CDS) (hsHTT-Q73) or with exon 1 only (hsHTTexon1-Q72), and performed RNA-IP experiments for the exogenously expressed hsHTT mRNAs (not the endogenous msHTT mRNA) to test whether the HuR protein can bind with them. The empty vector transfected control sample (mock) did not give any hsHTT signals (), confirming that the RNA-IP experiments and qPCR measurements only detect the binding to exogenously expressed hsHTT mRNAs. The HuR antibody- and IgG-conjugated beads pulled down similar levels of the hsHTTexon1-Q72 mRNA (), suggesting that the binding site(s) are probably not within the exon 1 region. In comparison, the HuR was able to bind with the mutant full-length hsHTT CDS (, hsHTT-Q73), suggesting that at least some of the binding site(s) are within the coding region. Interestingly, no binding with the wild-type full-length hsHTT CDS (, hsHTT-Q23) was detected, suggesting that the binding is dependent on the presence of mHTT, consistent with previous functional experiments (). To further test if this binding is functionally relevant, we knocked down HuR mRNA in these cells and tested if these exogenously expressed hsHTT mRNA levels were influenced. Consistent with the RNA-IP data, only the levels of full-length mutant HTT CDS mRNA (hsHTT-Q73) were significantly reduced by HuR knockdown, but not the ones of its exon 1 fragment mRNA (hsHTTexon1-Q72) or the wild-type full-length HTT CDS (hsHTT-Q23) (). Collectively, our data suggest that HuR functionally interacts with the HTT mRNA in its protein coding region, but not in its exon 1.
To map the precise binding site(s), we reviewed all the potential binding sites based on StarBase () and blasted these sequences with the msHTT mRNA to see whether these sites are conserved between human and mouse, because HuR was able to interact with both hsHTT and msHTT to regulate their stability ( & ). We found that only one candidate site, #105708, has conserved sequences between human and mouse (, highlighted). Thus, #105708 is likely a HuR-binding site that mediates HuR’s regulation of HTT mRNA stability. Since #105708 locates in exon 11 of the HTT mRNA, we tested if exon 11 is required for the interaction between HuR and the HTT mRNA. Thus, we constructed two plasmids expressing the first 10 (exon 1–10) or 11 exons (exon 1–11) of the mutant human HTT mRNA (hsHTT-Q73), respectively. The only difference between these two plasmids is the presence of exon 11. We then transfected the wild-type mouse STHdhQ7/Q7 cells with these plasmids to test whether HuR could bind to the mRNAs expressed by these two plasmids. RNA-IP experiment results showed that the HuR protein interacted with exon 1–11, but not exon 1–10 (), suggesting that the binding site is within the exon 11 region and likely to be #105708.
Figure 5. HuR interacts with #105708 site in exon 11 of the HTT mRNA.
(A) RT-qPCR quantifications of HuR-bound exogenously expressed HTT mRNA 5ʹ fragment (exon 1–10 or exon 1–11) levels in the wild-type mouse striatal cells (STHdhQ7/Q7) by RNA-IP (12 technical replicates from 3 biological replicates). IgG was used as a negative control for the IP, and the 18S level was quantified as a baseline control to normalize the signals. HuR interacted with exon 1–11 but not exon 1–10, suggesting that exon 11 contains the HuR-binding site.(B) Similar to (A), but in HD cells (STHdhQ7/Q111) transfected with cDNA plasmids expressing EGFP mRNAs containing different candidate binding sites in its 3ʹ UTR region (12 technical replicates from 3 biological replicates). The EGFP mRNA with #105708 site sequences (mouse or human) showed interaction with HuR, suggesting that #105708 (located in exon 11 of HTT mRNA) is a potential HuR-binding site.(C) A representative R-EMSA gel image of different Cy3-labelled RNA oligos of potential binding site sequences after co-incubation with purified HuR-MBP.His proteins (see Fig. S3). The first lanes in each group were loaded by the indicated RNA oligos alone. In the 2nd and 3rd lanes in each group, the indicated RNA oligos were incubated and loaded with purified HuR-MBP.His protein at different concentration ratios as indicated. Five repeats were performed, showing consistent results. HuR-binding with the RNA oligo leads to an apparent molecular weight shift to the top (dark arrows), due to mobility block caused by protein-binding.(D) Quantification of the ratio between firefly luciferase and renilla luciferase signals in HD mouse striatal cells (STHdhQ7/Q111) transfected with the pmirGLO reporter plasmids (12 technical replicates from 3 biological replicates). The firefly/renilla signal ratio is an indicator of the level of firefly luciferase mRNA, of which the 3ʹ UTR region contained different sequences of potential HuR-binding sites. The HuR knockdown by siRNA caused lowering of the firefly/renilla signal ratio only for #105708-containing plasmids, suggesting that #105708 is likely a functional binding site.(E) RT-qPCR quantifications of HuR-bound endogenous mouse HTT (msHTT) mRNA levels in HD mouse striatal cells (STHdhQ7/Q111) transfected with pmirGLO plasmids expressing firefly luciferase mRNAs containing different potential binding sites (12 technical replicates from 3 biological replicates). The mRNAs with functional binding sites may compete with endogenous msHTT mRNA for HuR binding (illustrated in the schematic picture in the left panel). Thus, the RNA-IP signals of the corresponding samples could be reduced (right panel). IgG was used as a negative control for the IP, and the 18S level was quantified as a baseline control to normalize the signals.For all plotted data, error bars represent mean and SEM. The statistical analysis was performed by two-tailed unpaired t tests, ****P < 0.0001; n.s.: P>0.05.
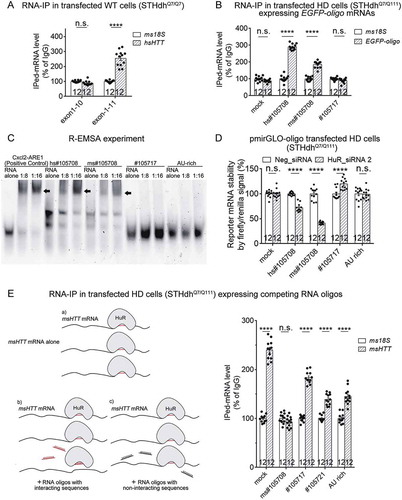
To confirm this, we cloned several candidate sequences (including #105708) into the EGFP construct, so that the 3ʹ UTR of EGFP mRNA contains these sequences. We then transfected the HD mouse STHdhQ7/Q111 cells with these plasmids and tested whether HuR may interact with the EGFP mRNAs expressed from these plasmids. RNA-IP experiments showed that the EGFP mRNAs with #105708 sequences (hs#105708 and ms#105708 for human and mouse sequences, respectively) but not the #105717 sequence exhibited interaction with HuR (). The experiment also suggests that #105708 is sufficient for HuR binding in the HD cells.
To further validate #105708 as the binding site, we performed RNA electrophoretic mobility shift assays (R-EMSA) experiments to test the direct interaction between HuR and #105708. We mixed Cy3-labelled RNA oligos of either the human #105708 (hs#105708) or the mouse #105708 (ms#105708) sequences with the HuR-MBP.His protein (with the human HuR amino acid sequence) purified from HEK293T cells (Fig. S3) at indicated ratios and ran the samples on native-PAGE gels (). We observed apparent molecular weight-shift of these RNA oligos due to mobility block caused by HuR binding (), suggesting that HuR interacts with both mouse and human #105708 sequences directly, consistent with the RNA-IP results (). As controls, we performed R-EMSA for an RNA oligo with known HuR-interacting sequence (, Cxcl 2) [Citation16], and observed expected apparent molecular weight shift as well. In addition, we tested two other candidate binding sites using R-EMSA. #105717 is a candidate binding site in the 3ʹ UTR region (). The ‘AU-rich’ oligo contains the only AU-rich element (ARE) in the HTT mRNA. Since AREs are the HuR preferred binding sites [Citation15], we anticipated that the ‘AU-rich’ oligo might interact with HuR as well. Surprisingly, we observed no apparent molecular weight shift of RNA oligos with either of these two sequences (), suggesting that #105708 is a relatively specific site in the HTT mRNA for HuR binding.
To investigate whether the HuR-#105708 interaction is functional in regulating mRNA levels in the cells, we cloned candidate binding site sequences into a firefly-renilla dual-luciferase plasmid (pmirGLO). The 3ʹUTR of its expressed firefly luciferase mRNA contains the sequence of every single candidate binding site, whereas the renilla luciferase was expressed by an independent promoter in the same plasmid as an internal control. Thus, the firefly luciferase signal divided by the renilla luciferase signal (firefly/renilla signal) reflects the relative level of firefly luciferase mRNA containing each individual candidate binding site, and this ratio serves as readout for mRNA stability. We thus transfected these plasmids into the mouse HD cells (STHdhQ7/Q111) and tested the effect of HuR knockdown. Among all the candidate sequences tested in this reporter assay, only the #105708 (hs#105708 and ms#105708) sequences caused significantly decreased firefly/renilla signal after HuR knockdown (), suggesting that the HuR-#105708 interaction is functional in regulating mRNA levels in the HD cells.
Finally, we tested if the #105708-containing mRNA may compete with endogenous HTT mRNA for HuR’s binding (, see the schematic picture in the left panel). If so, #105708 RNA oligos may serve as potential reagents to decrease mutant HTT RNA stability for HD research and possible therapeutic intervention. Exogenous expression of #105708 containing firefly luciferase mRNA largely blocked the interaction between endogenous HuR proteins and the HTT mRNA, based on the RNA-IP experimental results (, right panel), validating the competition effects of #105708. As controls, this phenomenon was not observed for firefly luciferase mRNAs containing other candidate HuR binding sequences (, right panel). Collectively, HuR interacts with the HTT mRNA directly at the #105708 site in its exon 11 region in vitro and in the HD cells, and this interaction is functional in regulating target mRNA levels.
HuR interacts with HTT mRNA in a mHTT-dependent manner via MAPK11
Interestingly, the HuR-HTT mRNA interaction was not detected in wild-type cells, where mHTT was not present (, mock; , STHdhQ7/Q7 & Q16; , hsHTT-Q23). This is consistent with the observation that HuR regulates both mutant and wild-type HTT mRNA in a mutant HTT dependent manner (). We thus tested if the HuR-HTT mRNA interaction is also dependent on the presence of mHTT. We transfected wild-type mouse cells (STHdhQ7/Q7) with plasmids expressing mHTT (hsHTT-Q73) or its exon 1 fragment (hsHTTexon1-Q72) or the empty control plasmid (mock), and then performed the RNA-IP experiments similar to previously presented in , but testing HuR’s interaction with the endogenous mouse HTT mRNA (msHTT) at this time (). Expression of mHTT or its exon 1 fragment restored the originally missing interaction between HuR and the wild-type HTT mRNA (, hsHTT-Q73 and hsHTTexon1-Q72 compared with mock), confirming the mHTT-dependence of HuR’s binding to the HTT mRNA.
Figure 6. HuR interacts with HTT mRNA in an mHTT-dependent manner.
(A) RT-qPCR quantifications of the levels of endogenous mouse HTT (msHTT) mRNA bound with HuR in WT mouse striatal cells (STHdhQ7/Q7) transfected with empty vector (mock) or cDNA plasmids expressing full-length (hsHTT-Q73) or exon 1 (hsHTTexon1-Q72) by RNA-IP (12 technical replicates from 3 biological replicates). IgG was used as a negative control for the IP, and the 18S level was quantified as a baseline control to normalize the signals.(B) Similar to , but in wild-type cells (STHdhQ7/Q7) (7 biological replicates).(C) Similar to , but in wild-type cells (STHdhQ7/Q7) transfected with hsHTTexon1-Q72 plasmid 24 hours before transfection of the pmirGLO reporter plasmids with the indicated potential binding sites (6 biological replicates).o-tailed unpaired t tests, ****P < 0.0001; n.s.: P > 0.05.For all plotted data, error bars represent mean and SEM. The statistical analysis was performed by two-tailed unpaired t tests. **P < 0.01; ****P < 0.0001; n.s., P > 0.05.
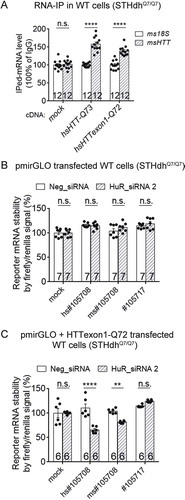
We further confirmed this by testing the functional consequence of the HuR-#105708 interaction using the dual-reporter assay. The firefly/renilla signal was not influenced by HuR knockdown in wild-type cells (STHdhQ7/Q7), even for #105708 containing pmirGLO plasmids (), suggesting that the HuR-#105708 interaction is absent in the wild-type cells, different from the one observed in HD cells (). However, when these cells were transfected with the mHTT exon 1 fragment (hsHTTexon1-Q72), the firefly/renilla signal in the #105708-containing group became sensitive to the HuR knockdown (), confirming that mHTT expression is sufficient to drive the HuR-#105708 interaction. Taken together, the interaction between HuR protein and HTT mRNA is dependent on the presence of mHTT.
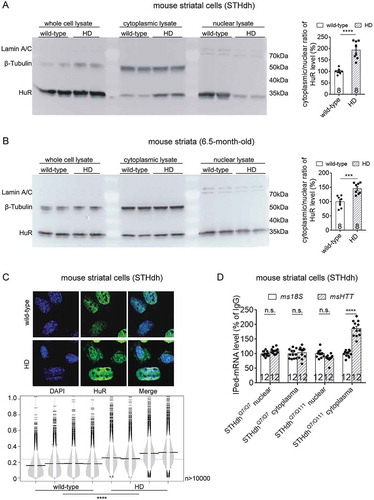
HuR locates in both the nuclei and cytoplasm, and shuttles between the two cellular compartments [Citation17]. Since HuR mainly locates in the nuclei but stabilize mRNA in the cytoplasm [Citation18], we hypothesize that it distributes more in the cytoplasm in the HD cells or the presence of mHTT, so that it interacts and regulates the HTT mRNA in an mHTT-dependent manner. Consistent with this hypothesis, the cytoplasmic localization of HuR is significantly higher in the mouse HD cells (STHdhQ7/Q111) compared to the one in wild-type cells (STHdhQ7/Q7), based on the Western-blots of cytoplasmic versus nuclear fractions of these cells (). A similar phenomenon was observed in vivo in the striatal tissue from 6.5-month-old HD versus wild-type mice (). To confirm the enhanced cytoplasmic localization of HuR, we immunostained the HuR protein in wild-type (STHdhQ7/Q111) versus HD (STHdhQ7/Q111) cells to investigate its cellular distribution by high-content confocal imaging technology. Imaging analysis of thousands of cells showed that the cytoplasmic to nuclear ratio of HuR protein is significantly higher in the HD than the wild-type cells (), consistent with the biochemical fractionation experiments (). Finally, to functional characterize the increased cytoplasmic localization of HuR in the HD cells, we performed RNA-IP in both wild-type and HD cells to test the HuR-bound HTT mRNA levels in nuclear versus cytoplasmic fractions. The cytoplasmic HuR bound HTT mRNA level was significantly higher in the HD cells than the wild-type ones (), consistent with the increased cytoplasmic localization of HuR in HD cells.
Figure 7. The nuclear/cytoplasmic distribution of HuR protein in wild-type versus HD cells or tissues.
(A) Representative Western-blots (from 4 biological replicates) of HuR protein levels in the nuclear or cytoplasmic fractions of wild-type (STHdhQ7/Q7) or HD (STHdhQ7/Q111) mouse striatal cells. β-Tubulin was blotted as a cytoplasmic loading control, and Lamin A/C as a nuclear loading control. These two control proteins were only detected in their localized fractions, confirming reliable nuclei/cytoplasm separation. The cytoplasmic/nuclear ratios of HuR signals were quantified and plotted (8 technical replicates from 4 biological replicates).(B) Similar to (A), but in the cytoplasmic and nuclear fractions of striatal tissue lysates from 6.5-month-old wild-type (HdhQ7/Q7) or HD (HdhQ140/Q140) mice (8 technical replicates from 4 biological replicates).(C) Representative images and quantifications of immunofluorescent staining of the distribution of HuR protein in wild-type (STHdhQ7/Q7) or HD (STHdhQ7/Q111) cells. The cytoplasmic/nuclear ratios of HuR signals were quantified for each cell and presented by beanplot (4 biological replicates with more than 10,000 cells for each replicate).(D) RT-qPCR quantifications of HuR-bound HTT mRNA levels in nuclear and cytoplasmic fractions of HD (STHdhQ7/Q111) or wild-type (STHdhQ7/Q7) cells detected by RNA-IP (12 technical replicates from 3 biological replicates). IgG was used as a negative control for the IP, and the 18S level was quantified as a baseline control to normalize the signals.For all plotted data, error bars represent mean and SEM. For all bean plots, the long black solid line represents the mean of each replicate, the grey dashed line represents the mean of all data points, the grey area represents the distribution of data, and short black solid lines represent the outliers of original data. The statistical analysis was performed by two-tailed unpaired t tests, ***P < 0.001, ****P < 0.0001; n.s., P > 0.05.
We then investigated the potential mechanism of the higher cytoplasmic localization of HuR in the HD cells. Our previous work identified MAPK11 as a positive modulator of HTT mRNA stability [Citation19] and it is also activated by mHTT [Citation20]. In addition, MAPK11 belongs to the p38 MAPK family, which are able to enhance HuR levels and activate cytoplasmic translocation of HuR [Citation21], although the exact member(s) of this family that makes a major contribution to this function is yet to be identified. We thus hypothesized that mHTT may activate MAPK11 to enhance the cytoplasmic localization of HuR, which may then interact with and stabilize the HTT mRNA.
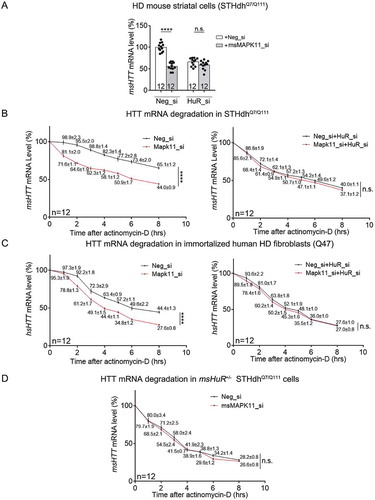
To test this hypothesis, we performed epistatic experiments to test if HuR is downstream of MAPK11 in regulating HTT mRNA levels. Consistent with our previous observation [Citation19], knockdown of mouse MAPK11 (msMAPK11) significantly reduced HTT mRNA levels in the HD cells (STHdhQ7/Q111) (). Meanwhile, knockdown of HuR largely abolished this effect, suggesting that HuR is downstream of MAPK11 in regulating HTT mRNA levels (). Consistently, lowering HuR by knockdown or heterozygous knockout significantly ameliorated MAPK11’s effects on the HTT mRNA stability in both mouse (STHdhQ7/Q111) and human HD cells (Q47) (), confirming that HuR is likely downstream of MAPK11 in stabilizing the HTT mRNA.
Figure 8. MAPK11 is an upstream modulator of HuR and controls HuR’s regulation of HTT mRNA levels in HD cells.
(A) RT-qPCR quantifications of endogenous msHTT mRNA levels in HD mouse striatal cells (STHdhQ7/Q111) transfected with the indicated siRNAs (12 technical replicates from 3 biological replicates). msMAPK11 siRNA (grey bars) or the non-targeting control siRNA (Neg_si, white bars) were co-transfected with HuR siRNA (HuR_si) or the non-targeting control siRNA (Neg_si). msHTT mRNA levels were measured 48 hours after transfection.(B) HTT mRNA degradation measurements in HD mouse striatal cells (STHdhQ7/Q111) transfected with the indicated siRNAs for 36 hours, and then treated with actinomycin D (12 technical replicates from 3 biological replicates). The quantification of HTT mRNA degradation was then performed the same as in . Data values (mean ± S.E.M.) have been indicated for each point.(C) Similar to (B), but in immortalized human HD fibroblast (Q47) (12 technical replicates from 3 biological replicates). Data values (mean ± S.E.M.) have been indicated for each point.(D) Similar to (B), but in pooled colonies of mouse HD cells (STHdhQ7/Q111) with the heterozygous knockout of HuR (12 technical replicates from 3 biological replicates). Data values (mean ± S.E.M.) have been indicated for each point.For all plotted data, error bars represent mean and SEM. The statistical analysis was performed by two-way ANOVA (B-D) or two-tailed unpaired t tests (A). ****P < 0.0001; n.s., P > 0.05.
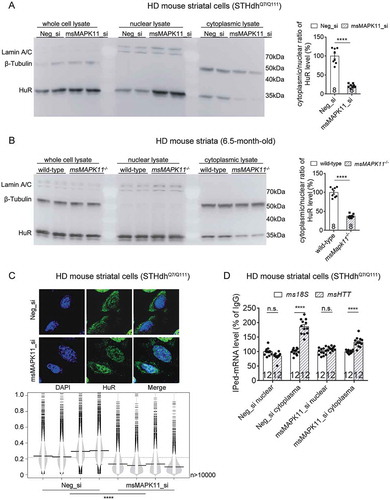
We thus further investigated whether MAPK11 influences HuR’s cytoplasmic localization. MAPK11 knockdown significantly reduced cytoplasmic localization of HuR based on the Western-blots of cytoplasmic versus nuclear fractions of mouse HD cells STHdhQ7/Q111 (), illustrating that MAPK11 regulates the nuclei-cytoplasm distribution of HuR. Consistently, knockout of the mouse MAPK11 (msMAPK11−/-) significantly reduced the cytoplasmic/nuclear ratio of HuR in the 6.5-month-old HD mouse (HdhQ140/Q140) striatal tissue as well, confirming that MAPK11 regulates HuR’s cytoplasmic localization as well (). Consistent with this, MAPK11 knockdown reduced HuR cytoplasmic localization () as well as the cytoplasmic HuR-bound HTT mRNA level (, in which the IPed msHTT mRNA in the cytoplasmic fraction of HD cells was obviously lowered by Mapk11_si transfection comparing to Neg_si transfected controls) in the HD cells. Taken together, the most likely interpretation of our data is that the presence of mHTT activates MAPK11 [Citation19,Citation20], which then induces cytoplasmic translocation of HuR. HuR then binds to the #105708 sequence in exon 11 of HTT mRNA and stabilizes it (). Thus, MAPK11 or HuR knockdown destabilizes the HTT mRNA in an mHTT-dependent manner.
Figure 9. MAPK11 regulated nuclei/cytoplasm distribution of HuR protein in HD cells or tissues.
(A) Representative Western-blots and quantifications of cytoplasmic versus nuclear HuR protein levels in the HD mouse striatal cells (STHdhQ7/Q111) transfected with the indicated siRNAs (8 technical replicates from 4 biological replicates).(B) Representative Western-blots and quantifications of cytoplasmic versus nuclear HuR protein levels in the lysates of striatal tissues from 6.5-month-old HD (HdhQ140/Q140) mice with or without msMAPK11 knockout (8 technical replicates from 4 biological replicates).(C) Similar as , but in HD (STHdhQ7/Q111) cells transfected with the indicated siRNAs (4 biological replicates).(D) Similar as in , but using HD cells transfected with the indicated siRNAs (12 technical replicates from 3 biological replicates). IgG was used as a negative control for the IP, and the 18S level was quantified as a baseline control to normalize the signals.For all plotted data, error bars represent mean and SEM. For all bean plots, the long black solid line represents the mean of each replicate, the grey dashed line represents the mean of all data points, the grey area represents the distribution of data, and short black solid lines represent the outliers of original data. The statistical analysis was performed by two-tailed unpaired t tests, ****P < 0.0001; n.s., P > 0.05.
Discussion
In this work, we identified HuR as a novel RBP that modulates HTT mRNA stability in HD cells. To our knowledge, it is the first reported RBP that interacting with and stabilizing the HTT mRNA.
The interaction between HuR and HTT mRNA is non-canonical in several ways, which may provide additional insights into HuR biology. It interacts with a non-AU-rich sequence in the coding region, instead of the canonical ARE in the 3ʹ-UTR (). This type of interaction could be loose, compared to canonical ones, so that it does not interfere with protein translation. Meanwhile, this coding-region and non-AU-rich type of HuR interaction is still functional in stabilizing the target mRNA, consistent with a very recent HuR study [Citation22].
Another interesting feature of HuR’s interaction with the HTT mRNA is that the interaction is dependent on the presence of mHTT DNA, RNA or protein (). The mHTT protein is likely the major contributor to activate HuR’s interaction with the HTT mRNA, because we utilized mixed CAGCAA sequences in the mHTT-expressing cDNA plasmids ( & , ), which lacked the expanded CAG repeat sequences required to induce DNA replication errors or RNA foci formation or RNA phase separation [Citation23,Citation24]. Thus, it is more likely that the mHTT protein enabled HuR’s capability in regulating the HTT mRNA, possibly via activating MAPK11 ( & ). Taken together, the mHTT-MAPK11-HuR-HTT mRNA pathway may form a positive feedback loop (), which may accelerate mHTT protein accumulation and contribute to HD progression over time.
Figure 10. A schematic model illustrating mHTT-dependent MAPK11-HuR mediated regulation of HTT mRNA stability.
The presence of mHTT activates MAPK11 (ref. [Citation19,Citation20]), leading to translocation of HuR protein to the cytoplasm (ref. [Citation21]) ( & ) and its interaction with HTT mRNA in its exon 11 at the #105708 site ( & ) to stabilize it in a mHTT-dependent manner (, & ).
![Figure 10. A schematic model illustrating mHTT-dependent MAPK11-HuR mediated regulation of HTT mRNA stability.The presence of mHTT activates MAPK11 (ref. [Citation19,Citation20]), leading to translocation of HuR protein to the cytoplasm (ref. [Citation21]) (Figs. 7 & 9) and its interaction with HTT mRNA in its exon 11 at the #105708 site (Figs. 4 & 5) to stabilize it in a mHTT-dependent manner (Figs. 2, 6 & 8B–D).](/cms/asset/78ed62cb-d2ac-445f-b0ca-ba3144e54add/krnb_a_1712894_f0010_oc.jpg)
The therapeutic potential of the pathway remains to be tested. While targeting the HTT mRNA by siRNA, shRNA or ASOs provides extremely promising strategies for HD treatment [Citation25], these approaches require delivery of large biomolecules or viral particles such as AAV, which is challenging and expensive. In addition, these exogenously delivered large biomolecules or viruses might activate certain biological responses such as the interferon response that might be risky in certain circumstances. A very recent study has also shown that the CAG repeat number rather than polyQ length might determine the age of onset in HD [Citation26], implying that the CAG repeat containing HTT mRNA may play a more important role than previously thought. Targeting the HTT mRNA by MAPK11/HuR provides a possible alternative approach for HD drug development. Both MAPK11 and HuR have reported inhibitors [Citation27,Citation28], which could be used to test the therapeutic potential of this pathway. In fact, our previous reports suggested that the MAPK11 inhibitor SB202190 may reduce mHTT levels to rescue HD-relevant phenotypes in the HD neurons [Citation19], although the HuR inhibitors are unavailable to us. Meanwhile, further optimization of MAPK11 and/or HuR inhibitors may provide small compound drugs lowering the HTT mRNA to treat HD.
Besides the involvement of mHTT in HD, the wild-type HTT protein is also an important protein that is essential during development and plays critical roles in various cellular functions [Citation29]. As an HTT mRNA modulator, HuR may participate in key biological functions via regulating the wild-type HTT as well. In our study, HuR-mediated modulation of HTT mRNA levels is dependent on the presence of mHTT, but it is possible that this regulation is activated under some other pathophysiological or even physiological conditions. In fact, MAPK11 and other p38 MAPKs are activated by various types of cellular stress such as oxidative, genotoxic, and osmotic stress and by proinflammatory cytokines occurring in many human diseases including neurodegenerative disorders other than HD [Citation30]. Thus, HuR might stabilize the wild-type HTT mRNA in those disease neurons to protect them, considering the neuroprotective function of wild-type HTT [Citation31]. Another interesting possibility is the HuR may play a role during development by modulating wild-type HTT. The p38 MAPK pathway is known to be activated during embryonic development [Citation32], whereas wild-type HTT is also an essential protein at this stage [Citation33,Citation34]. Thus, HuR might play an important role at this stage through regulating wild-type HTT levels.
Material and methods
Utilization of gene symbols
The official symbol of HuR is ‘ELAVL1’, but since most of publications in the literature used ‘HuR’, we also used HuR in this paper to be consistent with others. To clarify the species, we added ‘hs’ or ‘ms’ before the gene symbol to indicate human or mouse species, respectively. In a few places where the symbol stands for both human and mouse version, ‘hs’ or ‘ms’ was not added. We used the italic font for DNAs or RNAs, and the regular font for proteins.
Plasmid constructs
The human HTT-exon1 (hsHTTexon1-Q72) and full-length (hsHTT-Q23 & hsHTT-Q73) constructs were synthesized (Thermo Fisher Scientific) and subcloned into the pcDNA/Puro-CAG plasmid vector (The schematic models of inserted constructs are shown in Fig. S4A). The human HTT exon 1–10 and exon 1–11 constructs were cloned by PCR amplification from the hsHTT-Q73 and subcloned back to pcDNA/Puro-CAG plasmid vector. All these constructs have mixed CAGCAA sequences to express the polyQ sequence. The pFRT-to-eGFP-ELAVL1 (human HuR) (Addgene, #106105) construct was obtained from Addgene. The pmirGLO Dual-Luciferase miRNA target expression vector (Promega #E1330), and all of the dual-luciferase plasmids with specific HTT target site (#105708, # 105717, etc.) were constructed by ligating the annealed oligos to the original backbone at the 3ʹUTR region of the firefly luciferase (Fig. S4B). The HuR-MBP.His plasmid was cloned by PCR amplification of the HuR cDNA from pFRT-to-eGFP-ELAVL1 plasmid and then ligated it to the pMal-c2x (Addgene, #75286; we have inserted C-terminal poly-His and the TEV protease cleavage site expressing sequence to the 3ʹ) plasmid.
Cell culture
The patient-derived Q47 fibroblasts were described previously [Citation19]. Human fibroblasts cell lines (GM03868 – Q45; GM21757 – Q68) and STHdh (CH00097 – Q7/Q7 and CH00096 – Q111/Q7) were obtained from Coriell Cell Repositories. HEK293T and STHdh cells were cultured in DMEM (Thermo Fisher Scientific, #11965) with 10% FBS (Thermo Fisher Scientific, #10082), whereas human fibroblasts were cultured in DMEM with 15% FBS. All the STHdh cells were maintained at 33°C incubator with 5% CO2, and HEK293T and human fibroblasts were kept at 37°C with 5% CO2. The cells were tested for mycoplasma contamination every month.
The homogeneous time-resolved fluorescence (HTRF) assay
The HTRF assays were performed similarly to those previously described [Citation35]. Basically, the cells were lysed in the lysis buffer (1× PBS with 0.4% Triton X-100 and 1× protease inhibitor final) and the HTT protein was detected with 2B7/MW1 antibody pair. For all samples, the total protein concentration (by BCA, Beyotime, #P0009) was measured to correct the loadings. Different protein concentrations or cell numbers per well were tested to ensure that the signals were in the linear range. Background corrections were performed by subtracting the background signals from blank samples.
cDNA and siRNA transfection
All cDNAs were transfected with lipofectamine 3000 (Life Technologies, # L3000), whereas siRNAs were transfected into the STHdh cells with lipofectamine 2000 (Life Technologies, #11668). Transfection steps were performed according to the manufacturer’s protocol. Cells were collected at different timelines for diverse experimental purpose. Briefly, cells were collected 24 hours after plasmid transfection for dual-luciferase detection, or 48 hours after cDNA or siRNA transfection for qPCR or RNA-IP, or 72 hours after siRNA transfection for Western blot or HTRF. In some experiments (, & ), the cells were first transfected with siRNA and then with cDNA. In this case, the siRNA was first transfected for 24 hours. The culture medium was then changed, and the cDNA transfection was performed. The cells were then collected for experiments 48 hours after cDNA transfection. All siRNAs have been validated by qPCR and/or western blots for target knockdown. siRNA target sequences are shown as below: HuR_siRNA 1 (both human and mouse): AAGAGGCAATTACCAGTTTCA; HuR_siRNA 2 (both human and mouse): GGGATAAAGTAGCAGGACA; MAPK 11_siRNA (human): CACGCATGTATGCATGCACAA; Mapk 11_siRNA (mouse): CGCCAGAGATCATGCTAAA. Neg_siRNA was purchased commercially (QIAGEN, #1027280).
RT-qPCR and mRNA stability measurements
mRNA levels were determined by RT-qPCR, and the protocol was followed as previously described. Briefly, RNA from siRNA-transfected and/or actinomycin D-treated cells was extracted using RNAprep Pure Cell/Bacteria Kit according to the manufactory’s instruction (Tiangen, #DP430). cDNA was obtained by reverse transcription with random primers using the FastQuant RT Kit (Tiangen, #KR106). qPCR was then performed using SYBR Green Realtime PCR Master Mix (Toyobo, #QPK-201) with no reverse-transcriptase controls used to ensure the specificity of the signals. All the primers were tested with a standard curve (Fig. S5).
For mRNA stability measurements, the cultured cells were treated with actinomycin D (5 μg/ml, Sigma, #A9315) that inhibits transcription at time 0, and then collected at indicated time points for quantification of different mRNAs by RT-qPCR. The qPCR primer sequences are (note that STHdhQ7/Q111 cells express the mouse wild-type HTT mRNA, as well as a mouse-human chimera HTT mRNA with its exon1 and part of intron 1 replaced by the human version with expanded CAG repeats (111 CAG), but the msHTT qPCR primers targeting the 3ʹ sequences can detect both): hs18S-forward: GGTGGAGCGATTTGTCTGGTTA; hs18S-reverse (human): CGGACATCTAAGGGCATCACAG; ms18S -forward (mouse): CGCCGCTAGAGGTGAAATC; ms18S-reverse (mouse): CCAGTCGGCATCGTTTATGG; hsHTT-forward: CCTTTTTGAAGCAGCCCGTG; hsHTT-reverse: TGCAGCATCCCCAAACAGAT; msHTT-forward (mouse): GCAGAGATTTGGCTGCATTC; msHTT-reverse (mouse): CCATGGCATACAGCAGCA; hsHuR-forward (human): GGCTTGAGGCTCCAGTCAAA; hsHuR-Reverse (human): CCCTCTGGACAAACCTGTAGT; msHuR-forward (mouse): CACCACCAGGCACAGAGATT; msHuR-reverse (mouse): CGGGGACATTGACACCAGAA; EGFP-forward: TATATCATGGCCGACAAGCA; EGFP-reverse: GTTGTGGCGGATCTTGAAGT; exon 1–11 of mutant HTT-forward: CACCGCTGCTAAGGAGGA; exon 1–11 of mutant HTT-reverse: AGGAACCCCCTCCAGCTA; hsHTT exon 1-Q72-forward: CCTCAGCCACAACCTCCTC; hsHTT exon 1-Q72-reverse: CGTATGGGTATCGGTGCAG
RNA immunoprecipitation (RNA-IP)
To perform RNA-IP, protein G magnetic beads (Bio-rad, #161-4023) were first blocked with 5% BSA and 2 μg/mL tRNA on a roller for 1.5 hours at 4 °C. Then mixed the beads with antibody in RIP buffer (100 mM KCl + 25 mM Tris-HCl (pH = 7.4) + 1 mM EDTA + 0.5 mM DTT + 0.5% NP-40 + 1× RNase inhibitor (Thermo Fisher Scientific, #N8080119) + 1× Complete protease inhibitor (Roche, #04693132001)) overnight on a roller at 4 °C. The next day, cell samples were collected and lysed by the RIP buffer, and one-third of the sample was kept as the input control before pull-down by the antibody-conjugated beads. The lysates were incubated with the beads overnight on a roller at 4 °C, and then the beads were washed by cold RIP Buffer followed by 1× PBS once. The RNA pulled down by the beads was then isolated by RNA simple Total RNA kit (TIANGEN, #DP419). The mRNA level was then measured and analysed by RT-qPCR, as illustrated above.
RNA electrophoretic mobility shift assay (R-EMSA)
The human HuR protein was purified for the R-EMSA experiments. To obtain the recombinant purified HuR protein, HEK293T cells were transfected with the HuR-MBP.His plasmid, and then collected after 48h. The cellular protein was then extracted by NiA Buffer (20 mM imidazole + 5% glycerol + 150 mM NaCl + 100 mM Tris-HCl (pH = 7.4) + 1 × Complete protease inhibitor (Roche, #04693132001)), and then loaded onto the Ni-NTA superflow column (QIAGEN, #30622). Column bound proteins were then eluted by NiB Buffer (300 mM imidazole + 5% glycerol + 150 mM NaCl + 100 mM Tris-HCl (pH = 7.4) + 1 × Complete protease inhibitor (Roche, #04693132001)). Finally, the purified proteins were concentrated to approximately 2 mg/ml in 50 mM HEPES buffer with 100 mM NaCl for further analysis, and the purity of HuR protein was confirmed by coomassie brilliant blue staining. To perform RNA-EMSA, the Cy3-labelled RNA oligos (4μM) were first heated to 95 oC and cooled down to 25 oC at room temperature. Then the titrated HuR protein (2 mg/mL) and RNA oligos were dissolved in the EMSA interaction buffer (1 × R-EMSA Binding Buffer (Thermo Fisher Scientific, #20158) + 5% glycerol + 100 μg/mL tRNA+1× RNase inhibitor (Thermo Fisher Scientific, #N8080119)), and then incubated for 30 min at room temperature (25 °C). The reaction mixture was then loaded onto a 6% acrylamide native gel and run the electrophoresis at 4 °C. Shift band intensities were captured by Typhoone FLA-9000 (GE).
Nuclear/cytoplasmic protein extraction and Western-blots
The nuclear/cytoplasmic protein extraction was performed in the same way as previous reports [Citation36]. Cell pellets were collected and lysed on ice for 5 min in lysis buffer (50 mM Tris-HCl (pH = 7.4) + 10 mM NaCl + 0.5% NP-40 + 0.25% Triton X-100 + 1 mM EDTA + 1× Complete protease inhibitor (Roche, #04693132001)) and centrifuged at 3000 × g at 4 °C for 5 min. The nuclear compartments were in the precipitates, and the supernatants then contained cytoplasmic proteins. Then the precipitates were washed with the lysis buffer once and resuspended in the NET Buffer (20 mM HEPES + 1.5 mM MgCl2 + 0.5 M NaCl + 0.2 mM EDTA + 20% glycerol + 1% Triton X-100), sonicated for 0.8 minutes at 17 W, and then centrifuged at 10000 × g at 4 °C for 10 min. The supernatants collected at this time then contains specific proteins from the nuclei. Western-blots for the nuclei marker Lamin A/C and cytoplasmic marker β-Tubulin were performed to confirm the nuclei/cytoplasm separation (, , & , ). For whole cell lysates, 106 cells were collected and resuspended in 100 μl PBS-Triton X Buffer (1 × PBS + 1% Triton X-100) with 1× Complete protease inhibitor (Roche, #04693132001) on ice for 30 min. The suspension was sonicated for 0.8 minutes at 17 W and then centrifuged at 20000 × g at 4 °C for 10 min. The supernatant was kept as the total cell protein. All the protein samples were then loaded and transferred onto nitrocellulose membranes for Western-blots. Commercially purchased antibodies include anti-HuR (Proteintech, #11910-1-AP), anti-β-Tubulin (Abcam, #ab6046), anti-Lamin A/C (Proteintech, #10298-1-AP), anti-p38-β2 (Invitrogen, #33-8700). The specificity of all antibodies has been validated by previous reports or our knockdown or knockout experiments.
Immunofluorescence and high-content imaging
For immunofluorescence, cultured cells were washed three times with 1× PBS and then fixed in 4% paraformaldehyde (PFA) for 15 minutes, followed by permeablizing in 0.5% TritonX-100 for 10 min. The cells were then blocked in 4% BSA + 0.1% Triton X-100 in 1× PBS and incubated with primary antibodies at 4°C overnight, and then washed with blocking buffer and incubated with secondary antibody at room temperature for 1 h. The samples were then washed, 0.5 mg/ml DAPI (Beyotime Biotechnology, #C1002) was added for 5 min before the cells were final washed and stored in 1× PBS. The images of samples were scanned and then analysed by the high-content imaging system Operetta CLS (PerkinElmier) and its accompanying software Harmony 4.8. The bean plots of the analysed data were generated by online application: http://shiny.chemgrid.org/boxplotr, which was developed according to the description of Nature Method [Citation37].
Statistical analysis
Statistical comparisons between two groups were conducted by the two-tailed unpaired Student’s t-tests. Statistical comparisons among multiple groups were conducted by ANOVA tests. Significance was established at P < 0.05. In all graphs, error bars indicate SEM, and the biological replicate numbers are indicated on top of each bar and/or as the n numbers in the legends. The statistical powers for all analysis were calculated and confirmed to be > 80%. Data were excluded when there were clear indications of the artefact or experimental failures, such as contamination, transfection/infection failure, etc.
Supplemental Material
Download Zip (2.1 MB)Acknowledgments
We would like to thank Dr. Yang Li for kindly providing the pmirGLO plasmids. We would like to thank Drs. Jianhua Gan and Binglian Zheng for providing insightful discussion and technical supports.
Data availability
The CLIP-seq data analysis results were obtained from the previously published database StarBase, which is available at: http://starbase.sysu.edu.cn/;
The RBP expression was analyzed by the BioGPS database, which is available at: http://biogps.org/#goto=welcome
The accession numbers of the genes relevant to this study include:
HTT-huntingtin [Homo sapiens] (hsHTT): Gene ID 3064
Htt-huntingtin [Mus musculus] (msHTT): Gene ID 15194
ELAVL1-HuR [Homo sapiens] (hsHuR): Gene ID 1994
Elavl1-HuR [Mus musculus] (msHuR): Gene ID 15568
MAPK11 [Homo sapiens] (hsMAPK11): Gene ID 5600
Mapk11 [Mus musculus] (msMAPK11): Gene ID 19094
All the gene information is available at: https://www.ncbi.nlm.nih.gov/gene
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
The supplemantary data for this article can be accessed here.
Additional information
Funding
References
- TheHuntington’sDiseaseCollaborativeResearchGroup. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell. 1993;72:971–983.
- Rubinsztein DC, Carmichael J. Huntington’s disease: molecular basis of neurodegeneration. Expert Rev Mol Med. 2003;5:1–21.
- Feng X, Luo S, Lu B. Conformation polymorphism of polyglutamine proteins. Trends Biochem Sci. 2018;43:424–435.
- Rodriguez-Lebron E, Denovan-Wright EM, Nash K, et al. Intrastriatal rAAV-mediated delivery of anti-huntingtin shRNAs induces partial reversal of disease progression in R6/1 Huntington’s disease transgenic mice. Mol Ther. 2005;12:618–633.
- Harper SQ, Staber PD, He X, et al. RNA interference improves motor and neuropathological abnormalities in a Huntington’s disease mouse model. Proc Natl Acad Sci U S A. 2005;102:5820–5825.
- DiFiglia M, Sena-Esteves M, Chase K, et al. Therapeutic silencing of mutant huntingtin with siRNA attenuates striatal and cortical neuropathology and behavioral deficits. Proc Natl Acad Sci U S A. 2007;104:17204–17209.
- Kordasiewicz HB, Stanek LM, Wancewicz EV, et al. Sustained therapeutic reversal of Huntington’s disease by transient repression of huntingtin synthesis. Neuron. 2012;74:1031–1044.
- Tabrizi SJ, Leavitt BR, Landwehrmeyer GB, et al. Targeting Huntingtin expression in patients with Huntington’s disease. N Engl J Med. 2019;380:2307–2316.
- Chung DW, Rudnicki DD, Yu L, et al. A natural antisense transcript at the Huntington’s disease repeat locus regulates HTT expression. Hum Mol Genet. 2011;20:3467–3477.
- Ito D, Hatano M, Suzuki N. RNA binding proteins and the pathological cascade in ALS/FTD neurodegeneration. Sci Transl Med. 2017;9:eaah5436.
- Yu S, Liang Y, Palacino J, et al. Drugging unconventional targets: insights from Huntington’s disease. Trends Pharmacol Sci. 2014;35:53–62.
- Li JH, Liu S, Zhou H, et al. starBase v2.0: decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 2014;42:D92–97.
- Wu C, Jin X, Tsueng G, et al. BioGPS: building your own mash-up of gene annotations and expression profiles. Nucleic Acids Res. 2016;44:D313–316.
- Trettel F, Rigamonti D, Hilditch-Maguire P, et al. Dominant phenotypes produced by the HD mutation in STHdh(Q111) striatal cells. Hum Mol Genet. 2000;9:2799–2809.
- Peng SS, Chen CY, Xu N, et al. RNA stabilization by the AU-rich element binding protein, HuR, an ELAV protein. Embo J. 1998;17:3461–3470.
- Ke Y, Han Y, Guo X, et al. PARP1 promotes gene expression at the post-transcriptiona level by modulating the RNA-binding protein HuR. Nat Commun. 2017;8:14632.
- Fan XC, Steitz JA. Overexpression of HuR, a nuclear-cytoplasmic shuttling protein, increases the in vivo stability of ARE-containing mRNAs. Embo J. 1998;17:3448–3460.
- Fan XC, Steitz JA. HNS, a nuclear-cytoplasmic shuttling sequence in HuR. Proc Natl Acad Sci U S A. 1998;95:15293–15298.
- Yu M, Fu Y, Liang Y, et al. Suppression of MAPK11 or HIPK3 reduces mutant Huntingtin levels in Huntington’s disease models. Cell Res. 2017;27:1441–1465.
- Taylor DM, Moser R, Regulier E, et al. MAP kinase phosphatase 1 (MKP-1/DUSP1) is neuroprotective in Huntington’s disease via additive effects of JNK and p38 inhibition. J Neurosci. 2013;33:2313–2325.
- Joassard OR, Belanger G, Karmouch J, et al. HuR mediates changes in the stability of AChR beta-subunit mRNAs after skeletal muscle denervation. J Neurosci. 2015;35:10949–10962.
- Xiao L, Li X, Chung HK, et al. RNA-binding protein HuR regulates paneth cell function by altering membrane localization of TLR2 via posttranscriptional control of CNPY3. Gastroenterology. 2019;157:731–743.
- Jain A, Vale RD. RNA phase transitions in repeat expansion disorders. Nature. 2017;546:243–247.
- Swami M, Hendricks AE, Gillis T, et al. Somatic expansion of the Huntington’s disease CAG repeat in the brain is associated with an earlier age of disease onset. Hum Mol Genet. 2009;18:3039–3047.
- Tabrizi SJ, Ghosh R, Leavitt BR. Huntingtin lowering strategies for disease modification in Huntington’s disease. Neuron. 2019;102:899.
- Genetic Modifiers of Huntington’s Disease Consortium. Electronic address, g.h.m.h.e. and Genetic Modifiers of Huntington’s Disease, C. CAG repeat not polyglutamine length determines timing of Huntington’s disease onset. Cell. 2019;178:887–900 e814.
- Frantz B, Klatt T, Pang M, et al. The activation state of p38 mitogen-activated protein kinase determines the efficiency of ATP competition for pyridinylimidazole inhibitor binding. Biochemistry. 1998;37:13846–13853.
- Lang M, Berry D, Passecker K, et al. HuR small-molecule inhibitor elicits differential effects in adenomatosis polyposis and colorectal carcinogenesis. Cancer Res. 2017;77:2424–2438.
- Saudou F, Humbert S. The biology of Huntingtin. Neuron. 2016;89:910–926.
- Kim EK, Choi EJ. Compromised MAPK signaling in human diseases: an update. Arch Toxicol. 2015;89:867–882.
- Buren C, Wang L, Smith-Dijak A, et al. Region-specific pro-survival signaling and global neuronal protection by wild-type Huntingtin. J Huntingtons Dis. 2014;3:365–376.
- Bradham C, McClay DR. p38 MAPK in development and cancer. Cell Cycle. 2006;5:824–828.
- Zeitlin S, Liu JP, Chapman DL, et al. Increased apoptosis and early embryonic lethality in mice nullizygous for the Huntington’s disease gene homologue. Nat Genet. 1995;11:155–163.
- Nasir J, Floresco SB, O’Kusky JR, et al. Targeted disruption of the Huntington’s disease gene results in embryonic lethality and behavioral and morphological changes in heterozygotes. Cell. 1995;81:811–823.
- Lu B, Al-Ramahi I, Valencia A, et al. Identification of NUB1 as a suppressor of mutant Huntington toxicity via enhanced protein clearance. Nat Neurosci. 2013;16:562–570.
- Tollervey JR, Curk T, Rogelj B, et al. Characterizing the RNA targets and position-dependent splicing regulation by TDP-43. Nat Neurosci. 2011;14:452–458.
- Spitzer M, Wildenhain J, Rappsilber J, Tyers M. Boxplotr: a web tool for generation of box plots. Nature Methods. 2014;11:121–122. DOI: 10.1038/nmeth.2811