
ABSTRACT
Cellular mRNAs are exported from the nucleus as fully spliced RNAs. Proofreading mechanisms eliminate unprocessed and irregular pre-mRNAs to control the quality of gene expression. Retroviruses need to export partially spliced and unspliced full-length RNAs to the cytoplasm where they serve as templates for protein synthesis and/or as encapsidated RNA in progeny viruses. Genetically complex retroviruses such as HIV-1 use Rev-equivalent proteins to export intron-retaining RNA from the nucleus using the cellular CRM1-driven nuclear export machinery. By contrast, genetically simpler retroviruses such as murine leukaemia virus (MLV) recruit the NXF1 RNA export machinery. In this study, we reveal for the first time that MLV hijacks both NXF1 and CRM1-dependent pathways to achieve optimal replication capacity. The CRM1-pathway marks the MLV full-length RNA (FL RNA) for packaging, while NXF1-driven nuclear export is coupled to translation. Thus, the cytoplasmic function of the viral RNA is determined early in the nucleus. Depending on the nature of ribonucleoprotein complex formed on FL RNA cargo in the nucleus, the FL RNA will be addressed to the translation machinery sites or to the virus-assembly sites at the plasma membrane.
Introduction
In general, the therapeutic arsenal against viral infection targets viral components. However, this strategy could favour the emergence of resistant pathogenic strains. New strategies emerge targeting cellular functions, those manipulated by viruses to replicate. Identification of the cellular functions used by viruses has already led to major contributions to cell biology. Thus, viruses are invaluable tools for discovering and for further understanding key pathways: such as alternative splicing controlled by adenoviruses[Citation1], IRES-translation used by picornaviruses [Citation2] and nucleocytoplasmic transport machineries (NXF1/Tap [Citation3] and CRM1/exportin-1 [Citation4]) hijacked by retroviruses.
The Murine Leukaemia Virus (MLV) is classified in the simple retrovirus family since it encodes the three polyproteins common to all retroviruses: Gag, Pol and Env as well as the more recently discovered p50/p60 proteins [Citation5]. Whereas complex retroviruses (such as HIV) produce many alternatively spliced RNAs coding for accessory and regulatory proteins. Nevertheless, simple and complex retroviruses achieve similar life cycles. Thus, MLV, with its minimal coding capacity, is an excellent manipulator of cellular functions. This makes MLV an attractive model to study the molecular mechanisms underlying the relationship between viral infection and cellular pathways.
Like other retroviruses, MLV has a genome that is a capped and polyadenylated positive-sense RNA about 8Kb long and flanked by two long terminal repeats (LTR). In virions, the genome consists of two homologous RNA molecules forming a dimer. After virus entry, the genome is reverse transcribed by the viral reverse transcriptase and the cDNA imported into the nucleus is integrated into the host chromosome. After transcription by the cellular machinery, a subset of viral full-length (FL) transcripts escapes splicing and exits the nucleus despite the persistence of introns. Once in the cytoplasm, the FL RNA will be either translated to provide Gag and GagPol proteins or targeted to the plasma membrane to be packaged as a genome into assembling virions. Sorting mechanism of these two RNA fates (translation/packaging) remains unknown [Citation6]. Several stem-loops mapped in the 5ʹ UTR of the FL RNA drive the RNA packaging selection [Citation7–Citation9] and also influence the nuclear export of the FL RNA [Citation10,Citation11]. But when and where the RNA selection occurs remain open questions. Here we asked whether the RNA fate might depend to the pathway used by MLV to exit the nucleus.
The mechanism by which MLV hijacks the cellular mRNA export pathway remains largely unknown. In contrast to the family of complex lentiviruses (i.e. HIV-1), simple retroviruses do not encode homologues of the Rev protein, which mediates CRM1-dependent export of HIV-1 FL RNA [Citation12]. Instead, simple unspliced RNAs are exported due to the presence of a cis-acting sequence called the constitutive transport element (CTE) recognized by the cellular NXF1/Tap-dependent export machinery, as well described for the Mazon-Pfizer monkey virus (MPMV) [Citation3]. Recent studies show that MLV also uses the NXF1/Tap pathway to export both viral unspliced and spliced RNAs [Citation13–Citation16]. MLV sequences acting as CTE-like have been reported located in different positions in the 5ʹLTR, the packaging signal (Psi) and within the pol gene (reviewed in of [Citation17]). This multiplicity of signals implies a more complex mechanism for MLV than that for MPMV. The species context used to study MLV export should also be taken into account, since pathways may differ between murine and human cells [Citation18,Citation19 and personal data]. First data in 2014 revealed that the dependence of MLV export on the NXF1 machinery impacts FL RNA translation, since blocking NXF1 pathway reduces Gag protein levels in both murine [Citation13] and human [Citation15] cells. In contrast to HIV-1, spliced and unspliced MLV RNAs take the same route to reach the cytoplasm in order to be translated. However, they recruit different cellular co-factors since only the FL RNA harbours a CTE-like structure, named γ-CTE [Citation14], within the Post Transcription Element (PTE) region [Citation16] in pol intron. Some subunits from the TREX complex (THOC7, THOC5 and UAP56) and SRp20 proteins have been identified to promote the interaction between FL RNA and NXF1 coupled to p15 while only UAP6 interacts with NXF1-p15 for the export of MLV spliced RNA [Citation14,Citation15]. SRp20 links export and translation by promoting RNA association with polysomes.
In the present study, we investigated the possibility of a second nucleocytoplasmic export pathway used by MLV. We show for the first time that the cytoplasmic level of MLV FL RNA in murine cells depends on the CRM1 machinery. Strikingly, although blocking the CRM1 pathway retains the FL RNA in the nucleus, intracellular levels of Gag protein were not decreased and Gag continued in lower extent to assemble and release virus particles. However, analysis of virus particles content revealed decreased packaging of FL RNA. These results show that export by CRM1 machinery marks the FL RNA for packaging, while export by NXF1 pathway designates transcripts for translation. In conclusion, our study revealed that FL RNA forms different nuclear ribonucleoprotein complexes whose composition determines the cytoplasmic function of the viral RNA.
Results
MLV FL RNA uses the CRM1 pathway to exit the nucleus
In 2014, several studies demonstrated that MLV FL RNA nuclear export is NXF1-dependent [Citation13–Citation17,Citation20]. Interestingly, other retroviruses, such as HIV or Rous sarcoma virus (RSV), hijack the CRM1 pathway for this purpose (for review [Citation21,Citation22]). Because post-transcriptional RNA trafficking is a complex process, we aimed to determine if CRM1 plays any role in MLV FL RNA transport or if the process is exclusively governed by NXF1. Taking advantage that the Leptomycin B (LMB) drug inhibits the CRM1 pathway, we used single-molecule fluorescence in situ hybridization method (sm-FISH) to visualize FL RNA in murine cells treated with (5nM, 20 h) or without LMB (). Detection of MS2-FL RNA was achieved by sm-FISH using fluorescent DNA probes that specifically hybridize to 24 copies of the bacteriophage MS2 stem-loop (MLV-MS2-WT) inserted into the major intron so they are present in single FL RNA molecules but not in viral-spliced transcripts (). The MS2-tagged MLV produces Gag proteins and releases virions (). MLV-MS2-WT was stably expressed in NIH3T3 cells and FL RNAs are detected using the MS2 DNA probes conjugated to fluorophore AF488 (). As shown in , cells were imaged on a 63× NA 1.4 wide-field microscope (Zeiss AxioimagerZ1). Levels of nuclear FL RNA were determined for each condition for n = 86 cells based on fluorescence intensity measured using Image J software (). As shown in , the FL RNA signal was located in both nucleus and cytosol (nuclear average of microscopy fluorescence intensity (Avg. MFI) of 55 ± 2%), similar to what we have reported previously [Citation13]. Strikingly, however, treatment with LMB markedly increased levels of nuclear FL RNA fluorescence intensity (avg. MFI of 76 ± 2,5%), consistent with the hypothesis that CRM1 plays a role in FL RNA nuclear export.
To compare the relative contributions of CRM1 and NXF1 to MLV FL RNA nuclear export, we also compared MS2-tagged FL RNAs from ‘wild-type’ MLV to an MLV mutant virus harbouring a deletion of 512nt (position 2617–3129) within the Post Transcription Element (PTE) region, removing the entire γCTE (87nt) shown in human cells to be important for nuclear export of MLV FL RNA via the NXF1 pathway (MLV-MS2-∆γCTE, ). As anticipated, p65Gag levels were reduced for the ∆γCTE virus relative to the wild-type MLV construct when we expressed in mouse NIH3T3 cells (). Additionally, we observed a marked increase to the ∆γCTE FL RNA signal in the nucleus (avg. MFI of 72 ± 2%) relative to wild-type FL RNA (-). LMB treatment increased the nuclear signal for both wild-type and ∆γCTE FL RNAs, suggesting that MLV hijacks both CRM1 and NXF1 pathways.
Figure 1. Intracellular localization of FL RNA after NXF1 and/or CRM1 inhibition. (A) Schematic representation of the plasmids used in the experiments. MS2 stem loops insertion and γCTE deletion are shown. The fluorescent probes used to specifically detect the FL RNA are marked in green. (B) Western blotting of protein extracted from NIH3T3 cells transfected with the different constructs and from purified virions. The expressions of Gag (65 kDa) and Actin (42 kDa) are shown. (C) Representative images of MS2-tagged RNA (green) detected by sm-FISH in cells expressing WT or ∆γCTE MLV, treated or not with LMB. The nuclei are stained with DAPI (blue). For each cell, at least 11 stacks were taken and the image resulted from the sum of all stacks for each condition (D) Values of nuclear FL RNA levels are presented as the mean ± SEM of 86 cells from at least three independent transfections. The significance of differences between conditions was assessed using an unpaired Student’s t-test (****P ≤ 0.0001).
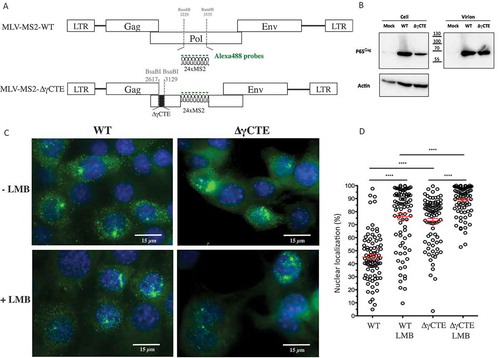
Figure 2. Impact of CRM1 on MLV translation. (A) Gag protein analysis. NIH3T3 cells chronically infected with MLV were treated or not with LMB. The expressions of Gag (65 kDa), Capsid (30 kDa) and Actin (42 kDa) are shown. The blot is a representative experiment among three independent assays. Band integrated densities were determined with the ImageJ software and normalized to Actin. Intensities are presented in the histogram as mean +/− SEM and the significance of differences with control (MLV-LMB) was assessed using an unpaired student’s t-test (*P ≤ 0.05). (B) Effect of LMB on MLV transcription in cells. Intracellular FL RNA levels were specifically determined by RT-qPCR and normalized to the copy numbers of GAPDH mRNA. Percentages are given in graph and are not significantly distinct between ± LMB as assessed by unpaired student’s t-test (nsP > 0.05).
Blocking CRM1 pathway did not prevent Gag translation
In a previous study, we demonstrated that NXF1 inhibition induced nuclear FL RNA retention and subsequent defect in Gag translation [Citation13]. As inhibition of CRM1 involves also a nuclear accumulation of FL RNA, we next investigate the impact of such RNA retention on Gag translation. NIH 3T3 cells chronically infected with MLV were treated with 5nM of LMB over 20 h [Citation23]. Total proteins were extracted and separated by PAGE to monitor the expression of Gag protein by western blot. Interestingly, LMB treatment did not decrease Gag translation. On the contrary, the quantifications of western blot revealed a slight increase (avg. of 25 ± 8%) of intracellular Gag production caused by LMB (). This effect is not due to an increase of FL RNA transcription since LMB did not alter levels of intracellular FL RNA as measured by RT-qPCR (). Despite higher levels of cell-associated Gag, particle production was actually decreased 27 ± 7% compared to untreated cells. In contrast to NXF1 [Citation13], CRM1-dependent export has little or no role in MLV translation.
CRM1 pathway regulates FL RNA packaging into virions
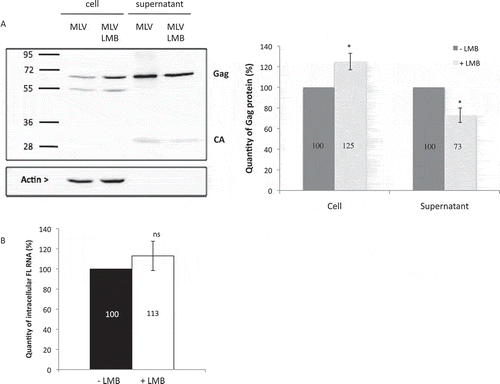
To further understand the importance of the CRM1 export pathway to MLV replication, we studied the packaging FL RNA into virus particles using fluorescence microscopy [Citation24]. Gag-GFP was expressed in murine cells with an MLV RNA reporter harbouring the 24xMS2 tag to be packaged inside GFP virions (). Two days after transfection, filtered supernatants were ultracentrifugated and RNA detected in virus particles using an MS2-Cy3 DNA probe, as previously described [Citation25]. Packaging was calculated as the ratio Gag-GFP particles with Cy3-labelled RNA to total Gag-GFP particles [Citation24]. The specificity of the RNA reporter was confirmed using a mutant FL RNA sequence wherein the MLV RNA packaging signal (Psi) was abolished, yielding a MS2-Cy3 signal ~10-fold lower than wild-type [Citation7,Citation10,Citation26] (-). Interestingly, when cells were treated with LMB, the level of the reporter RNA associated with virus particles was severely reduced (avg. of 55 ± 12%) relative to vehicle control.
Figure 3. Impact of CRM1 pathway on packaging of viral reporter RNA. (A) Schematic representation of the plasmids used in microscopy experiments of panel b. Reporter-MS2 plasmids express a viral MS2-tagged RNA reporter harbouring wild-type (WT) or deleted (∆Psi) MLV packaging signal. Cy3-labelled probes used in sm-FISH which interact specifically with MS2 stem loops, are shown. MLV splice donor and acceptor sites (SD and SA, respectively) are drawn. MLV Gag proteins fused to GFP are expressed in trans by the pCAG-GAG-GFP vector. (B) Representative image of virus sample obtained by sm-FISH. Bottom left image corresponds to zoom in area of the image (white square). Gag with RNA corresponds to red-green dots colocalization. The graph shows the calculated ratios of colocalized red-green dots/total green dots. Percentages are presented as mean ± SEM and the significance of differences was assessed using an unpaired student’s t-test (****P ≤ 0.0001 and ***P ≤ 0.001). More than 1500 viral like-particles were analysed per condition. (C) Effects of LMB on the packaging of native FL RNA in infectious context. Chronically infected NIH 3T3 cells were treated or not with LMB and RNA analyses conducted by RT-qPCR as described in materials and methods. The packaging efficiency was calculated as the ratio of cellular FL RNA copies/FL RNA copies in virus. Percentages are presented as mean ± SEM and the significance of differences with control (MLV WT) was assessed using an unpaired student’s t-test (****P ≤ 0.0001).
In parallel, packaging was assessed with LMB treatment for FL RNA using non-modified, replication-competent MLV using RT-qPCR. In this experiment, NIH3T3 cells were chronically infected with MLV prior to being treated with or without LMB for 20 h. RNA was extracted from cells and virions and reverse transcribed with oligo dT or MLV primer. Two sets of primers were used to quantify either GAPDH mRNA (for normalization) or FL RNA (see Methods section for sequences). FL RNA packaging values correspond to the ratio of FL RNA detected in virions relative to FL RNA detected in cells (FL RNAv/FL RNAc). Consistent with the results from our trans-packaging system () LMB caused a reduction of FL RNA packaging of 66 ± 10% ().
Altogether, these results, obtained by two complementary approaches showed that CRM1 is involved in the packaging process and suggest a link between packaging events and CRM1-dependent MLV FL RNA nuclear export.
Nuclear export pathway determines the fate of cytoplasmic FL RNA
The above experiments indicated that MLV exploits both pathways, NXF1 and CRM1, to export FL RNA from the nucleus to the cytoplasm. Accordingly, it was tempting to speculate that each of these pathways is linked to a different FL RNA function in the cytoplasm; either as mRNA encoding Gag or genome packaged into new viral particles. In a previous study, we showed that inhibiting the NXF1 pathway using a dominant-negative NXF1 mutant (Tap∆C) or competing for NXF1 by overexpressing a 4xCTE RNA construct abolished Gag translation [Citation17].
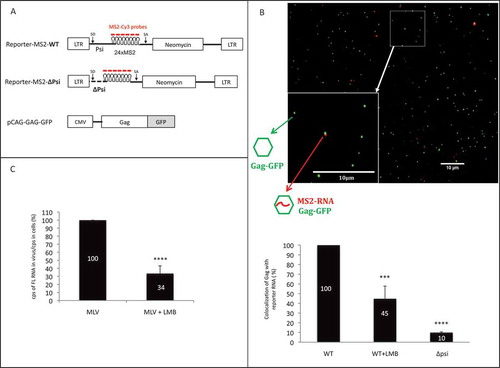
To test the relevance of NXF1 to genome packaging, we constructed an MLV molecular clone harbouring the 24xMS2 tag in which the gag reading frame was left intact with GFP inserted downstream of the Gag p12 domain. This construct allowed us to study virus particles visually wherein FL RNA and Gag-GFP proteins were expressed at native levels, and for viral constructs either encoding or lacking the γCTE (). The detection of viral particles with or without FL RNA is described in [Citation24]. Briefly, after virions purification, viral GFP particles harbouring FL RNA are tagged with mCherry, as the MS2 coat-mCherry protein interacts with MS2 stem loops on FL RNA. Inverted microscope with 100× oil objective is used for epifluorescence microscopy. Image analysis is performed with algorithms customized from ImageJ software to reduce the background intensities of the image and to achieve packaging efficiency calculation (ratio Gag-RNA/total Gag). As negative control, we used a Non-Viral RNA (NV-MS2), which harbour MS2 stem loops () [Citation25] but is not packaged in neo-particles.
Figure 4. Influence of CRM1 and/or NXF1 inhibition on FL RNA packaging. (A) Schematic maps of the MLV clones expressed into the NIH3T3 cells. The insertion of GFP in gag gene, is represented in grey and enables the production of Gag in cis in native level. The NV-MS2 construct producing a non-viral RNA was used as negative control as described in [Citation25]. The visualization of MS2-tagged RNA was based on the interaction with the MS2 coat protein fused to mCherry (dark grey) and co-transfected with the MLV plasmids. (B) The graph represents the packaging ability of MLV in each condition. The number of grey dots colocalized with dark grey dots was determined and packaging efficiencies were calculated as the ratios of Gag with RNA: total Gag. n ≥ 700 virions. Percentages are presented as mean ± SEM.
![Figure 4. Influence of CRM1 and/or NXF1 inhibition on FL RNA packaging. (A) Schematic maps of the MLV clones expressed into the NIH3T3 cells. The insertion of GFP in gag gene, is represented in grey and enables the production of Gag in cis in native level. The NV-MS2 construct producing a non-viral RNA was used as negative control as described in [Citation25]. The visualization of MS2-tagged RNA was based on the interaction with the MS2 coat protein fused to mCherry (dark grey) and co-transfected with the MLV plasmids. (B) The graph represents the packaging ability of MLV in each condition. The number of grey dots colocalized with dark grey dots was determined and packaging efficiencies were calculated as the ratios of Gag with RNA: total Gag. n ≥ 700 virions. Percentages are presented as mean ± SEM.](/cms/asset/bfae44f1-6e56-47f6-bfc6-2af354375ed1/krnb_a_1713539_f0004_b.gif)
Using this strategy, we determined FL RNA packaging efficiencies for three different contexts: (1) wherein CRM1 was blocked (+LMB); (2) wherein the FL RNA’s association with NXF1 was reduced (∆γCTE-RNA); and wherein both pathways were impaired simultaneously (∆γCTE+LMB) (). Whereas we observed no significant decrease to packaging efficiency when FL RNA was exported in the absence of LMB (comparison WT to ∆γCTE), there was a strong decrease in packaging for both wild-type and ∆γCTE viruses when LMB was added (e.g. decrease of 41% from WT to WT+LMB). These results reinforced that while the γCTE (and, by proxy, NXF1) regulates Gag synthesis, it has little to no role in modulating packaging events. By contrast, LMB (and, by proxy, CRM1) affects FL RNA packaging.
Discussion
In contrast to cellular mRNA, genomic RNA of retroviruses is required to be exported from cytoplasm to nucleus in an unspliced form. Indeed, the persistence of viral full-length RNA, generated by host transcriptional machinery, is crucial for the viral replication. Once in the cytoplasm, the FL RNA serves as template for Gag and Gag-Pol translation but also as genome for neo particles. Thus, nuclear export of FL RNA is a key step of viral life-cycle and it is important to gain insight into its mechanism. While MLV is considered a prototype for gamma retroviruses, until 2014 nothing was known about its export mechanism. Although we and others previously demonstrated that MLV FL RNA hijacks the NXF1 cellular pathway to go through the nuclear pore [Citation13–Citation17], here we show that MLV also exploits the CRM1-dependent pathway.
By using infectious MLV viruses, expressed in murine cells, we show that inhibition of either NXF1 or CRM1 increases FL RNA nuclear abundance by 27 ± 2% or 31 ± 2%, respectively (). Similar levels of nuclear retention for each pathway suggest that FL RNA intended for one export pathway is unable to access the other. Accordingly, we observed an additive effect when both NXF1 and CRM1 were inhibited (). Several explanations could account for this dual-dependence. First, both CRM1 and NXF1 are used by some cellular RNAs for their own nuclear export (for review [Citation26]), so that during infection only a subset of NXF1 or CRM1 molecules may be available for recruitment by individual FL RNA molecules. This hypothesis is supported by the fact that both NXF1 and CRM1 pathways are saturable [Citation17,Citation27–Citation29]. Another explanation could be that the MLV nuclear export pathway is more complex than originally anticipated. To date, retroviruses are thought to directly recruit NXF1 to defined cis-acting RNA elements [Citation3,Citation15,Citation17,Citation30–Citation32]. By contrast, CRM1 is not an RNA-binding protein and is thought to engage RNA only through activity of adaptor proteins that harbour a CRM1-specific nuclear export signal (NES). For genetically complex retroviruses (e.g. lentiviruses and deltaretroviruses that encode several accessory factors) this adaptor protein is the viral Rev or Rex protein, that homomultimerizes on FL RNA cis-acting Rev or Rex response elements, respectively; or, for a subset of genetically simple retroviruses such as RSV a ‘Rev-like’ role has been ascribed to the structural Gag protein [Citation33–Citation38]. For MLV there is no evidence yet that Gag is involved in CRM1-dependent RNA nuclear export although few Gag molecules was found in nucleus which could play the role of ‘Rev-like protein’[Citation39].
Our sm-FISH studies showed that MLV uses both CRM1 and NXF1 pathway to accumulate FL RNA in cytosol. What would be the benefit for MLV to use two export pathways? Classical experiments using inhibitors for transcription demonstrated that, for lentivirus (i.e. HIV), there is likely to be no functional separation between the packaged and translating pools of FL RNA [Citation6]. By contrast, it appears that for gammaretroviruses a subset of cytoplasmic FL RNAs is designated for packaging, leading to the existence of two pools of cytoplasmic FL RNA with different half-lives, one for translation and the other for packaging [Citation6,Citation40]. The existence of two different groups of cytoplasmic FL RNA implies that cellular translation machinery and viral assembly complexes compete for cytoplasmic utilization of FL RNA. How and when FL RNA is driven to translation or packaging is still unknown. One of tempting hypothesis is that discrimination is made earlier in the nucleus and is based on how the RNA arrived into the cytoplasm (). This hypothesis was well supported by the model of avian retroviruses (RSV) [Citation41,Citation42] and also highly suggested for another simple retrovirus: MPMV [Citation43]. Whereas CRM1 drives the FL RNA in cytosol in a non-localized fashion, NXF1 links FL RNA to microtubules to drive them until the microtubule-organizing centre (MTOC) in the cytoplasm [Citation44] to probably direct them to a polysome-rich environment [Citation14]. Accordingly, the cytoplasmic fate of FL RNA is pre-programmed by the nuclear export factors that interact with it ().
Figure 5. Current model of the MLV nuclear export and its impact on the outcome of the FL RNA in the cytoplasm. The model proposed is based on our present results and published data (arrow in full line) as well as hypothesis (dotted arrow). Briefly, the FL RNA is firstly driven to the NXF1 pathway probably because of its methylation. Because of cis element structures present in pol gene [Citation14,Citation16], NXF1 interacts with FL RNA and drives it through the nuclear pore until to reach the MTOC [Citation44]. Then, FL RNA is directed until polysome [Citation14] and is translated in Gag proteins. If a few amount of Gag proteins could reach the nucleus [Citation39], it could serve as viral partner for CRM1 interaction to export FL RNA. Then, FL RNA complexed to Gag targets the packaging sites at the plasma membrane to form neo particles.
![Figure 5. Current model of the MLV nuclear export and its impact on the outcome of the FL RNA in the cytoplasm. The model proposed is based on our present results and published data (arrow in full line) as well as hypothesis (dotted arrow). Briefly, the FL RNA is firstly driven to the NXF1 pathway probably because of its methylation. Because of cis element structures present in pol gene [Citation14,Citation16], NXF1 interacts with FL RNA and drives it through the nuclear pore until to reach the MTOC [Citation44]. Then, FL RNA is directed until polysome [Citation14] and is translated in Gag proteins. If a few amount of Gag proteins could reach the nucleus [Citation39], it could serve as viral partner for CRM1 interaction to export FL RNA. Then, FL RNA complexed to Gag targets the packaging sites at the plasma membrane to form neo particles.](/cms/asset/f1fb1160-31e6-409e-9bfd-6114fdcae53d/krnb_a_1713539_f0005_oc.jpg)
Up to now, there was no direct evidence linking the fate of FL RNA and its export mode. In this study, we discriminated both MLV nuclear exports by deleting the cis-element sequences important for NXF1 action on FL RNA and by using CRM1 inhibitor with regards to the RNA cytoplasmic functions (–). The role of the γCTE (nt 2918–3016) alone or within a larger structure named PTE (nt 2238–3705) in recruiting NXF1 and favouring polysomes loading on the FL RNA was previously identified in human cells [Citation14]. But, this is the first time that the roles of γCTE and PTE in export and Gag translation were studied in the context of an MLV genome and in murine cells. In our system, however, we did not observe a complete abolishment of Gag production as reported by both the Luban and Felber groups [Citation14,Citation16]. This difference may be attributable to the different species context, since mechanisms of cellular pathways can differ between human and murine cells [Citation18,Citation19,Citation45]. It can be considered that under NXF1 inhibition, a part of the CRM1-exported RNA could be translated. Also, we showed that NXF1 inhibition does not perturb FL RNA packaging (), suggesting that NXF1 pathway is preferentially used by MLV to drive the FL RNA to the translation of Gag and Gag-Pol proteins. In contrast, an ~50% decrease of packaging was observed when treating murine cells infected with MLV with LMB ( and ). These results revealed, for the first time, a role of CRM1 in MLV RNA packaging and suggested that the export pathway used determines the cytoplasmic fate of MLV FL RNA.
The molecular mechanism that regulates the switch between the two export pathways is unknown. Several elements could account to this choice. First, several cis-acting sequences throughout the FL RNA might interfere with CRM1 and NXF1 since multiple cis-acting sequences have been identified [Citation10,Citation11,Citation16,Citation20,Citation46,Citation47]. Except for γCTE [Citation14] for the majority of these elements, their role is undeciphered. We do not know yet which viral or cellular partners they might recruit and it is likely that they act under a fine spatiotemporal regulation. Second, it is possible that MLV Gag or another, yet to be identified, viral element is important to this dual-pathway. Indeed, the alphatretrovirus RSV is purported to use its Gag protein to transfer FL RNA from the nucleus to the cytosol via the CRM1 pathway [Citation41]. In the nucleus, RSV Gag recruits the FL RNA via an interaction with the packaging signal located in its 5ʹ end [Citation42]. This then forms a Gag/FL RNA complex that recruits CRM1 using Gag’s NES [Citation34,Citation48]. Concerning MLV, despite Gag harbours putative NLS and NES, its nuclear presence is controversial [Citation49]. This is likely due to the amount of MLV Gag trafficking in the nucleus being significantly lower than that observed for RSV [Citation49]. FL RNA packaging is highly facilitated by FL RNA dimerization and by Gag (for review [Citation50,Citation51]). MLV Gag interaction with the FL RNA is mainly mediated by the nucleocapsid domain (NC) [Citation52–Citation54]. There are several indirect evidences that MLV RNA dimerization, unlike HIV-1, occurs in nucleus and is coupled to transcription and splicing processes [Citation55,Citation56]. Indeed, MLV RNA transcribed from the same locus form dimers at higher frequencies than RNA transcribed from different loci [Citation57]. Based on these data, it is plausible that the selection of MLV FL RNA for CRM1 export, and then packaging, is made by the recognition of dimeric FL RNA by the nuclear Gag (). This correlates with the multiple roles ascribed to the Psi packaging signal in dimerization, packaging and nuclear export [Citation7,Citation10,Citation11,Citation57].
Finally, another way that specific pools of FL RNA are designated in the nucleus could rely on RNA modifications. Interestingly, epitranscriptomic methylation of A and C residues of MLV have been shown to enhance viral gene expression [Citation58]. These methylations are made by the “writer” proteins METTL3, METTL14, and WTAP in nucleus. Since Cullen and coll demonstrated a difference of methylation level between packaged FL RNA and cellular FL RNA, it is tempting to link the level of methylation of FL RNA and its recognition by NXF1 protein, leading to translation ().
In summary, we demonstrate that MLV exits the nucleus by two different pathways, NXF1 and CRM1. These two different RNP formed in nucleus mediates the cytoplasmic function of their respective FL RNA cargo. Our data indicated that NXF1 targets the FL RNA to the translation whereas CRM1 addresses the FL RNA to its packaging into viral particles (see model in ). This model considers the present results and published data as well as hypothesis. Following transcription, a pool of FL transcripts interacts with the NXF1 nuclear export machinery in part via the γCTE stem loop located in pol gene. The level of FL RNA methylation is potentially important to facilitate this interaction. The FL RNA/NXF1 complexes go through the nuclear pore and reach the microtubules-organizing centre (MTOC). Then, polysomes are loading on the FL RNA for translation and Gag proteins were synthesized. While most of Gag proteins serve for assembly of neoparticles at the plasma membrane, a minor subset of Gag molecules would reach the nucleus. Nuclear Gag could bind the FL RNA dimers, stabilizing the dimer structures. Then, this is under the form of Gag-RNA complexes that the FL RNA would use, directly or indirectly, CRM1 to exit the nucleus. Once in the cytoplasm, Gag-RNA complexes joined the virus-assembling sites at the plasma membrane to be incorporated inside new particles. The confirmation of this model requires further investigation.
Materials and methods
Plasmid constructions
Plasmids were constructed by using common cloning techniques [Citation59] and propagated in the E. coli STBL2 strain at 30°C to prevent any recombination. The pMov9.1 [Citation13] et pBSK-Eco [Citation60] plasmids correspond to the entire molecular clone of Moloney MLV (NCBI AF033811) cloned in different vectors. The plasmids pMov9.1-MS2 (MLV-MS2-WT) and pMov9.1∆ɣCTE-MS2 (MLV-MS2-∆ɣCTE) were constructed from the parental pMov9.1 with the substitution of a portion of the pol gene (positions 3235–3535 of MLV-WT) by 24 xMS2 hairpins by using BamHI digestion (). The portion of the pol gene (position 2617–3129) containing the γCTE sequence [Citation14] was deleted by the BsaBI digestion to give the pMov9.1∆ɣCTE-MS2 (). The MLV reporter plasmid (Reporter-MS2-PsiWT) was previously described in a previous study. For the reporter-MS2-∆Psi mutant, we inserted the 24-MS2 tag in the vector Pina10∆Psi [Citation7] by opening the vector at the BamHI site (nt 1070) (). The pCAG-Gag-GFP, expressing Gag fused to the GFP was described in [Citation61] (). pMCP-Cherry expressing the MS2-coat protein fused to the mCherry and non-viral MS2 reporter (NV-MS2) were previously described in [Citation25,Citation62]. The pBSKEco-p12-GFP-MS2 WT (MLV-GFP-MS2-WT) and ɣCTE (MLV-GFP-MS2-∆ɣCTE) version were made by fragment exchange between the MS2 hairpins and ɣCTE mutation of pMov9.1-MS2 or pMov9.1∆ɣCTE-MS2 and the pBSK-Eco, respectively, by using XhoI and HindIII sites. The Gag region encompassing the GFP comes from the plasmid pcDNA.MLVgp.EGFP.p12 (named GFP-p12) previously described in [Citation63]. The AflII fragment containing Gag-GFP of the plasmid GFP-p12 was introduced into pBSK-Eco-MS2 or pBSK-Eco-MS2-∆ɣCTE opened with AflII ().
Cell culture and transfection
NIH-3T3 and NIH3T3 chronically infected by Moloney MLV strain 8.2 [Citation64] were maintained in DMEM medium supplemented with penicillin-streptomycin (100 U/mL) and 10% heat-inactivated foetal bovine serum (FBS) at 37°C and 5% CO2. To establish the stable cell-line, NIH3T3 were co-transfected in a ratio of 50/1 with pMov9.1∆ɣCTE MS2 or pMov9.1 MS2 plasmids and the pGkNeo plasmid, bringing the neomycin resistance gene. The transfection was made with JetPei method according to the manufacturer’s instructions (Polyplus Transfection). Two days after transfection, G418 drug was added (100 mg/mL). After 1-month, total clones were trypsinized and transferred in a new dish to be further analysed by RTq-PCR and Western Blot.
Treatment with the Leptomycin B drug (LC Laboratories) was achieved as followed: 1.5 × 106 cells were seeded in 10-cm dishes and the next day, medium was removed and replaced by fresh medium ± LMB and incubation followed for 20 h. LMB was added to the final concentration of 5 nM [Citation23].
For MLV reporter, NIH 3T3 cells were transfected with 10 µg of plasmid (Pina10-MS2-Psi) and 40 h post transfection, the medium was changed and LMB was added or not for 8 h at 20 nM [Citation13]
Western blotting
Protein analysis was performed by western-blotting. Cells were scraped and lysed in the presence of protease inhibitor cocktail with the Complete lysis-M kit (Sigma) according to the manufacturer’s instructions. Total protein concentration was determined by Bradford assay using a BSA standard set (Fermentas). Virions were purified from culture medium by centrifugation at 1500xg for 5 min at 4°C and filtered through a 0.45-µm pore-size filter to eliminate cell debris before ultracentrifugation on a 20% sucrose/phosphate-buffered saline (PBS) cushion at 100 000 × g for 1.5 h at 4°C. Virus-containing pellets were then resuspended in 160 µL of PBS. Proteins (100 μg of cell lysate or 12.5% of virus lysate) were loaded on 12% SDS-PAGE and were electro-transferred onto nitrocellulose membrane. Gag was detected with a rat anti-capsid (CA) antibody (1/500, hybridoma H187, from B. Chesebro). Actin was detected with an anti-actin (1/500, Sigma). After incubation with a peroxidase-conjugated (HRP) secondary antibody, ECL fluorescence was recorded by a CCD chemiluminescence camera system (ChemiDoc™ MP Imaging System, Bio-Rad) and quantified by Image J software.
RNA analysis by RT-qPCR
Aliquots of cells and viral particle samples (respectively half and 2/3rd) were subjected to RNA analysis by RT-qPCR as previously described in [Citation52]. RNA was extracted from cells with TriReagent (MRC) according to the manufacturer’s instructions. RNA was extracted from virions by phenol extraction in the presence of 20 μg of tRNA carrier. All RNA samples were treated with RQ1 DNase (Promega) in the presence of RNasin (Promega) for 25 min at 37°C. RNA was extracted with phenol–chloroform then chloroform and finally precipitated with 100% ethanol. RNA pellets were washed with 70% ethanol and dissolved in water. RNAs were quantified by measuring optical absorption at 260 nm. For RT-qPCR, RNAs were reverse transcribed with Expand reverse transcriptase (Roche) either by oligo (dT) primer or by the same antisense primer used for the qPCR reaction. Quantitative PCR reactions were performed with 1/10th of the RT reaction as described in [Citation52]. Primers of the following oligonucleotides pairs were used to specifically amplify viral MLV FL RNA, and GAPDH mRNA for normalization: for FL aM275 5ʹ-GCAGGCGCATAAAATCAGTCATAG antisense and sFM76 5ʹ-GTGGTCTCGCTGTTCCTTGGGA sense; for GAPDH sGAPDH721 5ʹ-GCTCACTGGCATGGCCTTCCGTGT sense and aGAPDH921 5ʹ-TGGAAGAGTGGGAGTTGCTGTTGA antisense.
Single-molecule fluorescence in situ hybridization (sm-FISH)
NIH3T3 cells were grown on gelatin-treated coverslip while purified virions were loaded on gelatin-treated coverslip for 2 h @37°C [Citation25]. Cells and virions were fixed with Formaldehyde (3.7%formaldehyde/PBS) for 10 min at RT. After two washes in PBS, ethanol 70% was used for cell permeabilization 10 min at RT. The cells were incubated in FISH Wash solution (1× SSC/20% formamide (Sigma)) 15 min RT and then hybridized O/N at 37°C with the hybridization mix containing the fluorescent probe (1× SSC, 20% formamide, 10% dextran sulphate, 0.02% RNA grade BSA, 2 mM Vanadyl-R-C, 40 µg tRNA, MS2-Alexa488 probe (for MLV MS2 constructs)) or MS2-Cy3 probe (MLV reporter analysis). The probe was denatured in formamide for 1 min at 80°C, put in ice and then added to the hybridization mix. After overnight incubation coverslips were washed twice for 30 min at 37◦C in 1× SSC/20% formamide and then in PBS for 30 min at 37°C [Citation25]. Sequence of MS2 probe was described previously [Citation62]. Labelling of aminoallyl-T modified oligonucleotides was performed with Alexa Fluor® 488 Reactive Dye Decapack (Thermo Fisher Scientific) or Cy3 mono-Reactive Dye Pack (GE Healthcare) following the manufacturer’s instructions. Coverslips with virions were mounted with Vectashield (Vectorlabs) and coverslips with cells with vectashield+DAPI (diluted 1/2000) (Thermo Fisher Scientific). FISH-stained viral particles and cells were imaged using a wide-field DM6000 upright microscope with 100× and 63x oil objectives (Leica), respectively, and a Coolsnap HQ charge-coupled device camera (Roper Scientific) at Montpellier Rio Imaging (MRI) facilities. For cells imaging, stack images were taken by axial shifting of focal plane by 300 nm step from the bottom of the cell (cell-surface interface) to equatorial plane of the nucleus (situated to approximatively 5 microns from the substrate).
For virions analysis, dual-colour images were acquired with the excitation and emission filter sets of green fluorescent protein (GFP) and Cy3. For colocalization analysis, the centre positions of fluorescent spots were identified by their local maxima using a manually defined threshold in ImageJ software. To calculate the percentage of MS2 FL RNA (red spots) that colocalized with a Gag-GFP (green spots), the number of red spots that colocalized with a green spot was divided by the total number of Gag-GFP
Direct RNA imaging with MCP-cherry protein
1.6 × 106 NIH3T3 cells were co-transfected with a ratio (20:1) of MS2-tagged MLV-GFP or NV plasmids and the plasmid encoding the fluorescent coat proteins (MCP-mCherry) (). 24 h post-transfection, cells were trypsinized, washed with PBS and transferred in new plates with fresh medium with or without LMB 5nM and incubated for 20 h. Then, the supernatants were collected and virions were purified as described above for FISH. Two µL of virions samples are directly mounted between slide and coverslip and sealed with nail polish.
Samples were imaged by a wide-field Epifluorescence Nikon Ti2 inverted microscope controlled by the NIS Element software; the microscope is equipped with a 100x PL APO 1.4 NA oil-objective and a CMOS camera (back-illuminated Prime95B Photometrics) at Montpellier Rio Imaging (MRI) facilities. A Lumencor system with a QUAD LED was employed to acquire dual-colour images of the virions samples with the excitation and emission filter sets of green fluorescent protein (GFP) and Cy3. Different exposure times were used, 150 ms for GFP and 800 ms for Cy3. Images were recorded, covering decades of fields, in order to visualize more than 650 Gag-GFP green spots for each case. The image analysis was done by using the Fiji software. The masks of green spots were superposed with the corresponding image of red spots and the colocalized points were counted by ‘Analyse particle’ procedure.
Statistics
The statistical significance of data was evaluated using the Student’s-t-test in GraphPad Prism. P values are represented as follows: *P≤ 0.05, **P≤ 0.01, ***P≤ 0.001, ****P≤ 0.0001.
Disclosure of potential conflicts of interest
No potential conflict of interest was reported by the authors.
Acknowledgments
We are grateful to the Montpellier RIO Imaging staff for technical assistance. We thank C. Baum for the gifts of the pGFP-p12 plasmid and P. Bieniasz for the gifts of the pCag-GAG-GFP. Many thanks to Dr N. Sherer for critical reading of the manuscript. This work was supported by institutional grants of the CNRS, the University of Montpellier and the Ligue Régionale contre le Cancer. CA and LPV were supported by fellowships from the Palestinian Students Fund and the Ligue Nationale contre le cancer, respectively.
Additional information
Funding
References
- Kanopka A, Muhlemann O, Akusjarvi G. Inhibition by SR proteins splicing of a regulated adenovirus pre-mRNA. Nature. 1996;381:535–538.
- Jang SK, Wimmer E. Cap-independent translation of encephalomyocarditis virus RNA: structural elements of the internal ribosomal entry site and involvement of a cellular 57-kD RNA-binding protein. Genes Dev. 1990;4:1560–1572.
- Bray M, Prasad S, Dubay JW, et al. A small element from the Mason-Pfizer monkey virus genome makes human immunodeficiency virus type 1 expression and replication Rev- independent. Proc Natl Acad Sci U S A. 1994;91:1256–1260.
- Fornerod M, Ohno M, Yoshida M, et al. CRM1 is an export receptor for leucine-rich nuclear export signals. Cell. 1997;90:1051–1060.
- Houzet L, Battini JL, Bernard E, et al. A new retroelement constituted by a natural alternatively spliced RNA of murine replication-competent retroviruses. Embo J. 2003;22:4866–4875.
- Dorman N, Lever A. Comparison of viral genomic RNA sorting mechanisms in human immunodeficiency virus type 1 (HIV-1), HIV-2, and moloney murine leukemia virus. J Virol. 2000;74:11413–11417.
- Mougel M, Zhang Y, Barklis E. cis-Active structural motifs involved in specific encapsidation of Moloney murine leukemia virus RNA. J Virol. 1996;70:5043–5050.
- D’Souza V, Summers MF. Structural basis for packaging the dimeric genome of Moloney murine leukaemia virus. Nature. 2004;431:586–590.
- Mougel M, Barklis E. A role for two hairpin structures as a core RNA encapsidation signal in murine leukemia virus virions. J Virol. 1997;71:8061–8065.
- Smagulova F, Maurel S, Morichaud Z, et al. The highly structured encapsidation signal of MuLV RNA is involved in the nuclear export of its unspliced RNA. J Mol Biol. 2005;354:1118–1128.
- Basyuk E, Boulon S, Skou Pedersen F, et al. The packaging signal of MLV is an integrated module that mediates intracellular transport of genomic RNAs. J Mol Biol. 2005;354:330–339.
- Malim MH, Hauber J, Le SY, et al. The HIV-1 rev trans-activator acts through a structured target sequence to activate nuclear export of unspliced viral mRNA. Nature. 1989;338:254–257.
- Pessel-Vivares L, Ferrer M, Lainé S, et al. MLV requires Tap/NXF1-dependent pathway to export its unspliced RNA to the cytoplasm and to express both spliced and unspliced RNAs. Retrovirology. 2014;11:21.
- Bartels H, Luban J. Gammaretroviral pol sequences act in cis to direct polysome loading and NXF1/NXT-dependent protein production by gag -encoded RNA. Retrovirology. 2014;11:73.
- Sakuma T, Davila JI, Malcolm JA, et al. Murine leukemia virus uses NXF1 for nuclear export of spliced and unspliced viral transcripts. J Virol. 2014;88:4069–4082.
- Pilkington GR, Purzycka KJ, Bear J, et al. Gammaretrovirus mRNA expression is mediated by a novel, bipartite post-transcriptional regulatory element. Nucleic Acids Res. 2014;42:11092–11106.
- Pessel-Vivares L, Houzet L, Lainé S, et al. Insights into the nuclear export of murine leukemia virus intron-containing RNA. RNA Biol. 2015;12:942–949.
- Sherer NM, Swanson CM, Papaioannou S, et al. Matrix mediates the functional link between human immunodeficiency virus type 1 RNA nuclear export elements and the assembly competency of Gag in murine cells. J Virol. 2009;83:8525–8535.
- Swanson CM, Puffer BA, Ahmad KM, et al. Retroviral mRNA nuclear export elements regulate protein function and virion assembly. Embo J. 2004;23:2632–2640.
- Volkova NA, Fomina EG, Smolnikova VV, et al. The U3 region of Moloney murine leukemia virus contains position-independent cis-acting sequences involved in the nuclear export of full-length viral transcripts. J Biol Chem. 2014;289:20158–20169.
- Jouvenet N, Lainé S, Pessel-Vivares L, et al. Cell biology of retroviral RNA packaging. RNA Biol. 2011;8:1–9.
- Jouvenet N, Simon SM. Viral houseguests undertake interior redesign. Cell. 2011;141:754–756.
- Popa I, Harris ME, Donello JE, Hope TJ. CRM1-Dependent Function of a cis-acting RNA export element. Mol Cell Biol. 2002;22:2057–2067.
- Ferrer M, Henriet S, Chamontin C, et al. From cells to virus particles: quantitative methods to monitor RNA packaging. Viruses. 2016;8:1–16.
- Ferrer M, Clerté C, Chamontin C, et al. Imaging HIV-1 RNA dimerization in cells by multicolor super-resolution and fluctuation microscopies. Nucleic Acids Res. 2016;44:7922–7934.
- Okamura M, Inose H, Masuda S. RNA export through the NPC in Eukaryotes. Genes. 2015; 6:124–149.
- Saavedra C, Felber B, Izaurralde E. The simian retrovirus-1 constitutive transport element, unlike the HIV- 1 RRE, uses factors required for cellular mRNA export. Curr Biol. 1997;7:619–628.
- Askjaer P, Bachi A, Wilm M, et al. RanGTP-regulated interactions of CRM1 with nucleoporins and a shuttling DEAD-box helicase. Mol Cell Biol. 1999;19:6276–6285.
- Fischer U, Huber J, Boelens WC, et al. The HIV-1 Rev activation domain is a nuclear export signal that accesses an export pathway used by specific cellular RNAs. Cell. 1995;82:475–483.
- Ernst RK, Bray M, Rekosh D, et al. A structured retroviral RNA element that mediates nucleocytoplasmic export of intron-containing RNA. Mol Cell Biol. 1997;17:135–144.
- Pasquinelli AE, Ernst RK, Lund E, et al. The constitutive transport element (CTE) of Mason-Pfizer monkey virus (MPMV) accesses a cellular mRNA export pathway. Embo J. 1997;16:7500–7510.
- LeBlanc JJ, Uddowla S, Abraham B, et al. Tap and Dbp5, but not Gag, are involved in DR-mediated nuclear export of unspliced Rous sarcoma virus RNA. Virology. 2007;363:376–386.
- Kemler I, Saenz D, Poeschla E. Feline immunodeficiency virus Gag is a nuclear shuttling protein. J Virol. 2012;86:8402–8411.
- Scheifele LZ, Garbitt RA, Rhoads JD, et al. Nuclear entry and CRM1-dependent nuclear export of the Rous sarcoma virus Gag polyprotein. Proc Natl Acad Sci. 2002;99:3944–3949.
- Renault N, Tobaly-Tapiero J, Paris J, et al. A nuclear export signal within the structural Gag protein is required for prototype foamy virus replication. Retrovirology. 2011;8:6.
- Hanly SM, Rimsky LT, Malim MH, et al. Comparative analysis of the HTLV-I Rex and HIV-1 Rev trans-regulatory proteins and their RNA response elements. Genes Dev. 1989;3:1534–1544.
- Hidaka M, Inoue J, Yoshida M, et al. Post-transcriptional Regulator (rex) of HTLV-I initiates expression of viral structural proteins but suppresses expression of regulatory proteins. EMBO. 1988;7:519–523.
- Mertz JA, Simper MS, Lozano MM, et al. Mouse mammary tumor virus encodes a self-regulatory RNA export protein and is a complex retrovirus. J Virol. 2005;79:14737–14747.
- Nash MA, Meyer MK, Decker GL, et al. A subset of Pr65gag is nucleus associated in murine leukemia virus-infected cells. J Virol. 1993;67:1350–1356.
- Levin JG, Grimley PM, Ramseur JM, et al. Deficiency of 60 to 70S RNA in murine leukemia virus particles assembled in cells treated with actinomycin D. J Virol. 1974;14:152–161.
- Garbitt-Hirst R, Kenney SP, Parent LJ. Genetic evidence for a connection between Rous Sarcoma virus Gag nuclear trafficking and genomic RNA packaging. J Virol. 2009;83:6790–6797.
- Gudleski N, Flanagan JM, Ryan EP, et al. Directionality of nucleocytoplasmic transport of the retroviral gag protein depends on sequential binding of karyopherins and viral RNA. Proc Natl Acad Sci. 2010;107:9358–9363.
- Bohl CR, Brown SM, Weldon RA. The pp24 phosphoprotein of Mason-Pfizer monkey virus contributes to viral genome packaging. Retrovirology. 2005;2:1–14.
- Pocock GM, Becker JT, Swanson CM, et al. HIV-1 and M-PMV RNA nuclear export elements program viral genomes for distinct cytoplasmic trafficking behaviors. PLoS Pathog. 2016;12:1–30.
- Zheng Y-H, Yu H-F, Peterlin BM. Human p32 protein relieves a post-transcriptional block to HIV replication in murine cells. Nat Cell Biol. 2003;5:611–618.
- Cupelli L, Okenquist SA, Trubetskoy A, et al. The secondary structure of the R region of a murine leukemia virus is important for stimulation of long terminal repeat-driven gene expression. J Virol. 1998;72:7807–7814.
- Trubetskoy AM, Okenquist SA, Lenz J. R region sequences in the long terminal repeat of a murine retrovirus specifically increase expression of unspliced RNAs. J Virol. 1999;73:3477–3483.
- Scheifele LZ, Ryan EP, Parent LJ. Detailed mapping of the nuclear export signal in the Rous Sarcoma virus Gag protein. J Virol. 2005;79:8732–8741.
- Baluyot MF, Grosse SA, Lyddon TD, et al. CRM1-dependent trafficking of retroviral Gag proteins revisited. J Virol. 2012;86:4696–4700.
- D’Souza V, Summers MF. How retroviruses select their genomes. Nat Rev Microbiol. 2005;3:643–655.
- Mailler E, Bernacchi S, Marquet R, et al. The life-cycle of the HIV-1 Gag – RNA complex. Viruses. 2016;1–19.
- Chamontin C, Yu B, Racine PJ, et al. MoMuLV and HIV-1 nucleocapsid proteins have a common role in genomic RNA packaging but different in late reverse transcription. PLoS One. 2012;7:e51534.
- Mark-Danieli M, Laham N, Kenan-Eichler M, et al. Single point mutations in the zinc finger motifs of the human immunodeficiency virus type 1 nucleocapsid alter RNA binding specificities of the gag protein and enhance packaging and infectivity. J Virol. 2005;79:7756–7767.
- Jouvenet N, Simon SM, Bieniasz PD. Imaging the interaction of HIV-1 genomes and Gag during assembly of individual viral particles. Proc Natl Acad Sci U S A. 2009;106:19114–19119.
- Maurel S, Houzet L, Garcia EL, et al. Characterization of a natural heterodimer between MLV genomic RNA and the SD’ retroelement generated by alternative splicing. RNA. 2007;13:2266–2276.
- Rasmussen SV, Pedersen FS. Co-localization of gammaretroviral RNAs at their transcription site favours co-packaging. J Gen Virol. 2006;87:2279–2289.
- Maurel S, Mougel M. Murine leukemia virus RNA dimerization is coupled to transcription and splicing processes. Retrovirology. 2010;7:1–8.
- Courtney DG, Chalem A, Bogerd HP, et al. Extensive epitranscriptomic methylation of A and C residues on murine leukemia virus transcripts enhances viral gene expression. MBio. 2019;10:1–12.
- Sambrook J, Russell DW. Molecular cloning: a laboratory manual. 3th ed. New York: Cold Spring Harbor Laboratory; 2001.
- Shinnick TM, Lerner RA, Sutcliffe JG. Nucleotide sequence of Moloney murine leukaemia virus. Nature. 1981;293:543–548.
- Jouvenet N, Neil S, Zhadina M, et al. Hatziioannou T and Bieniasz P. Broad-spectrum inhibition of retroviral and filoviral particle release by tetherin. J Virol. 2009;83(4):1837–1844.
- Boireau S, Maiuri P, Basyuk E, et al. The transcriptional cycle of HIV-1 in real-time and live cells. J Cell Biol. 2007;179:291–304.
- Voelkel C, Galla M, Maetzig T, et al. Protein transduction from retroviral Gag precursors. Proc Natl Acad Sci. 2010;107:7805–7810.
- Dejardin J, Bompard-Marechal G, Audit M, et al. A Novel subgenomic murine leukemia virus RNA transcript results from alternative splicing. J Virol. 2000;74:3709–3714.