
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
Assembly of eukaryotic ribosomal subunits is a complex and dynamic process involving the action of more than 200 trans-acting assembly factors. Although recent cryo-electron microscopy structures have provided information on architecture of several pre-ribosomal particles and the binding sites of many AFs, the RNA and protein interactions of many other AFs not captured in these snapshots still remain elusive. RNA helicases are key regulators of structural rearrangements within pre-ribosomal complexes and here we have analysed the eIF4A-like RNA helicase Fal1 and its putative cofactor Sgd1. Our data show that these proteins interact directly via the MIF4G domain of Sgd1 and that the MIF4G domain of Sgd1 stimulates the catalytic activity of Fal1 in vitro. The catalytic activity of Fal1, and the interaction between Fal1 and Sgd1, are required for efficient pre-rRNA processing at the A0, A1 and A2 sites. Furthermore, Sgd1 co-purifies the early small subunit biogenesis factors Lcp5 and Rok1, suggesting that the Fal1-Sgd1 complex likely functions within the SSU processome. In vivo crosslinking data reveal that Sgd1 binds to helix H12 of the 18S rRNA sequence and we further demonstrate that this interaction is formed by the C-terminal region of the protein, which is essential for its function in ribosome biogenesis.
Introduction
Ribosomes are responsible for the production of all cellular proteins and in eukaryotes, are composed of four ribosomal RNAs (rRNA) and 79 (Saccharomyces cerevisiae (yeast))/80 (human) ribosomal proteins (RPs) [Citation1,Citation2]. Assembly of these large ribonucleoprotein (RNP) complexes is a complex and hierarchical process that requires the action of in excess of 200 trans-acting assembly factors (AFs) that associate with different pre-ribosomal complexes but are not present in mature ribosomes [Citation3,Citation4]. In yeast, transcription of rDNA by RNA polymerase I in the nucleolus generates the initial 35S pre-rRNA transcript. During its synthesis, the 35S pre-rRNA transcript undergoes a plethora of rRNA modifications guided by small nucleolar RNPs (snoRNPs) and is also cleaved at a number of pre-rRNA cleavage sites [Citation5–Citation8]. In addition, numerous RPs and AFs are recruited already co-transcriptionally to the nascent pre-rRNA transcript giving rise to the terminal knob structures observed in Miller spreads [Citation9,Citation10]. One of the earliest stable pre-ribosomal particles is the small subunit (SSU) processome, which is assembled on the 5ʹ end of the nascent pre-rRNA transcript containing the 5ʹ external transcribed spacer (5ʹ ETS) and the 18S rRNA sequence, as well as the U3 snoRNA [Citation11,Citation12]. Although many protein components of the SSU processome are recruited as pre-assembled subcomplexes, such as the Utp-A, Utp-B and Utp-C complexes, others associate individually (reviewed in [Citation13]). As ribosomal subunit assembly progresses, structural rearrangements and pre-rRNA cleavage steps induce separation of the pre-SSU and precursor large subunit (LSU) particles. These complexes follow independent assembly pathways; pre-SSU complexes transit quickly through the nucleoplasm and are rapidly exported to the cytoplasm whereas pre-LSU particles undergo numerous maturation steps in the nucleolus and nucleoplasm before finally achieving export competence [Citation3,Citation14]. Both pre-SSU and pre-LSU complexes undergo final maturation steps, including the release of remaining AFs and various quality control checks, in the cytoplasm before the mature subunits can engage in translation.
Recent structural snapshots of the architecture of several pre-ribosomal complexes, including the yeast SSU processome and its precursor the ‘5ʹ ETS RNP’ (see for example [Citation11,Citation12,Citation15–Citation18], have provided a wealth of information on the compositions of different pre-ribosomal particles and the interactions that individual AFs form within these structures. These structures have also underscored how dynamic the process of ribosome assembly is by providing many insights into the structural transitions that pre-ribosomal complexes undergo during their maturation. Such remodelling events often serve as checkpoints along the assembly pathway and several types of energy-dependent proteins, including AAA-ATPases, kinases, ATP-binding cassette proteins, GTPases and ATP-dependent RNA helicases have emerged as important regulators of these events [Citation19].
To date, 21 RNA helicases have been suggested to act during the process of ribosome biogenesis in yeast with eight implicated in SSU maturation, ten in LSU biogenesis and three in the assembly of both subunits [Citation20]. A wealth of research has provided information on the binding sites and potential functions of many of these RNA helicases [Citation21–Citation33], with several RNA helicases implicated in promoting the release of specific snoRNPs from early pre-ribosomal particles [Citation21,Citation22,Citation26,Citation27,Citation30,Citation31] and others suggested to catalyse pre-rRNA rearrangements that induce formation of mature rRNA conformations facilitating the recruitment of RPs and/or AFs [Citation23,Citation32,Citation34]. However, in the case of other RNA helicases that have been found associated with pre-ribosomal particles, little is known about the timing of their association, the interactions they make within pre-ribosomes and what contribution their activity makes to the assembly process. In addition to these factors, a key aspect of understanding the functions of RNA helicases in any aspect of RNA metabolism is to understand how their catalytic activity is regulated [Citation35]. Crystallographic and biochemical studies of different RNA helicases (reviewed in [Citation36]) have revealed that the helicase core domain predominantly interacts with the backbone of their RNA substrates meaning that this typically lacks specificity. Moreover, many of these enzymes have intrinsically low catalytically activity, likely to limit their promiscuity until the correct substrate is encountered. In line with this, several RNA helicases only achieve substrate specificity and robust catalytic activity by associating with protein cofactors. Within the pathway of ribosome biogenesis, the activities of the RNA helicases Rok1, Dbp8 and Dhr1 have been shown to be regulated by the AFs Rrp5, Esf2 and Utp14 respectively [Citation37–Citation39]. Interestingly, alongside these dedicated RNA helicase-cofactor partnerships, several cofactor proteins containing conserved domains, and therefore described to belong to cofactor families, have emerged. The most prominent example is the G-patch proteins, which contain a glycine-rich domain via which they associate with and regulate the activity of the DEAH box helicases Prp43 and Prp2 [Citation40–Citation44]. During ribosome assembly, Prp43 mediates the release of a specific subset of snoRNPs from early pre-LSU particles, a function which has been linked to the G-patch protein Gno1 (Pxr1) [Citation27,Citation45]. In addition, Prp43 is suggested to remodel cytoplasmic pre-SSU particles to facilitate cleavage at the 3ʹ end of the 18S rRNA and the G-patch protein Pfa1 (Sqs1) has been linked to this function [Citation27,Citation28,Citation46]. Alongside the G-patch proteins, a family of cofactors dedicated to regulation of eIF4A-like RNA helicases and characterized by the presence of a ‘middle of eIF4G’ (MIF4G) domain has been described ([Citation47] and reviewed in [Citation35,Citation48]). RNA helicase-cofactor complexes of this family bear structural similarity to the eIF4A-eIF4G complex, where eIF4G stimulates the action of eIF4A in resolving structured 5ʹ untranslated regions (UTRs) during translation initiation ([Citation49–Citation51]). Interestingly, the yeast eIF4A-like RNA helicase Fal1 (related to eIF4A-III in humans) and the MIF4G domain-containing protein Sgd1 (NOM1 in humans) have both been implicated in SSU biogenesis [Citation50,Citation52] but the functions of these proteins and whether Sgd1 serves as a cofactor for Fal1 have remained unknown.
Here we show that both Sgd1 and the catalytic activity of Fal1 are required for efficient pre-rRNA cleavage at the A0, A1 and A2 processing sites, which are necessary for early steps in maturation of the 18S rRNA of the SSU. We also reveal a direct interaction between Fal1 and Sgd1 that requires the MIF4G domain of Sgd1, and show that this domain of Sgd1 stimulates the ATPase activity of Fal1 in vitro. Furthermore, using in vivo protein-RNA crosslinking, we identify the binding site of Sgd1 on the 18S pre-rRNA sequence and demonstrate that the C-terminal region of Sgd1 is responsible for RNA binding. Together, our data suggest that Sgd1 and Fal1 likely associate transiently with SSU processomes containing the AF Lcp5, where Sgd1 stimulates pre-ribosome remodelling by Fal1.
Results
Depletion of Fal1 or Sgd1 inhibits pre-rRNA processing at sites A0, A1 and A2
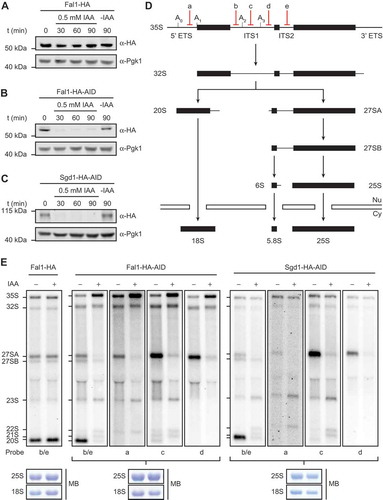
The putative eIF4A-like RNA helicase Fal1 and the MIF4G domain-containing protein Sgd1 have both been implicated in yeast ribosome assembly but their precise roles remain unknown. As other MIF4G domain-containing proteins have been shown to function as cofactors that regulate the activity of specific eIF4A-like RNA helicases, it was speculated that Fal1 and Sgd1 may act together in ribosome assembly. To analyse the aspects of the ribosome assembly pathway that require Fal1 or Sgd1, auxin-dependent systems [Citation53,Citation54] for depletion of these proteins were established. In a yeast strain expressing the Oryza sativa TIR1 complex, FAL1 or SGD1 were C-terminally HA-auxin-inducible degron (AID) tagged, enabling their rapid degradation upon exposure to auxin. Compared to classic promoter exchange-based depletion systems, this approach has the advantages of allowing efficient and specific depletion without altering endogenous expression levels or requiring changes of carbon source. A yeast strain expressing C-terminally HA tagged Fal1 in this background was also generated as a control. Exponentially growing cells of these strains were treated with auxin for 0, 30, 60 or 90 min, or left untreated. Analysis of protein levels revealed that the addition of the AID tag to Fal1 had minimal effect on the basal expression level of the protein while addition of auxin to either the Fal1-HA-AID or Sgd1-HA-AID strains, but not the Fal1-HA strain, lead to complete depletion of the tagged protein within 30 min (). Next, pre-rRNA processing () was examined in these strains to determine which step(s) of ribosome assembly Fal1 and Sgd1 are required for. Total RNA prepared from auxin-treated, or untreated cells was separated by denaturing agarose gel electrophoresis and pre-rRNAs were detected by northern blotting using numerous probes hybridizing to different regions of the pre-rRNA transcript (). The levels of all pre-rRNA intermediates detected in the Fal1-HA strain, and the Fal1-HA-AID and Sgd1-HA-AID strains before treatment with auxin were similar, confirming the lack of effects on pre-rRNA processing caused by AID-tagging these proteins. Upon treatment of either the Fal1-HA-AID or Sgd1-HA-AID strains with auxin, increased levels of the initial 35S pre-rRNA transcript and concomitant decreases in the levels of the 20S and 27SA pre-rRNA species were observed (). Furthermore, aberrant intermediates not normally detected in yeast were present in both the auxin-treated Fal1-HA-AID and Sgd1-HA-AID strains (). Mapping of these aberrant intermediates (Supplementary Figure S1) with additional probes hybridizing to different regions of the transcript between specific pre-rRNA cleavage sites, enabled these intermediates to be confirmed as the 21S, 22S and 23S pre-rRNAs that are generated when processing at the A0, A1 and A2 sites is impaired. This is consistent with the reduced levels of the 20S pre-rRNA, the immediate precursor of the 18S rRNA, as well as the 27SA (predominantly 27SA2), pre-rRNA. Extended northern blot analyses did not reveal the accumulation of excised fragments of the 5ʹ ETS (5ʹ-A0 or A0-A1), suggesting that these fragments, which are likely produced at lower levels upon depletion of either Fal1 or Sgd1, are still efficiently degraded. The finding that both Fal1 and Sgd1 are required for the same pre-rRNA processing steps supports the model that they may function together during ribosome assembly and, furthermore, our pre-rRNA processing analyses suggests that they act during the early assembly steps of the SSU.
Figure 1. Fal1 and Sgd1 are required for pre-rRNA cleavages at sites A0, A1 and A2. (A-C) Exponentially growing yeast cells expressing Fal1-HA (A), Fal1-HA-AID (B) or Sgd1-HA-AID (C) were treated with IAA for the indicated times (t) or left untreated (-IAA) before harvesting. Total proteins were separated by SDS-PAGE followed by western blotting using antibodies against the HA tag or Pgk1. (D) Simplified schematic overview of pre-rRNA processing in S. cerevisiae. Mature rRNA sequences are indicated by black rectangles, and internal transcribed spacers (ITS1 and ITS2) and external transcribed spacers (5ʹ ETS and 3ʹ ETS) are represented by black lines. Selected pre-rRNA cleavage sites and the hybridization positions of probes used for northern blotting are marked on the 35S pre-rRNA transcript. (E) Exponentially growing yeast cells expressing Fal1-HA, Fal1-HA-AID or Sgd1-HA-AID were treated with IAA (+) for 60 min or left untreated (-) before harvesting. Total RNA was extracted, separated by denaturing agarose gel electrophoresis and transferred to a nylon membrane. The mature 25S and 18S rRNAs were visualized by methylene blue staining (MB) and pre-rRNAs were detected by northern blotting using probes hybridizing to different regions of the pre-rRNA transcript.
Fal1 is an active ATPase and its catalytic activity is required for SSU biogenesis
Based on the presence of two RecA-like domains, Fal1 is a putative RNA helicase, although such activity has not yet been demonstrated. Therefore, to investigate whether the catalytic activity of Fal1 is important for its function(s) in ribosome assembly, it was first necessary to determine if Fal1 is catalytically active and identify a mutation that impairs such activity.
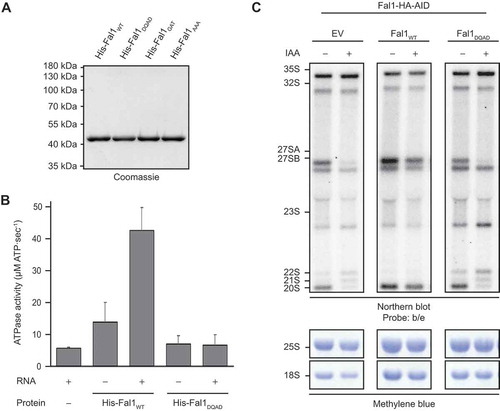
N-terminally His tagged wild type Fal1 (His-Fal1WT), or mutant versions carrying a glutamate to glutamine substitution at position 174 within the conserved DEAD motif (His-Fal1DQAD), a lysine to alanine substitution at position 73 within the GKT motif (His-Fal1GAT) or substitutions of serine 206 and threonine 208 within the SAT motif to alanine (His-Fal1AAA) were overexpressed in E. coli and the recombinant proteins were purified (). To monitor catalytic activity, in vitro ATPase assays were performed using NADH-coupled reactions [Citation55,Citation56]. Compared to a control reaction where no protein was added, His-Fal1WT showed minimal ATPase activity in the absence of RNA, but this activity was stimulated > 3-fold in the presence of RNA (). In contrast, His-Fal1DQAD displayed almost no specific ATP hydrolysis in either the presence or absence of RNA, indicating that this mutation largely abolishes the catalytic activity of Fal1 (). Similarly, His-Fal1AAA did not hydrolyse ATP above the background observed in the control and His-Fal1GAT only minimally, and neither protein was stimulated by the presence of RNA (Supplementary Figure S2). The requirement for the catalytic activity of Fal1 for ribosome assembly was then addressed by generating a yeast complementation system. The Fal1-HA-AID strain was transformed with either an empty pRS415 plasmid (EV) or pRS415-based plasmids for expression of Fal1WT or Fal1DQAD from the endogenous FAL1 promoter. Total RNA extracted from untreated cells or cells treated with auxin to deplete endogenous Fal1 was separated by denaturing agarose gel electrophoresis and pre-rRNAs were analysed by northern blotting using probes hybridizing within the internal transcribed spacers (ITS1 and ITS2) of the pre-rRNA sequence. As previously, in the presence of the empty plasmid, treatment with auxin caused accumulation of the 35S, 23S, 22S and 21S pre-RNAs as well as impaired production of the 27SA and 20S pre-RNAs ( and , left panel). Importantly, expression of plasmid-derived Fal1WT rescued all these pre-rRNA processing defects, confirming that they specifically arise due to a lack of this protein (, middle panel). In contrast, exogenous expression of Fal1DQAD lead to increased levels of the 35S, 23S, 22S and 21S pre-RNAs, and reduced amounts of the 27SA and 20S species (, right panel), confirming that the catalytic activity of Fal1 is required for efficient pre-rRNA cleavages at the A0, A1 and A2 sites.
Figure 2. The catalytic activity of Fal1 is required for biogenesis of the small ribosomal subunit. (A) N-terminally His tagged Fal1WT or Fal1 carrying a substitution of glutamate 174 for glutamine (His-Fal1DQAD), lysine 73 for alanine (His-Fal1GKT) or serine 206 and threonine 208 for alanines (His-Fal1AAA) were recombinantly expressed in E. coli and purified using an Ni2+ matrix. After elution and dialysis, recombinant proteins were separated by SDS-PAGE and detected by Coomassie staining. (B) The ATPase activity of His-Fal1WT and His-Fal1DQAD in the presence (+) and absence (-) of RNA was determined using in vitro NADH-coupled assays. The rate of ATP hydrolysis observed in the absence of protein was also monitored. Three independent experiments were performed and the data are presented as mean ± standard deviation. (C) The Fal1-HA-AID yeast strain was transformed with an empty pRS415 plasmid (EV) or pRS415-based plasmids for the expression of Fal1WT or Fal1DQAD from their endogenous promoters. Exponentially growing yeast cells were treated with auxin (IAA; +) for 60 min or left untreated (-). Total RNA was extracted, separated by denaturing agarose gel electrophoresis and transferred for northern blotting using probes hybridizing within ITS1 and ITS2. Mature rRNAs were visualized by methylene blue staining.
The direct interaction between Fal1 and Sgd1 requires the MIF4G domain of Sgd1
Various eIF4A-like RNA helicases have been shown to interact with cofactors containing MIF4G domains, suggesting that Fal1 and Sgd1 may associate in a similar manner. A possible interaction between these proteins was first explored using a binding assay in which MBP-Sgd1FL-His (Supplementary Figure S3A) or the MBP-His tag was immobilized on amylose resin and incubated with yeast cell extracts prepared from strains expressing HA tagged Fal1, or as a control, HA tagged Rok1, that had either been pre-treated with RNase or not. Western blot analysis of co-precipitated proteins revealed that, in the absence of RNase treatment, both Fal1-HA and HA-Rok1 were retrieved with MBP-Sgd1-His but not empty amylose beads or the MBP-His tag (). Upon RNase treatment, however, only Fal1-HA was co-purified with MBP-Sgd1-His, indicating that Sgd1, Fal1 and Rok1 all co-exist in pre-ribosomal particles that are stable in the absence of RNase treatment, but that Fal1 and Sgd1 also form an RNA-independent interaction.
Figure 3. Sgd1 interacts directly with Fal1 and stimulates its ATPase activity. (A) Soluble whole cell extracts were prepared from exponentially growing yeast cells expressing HA tagged Fal1 or Rok1 from their endogenous promoters and either treated with RNase (+) or left untreated (-). Recombinant MBP-Sgd1-His or the MBP-His tag alone purified from E. coli were immobilized on amylose resin and unbound proteins were washed away. The cell extracts were incubated with the Sgd1-bound beads or empty amylose resin (A) and co-purified complexes were retrieved. Input samples (25%) and recovered proteins (Eluate) were separated by SDS-PAGE and transferred for western blotting using antibodies against the MBP and HA tags. (B) Schematic views of N-terminally MBP, C-terminally His tagged fragments of Sgd1 showing the positions of the predicted MIF4G and MA3 domains. Amino acid numbers corresponding to the boundaries of domains and fragments are given above each scheme. (C) MBP-His tagged full length Sgd1 (Sgd1FL) or its fragments were recombinantly expressed in E. coli and purified. Proteins were separated by SDS-PAGE and detected by Coomassie staining. (D) Recombinant MBP-His tagged Sgd1FL or its fragments, or the MBP-His tag, were immobilized on amylose resin. Excess, unbound proteins were removed before addition of His-Fal1. After thorough washing steps, co-purified proteins were eluted. Eluates were separated by SDS-PAGE and proteins were detected by Coomassie staining. The band corresponding to His-Fal1 is indicated by an arrow head. (E) The ATPase activity of Fal1 alone or together with the His-ZZ-tagged MIF4G domain of Sgd1 (Sgd1MIF4G) in the presence or absence of RNA was monitored in vitro using NADH-coupled assays. The ATPase activity of Sgd1MIF4G in the presence or absence of RNA and background (no protein) ATP hydrolysis were also monitored. Data from three independent experiments are presented as mean ± standard deviation.
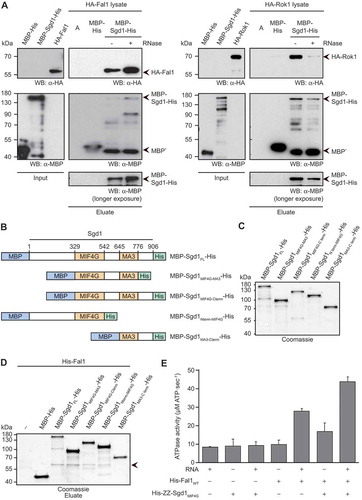
Next, to determine if Fal1 and Sgd1 interact directly full length MBP-His tagged Sgd1 (MBP-Sgd1FL-His; ) or the MBP-His tag were immobilized on amylose resin and in vitro binding assays were then performed with His-Fal1. Analysis of co-purified proteins revealed a direct interaction between MBP-Sgd1FL-His and His-Fal1 (). As this interaction was relatively weak, an alternative binding assay was also performed in which MBP-Sgd1FL-His was incubated with His-ZZ-Fal1 or the His-ZZ tag immobilized on IgG sepharose or the empty beads. MBP-Sgd1FL-His was specifically retrieved with the His-ZZ-Fal1, confirming the direct interaction between these two proteins (Supplementary Figure 3B). Further binding assays were also performed in the presence of ATP, the non-hydrolysable ATP analogue ADPNP and RNA to determine if the presence of any of these components promotes or inhibits the interaction between Fal1 and Sgd1, but this was not the case (Supplementary Figure 4).
To identify which region(s) of Sgd1 contribute to the interaction with Fal1, a series of truncated versions of MBP-Sgd1-His were recombinantly expressed in E. coli and purified (). These fragments, immobilized on amylose resin, were incubated with His-Fal1. While an Sgd1 fragment containing the MA3 domain and C-terminal region of the protein did not interact with His-Fal1, His-Fal1 was co-purified with all Sgd1 fragments containing the MIF4G domain (). Although lack of either the N- or C-terminal regions of Sgd1 decreased the amount of His-Fal1 recovered ( and Supplementary Figure 3C), neither of these regions were essential for Sgd1-Fal1 association, implying that only the MIF4G domain is critical for the direct interaction of Sgd1 with Fal1. It was, however, not possible to specifically recover a His-ZZ tagged version of the MIF4G domain of Sgd1 alone (Supplementary Figure S3D) with immobilized Fal1 or to retrieve Fal1 with an immobilized MBP-His tagged version of the MIF4G domain under similar conditions, further supporting the notion that other regions of Sgd1 likely help stabilize this interaction.
The direct interaction of Fal1 with Sgd1 via its MIF4G domain suggests that this interaction may influence the catalytic activity of Fal1. In vitro ATPase assays were performed using His-Fal1 and an N-terminally His-ZZ tagged version of the MIF4G domain of Sgd1. The MIF4G domain of Sgd1 showed no significant ATPase activity in the absence or presence of RNA and, as previously, in the absence of RNA, Fal1 itself showed minimal activity ( and ). ATP hydrolysis by Fal1 was stimulated by the presence of RNA and addition of Sgd1 even further increased the rate of ATP hydrolysis () indicating that the MIF4G domain of Sgd1 serves as a positive regulator of Fal1 activity. It is possible that the extent of stimulation of the ATPase activity of Fal1 by full length Sgd1 may be even greater than that observed with the MIF4G domain alone, however, for technical reasons, it was not possible to analyse this.
Sgd1 crosslinks to helix H12 of the 18S rRNA sequence
Together, the findings that Fal1 and Sgd1 act as a helicase-cofactor partnership and that both proteins are required for early steps during maturation of the SSU, raise the question of where this complex binds to pre-ribosomal particles. The crosslinking and analysis of cDNA (CRAC) approach [Citation57,Citation58] was therefore applied with the aim of determining the binding sites of Fal1 and Sgd1 on pre-rRNAs. Wild type yeast or strains expressing C-terminally His-TEV protease cleavage site-Protein A (HTP) tagged Fal1 or Sgd1 from their genomic loci were crosslinked in culturo and protein-RNA complexes were retrieved on IgG sepharose. After partial RNase digestion to leave a footprint of the proteins on their target RNAs, complexes were enriched under denaturing conditions by nickel affinity chromatography, and RNA fragments were 5ʹ labelled with [32P] and ligated to sequencing adaptors. Complexes were separated by denaturing polyacrylamide gel electrophoresis (PAGE), transferred to a nitrocellulose membrane and radioactive RNAs were detected by autoradiography. No crosslinking of Fal1 to RNA was detected under these conditions, which likely reflects the fact that only a small portion of the helicase is associated with pre-ribosomal particles at any given time (Supplementary Figure S5A,B). However, for Sgd1, which more stable associates with pre-ribosomes (Supplementary Figure S5C), a specific signal corresponding to protein with crosslinked RNA fragments, which was not present in the sample derived from wild type cells, was observed (). The region of the membrane carrying RNA-Sgd1-HTP complexes was excised alongside a corresponding region of the wild type lane and RNAs were released from the membrane by protease-mediated digestion. RNA fragments were isolated, reverse transcribed and, after amplification, the resulting cDNA library was subjected to deep sequencing. The obtained sequencing reads were mapped to the S. cerevisiae genome and analysis of the relative proportions of reads mapping to genes encoding different types of RNAs revealed an enrichment of reads derived from (pre-)rRNAs in the Sgd1-HTP dataset compared to the wild type control (). Inspection of the distribution of sequencing reads mapping to RND37, which encodes the full length 35S pre-rRNA transcript, showed that the majority of sequencing reads mapped to a particular region of the 18S rRNA sequence that was not over-represented in the dataset from the wild type control cells (). A heatmap of the Sgd1-HTP CRAC data on the secondary structure of the mature 18S rRNA [Citation59] revealed that Sgd1 crosslinks to helix 18S-H12 within the 5ʹ domain of the pre-SSU (). As our pre-rRNA processing analyses indicated that Sgd1 is required for co-transcriptional pre-rRNA processing events at sites A0, A1 and A2, the Sgd1-HTP CRAC data was also mapped onto the tertiary structure of the rRNA within the SSU processome, one of the earliest known pre-ribosomal complex ([Citation12]; PBD 5WLC). In this structure, 18S-H12 is accessible within a pocket composed of the RPs Rps4, Rps8, Rps9, Rps11 and Rps22, and the AF Lcp5 (). While Sgd1 is not present in this cryo-electron microscopy (cryoEM) structure, an interaction between Sgd1 and Lcp5 was detected by mass spectrometry of the purified particles after protein crosslinking [Citation12]. Furthermore, Lcp5-HA co-purifies with Sgd1-HTP from yeast cells (Supplementary Figure 6) and sucrose density centrifugation revealed that Sgd1-HTP partially co-migrates with Lcp5-HA in fractions containing (pre-)40S particles (Supplementary Figure S5C). Together, these findings support 18S-H12 as a bona fide binding site of Sgd1 and suggest that Sgd1 likely associates with this region in the context of the SSU processome.
Figure 4. Sgd1 crosslinks to helix H12 of the 18S rRNA sequence. (A) Wild type yeast (WT) or a yeast strain expressing C-terminally His-TEV protease cleavage site-Protein A (HTP) tagged Sgd1 from its genomic locus were crosslinked in culturo using irradiation at 254 nm. The tagged protein and crosslinked RNAs were retrieved under native conditions on IgG sepharose and subjected to a partial RNase digest. Complexes were then enriched under denaturing conditions on NiNTA, and co-purified RNA fragments were [32P] labelled and ligated to sequencing adaptors. Protein-RNA complexes were separated by denaturing PAGE, transferred to a nitrocellulose membrane and radiolabelled RNAs were detected by autoradiography. A non-specific signal is indicated by an asterisk. (B) The region of the membrane containing crosslinked Sgd1-HTP complexes, and a corresponding region of the lane containing the WT sample, were excised and RNAs were release by protease treatment. Purified RNAs were converted to a cDNA library that was subjected to deep sequencing. The obtained sequencing reads were mapped to the S. cerevisiae genome and the relative proportions of reads mapped to gene features encoding different classes of RNA was determined. Abbreviations – ribosomal RNA (rRNA), transfer RNA (tRNA), small nucleolar RNA (snoRNA), non-coding RNA (ncRNA), small nuclear RNA (snRNA). (C) After normalization, the number of reads mapping to each nucleotide of RDN37, which encodes the 35S pre-rRNA transcript, was determined for the WT and Sgd1-HTP samples and is shown above a schematic view of the pre-rRNA transcript. The number of mutations, induced by the presence of crosslinked nucleotides, mapping to each nucleotide is also shown. (D) The number of sequencing reads in the Sgd1-HTP CRAC dataset mapping to each nucleotide of the 18S rRNA sequence is shown on the secondary structure of the mature rRNA using a colour scale in which the maximum number of reads (100%) is highlighted in red and lesser numbers of reads (>20%) are shown in yellow. A magnified view of the region of the 18S rRNA to which Sgd1 binds is presented. (E) The number of sequencing reads mapping to each nucleotide of the 18S rRNA sequence was mapped onto the tertiary structure of the SSU processome (PBD: 5WLC) using a colour scale as in (D). Pre-rRNA sequences are shown in cartoon mode, and ribosomal proteins and ribosome assembly factors are shown in surface view in pale cyan. The densities corresponding to specific ribosomal proteins and ribosome assembly factors that are present in close proximity to the crosslinking site of Sgd1 are highlighted.
![Figure 4. Sgd1 crosslinks to helix H12 of the 18S rRNA sequence. (A) Wild type yeast (WT) or a yeast strain expressing C-terminally His-TEV protease cleavage site-Protein A (HTP) tagged Sgd1 from its genomic locus were crosslinked in culturo using irradiation at 254 nm. The tagged protein and crosslinked RNAs were retrieved under native conditions on IgG sepharose and subjected to a partial RNase digest. Complexes were then enriched under denaturing conditions on NiNTA, and co-purified RNA fragments were [32P] labelled and ligated to sequencing adaptors. Protein-RNA complexes were separated by denaturing PAGE, transferred to a nitrocellulose membrane and radiolabelled RNAs were detected by autoradiography. A non-specific signal is indicated by an asterisk. (B) The region of the membrane containing crosslinked Sgd1-HTP complexes, and a corresponding region of the lane containing the WT sample, were excised and RNAs were release by protease treatment. Purified RNAs were converted to a cDNA library that was subjected to deep sequencing. The obtained sequencing reads were mapped to the S. cerevisiae genome and the relative proportions of reads mapped to gene features encoding different classes of RNA was determined. Abbreviations – ribosomal RNA (rRNA), transfer RNA (tRNA), small nucleolar RNA (snoRNA), non-coding RNA (ncRNA), small nuclear RNA (snRNA). (C) After normalization, the number of reads mapping to each nucleotide of RDN37, which encodes the 35S pre-rRNA transcript, was determined for the WT and Sgd1-HTP samples and is shown above a schematic view of the pre-rRNA transcript. The number of mutations, induced by the presence of crosslinked nucleotides, mapping to each nucleotide is also shown. (D) The number of sequencing reads in the Sgd1-HTP CRAC dataset mapping to each nucleotide of the 18S rRNA sequence is shown on the secondary structure of the mature rRNA using a colour scale in which the maximum number of reads (100%) is highlighted in red and lesser numbers of reads (>20%) are shown in yellow. A magnified view of the region of the 18S rRNA to which Sgd1 binds is presented. (E) The number of sequencing reads mapping to each nucleotide of the 18S rRNA sequence was mapped onto the tertiary structure of the SSU processome (PBD: 5WLC) using a colour scale as in (D). Pre-rRNA sequences are shown in cartoon mode, and ribosomal proteins and ribosome assembly factors are shown in surface view in pale cyan. The densities corresponding to specific ribosomal proteins and ribosome assembly factors that are present in close proximity to the crosslinking site of Sgd1 are highlighted.](/cms/asset/3bc0de2d-db14-4cd4-a965-0276a0cf7d8e/krnb_a_1716540_f0004_oc.jpg)
The C-terminal region of Sgd1 binds RNA
Sgd1 is a large protein (102 kDa) for which structural information is currently lacking and beyond the MIF4G (and associated MA3) domain, which is generally implicated in protein-protein interactions, no other conserved domains have been identified. The finding that Sgd1 can be efficiently crosslinked to RNA therefore raised the question of which region of the protein is responsible for RNA binding. To address this, a series of HTP tagged Sgd1 fragments () were expressed in the Sgd1-HA-AID yeast strain. After auxin-mediated depletion of endogenous Sgd1, cells were subjected to crosslinking using irradiation at 254 nm. Protein-RNA complexes were immobilized on IgG sepharose and a partial RNase digestion was performed. Affinity purification of complexes on NiNTA under denaturing conditions was followed by 5ʹ [32P] labelling of RNAs and separation of complexes by denaturing PAGE. Proteins were detected by western blotting to confirm similar expression and enrichment of full length Sgd1 and each of the fragments (, upper panel). Detection of radiolabelled RNAs by autoradiography demonstrated that only Sgd1FL-HTP, HTP-Sgd1MIF4G-C-term and HTP-Sgd1MA3-Cterm specifically crosslinked to RNA in vivo, while no RNA was specifically recovered with any of the other Sgd1 fragments tested (, lower panel). To confirm that the C-terminal region of Sgd1 is responsible for RNA binding, fluorescence anisotropy experiments were performed using a fluorescently labelled 11 nt RNA substrate and recombinant MBP-Sgd1FL-His and truncated fragments purified from E. coli. Neither the MIF4G domain alone nor a fragment encompassing the N-terminal region and the MIF4G domain bound to the RNA whereas the fragments containing either the MIF4G domain, MA3 domain and C-terminal region or the MA3 domain and C-terminal region showed increasing anisotropy as the concentration of protein was raised, indicating that they bind to RNA (). These results are consistent with the in vivo crosslinking data and confirm that the C-terminal region of Sgd1 binds RNA.
Figure 5. The C-terminal region of Sgd1 binds RNA. (A) Schematic view of HTP tagged fragments of Sgd1 showing the positions of the predicted MIF4G and MA3 domains. Amino acid numbers corresponding to the boundaries of domains and fragments are given above each scheme. (B) The Sgd1-HA-AID yeast strain was transformed with plasmids for expression of the fragments depicted in (A). Exponentially growing cells were depleted of endogenous Sgd1 by auxin treatment before crosslinking in culturo using irradiation of 254 nm. Protein-RNA complexes were enriched by tandem affinity purification under native and denaturing conditions, and RNAs were partially digested and 5ʹ labelled with [32P]. Complexes were separated by denaturing PAGE and transferred to nitrocellulose membranes. Proteins were detected by western blotting using an anti-PAP antibody (upper panel) and radiolabelled RNAs were detected by autoradiography (lower panel). Signals corresponding to specifically crosslinked RNAs are indicated by boxes and non-specific signals are indicated with asterisks. (C) Anisotropy measurements of an 11 nt, Atto647 labelled RNA in the presence of different amounts of purified, MBP/His tagged Sgd1 fragments were performed. The data from three independent experiments is presented as mean ± standard deviation.
![Figure 5. The C-terminal region of Sgd1 binds RNA. (A) Schematic view of HTP tagged fragments of Sgd1 showing the positions of the predicted MIF4G and MA3 domains. Amino acid numbers corresponding to the boundaries of domains and fragments are given above each scheme. (B) The Sgd1-HA-AID yeast strain was transformed with plasmids for expression of the fragments depicted in (A). Exponentially growing cells were depleted of endogenous Sgd1 by auxin treatment before crosslinking in culturo using irradiation of 254 nm. Protein-RNA complexes were enriched by tandem affinity purification under native and denaturing conditions, and RNAs were partially digested and 5ʹ labelled with [32P]. Complexes were separated by denaturing PAGE and transferred to nitrocellulose membranes. Proteins were detected by western blotting using an anti-PAP antibody (upper panel) and radiolabelled RNAs were detected by autoradiography (lower panel). Signals corresponding to specifically crosslinked RNAs are indicated by boxes and non-specific signals are indicated with asterisks. (C) Anisotropy measurements of an 11 nt, Atto647 labelled RNA in the presence of different amounts of purified, MBP/His tagged Sgd1 fragments were performed. The data from three independent experiments is presented as mean ± standard deviation.](/cms/asset/c4a1346c-6335-41ce-91d9-b945eb97938b/krnb_a_1716540_f0005_oc.jpg)
Both the RNA binding region of Sgd1 and the interaction of Sgd1 with Fal1 are required for SSU biogenesis
Our data suggest that the C-terminal region of Sgd1 contributes to the association of Sgd1 with the 18S rRNA sequence while the MIF4G domain mediates an interaction with Fal1 that stimulates the catalytic activity of the helicase. To further consolidate this model and demonstrate the functional importance of these interactions for ribosome biogenesis, a yeast complementation system was generated. The Sgd1-HA-AID strain was transformed with constructs for expression of Sgd1FL or fragments of Sgd1 from a MET25 promoter to allow the ability of each fragment to rescue pre-rRNA processing defects caused by lack of the endogenous protein to be monitored. When cells carrying an empty plasmid backbone (EV) were depleted of endogenous Sgd1, the 35S, 23S, 22S and 21S pre-rRNA accumulated while the 20S and 27SA pre-rRNAs were reduced, as previously observed ( and ). Importantly, expression of plasmid-derived Sgd1FL rescued all these defects confirming that they arise due to the specific depletion of Sgd1. Expression of each of the fragments of Sgd1, with the exception of a fragment encompassing both the MIF4G domain and the
Figure 6. The RNA binding domain of Sgd1 and the interaction with Fal1 are required for the function of Sgd1 in SSU biogenesis. The Sgd1-HA-AID yeast strain was transformed with a pRS415 plasmid for expression of proteins from a MET25 promoter (EV) or derivative plasmids for the expression of full length Sgd1 (Sgd1FL) or different fragments of Sgd1. Exponentially growing cells were treated with auxin (IAA) to deplete endogenous Sgd1 and then total RNA was isolated. RNAs were separated by denaturing agarose gel electrophoresis then transferred for northern blotting using [32P] labelled probes hybridizing within ITS1 and ITS2 of pre-rRNA transcripts. Pre-rRNAs were detected using a phosphorimager and mature rRNAs were visualized by methylene blue staining.
![Figure 6. The RNA binding domain of Sgd1 and the interaction with Fal1 are required for the function of Sgd1 in SSU biogenesis. The Sgd1-HA-AID yeast strain was transformed with a pRS415 plasmid for expression of proteins from a MET25 promoter (EV) or derivative plasmids for the expression of full length Sgd1 (Sgd1FL) or different fragments of Sgd1. Exponentially growing cells were treated with auxin (IAA) to deplete endogenous Sgd1 and then total RNA was isolated. RNAs were separated by denaturing agarose gel electrophoresis then transferred for northern blotting using [32P] labelled probes hybridizing within ITS1 and ITS2 of pre-rRNA transcripts. Pre-rRNAs were detected using a phosphorimager and mature rRNAs were visualized by methylene blue staining.](/cms/asset/aab1295b-b577-4d74-87b5-6c2754560476/krnb_a_1716540_f0006_oc.jpg)
Discussion
Many of the RNA helicases expressed in eukaryotes associate with large RNP complexes, such as spliceosomes or pre-ribosomes where they contribute to diverse aspects of RNP remodelling, such as rearranging RNA-RNA interactions to help establish mature folds or resolving aberrant structures as well as displacing proteins from their binding sites on target RNAs. Several of the RNA helicases implicated in ribosome assembly have remained largely uncharacterized and little is known about the interactions they form within pre-ribosomal complexes or the functional importance of their catalytic activity and how this is regulated.
Here we have analysed the role of the eIF4A-like RNA helicase Fal1 and its putative cofactor, the MIF4G domain-containing protein Sgd1, in small subunit maturation. We uncover a direct interaction between Fal1 and Sgd1, and demonstrate that the MIF4G domain of Sgd1 is required for formation of this complex. Interestingly, while several MIF4G domain-containing cofactors have been shown to stimulate the activity of their cognate helicases (e.g. eIF4G and eIF4A, Cnot1 and Dhh1, and PAIP1and eIF4AII/III [Citation60–Citation62]), the MIF4G domain-containing protein CWC22 inhibits the catalytic activity of eIF4A-III, converting it to an RNA clamp that locks the exon-junction complex (EJC) onto its substrate RNAs [Citation63,Citation64]. Structurally, the MIF4G domain is composed of ten anti-parallel α helices arranged into five HEAT repeats that together form an arc. Notably, in comparison to the MIF4G domain of Sgd1 and most other MIF4G domain-containing proteins, CWC22 contains an additional α helix that folds back across the MIF4G domain, altering its interactions with the RecA-like domains of eIF4A-III such that the RecA1 and RecA2 domains are orientated away from each other inhibiting ATP binding and hydrolysis [Citation65]. The finding that the MIF4G domain of Sgd1 stimulates the ATPase activity of Fal1 implies that it forms a more classical structure in which binding of the MIF4G domain to the RecA-like domains of Fal1 stabilizes the closed conformation of the helicase such that the conserved sequence motifs involved in ATP binding and hydrolysis, and substrate binding are in closer proximity, leading to a higher rate of catalytic activity.
Using in vivo crosslinking, we identify 18S-H12 as a pre-ribosomal binding site of Sgd1 and our data suggest that this pre-rRNA region is contacted by the C-terminal region of Sgd1. Interestingly, this region does not contain any known RNA binding domains/motifs and although there are some patches of basic amino acids present, these are not extensive, so the mode of RNA binding by Sgd1 remains elusive. In contrast to Sgd1, it was, unfortunately, not possible to identify a crosslinking site of Fal1 on pre-rRNAs. While it is possible that the lack of RNAs crosslinked to Fal1 reflects the nature of the contact site (UV254-induced RNA-protein crosslinks predominantly form between uridine nucleotides and the side chains of aromatic amino acids [Citation57]), it is more likely due to the transient nature of the interactions of Fal1 with pre-ribosomal complexes as, under steady-state conditions, the vast majority of Fal1 is not pre-ribosome associated. This is consistent with the absence of Fal1 from any of the recently isolated early pre-ribosomal complexes (see for example [Citation11,Citation12]). It remains unknown if Fal1 and Sgd1 are recruited to pre-ribosomal complexes together, but the finding that Sgd1 is more stably associated with pre-ribosomal complexes than Fal1 rather suggests a model in which the interaction between these proteins is formed within the context of pre-ribosomes. This notion is further supported by the finding that depletion of Fal1 does not influence the amount of Sgd1 co-migrating with pre-ribosomal complexes (Supplementary Figure S7). Together with the low intrinsic activity of Fal1 in the absence of RNA, a transient interaction with a cofactor-bound substrate is likely to be an effective means of minimizing non-specific activity of this helicase. As structural information on Sgd1 is not currently available, it remains to be determined whether the MIF4G domain that mediates interaction with Fal1 is also in close proximity to the 5ʹ domain of the 18S rRNA and therefore how Fal1 is placed relative to Sgd1. Notably, the crosslinking site of Sgd1 is adjacent to the binding site of the AF Lcp5 [Citation12] and genetic interactions between Fal1 and Lcp5 have previously been reported [Citation66], supporting the model that Fal1 also associates with this region of the pre-ribosome.
The identification of crosslinking sites of RNA helicases and/or their cofactors on pre-rRNAs can drive hypotheses on the functions of these complexes during ribosome assembly. Nevertheless, in the absence of cryoEM structures of pre-ribosomal particles containing the helicase/cofactor of interest, identification of the precise molecular targets of the remodelling activity of RNA helicases within such RNPs remains challenging. In the case of Fal1-Sgd1, our data strongly suggest that they function during the early stages of ribosome assembly and likely within the context of the SSU processome. Surprisingly, although most snoRNA-guided rRNA modifications occur on such early pre-ribosomal particles, depletion of Fal1 does not significantly alter the levels of any specific snoRNAs on pre-ribosomes [Citation26], implying that Fal1-Sgd1 do not play roles in either promoting the release of snoRNAs or facilitating rRNA modifications. When either Fal1 or Sgd1 is lacking, or the interaction between these proteins is disrupted, pre-rRNA cleavages at sites A0, A1 and A2 are impeded. Processing at these sites is spatially and temporally co-ordinated and cleavage at the A0 site has recently been shown to only occur upon compaction of the SSU processome, triggered by conformational changes within the Utp-B complex [Citation18]. Impaired cleavage at the A0, A1 and A2 sites is a pre-rRNA processing phenotype that is also observed upon depletion of various other SSU processome components (see for example [Citation67–Citation69]), implying that it can arise due to impaired assembly of this complex when key factors are lacking. However, the fact that stable core SSU processomes that lack Fal1 or Sgd1 can be isolated [Citation11,Citation12], suggests that neither of these proteins is critical for the integrity of these particles. Nevertheless, in the available cryoEM structures of SSU processomes [Citation12] and 5ʹ ETS RNPs [Citation18], the pre-rRNA sequences containing the A0 and A1 sites are distant from the Sgd1 crosslinking site, implying that Fal1-Sgd1 do not directly facilitate processing at these sites. While the molecular function of Fal1-Sgd1 in small subunit maturation remains elusive, it is tempting to speculate that Sgd1 stimulates the catalytic activity of Fal1 to re-arrange rRNA-rRNA interactions in the 5ʹ domain of the 18S rRNA sequence within the SSU processome to help establish the mature rRNA fold, which may in turn promote recruitment or correct positioning of specific RPs or AFs. Taken together, our data provide spatial, temporal and catalytic insights into AFs not captured within the cryoEM structures of SSU processomes.
Materials and methods
Yeast strains and protein depletion
Yeast strains used in this study are based on Saccharomyces cerevisiae BY4171 (Supplementary Table S1) and were grown at 30°C in YPD (1% yeast extract, 2% peptone/tryptone, 2% glucose) or synthetic glucose media lacking specific amino acids (1.9 g/L yeast nitrogen base without amino acids, 5 g/L ammonium sulphate, 2% dextrose, 0.77 g/L complete supplemented drop out -His/-Ura or 0.69 g/L complete supplemented drop out -Leu (Formedium)). Cells were maintained in exponential phase for >12 h before harvesting. Yeast strains for genomic expression of C-terminally His6-TEV protease cleavage site-ProteinA (C-HTP) tagged Sgd1 or Fal1, or C-terminally 6xHA tagged Fal1, were generated according to standard protocols using the primers listed in Supplementary Table S2. To generate strains for the auxin-dependent depletion of Fal1 or Sgd1 from yeast, strains expressing C-terminally auxin-induced degron (AID)-6x HA tagged Fal1 or Sgd1 from their genomic loci were generated in a parental strain expressing the Oryza sativa TIR1 gene (Supplementary Table S1 [Citation54]). For the transient depletion of Fal1 or Sgd1, exponentially growing yeast expressing Fal1-AID-HA or Sgd1-AID-HA were treated with 0.5 mM 3-indoleacetic acid (IAA; Sigma-Aldrich) for 30–90 min before harvesting. To generate a yeast complementation system, the coding sequence of Fal1 with 500 bp upstream and downstream was amplified and cloned in the pRS415 plasmid to enable expression of wild type or mutant Fal1 at the endogenous level in yeast. Site-directed mutagenesis, using the primers listed in Supplementary Table S2, was performed to induce a substitution of glutamate 174 with glutamine in the expressed protein (Fal1DQAD). For expression of C-terminally His6-TEV protease cleavage site-ProteinA (C-HTP) tagged full length Sgd1, N-terminally ProteinA-TEV protease cleavage site-His6 (N-HTP) tagged fragments of Sgd1 or untagged versions of Sgd1 fragments in yeast, the coding sequence of Sgd1 (or fragments thereof) were cloned into pRS415-derived plasmids for expression of (un-)tagged proteins from a Met25 promoter. All constructs generated in this study are listed in Supplementary Table S3. Yeast strains were transformed with pRS415-derived plasmids as listed in Supplementary Table S3. Analysis of proteins from yeast was performed by lysis of cells using glass beads, precipitation of proteins using 15% trichloroacetic acid (TCA) followed by SDS-PAGE and western blotting using the antibodies listed in Supplementary Table S4.
Expression of recombinant proteins in escherichia coli and their purification
For recombinant expression of Fal1 in E. coli, the Fal1 coding sequence was amplified from yeast genomic DNA with appropriate restriction enzyme cleavage sites and cloned into pQE80-derived plasmid for the IPTG-inducible expression of a protein with an N-terminal His10 tag (Fal1) using the primers listed in Supplementary Table S2. Using the primers listed in Supplementary Table S2, site-directed mutagenesis was performed to generate a construct for expression of Fal1DQAD as described above. For recombinant expression of Sgd1 in E. coli, a codon-optimized version of the Sgd1 coding sequence was synthesized by MWG Eurofins and sub-cloned into a pQE80-derived plasmid for the expression of an N-terminally MBP-TEV protease cleavage site, C-terminally His tagged protein. Fragments of the codon-optimized Sgd1 coding sequence (amino acids 1–541 (Sgd1Nterm-MIF4G); 330–775 (Sgd1MIF4G-MA3); 330–900 (Sgd1MIF4G-Cterm); 645–900 (Sgd1MA3-Cterm)) were similarly cloned for expression with N-terminal MBP-TEV protease cleavage site and C-terminal His10 tags or alternatively, into a plasmid for expression of N-terminally His-ZZ-tagged proteins.
Expression of His-Fal1, MBP-Sgd1-His or MBP-His tagged fragments of Sgd1 were induced in BL21 codon+ or pLysS E. coli cells by addition of 1 mM IPTG for 16 h at 18°C. After addition of 0.1 mM phenylmethylsulfonylfluoride (PMSF), cells were pelleted, resuspended in Lysis Buffer (50 mM Tris-HCl pH 7.4, 500 mM NaCl, 1 mM MgCl2, 10% glycerol, 1 mM PMSF and 5 mM imidazole) and disrupted by sonication. The cell lysate was cleared by centrifugation at 20,000 g for 20 min at 4°C. To remove protein-associated nucleic acids, 0.05% (v/v) polyethyleneimine (Sigma-Aldrich) was added and the lysate was incubated at 4°C for 15 min with agitation before centrifugation at 33,000 g for 30 min at 4°C. The soluble fraction was incubated with cOmplete His-tag purification resin (Roche) that had been pre-equilibrate in Wash Buffer I (50 mM Tris-HCl pH 7.4, 500 mM NaCl, 1 mM MgCl2, 10% glycerol and 30 mM imidazole,) for 1 h rotating at 4°C. The matrix was then washed sequentially with Wash Buffer I, Wash Buffer II (50 mM Tris-HCl pH 7.4, 1 M NaCl, 1 mM MgCl2, 10% glycerol and 30 mM imidazole) and Wash Buffer I. Bound proteins were eluted by addition of Elution Buffer (50 mM Tris-HCl pH 7.4, 500 mM NaCl, 1 mM MgCl2, 10% glycerol and 300 mM imidazole) for 5 min. The eluate was dialysed against a buffer containing 50 mM Tris-HCl pH 7.4, 120 mM NaCl, 2 mM MgCl2 and 20% glycerol. The concentration of proteins was determined using a Bradford assay and purity was assessed by SDS-PAGE followed by Coomassie staining.
In vitro assays
NADH-coupled ATPase assay
ATP hydrolysis was monitored used an NADH-coupled assay [Citation55,Citation70]. Reactions contained 50 mM Tris-HCl pH 7.4, 25 mM NaCl, 2 mM MgCl2, 1.5 mM phosphoenolpyruvate, 450 μM NADH, 20 U/ml pyruvate kinase/lactic dehydrogenase (PK/LDH; Sigma-Aldrich) and 4 mM ATP, and were supplemented with 1.5 μM RNA (5ʹ-GUAAUGCAAGUGAACGUACACACACACACACA-3ʹ), 1.5 μM recombinant His tagged Fal1/Fal1DQAD and/or 1.5 μM recombinant His- and ZZ-tagged Sgd1MIF4G. The decrease in absorbance at 340 nm was monitored using a BioTEK Synergy plate reader and used to calculate the ATP hydrolysis using the following equation.
The molar absorption coefficient for a given optical pathlength (Kpath = 2.1515 mM−1) was determined by monitoring the background NADH decomposition.
Protein-protein interactions
Interactions between purified recombinant proteins were analysed using an in vitro binding assay [Citation71]. 300 pmol recombinant MBP/His tagged full length Sgd1 or Sgd1 fragments in Binding Buffer (50 mM Tris-HCl pH 7.8, 300 mM NaCl, 1.5 mM MgCl2, 0.1% NP-40, 1 mM DTT, 66 ng/μL RNase A, supplemented with protease inhibitors) were incubated with amylose beads that had been pre-equilibrated in Binding Buffer for 1 h at 4°C rotating. The supernatant was removed and the beads were then washed 5x with Binding Buffer to remove unbound bait protein. 300 pmol His-Fal in Binding Buffer was added and samples were incubated for 2 h at 4°C rotating. After 5x washing steps with Binding Buffer, proteins were eluted using 1x SDS Buffer (60 mM Tris/HCl pH 6.8, 2% SDS, 0.01% bromophenol blue, 1.25% β-mercaptoethanol) at 95°C for 5 min. Eluted proteins were visualized by SDS-PAGE followed by Coomassie staining.
Alternatively, proteins were retrieved from cells extract using immobilized recombinant proteins [Citation72]. Recombinant MBP-TEV-Sgd1-His was immobilized on amylose beads in Yeast lysis buffer (50 mM Tris-HCl pH 7.8, 150 mM NaCl, 1.5 mM MgCl2, 0.05% NP-40, 5 mM β-mercaptothanol, supplemented with protease inhibitors). Yeast cells from strains expressing pTetO7-HA-Rok1 or Fal1-HA were lysed and cell debris were pelleted by centrifugation at 4,000 g for 10 min. The soluble extract was either left untreated or treated with 30 μg RNase A at 16°C for 15 min. The lysate was incubated with the MBP-TEV-Sgd1-His-bound beads before thorough washing steps. Sgd1-containing complexes were eluted with TEV protease overnight at 4°C and proteins were analysed by SDS-PAGE and western blotting.
Fluorescence anisotropy
Fluorescence anisotropy measurements were performed as previously described [Citation73], proteins were first dialysed against a buffer containing 30 mM Tris-HCl pH 7.5, 50 mM NaCl) overnight. Reactions containing 0.1–4 μM protein and 20 nM Atto647-labelled RNAs (5ʹ-GUAAUGAAAGU-ATTO647NN −3ʹ) in 30 mM Tris-HCl pH 7.5, 50 mM NaCl were incubated at RT for 1 min. Anisotropy measurements were then performed using a FluoroMax-4 spectrofluorometer (Horiba Scientific).
RNA extraction and northern blotting
Total RNA was extracted from yeast cells as previously described [Citation23]. For detection of pre-rRNAs by northern blotting 6 μg of total RNA was separated on a 1.2% denaturing (glyoxal) agarose gel and transferred to a Hybond-N+ nylon membrane by vacuum blotting in 6x SSC Buffer (900 mM NaCl, 100 mM sodium citrate pH 7.0). Membranes were stained with methylene blue solution (0.3 M sodium acetate pH 5.2, 0.1% methylene blue) to enable visualization of abundant RNAs, then pre-hybridized in SES1 Buffer (0.5 M sodium phosphate pH 7.0, 7% SDS (w/v), 1 mM EDTA) before incubation with 5ʹ [32P]-labelled DNA oligonucleotides (Supplementary Table S2) diluted in SES1 buffer at 37°C overnight. Membranes were washed sequentially in 6x SSC and 2x SSC supplemented with 0.1% SDS for 30 min each at 37°C. Radioactive signals were detected using a Typhoon FLA9500 phosphorimager. For re-probing, membranes were stripped using Stripping Buffer (0.1x SSC, 0.1% SDS, previously heated to 100°C) at 70°C for 1 h.
In vivo protein-RNA crosslinking techniques
UV crosslinking and analysis of cDNA (CRAC) was performed as previously described [Citation57,Citation58,Citation74]. In brief, exponentially growing yeast cells expressing C-terminally HTP tagged Fal1 or Sgd1 from their genomic loci, or wild type yeast as a control, were crosslinked using UV light at 254 nm in culturo. Cells were harvested, lysed and HTP tagged proteins and their associated RNAs were retrieved by sequential enrichment on IgG sepharose and Ni-NTA under native and denaturing (6 M guanidium hydrochloride) conditions respectively. Co-purified RNAs were partially digested using RNace-IT (Agilent Technologies), 5ʹ and 3ʹ adaptors were ligated and the RNAs were 5ʹ [32P]-labelled. Crosslinked protein-RNA complexes were separated by denaturing PAGE and transferred to a nitrocellulose membrane by wet blotting. Radioactive signals were visualized by autoradiography and regions of the membrane containing specific radioactive signals were excised. RNAs were eluted by Proteinase K digestion, isolated and reverse transcribed. PCR was used to amplify cDNA libraries that were subjected to Illumina sequencing. The obtained sequencing reads were trimmed and quality controlled using Flexbar then mapped to the S. cerevisiae genome (version 2.2.4) using Bowtie 2. The proportions of reads mapping to different types of RNA were determined using pyCRAC read counting [Citation75] and python scripts were used to map the data onto the available secondary [Citation59] and tertiary (PDB: 5WLC [Citation12];) structures of the rRNA as previously described [Citation76].
To detect RNA crosslinking to HTP-tagged proteins, cells expressing plasmid-encoded C-terminally HTP tagged full length Sgd1 or N-terminally HTP-tagged Sgd1 fragments were UV crosslinked as described above. After tandem affinity purification of crosslinked protein-RNA complexes and partial RNase digestion, RNA fragments were 5ʹ [32P]-labelled. Protein-RNA complexes were separated by denaturing PAGE, transferred to a nitrocellulose membrane and visualized by autoradiography.
Supplemental Material
Download Zip (6.8 MB)Acknowledgments
We thank the lab of Blanche Schwappach for assistance establishing the auxin-mediated depletion systems, Sven Vanselow for molecular cloning and Jens Kretschmer for bioinformatics analysis. This work was supported by the Deutsche Forschungsgemeinschaft (BO3442/1-2 to M.T.B) and the University Medical Centre Göttingen (to M.T.B.).
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
References
- Ben-Shem A, Garreau de Loubresse N, Melnikov S, et al. The structure of the eukaryotic ribosome at 3.0 Å resolution. Science. 2011;334:1524–1529.
- Anger AM, Armache J-P, Berninghausen O, et al. Structures of the human and drosophila 80S ribosome. Nature. 2013;497:80–85.
- Klinge S, Woolford JLJ. Ribosome assembly coming into focus. Nat Rev Mol Cell Biol. 2019;20:116–131.
- Bohnsack KE, Bohnsack MT. Uncovering the assembly pathway of human ribosomes and its emerging links to disease. Embo J. 2019;38:e100278.
- Koš M, Tollervey D. Yeast pre-rRNA processing and modification occur cotranscriptionally. Mol Cell. 2010;37:809–820.
- Watkins NJ, Bohnsack MT. The box C/D and H/ACA snoRNPs: key players in the modification, processing and the dynamic folding of ribosomal RNA. Wiley Interdiscip Rev RNA. 2012;3:397–414.
- Sloan KE, Warda AS, Sharma S, et al. Tuning the ribosome: the influence of rRNA modification on eukaryotic ribosome biogenesis and function. RNA Biol. 2017;14:1138–1152.
- Henras AK, Plisson-Chastang C, O’Donohue MF, et al. An overview of pre-ribosomal RNA processing in eukaryotes. Wiley Interdiscip Rev RNA. 2015;6:225–242.
- Miller OLJ, Beatty BR. Visualization of nucleolar genes. Science. 1969;164:955–957.
- Osheim YN, French SL, Keck KM, et al. Pre-18S ribosomal RNA is structurally compacted into the SSU processome prior to being cleaved from nascent transcripts in Saccharomyces cerevisiae. Mol Cell. 2004;16:943–954.
- Chaker-Margot M, Barandun J, Hunziker M, et al. Architecture of the yeast small subunit processome. Science. 2017;355:eaal1880.
- Barandun J, Chaker-Margot M, Hunziker M, et al. The complete structure of the small-subunit processome. Nat Struct Mol Biol. 2017;24:944–953.
- Phipps KR, Charette JM, Baserga SJ. The small subunit processome in ribosome biogenesis-progress and prospects. Wiley Interdiscip Rev RNA. 2011;2:1–21.
- Sloan KE, Gleizes PE, Bohnsack MT. Nucleocytoplasmic transport of RNAs and RNA-protein complexes. J Mol Biol. 2016;428:2040–2059.
- Wu S, Tutuncuoglu B, Yan K, et al. Diverse roles of assembly factors revealed by structures of late nuclear pre-60S ribosomes. Nature. 2016;534:133–137.
- Kater L, Thoms M, Barrio-Garcia C, et al. Visualizing the assembly pathway of nucleolar Pre-60S ribosomes. Cell. 2017;171:1599–1610.
- Sanghai ZA, Miller L, Molloy KR, et al. Modular assembly of the nucleolar pre-60S ribosomal subunit. Nature. 2018;556:126–129.
- Hunziker M, Barandun J, Buzovetsky O, et al. Conformational switches control early maturation of the eukaryotic small ribosomal subunit. Elife. 2019;8:e45185.
- Kressler D, Hurt E, Bassler J. Driving ribosome assembly. Biochim Biophys Acta. 2010;1803:673–683.
- Martin R, Straub AU, Doebele C, et al. DExD/H-box RNA helicases in ribosome biogenesis. RNA Biol. 2013;10:4–18.
- Koš M, Tollervey D. The putative RNA helicase Dbp4p is required for release of the U14 snoRNA from preribosomes in Saccharomyces cerevisiae. Mol Cell. 2005;20:53–64.
- Liang X-H, Fournier MJ. The helicase Has1p is required for snoRNA release from pre-rRNA. Mol Cell Biol. 2006;26:7437–7450.
- Bohnsack MT, Kos M, Tollervey D. Quantitative analysis of snoRNA association with pre-ribosomes and release of snR30 by Rok1 helicase. EMBO Rep. 2008;9:1230–1236.
- Bohnsack MT, Martin R, Granneman S, et al. Prp43 bound at different sites on the Pre-rRNA performs distinct functions in ribosome synthesis. Mol Cell. 2009;36:583–592.
- Pertschy B, Schneider C, Gnadig M, et al. RNA helicase Prp43 and its co-factor Pfa1 promote 20 to 18 S rRNA processing catalyzed by the endonuclease Nob1. J Biol Chem. 2009;284:35079–35091.
- Dembowski JA, Kuo B, Woolford JLJ. Has1 regulates consecutive maturation and processing steps for assembly of 60S ribosomal subunits. Nucleic Acids Res. 2013;41:7889–7904.
- Martin R, Hackert P, Ruprecht M, et al. A pre-ribosomal RNA interaction network involving snoRNAs and the Rok1 helicase. RNA. 2014;20:1173–1182.
- Sardana R, Liu X, Granneman S, et al. The DEAH-box helicase Dhr1 dissociates U3 from the pre-rRNA to promote formation of the central pseudoknot. PLoS Biol. 2015;13:e1002083.
- Khoshnevis S, Askenasy I, Johnson MC, et al. The DEAD-box protein Rok1 Orchestrates 40S and 60S Ribosome assembly by promoting the release of Rrp5 from Pre-40S ribosomes to allow for 60S maturation. PLoS Biol. 2016;14:e1002480.
- Sharma S, Langhendries JL, Watzinger P, et al. Yeast Kre33 and human NAT10 are conserved 18S rRNA cytosine acetyltransferases that modify tRNAs assisted by the adaptor Tan1/THUMPD1. Nucleic Acids Res. 2015;43:2242–2258.
- Bruning L, Hackert P, Martin R, et al. RNA helicases mediate structural transitions and compositional changes in pre-ribosomal complexes. Nat Commun. 2018;9:5383.
- Neumann B, Wu H, Hackmann A, et al. Nuclear export of pre-ribosomal subunits requires Dbp5, but not as an RNA-helicase as for mRNA export. PLoS One. 2016;11:e0149571.
- Gnanasundram SV, Kos-Braun IC, Kos M. At least two molecules of the RNA helicase Has1 are simultaneously present in pre-ribosomes during ribosome biogenesis. Nucleic Acids Res. 2019;47:10852–10864.
- Dembowski JA, Kuo B, Woolford JL. Has1 regulates consecutive maturation and processing steps for assembly of 60S ribosomal subunits. Nucleic Acids Res. 2013;41:7889–7904.
- Sloan KE, Bohnsack MT. Unravelling the mechanisms of RNA helicase regulation. Trends Biochem Sci. 2018;43:237–250.
- Linder P, Jankowsky E. From unwinding to clamping - the DEAD box RNA helicase family. Nat Rev Mol Cell Biol. 2011;12:505–516.
- Granneman S, Lin C, Champion EA, et al. The nucleolar protein Esf2 interacts directly with the DExD/H box RNA helicase, Dbp8, to stimulate ATP hydrolysis. Nucleic Acids Res. 2006;34:3189–3199.
- Young CL, Khoshnevis S, Karbstein K. Cofactor-dependent specificity of a DEAD-box protein. Proc Natl Acad Sci USA. 2013;110:2668–2676.
- Zhu J, Liu X, Anjos M, et al. Utp14 recruits and activates the RNA helicase Dhr1 to undock U3 snoRNA from the preribosome. Mol Cell Biol. 2016;36:965–978.
- Aravind L, Koonin EV. G-patch: a new conserved domain in eukaryotic RNA-processing proteins and type D retroviral polyproteins. Trends Biochem Sci. 1999;24:342–344.
- Robert-Paganin J, Réty S, Leulliot N. Regulation of DEAH/RHA helicases by G-patch proteins. Biomed Res Int. 2015;2015:1–9.
- Christian H, Hofele RV, Urlaub H, et al. Insights into the activation of the helicase Prp43 by biochemical studies and structural mass spectrometry. Nucleic Acids Res. 2014;42:1162–1179.
- Heininger AU, Hackert P, Andreou AZ, et al. Protein cofactor competition regulates the action of a multifunctional RNA helicase in different pathways. RNA Biol. 2016;13:320–330.
- Fourmann J-B, Tauchert MJ, Ficner R, et al. Regulation of Prp43-mediated disassembly of spliceosomes by its cofactors Ntr1 and Ntr2. Nucleic Acids Res. 2017;45:4068–4080.
- Robert-Paganin J, Halladjian M, Blaud M, et al. Functional link between DEAH/RHA helicase Prp43 activation and ATP base binding. Nucleic Acids Res. 2017;45:1539–1552.
- Lebaron S, Papin C, Capeyrou R, et al. The ATPase and helicase activities of Prp43p are stimulated by the G-patch protein Pfa1p during yeast ribosome biogenesis. Embo J. 2009;28:3808–3819.
- Ponting CP. Novel eIF4G domain homologues linking mRNA translation with nonsense-mediated mRNA decay. Trends Biochem Sci. 2000;25:423–426.
- Ozgur S, Buchwald G, Falk S, et al. The conformational plasticity of eukaryotic RNA-dependent ATPases. Febs J. 2015;282:850–863.
- Schutz P, Bumann M, Oberholzer AE, et al. Crystal structure of the yeast eIF4A-eIF4G complex: an RNA-helicase controlled by protein-protein interactions. Proc Natl Acad Sci USA. 2008;105:9564–9569.
- Alexandrov A, Colognori D, Steitz JA. Human eIF4AIII interacts with an eiF4G-like partner, NOM1, revealing an evolutionarily conserved function outside the exon junction complex. Genes Dev. 2011;25:1078–1090.
- Andreou AZ, Klostermeier D. The DEAD-box helicase eIF4A: paradigm or the odd one out? RNA Biol. 2013;10:19–32.
- Kressler D, de la Cruz J, Rojo M, et al. Fal1p is an essential DEAD-box protein involved in 40S-ribosomal-subunit biogenesis in Saccharomyces cerevisiae. Mol Cell Biol. 1997;17:7283–7294.
- Nishimura K, Fukagawa T, Takisawa H, et al. An auxin-based degron system for the rapid depletion of proteins in nonplant cells. Nat Methods. 2009;6:917–922.
- Morawska M, Ulrich HD. An expanded tool kit for the auxin-inducible degron system in budding yeast. Yeast. 2013;30:341–351.
- Kiianitsa K, Solinger JA, Heyer W-D. NADH-coupled microplate photometric assay for kinetic studies of ATP-hydrolyzing enzymes with low and high specific activities. Anal Biochem. 2003;321:266–271.
- Memet I, Doebele C, Sloan KE, et al. The G-patch protein NF-kappaB-repressing factor mediates the recruitment of the exonuclease XRN2 and activation of the RNA helicase DHX15 in human ribosome biogenesis. Nucleic Acids Res. 2017;45:5359–5374.
- Bohnsack MT, Tollervey D, Granneman S. Identification of RNA helicase target sites by UV cross-linking and analysis of cDNA. Methods Enzymol. 2012;511:275–288.
- Jungfleisch J, Nedialkova DD, Dotu I, et al. A novel translational control mechanism involving RNA structures within coding sequences. Genome Res. 2017;27:95–106.
- Petrov AS, Bernier CR, Gulen B, et al. Secondary structures of rRNAs from all three domains of life. PLoS One. 2014;9:e88222.
- Craig AW, Haghighat A, Yu AT, et al. Interaction of polyadenylate-binding protein with the eIF4G homologue PAIP enhances translation. Nature. 1998;392:520–523.
- Mugler CF, Hondele M, Heinrich S, et al. ATPase activity of the DEAD-box protein Dhh1 controls processing body formation. Elife. 2016;5:e18746.
- Hilbert M, Kebbel F, Gubaev A, et al. eIF4G stimulates the activity of the DEAD box protein eIF4A by a conformational guidance mechanism. Nucleic Acids Res. 2011;39:2260–2270.
- Alexandrov A, Colognori D, Shu M-D, et al. Human spliceosomal protein CWC22 plays a role in coupling splicing to exon junction complex deposition and nonsense-mediated decay. Proc Natl Acad Sci USA. 2012;109:21313–21318.
- Steckelberg A-L, Boehm V, Gromadzka AM, et al. CWC22 connects pre-mRNA splicing and exon junction complex assembly. Cell Rep. 2012;2:454–461.
- Buchwald G, Schussler S, Basquin C, et al. Crystal structure of the human eIF4AIII-CWC22 complex shows how a DEAD-box protein is inhibited by a MIF4G domain. Proc Natl Acad Sci USA. 2013;110:4611–4618.
- Wiederkehr T, Pretot RF, Minvielle-Sebastia L. Synthetic lethal interactions with conditional poly(A) polymerase alleles identify LCP5, a gene involved in 18S rRNA maturation. RNA. 1998;4:1357–1372.
- Bernstein KA, Gallagher JEG, Mitchell BM, et al. The small-subunit processome is a ribosome assembly intermediate. Eukaryot Cell. 2004;3:1619–1626.
- Wells GR, Weichmann F, Colvin D, et al. The PIN domain endonuclease Utp24 cleaves pre-ribosomal RNA at two coupled sites in yeast and humans. Nucleic Acids Res. 2016;44:5399–5409.
- Lee SJ, Baserga SJ. Functional separation of pre-rRNA processing steps revealed by truncation of the U3 small nucleolar ribonucleoprotein component, Mpp10. Proc Natl Acad Sci USA. 1997;94:13536–13541.
- Choudhury P, Hackert P, Memet I, et al. The human RNA helicase DHX37 is required for release of the U3 snoRNP from pre-ribosomal particles. RNA Biol. 2019;16:54–68.
- Sloan KE, Knox AA, Wells GR, et al. Interactions and activities of factors involved in the late stages of human 18S rRNA maturation. RNA Biol. 2019;16:196–210.
- Warda AS, Freytag B, Haag S, et al. Effects of the Bowen-Conradi syndrome mutation in EMG1 on its nuclear import, stability and nucleolar recruitment. Hum Mol Genet. 2016;25:5353–5364.
- Kretschmer J, Rao H, Hackert P, et al. The m(6)A reader protein YTHDC2 interacts with the small ribosomal subunit and the 5ʹ-3ʹ exoribonuclease XRN1. RNA. 2018;24:1339–1350.
- Braun CM, Hackert P, Schmid CE, et al. Pol5 is required for recycling of small subunit biogenesis factors and for formation of the peptide exit tunnel of the large ribosomal subunit. Nucleic Acids Res. 2019;48:405–420.
- Webb S, Hector RD, Kudla G, et al. PAR-CLIP data indicate that Nrd1-Nab3-dependent transcription termination regulates expression of hundreds of protein coding genes in yeast. Genome Biol. 2014;15:R8.
- Sloan KE, Leisegang MS, Doebele C, et al. The association of late-acting snoRNPs with human pre-ribosomal complexes requires the RNA helicase DDX21. Nucleic Acids Res. 2015;43:553–564.