
ABSTRACT
Enteroviruses, which may cause neurological complications, have become a public health threat worldwide in recent years. Interactions between cellular proteins and enteroviral proteins could interfere with cellular biological processes to facilitate viral replication in infected cells. Enteroviral RNA-dependent RNA polymerase (RdRP), known as 3D protein, mainly functions as a replicase for viral RNA synthesis in infected cells. However, the 3D protein encoded by enterovirus A71 (EV-A71) could also interact with several cellular proteins to regulate cellular events and responses during infection. To globally investigate the functions of the EV-A71 3D protein in regulating biological processes in host cells, we performed immunoprecipitation coupled with liquid chromatography−tandem mass spectrometry (LC-MS/MS) to identify host proteins that may associate with the 3D protein. We found that the 3D protein interacts with factors involved in translation-related biological processes, including ribosomal proteins. In addition, polysome profiling analysis showed that the 3D protein cosediments with small and large subunits of ribosomes. We further discovered that the EV-A71 3D protein could enhance EV-A71 internal ribosome entry site (IRES)-dependent translation as well as cap-dependent translation. Collectively, this research demonstrated that the RNA polymerase encoded by EV-A71 could join a functional ribosomal complex and positively regulate viral and host translation.
Introduction
Interactions between a pathogen and its host cells may facilitate pathogen multiplication or dysregulate the biological processes of the host cell. Accordingly, the pathogen–host interactions could induce pathogenesis during infection. Because enterovirus A71 (EV-A71) causes severe neurological complications in children, this virus has been considered a significant threat to public health [Citation1–Citation4]. Many cellular factors have been revealed to interact with EV-A71 RNA or proteins to promote viral replication during infection. In addition, the interactions may regulate host cellular processes and responses upon infection [Citation4–Citation6].
EV-A71 belongs to the picornavirus family. The virion contains a positive-stranded RNA genome, which also serves as an mRNA for translating a viral precursor polyprotein. The precursor polyprotein can be further processed by virus-encoded proteases to form functional viral proteins. Among the viral proteins, the RNA-dependent RNA polymerase (RdRP), known as the 3D protein, is involved in replicating the viral RNA genome. Previous studies have revealed that in addition to its RNA polymerase function, the enteroviral 3D protein could interact with the splicing factor Prp8 to regulate host splicing events or associate with the ER protein UGGT1 to facilitate viral RNA synthesis and be involved in pathogenesis [Citation7,Citation8]. Moreover, the 3D protein of EV-A71 could also be involved in the regulation of host innate immune responses. For example, the 3D protein binds to NLRP3 to support inflammasome formation and activate the inflammatory response [Citation9]. In addition, the 3D protein decreases STAT1 expression to attenuate IFN-γ signalling [Citation10] and interacts with the cytoplasmic pathogen sensor MDA5 to inhibit IFN-β promoter activation [Citation11]. Collectively, the 3D protein of EV-A71 might broadly associate with cellular factors to regulate host cellular processes.
To fulfil the requirement for productive infection, enterovirus also hijacks the host translation machinery by subverting cellular cap-dependent translation, which is the primary process for protein synthesis of host cells. The translation is carried out by ribosomes consisting of small (40S) and large (60S) subunits that are large assembly of ribosomal RNAs with a different set of ribosomal proteins, i.e. small (RPSs) and large (RPLs) ribosomal proteins for 40S and 60S subunits, respectively. Assembly of the 40S and 60S subunits forms 80S monosomes, the building blocks of polysomes [Citation12–Citation14]. Translation in eukaryotic cells is initiated by the association of the small ribosome subunit, the initiator tRNA, translation initiation factors, and mRNA. The translation initiation factor eIF4G interacts with several initiation factors, including eIF4E, which is the cap-binding initiation factor; hence, it links cellular capped mRNA to other translation initiation components for the loading of the small ribosome subunit to form the 48S preinitiation complex for translation initiation. The 2A protease encoded by picornavirus cleaves eIF4G [Citation15–Citation17]. The cleaved eIF4G loses the eIF4E-binding domain and consequently shut off cap-dependent translation in picornavirus-infected cells. Unlike cellular capped mRNA, the 5ʹ end of picornavirus mRNA is linked with the viral VPg protein, and its 5ʹ untranslated region forms a complex RNA secondary structure, known as the internal ribosome entry site (IRES), to initiate its translation [Citation18–Citation20]. However, loading of ribosomes onto IRES-containing mRNA also depends on binding of translation initiation factors, and the cleaved eIF4G recognizes picornavirus IRES and recruits ribosomes through the association with initiation factor eIF3. Therefore, the translation of the viral RNA can be maintained during enterovirus infection. In addition, several cellular proteins serve as IRES-specific trans-acting factors (ITAFs), such as FUBP1, FUBP2, and hnRNP A1, which could also interact with the EV-A71 IRES and regulate the translation efficiency in infected cells [Citation21–Citation23]. Still, further investigation is required to determine whether enteroviral proteins other than the 2A protein are involved in IRES- or cap-dependent translation regulation.
Interactome analysis has been widely applied to explore virus–host interactions. Previous studies have used yeast-two-hybrid assays to screen host factors that may interact with enteroviral proteins [Citation24,Citation25]. In addition, proteomic approaches based on MALDI-TOF mass spectrometry were used to identify the interacting proteins of EV-A71 3D protein in infected cells [Citation7,Citation8]. Nevertheless, advanced approaches using immunoprecipitation coupled with liquid chromatography−tandem mass spectrometry (LC−MS/MS) can be practical to broadly detect cellular proteins that associate with viral proteins [Citation26]. In the present study, we applied this approach to investigate the interacting partners in cells overexpressing the EV-A71 3D protein. Through a bioinformatics analysis, we discovered that the cellular proteins involved in the translation process, such as the ribosomal protein RPS6, could interact with the EV-A71 3D protein. In addition, we found that EV-A71 3D protein could be cosedimented with the small and large subunits of ribosomes in the cells transfected with the 3D protein or infected with EV-A71 and that the viral 3D protein positively regulates either cap- or EV-A71 IRES-dependent translation. This research not only provides a potential approach to examine the cellular biological processes that are regulated by the EV-A71 3D protein but also reveals the role of the EV-A71 3D protein in translational regulation.
Results
Identification of the host factors that interact with the EV-A71 3D protein
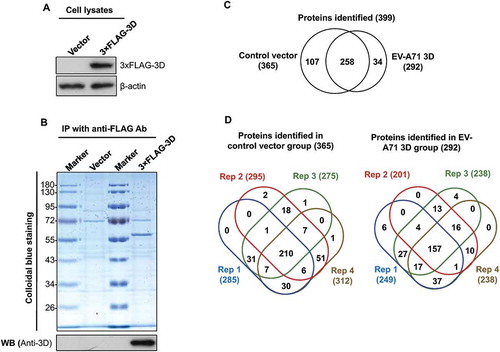
The EV-A71 3D protein, which mainly functions as an RNA polymerase, interacts with several cellular proteins, such as the key factor for spliceosome assembly, Prp8, to downregulate host protein expression in infected cells [Citation7], suggesting that profiling of the EV-A71 3D interactome can lead to the global discovery of EV-A71 3D functions. However, the aforementioned studies were conducted by immunoprecipitation of EV-A71-infected cell extracts with anti-3D antibodies that also recognize precursor proteins, and whether the identified proteins were 3D-specific is unclear. In addition, these results indicated that enterovirus may regulate the gene expression of host cells through a non-proteolytic mechanism [Citation27]. Therefore, we used overexpression rather than an infection system to prevent the interference of viral proteases and specifically analysed the 3D-interacting proteins. To systemically identify host proteins that interact with EV-A71 3D alone, we transfected HEK293T cells with plasmids expressing 3× FLAG-tagged 3D encoded by EV-A71. At 48 h post-transfection, the cell extracts were collected and subjected to anti-FLAG immunoprecipitation (). The precipitates were separated by SDS-PAGE and visualized with Colloidal Blue staining (). The gel lanes were then divided into 10 fractions and analysed by LC-MS/MS after in-gel tryptic digestion. To provide technical replicates, we sliced each fraction into four parts for the LC-MS/MS analysis. The analysis resulted in the identification of 399 non-redundant proteins with at least two unique peptides (Supplemental Table 1). Among the 399 proteins, 34 (8.52%) and 107 (26.82%) were only detected in the 3D and control groups, respectively, whereas 258 (64.66%) were found in both groups (Supplemental Table 2 and ). To evaluate the reproducibility of the proteomic analyses, we examined the proteins detected in four replicates for overlap. In total, 157 (53.77%) and 210 (57.53%) proteins were identified in all replicates of the 3D and control groups, respectively (Supplemental and ). Approximately 97% and 65% of the proteins were detected in at least two and three replicates of the experiments, respectively, whereas only approximately 3% were unique to single replicates (). Furthermore, using a decoy database, the false discovery rate (FDR) of peptide identification in each group was evaluated. All FDRs were less than 0.05%. The results collectively demonstrated that the proteome analysis was appropriately conducted.
Table 1. Spectral counting-based identification of proteins co-immunoprecipitating with EV-A71 3D protein.
Table 2. Enrichment analysis of biological processes for proteins interacted with the EV-A71 3D protein.
Figure 1. Identification of cellular proteins that interact with EV-A71 3D. HEK293T cells were transfected with control vectors and plasmids that express the 3× FLAG-tagged EV-A71 3D, respectively. At 48 h posttransfection, the lysates of transfected cells were examined by immunoblotting with anti-FLAG and anti-β-actin antibodies (A). The lysates were then subjected to immunoprecipitation with anti-FLAG resin, and the precipitated proteins were separated by SDS-PAGE following by a Colloidal Blue stain or an immunoblot with anti-EV-A71 3D antibody (B). The precipitated proteins were identified with LC-MS/MS. (C) Venn diagrams show overlaps between the proteins identified in the control and the 3D groups. (D) Venn diagrams display overlaps between the proteins identified in the four replicates. The total numbers of identified proteins are listed in brackets.
Spectral counting-based quantification to identify EV-A71 3D-interacting proteins
To identify proteins potentially interacting with the 3D protein, we determined the relative amounts of proteins identified in the immunoprecipitants with spectral counting-based protein quantification (Supplemental Table 2). The fold change for an individual protein was a ratio of the average spectral count (SC) of the protein in the 3D group versus that in the control group. Proteins with fold change greater than one standard deviation (SD) above the mean ratio (the fold changes were above 2.196) and observed in at least 2 replicates of the 3D group were considered proteins interacting with the 3D protein. As shown in , 50 proteins were observed based on these cut-off values. Among these proteins, 24 proteins in the 3D group had levels that were relatively higher than those in the control group, while the others were 3D group-specific proteins, including the COPI and ARF3 proteins that are involved in the generation of picornaviral replication organelles [Citation28,Citation29].
Involvement of EV-A71 3D in protein translation revealed by bioinformatics analysis
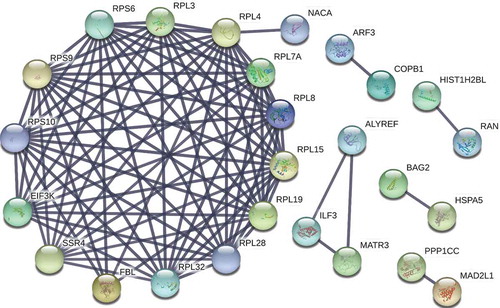
To determine the biological processes that are most likely affected by the presence of EV-A71 3D-associated complexes, we used DAVID to annotate the functions of the 50 proteins (). As shown in , enriched biological processes included protein translation, translational initiation, SRP-dependent cotranslational protein targeting to membrane, viral transcription, rRNA processing, and cell-cell adhesion. Moreover, pathway analysis of these proteins using the KEGG database revealed that the proteins most likely engage in ribosome and protein processing in the endoplasmic reticulum (). We further used the STRING online database to establish a network of protein–protein interactions (PPIs) between the 50 proteins, and 90 strong interaction links between individual nodes/proteins were depicted in the PPI network (). Consistent with the results from DAVID and KEGG analyses ( and ), one module was identified in the STRING analysis that depicted the interactions between ribosomal proteins associated with the biological processes of protein translation, translational initiation, and SRP-dependent cotranslational protein targeting to the membrane ().
Table 3. Pathway analysis of the proteins interacted with the EV-A71 3D protein.
Figure 2. Protein–protein interaction (PPI) network of the proteins identified in co-immunoprecipitate of EV-A71 3D. A PPI network of the 50 proteins listed in was constructed using the STRING v10 database (http://string-db.org/), depicting 90 interaction links between individual nodes/proteins (solid lines). One module was identified in STRING analysis that depicted the interactions of RPS6 with proteins involved in biological processes of protein translation, translational initiation, and SRP-dependent cotranslational protein targeting to membrane.
The EV-A713D interacts with the RPS6
To validate the interaction between EV-A71 3D and ribosomal proteins, we analysed the aforementioned anti-FLAG immunoprecipitates by immunoblotting. In addition, because EV-A71 strains circulating recently might carry 3D variants due to recombination [Citation30], 3D proteins from two different strains – the C2 and C4 subgenotypes – were used in this study, and we chose RPS6 as the candidate for further validation since it has been proven to specifically regulate IRES-dependent translation of poliovirus (PV) and Hepatitis C virus as well as cap-independent translation of Turnip mosaic virus and Tomato bushy stunt virus [Citation31–Citation33]. As shown in , RPS6 was coprecipitated with the 3× FLAG-tagged EV-A71 3D protein. The results confirmed that the 3D protein of EV-A71 could interact with the ribosomal protein RPS6. Moreover, RNase A treatment of the lysates did not disrupt this interaction (, middle panel), indicating that the association of EV-A71 and RPS6 is RNA-independent. We next determined whether EV-A71 3D can interact directly with RPS6 by using glutathione S-transferase (GST) pull-down assay. However, neither GST nor GST-RPS6 can pull down 3D protein (, Pull-down), suggesting that EV-A71 3D may interact with RPS6 indirectly. Given that many other ribosomal proteins were also identified in the current EV-A71 3D interactome, we hypothesized that EV-A71 3D might associate with ribosomes consisting of RPS6.
Figure 3. Validation of the interaction between the EV-A71 3D and ribosomal protein RPS6. (A) HEK293T cells were transfected with plasmids expressing the 3× FLAG-tagged EV-A71 3D from either C2 or C4 subgenotype. At 24 h posttransfection, the cell extracts were collected for anti-FLAG immunoprecipitation without (upper panel) or with RNase pretreatment (middle panel, 1 μg/μL at 27°C for 10 min). (B) Recombinant EV-A71 3D proteins were incubated with GST or GST-tagged RPS6 recombinant proteins and then selected by glutathione-Sepharose resin. Pull-down samples were subjected to immunoblotting using anti-EV-A71 3D and anti-GST antibodies. Input: 10% of the mixture composed of recombinant 3D and RPS6 proteins. (C) RD cells were infected with EV-A71 strain of 4643 at MOI 40 for 8 h. The cell extracts were subjected to immunoprecipitation with anti-3D antibody. (D) RD cells were transfected with the 3× FLAG-tagged RPS6-expressing plasmid for 20 h, and then infected with EV-A71 strain of 4643 at MOI 40 for 8 h. The cell extracts were then subjected to anti-FLAG immunoprecipitation. The extracts and immunoprecipitates were analysed by immunoblotting with anti-FLAG, anti-EV-A71 3D, anti-RPS6, and anti-β-actin antibodies.
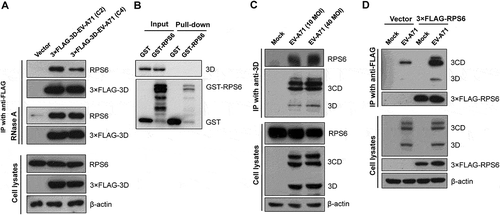
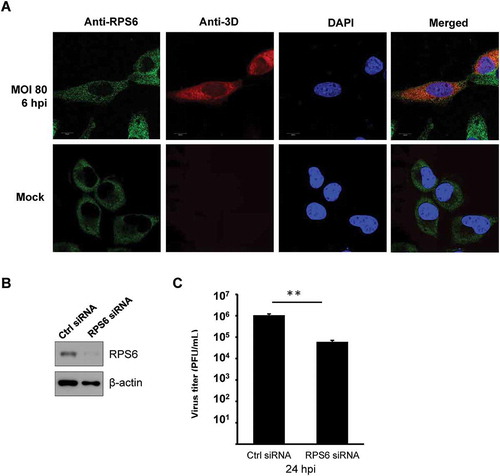
To further confirm the interaction in EV-A71-infected cells, we subjected the extracts of EV-A71-infected RD cells to immunoprecipitation with anti-EV-A71 3D antibody, which also recognizes the precursor protein 3CD (). The immunoprecipitates were then examined by immunoblotting with an anti-RPS6 antibody. We found that RPS6 was detected in the precipitates of anti-EV-A71 3D from the extracts of the infected cells (). We also verified the reciprocal interaction. FLAG-tagged RPS6 was overexpressed in HEK293T cells, which in turn were infected by EV-A71. Lysates of the infected cells were subjected to immunoprecipitation using an anti-FLAG antibody. Although a non-specific binding of 3D/3CD to anti-FLAG resin was detected (, Vector), a much stronger signal of 3D/3CD was identified in the immunoprecipitates of 3× FLAG-RPS6, suggesting that both EV-A71 3D and 3CD can be coimmunoprecipitated by 3× FLAG-RPS6 (). In addition, the localization of EV-A71 3D and RPS6 was examined in infected cells. RPS6 was exclusively localized in the cytoplasm with some dot-like foci in mock- or EV-A71-infected cells, while EV-A71 3D mainly localized in the cytoplasm, and weak nuclear signals were observed (). We found that infection had no obvious effect on the intracellular distribution of RPS6, and the protein partly colocalized with EV-A71 3D in the cytoplasm of the infected cells, especially in certain dots around the nuclei (). Thus, EV-A71 3D could interact with the ribosomal protein RPS6 during EV-A71 infection. Previous studies have revealed the importance of RPS6 to the replication of several RNA viruses [Citation31–Citation33]; hence, we also verified the RPS6-dependence of EV-A71 by knockdown experiment. Due to the abundance of endogenous ribosomal proteins, only 70% reduction of RPS6 protein level can be achieved (). However, under the circumstances, EV-A71 replication was severely impaired and a reduction of ~20 folds can be observed at 24 h postinfection (). Therefore, like other IRES-containing RNA viruses [Citation31,Citation33], EV-A71 replication is also sensitive to the protein level of RPS6.
Figure 4. EV-A71 3D protein partially colocalizes with RPS6 during infection and knockdown of RPS6 inhibits viral replication. (A) HeLa cells were infected with EV-A71 at MOI of 80 for 6 h. The cells were fixed and reacted with rabbit anti-RPS6 and mouse anti-EV-A71 3D antibodies. After washing, the cells were incubated with the species-specific secondary antibodies conjugated with fluorescence dyes, stained with Hoechst 33,258, and then examined under confocal microscopy. (B) RD cells were transfected with either control siRNA (Ctrl) or siRNA against RPS6. Knockdown efficiency was evaluated by immunoblotting with anti-RPS6 antibody. β-actin served as the loading control. (C) siRNA-transfected RD cells were infected with EV-A71 at MOI of 0.001. Infected cells were harvested at 24 hpi and the viral yield was estimated by plaque formation assay. The experiment was triplicated. The result represents one of the experiments. The error bars represented the standard deviation (SD). Statistical analysis was performed by the Student’s t test. **: p < 0.01. Bar, 10 µm. hpi, hour postinfection. PFU: plaque-forming unit.
The EV-A71 3D protein cofractionates with the small and large subunits of ribosomes
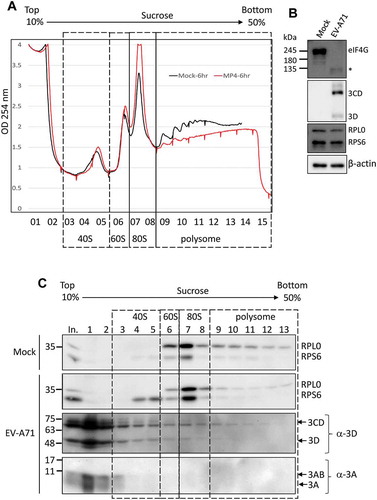
Upon PV infection, the cellular proteins SRSF3 and PCBP2 were relocalized to the cytoplasm and might be involved in the translation initiation complex assembly of viral RNA, as evidenced by polysome profiles [Citation34]. Since we validated the interactome analysis of the EV-A71 3D protein and demonstrated that the 3D protein associates with the ribosomal protein RPS6 in an RNA-independent manner, we next investigated whether overexpressed EV-A71 3D could form complexes with intact ribosomes or whether it just binds to free ribosomal proteins. We performed polysome profiling using a 10% to 50% sucrose gradient for EV-A71 3D-expressing cells and detected the distribution of EV-A71 3D by immunoblotting. The polysome profiles showed that overexpression of the 3D protein encoded by either the C2 or C4 subgenotype of EV-A71 did not change the distributions of the 40S and 60S subunits, 80S monosomes, and polysomes (, upper panel). We also analysed the distribution of 18S and 28S rRNAs in fractions prepared from the gradient, and no obvious difference was found (, lower panel). These results suggested that the steady-state translation status might not be altered by the overexpressed EV-A71 3D proteins. Next, the distribution of EV-A71 3D in each fraction was examined by western blotting using an anti-FLAG antibody. The EV71-A71 3D protein was not only enriched at the top of the fractions but also spread into the 40S, 60S, and 80S fractions (, fractions 1–9). In addition, a faint but detectable signal was observed in the polysome fractions (, fractions 10–13), suggesting that EV-A71 3D might not only be involved in translation initiation but may also play a role in the elongation step. To further verify whether EV-A71 3D could be the bona fide component of the ribosome during infection, we conducted polysome profiling of RD cells that were infected with EV-A71 for 6 h. The results showed that infection with EV-A71 caused a dramatic loss of polysomes and an accumulation of the 80S monosome (). The disruption of the polysome might be due to the cleavage of eIF4G upon EV-A71 infection (). Using anti-EV-A71 3D immunoblotting for fractions of the sucrose gradient, we found that the EV-A71 3D and 3CD proteins could be detected not only in fractions containing the 40S, 60S, and 80S monosomes but also in the polysomal fractions (). Thus, consistent with the results using the overexpression system, EV-A71 3D was cosedimented with ribosomal proteins during infection. In contrast, other viral proteins, such as 3A and 3AB, were enriched in the free mRNP fraction rather than the ribosomal fraction, indicating that the cofractionation of 3D/3CD with ribosomes is specific. Collectively, we provided evidence that EV-A71 3D cosedimented with ribosomes in cells either transfected with the EV-A71 3D-expressing plasmid or infected with EV-A71 and might be a bona fide component of the ribosome during infection.
Figure 5. The overexpressed EV-A71 3D cosediments with ribosome complexes. (A) Polysome profiling of HEK293T cells transfected with empty vector, or expression vector encoding the EV-A71 3D of either C2 (marked in red) or C4 (blue) subgenotype was analysed by 10–50% sucrose gradient. Peaks representative of 40S, 60S subunits, 80S monosomes and polysomes were indicated (upper panel). 18S and 28S rRNA of the 15 fractions corresponding to each ribosomal assembly were shown below. (B) Distribution of EV-A71 3D proteins in the factions described in (A) was analysed by immunoblotting using anti-FLAG antibody. Ribosomal protein RPS6 and RPL0 that served as the marker of small and large ribosomal subunit were also detected.
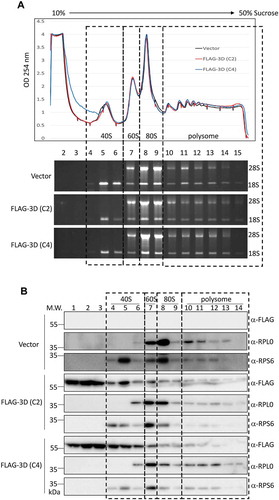
Figure 6. The EV-A71 3D/3CD in infected cell associates with ribosome complex despite polysome disruption. (A) Polysome profiling of RD cells infected with EV-A71 MP4 strain at MOI of 10 was analysed as described in Fig. 6. MP4 infection (marked in red) disrupted the polysome peaks while the 80S monosome peak increased when compared with the profile of mock-infected cell (black). (B) Cleavage of eIF4G upon EV-A71 infection was examined by immunoblotting using anti-eIF4G antibody. Expression of EV-A71 3D and 3CD proteins was analysed by using anti-EV-A71 3D antibody. Proteins level of RPS6, RPL0, and β-actin served as the loading control. (C) Immunoblotting was performed to analyse the distributions of EV-A71 3D/3CD and 3A/3AB in the sucrose gradient using antibody against EV-A71 3D and 3A, respectively. Fractions representative of different ribosomal assemblies were verified by antibodies against RPS6 and RPL0. In. represented 5% lysates layered onto the gradient in the beginning.
The EV-A71 3D protein enhances IRES-driven translation
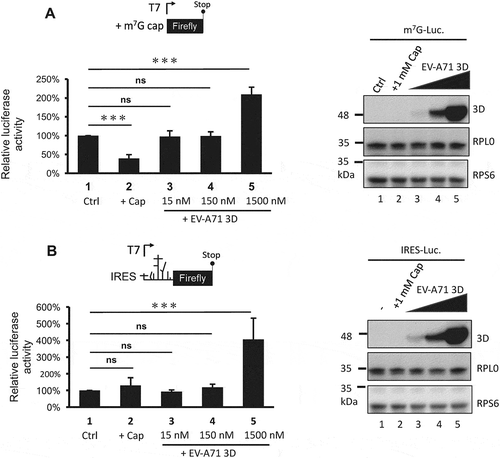
Previous results showed that the EV-A71 3D protein interacts with ribosomal components and cosediments with both 80S monosomes and polysomes. Therefore, we hypothesized that EV-A71 3D could be involved in the regulation of viral and/or host protein translation. To test this hypothesis, we determined the EV-A71 IRES-dependent translation activity by cotransfection of an EV-A71 3D-expressing construct and a reporter plasmid that expresses a bicistronic mRNA containing Renilla and firefly luciferase open reading frames whose translation is driven by the 5ʹ cap and EV-A71 IRES, respectively (). IRES activity was defined by normalization of the firefly luciferase activity with that of Renilla luciferase, and the results from the luciferase reporter assay showed that overexpression of the EV-A71 3D protein could enhance EV-A71 IRES-dependent translation by approximately 20% to 40% compared with that of the vector control (). To ensure the involvement of EV-A71 3D in the regulation of IRES-dependent translation, we assessed the IRES activity under RPS6-knockdown condition that has been shown to specifically inhibit IRES – but not cap-dependent translation [Citation31,Citation33]. Knockdown of RPS6 significantly inhibited the EV-A71 IRES-driven translation even when EV-A71 3D was overexpressed (). Intriguingly, when we compared the IRES activity solely in the RPS6-knockdown group, we still observed an elevated IRES activity caused by overexpressed EV-A71 3D (an increase of 60%). The results suggested that an RPS6-independent translation might be operated by EV-A71 3D. In addition, the activation might be underestimated and biased if the EV-A71 3D protein also activated cap-dependent translation. To clarify this possibility, we performed an in vitro translation assay with model RNA for cap- or IRES-dependent translation encoding firefly luciferase using S10 lysates of HEK293T cells in the presence of recombinant EV-A71 3D protein. To examine cap-dependent translation, we treated the lysates with micrococcal nuclease prior to the translation assay to release ribosomes from cellular mRNAs. Pretreatment of the lysates with 1 mM m7G-cap, which has been shown to efficiently compete with cellular cap-binding proteins [Citation35], reduced the cap-dependent translation to 30% of the control (, lanes 1 and 2). However, the addition of 1.5 μM recombinant 3D protein of EV-A71 (at a protein:RNA molar ratio of 100:1) significantly increased the translation activity (, lanes 1 and 5). Therefore, EV-A71 3D can activate cap-dependent translation in vitro. The effect of the EV-A71 3D protein on IRES-dependent translation was also evaluated using a similar strategy. Consistent with previous findings that IRES activity is sensitive to the status of cap-dependent translation [Citation36], m7G-cap treatment slightly increased IRES activity (, lanes 1 and 2). Furthermore, the efficiency of IRES-dependent translation was significantly enhanced in the presence of excess EV-A71 3D protein (, lanes 1 and 5). Overall, these results indicate that an RNA polymerase encoded by EV-A71 can function as a novel translation factor and positively regulates cap- and IRES-dependent translation.
Figure 7. The overexpressed EV-A71 3D elevates IRES-driven translation in vivo. (A) Scheme shows the bicistronic RNA produced from pRHF-EV-A71 reporter plasmid. Translation of firefly and Renilla luciferase are dependent on 5ʹ end cap and EV-A71 IRES, respectively. (B) Reporter plasmid was co-transfected with effector plasmid encoding EV-A71 3D proteins into HEK293T cells. The overexpressed 3D of EV-A71 C2 (lane2) and C4 (lane3) strain enhanced the EV-A71 IRES activity. Expression level of 3D proteins was examined by immunoblotting using anti-FLAG antibody. β-actin served as the loading control. (C) Similar to the experiment described in (B) but control siRNA (Ctrl) or siRPS6 was included. Knockdown of RPS6 was confirmed by immunoblotting with anti-RPS6 antibody. The error bars represented the standard deviation (SD). Statistical analysis was performed by the Student’s t test. ***: p <0.001; **: p <0.01. ns: not significant.
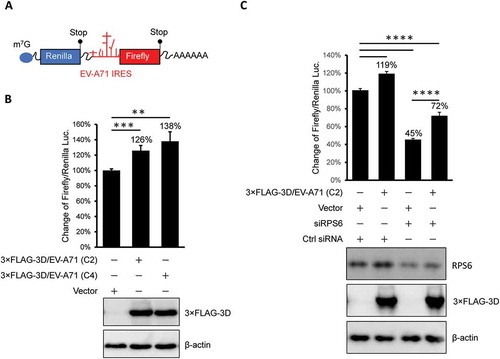
Figure 8. The 3D protein directly activates both cap- and IRES-dependent translation in vitro. In vitro translation was performed in 60% S10 extracts prepared from HEK293T cells in the presence of the recombinant EV-A71 3D proteins (lane 3–5) or not (lane 1–2) to analyse (A) cap- and (B) IRES-dependent translation. Ctrl: control group, +Cap: lysates pretreated with 1mM m7G cap analogue. The recombinant EV-A71 3D proteins in each reaction were examined by immunoblotting using anti-EV-A71 3D antibody. Ribosomal proteins RPS6 and RPL0 served as the loading controls. Statistical analysis was performed by the two-way ANOVA. ***: p <0.001; ns: not significant.
Discussion
Enterovirus RdRP, denoted as 3D protein, is vital for replication of viral RNA in infected cells. However, previous studies have demonstrated that enteroviral 3D protein could interact with the splicing factor Prp8 and interfere with cellular splicing events [Citation7]. In addition, we recently found that the 3D protein encoded by EV-A71 plays a role in the regulation of IFN-β transcription [Citation11]. Therefore, the enteroviral 3D protein may not only function in viral RNA replication but may also be involved in the regulation of cellular biological processes. To globally explore the functions of the viral 3D protein in the regulation of biological processes in host cells, we conducted immunoprecipitation coupled with LC-MS/MS to identify host proteins that may interact with the EV-A71 3D protein. The results showed that the 3D protein of EV-A71 could associate with factors involved in translation, such as RPS6. We further demonstrated that the 3D protein of EV-A71 could interact and be partly colocalized with RPS6 during EV-A71 infection. In addition, sucrose gradient analysis showed that the 3D protein could be cosedimented with small and large subunits of ribosomes during 3D protein overexpression or EV-A71 infection. These results provide biochemical evidence that the 3D protein of EV-A71 interacts with the components of the ribosome. Moreover, we used an in vitro translation assay to prove that the EV-A71 3D protein directly increases EV-A71 IRES-dependent translation as well as cap-dependent translation. Taken together, this research demonstrated that the 3D protein encoded by EV-A71 could interact with ribosomal proteins to form complexes with small and large subunits of ribosomes and activate viral and host translation.
Infection of enteroviruses shuts off host cap-dependent translation due to the cleavage of the translation initiation factor eIF4G by the viral 2A protease [Citation17,Citation37,Citation38]. Because the viral protease-cleaved eIF4G still binds to viral IRES to mediate enteroviral protein translation, the cleavage of eIF4G does not affect viral protein synthesis. Nevertheless, cellular factors known as ITAFs may associate with viral IRESs to regulate its translation initiation [Citation6,Citation39]. We found that the EV-A71 3D protein may interact with ribosomal proteins in an RNA-independent manner, indicating that the regulatory function of the 3D protein in translation is not a result of non-specific interactions between the 3D protein and viral or cellular mRNA. In addition, the aforementioned results demonstrated that the 3D protein positively regulates host translation, suggesting that the translation of cellular mRNA is coordinated by the viral proteins 2A and 3D during EV-A71 infection. Interestingly, our previous proteome-wide profiling of EV-A71-infected cells showed that the expression of several host proteins was increased during EV-A71 infection [Citation40]. These results may indicate that infection with EV-A71 could enhance cellular protein translation to a certain extent by the viral 3D protein.
Although the viral positive-stranded RNA genome serves as an mRNA for viral polyprotein translation, it is also the template for negative-stranded viral RNA synthesis. The replication of viral RNA is initiated by the uridylylation of the VPg protein and followed by elongation from the 3ʹ terminus of the template RNA to produce a minus-stranded RNA, which in turn is utilized to synthesize more viral genomes [Citation41,Citation42]. Both of these events are catalysed by the 3D protein in distinct membraned organelles connected to the endoplasmic reticulum (ER) [Citation43], where protein translation also occurs.
Despite the opposite action of translation and minus-stranded RNA synthesis, coreplicational translation (cis-translation) of positive-stranded RNAs has been proposed as a quality control mechanism of PV to ensure the coding accuracy and genome integrity of progeny RNAs [Citation44,Citation45]. Since our results demonstrated that the EV-A71 3D protein facilitates viral and host translation in cells, a proportion of the 3D protein and the de novo synthesized viral mRNA may be transported to the nearby ER, or coreplicational translation might occur within replication organelles. The close linkage between viral replication and translation is also reported in hepatitis C virus infection, whose translation depends on the active replication and occurs in close proximity to the replication loci [Citation46,Citation47]. Given that the coupling is widely used for regulating gene expressions of eukaryotic cells [Citation48], coreplicational translation not only can increase the replication efficiency but might also protect the progeny genomes from nuclease attack. On the other hand, since most surveillance mechanisms for cytoplasmic mRNAs of eukaryotic cell are translation-dependent [Citation49], replication-coupled translation could prevent the accumulation of defective genomes. Nevertheless, the impact of the 3D-mediated translation in infected cells requires further investigation.
Previous studies conducted by anti-3D immunoprecipitation of EV-A71-infected cell extracts and MALDI-TOF mass spectrometry showed that EV-A71 3D protein could interact with cellular factors involved in translation [Citation7,Citation8]. However, the factors identified may be associated with the precursor of 3D protein, 3CD, and not the 3D protein alone. Therefore, an interactome analysis was conducted by overexpressing the EV-A71 3D protein. In addition to the replication and translation-related proteins, the present interactome analysis of the EV-A71 3D protein also identified RAN as one of the interacting host proteins. RAN is a GTPase that participates in the nucleocytoplasmic transport of proteins and RNA [Citation50,Citation51]. The L protein of cardiovirus, belonging to the picornavirus family, was reported to interact with RAN GTPase and interfere with nucleocytoplasmic trafficking [Citation52]. In addition, EV-A71 infection downregulates miR-197, which targets RAN translation. The increase in RAN expression could enhance EV-A71 replication by supporting nuclear transport of the viral 3D and 3CD and host hnRNP K proteins [Citation53]. Hence, the interaction of viral 3D protein and RAN may also impact EV-A71 replication. In addition, by the enrichment analysis of biological processes for proteins interacting with the EV-A71 3D protein, we found that the process of cell-cell adhesion was identified, along with events correlated with translation and RNA processing. Among the proteins identified in this process, CCT8, DBN1, IQGAP1, and ELMO2 are involved in the regulation of cytoskeleton assembly. These results indicate that the viral 3D protein could play a role in cytoskeleton rearrangement during infection. Since cells infected with enterovirus form distinct replication organelles for viral replication, the redistribution and rearrangement of cytoplasmic organelles might be regulated by the viral 3D protein. Collectively, this research demonstrated that the interactome analysis of viral proteins is an applicable strategy to explore host responses regulated by viral proteins during infection. The interaction network could be applied to antiviral drug development in the future [Citation54].
Materials and methods
Cells, viruses and plasmids
HEK293T, RD, and HeLa cells were cultivated with Dulbecco’s modified Eagle’s medium (DMEM) with 10% FBS at 37°C, as described previously [Citation11]. The EV-A71 strains of Tainan/4643/98 (denoted as 4643) and MP4, which are C2 subgenotypes, were provided by Dr. Jen-Ren Wang at the National Cheng-Kung University, Taiwan. Plasmids expressing the 3D protein of EV-A71 4643 (C2 subgenotype) and TW-00184-2012 (C4 subgenotype) were constructed with the vectors p3× FLAG-Myc-CMV25 and p3× FLAG-Myc-CMV26 (Sigma-Aldrich, USA), respectively. The RPS6-expressing plasmid was constructed with cDNA of RPS6 amplified from total RNA of HEK293T cells by reverse-transcription and PCR using the p3× FLAG-Myc-CMV26 vector. To construct bacterial expression plasmid of RPS6, cDNA of RPS6 was amplified by PCR and in-frame inserted into the pGEX-6P-1 GST expression vector through BamHI and EcoRI. Dicistronic reporters without (pRHF) or with EV-A71 IRES (pRHF-EV-A71) were described previously [Citation22]. pGL3-EV71-5ʹ-UTR-Fluc used to synthesize IRES-containing luciferase mRNA in vitro was described by Huang et al. [Citation23]. ON TARGETplus SMARTpool siRNA against RPS6 were purchased from Dharmacon (Horizon Discovery) and their sense-strand sequences were as follows: siRPS6-J9, 5ʹ-GGACGAUGAACGCAAACUU; siRPS6-J10, 5ʹ-UAAAGAAGAUGAUGUCCGC-3ʹ; siRPS6-J11, 5ʹ-UGGACUGACUGAUACUACA-3ʹ; siRPS6-J12, 5ʹ-GAUGCAAAUCUGAGCGUUC-3ʹ.
Immunoprecipitation, in-gel digestion and mass spectrometry analysis
For 3D immunoprecipitation, lysates of HEK293T cells transfected with the 3× FLAG-tagged EV-A71 3D-expressing plasmid for 48 h were mixed and rotated with anti-FLAG-M2 affinity resin (Sigma-Aldrich) at 4°C for 12 h. In-gel digestion of proteins in the immunoprecipitates was performed as described previously [Citation26,Citation55,Citation56]. In brief, the immunoprecipitates were separated by SDS-PAGE. Each gel lane was cut into 10 pieces and further separated into four replicates. Proteins in each gel piece were reduced with dithiothreitol (10 mM; Biosynth AG, Switzerland), alkylated with iodoacetamide (55 mM; Amersham Biosciences, UK), digested by trypsin (20 μg/ml; Promega, USA), and then extracted by acetonitrile.
Peptide identification was performed as described previously [Citation26,Citation57,Citation58]. In brief, the extracted peptides were reconstituted in formic acid (0.1%; Sigma-Aldrich), separated on a resolving 100 mm analytical C18 column (inner diameter, 75 μm) using a 15 μm tip (New Objective, Woburn, MA), and identified by LTQ-Orbitrap Discovery (Thermo Fisher, Waltham, MA). The LTQ Orbitrap was operated using Xcalibur 2.0 software (Thermo Fisher). A data-dependent mode containing one MS scan in the Orbitrap at a resolution of 30,000 and 10 MS/MS scans in the ion trap for the 10 most abundant precursor ions was used to acquire data. The precursor ions selected for MS/MS analysis were dynamically excluded for 180 s.
Database searching and protein identification
The spectra were searched with the Mascot algorithm (Version 2.1, Matrix Science, Boston, MA) against the Swiss-Prot human sequence database (released March 2018, selected for Homo sapiens, 20198 entries) of the European Bioinformatics Institute. The peak list was generated using Thermo ExtractMSn software (Version 1.0.0.8, May 2012 release). The mass tolerances for parent and fragment ions were set as 10 ppm and 0.5 Da, respectively. The oxidation on methionine (+15.99 Da) and carbamidomethylation on cysteine (+57 Da) were set as variable and fixed modifications, respectively. The enzyme was set as trypsin, and up to one missed cleavage was allowed. The random sequence database was used to estimate false-positive rates for protein matches. The resulting files were further integrated by Scaffold software (Version 4.2.1, Proteome Software, USA), which included the PeptideProphet algorithm [Citation59] and the ProteinProphet algorithm [Citation60]. The thresholds for PeptideProphet and ProteinProphet probability were set as 0.95 to ensure an overall FDR below 0.5%. Furthermore, only proteins with two unique peptides were retained in the present study.
Spectral counting-based protein quantification and bioinformatics analysis
To identify associated partners of the EV-A71 3D protein, we compared the protein levels between the control and EV-A71 3D groups with a spectral counting-based quantification as previously described [Citation26,Citation56,Citation58]. Briefly, the SC of each identified protein was exported from the Scaffold software in Excel format. To reduce the difference between analyses, we calculated the normalized SC (NSC) of each protein in the analysis by the SC for a given protein divided by the total SC of the analysis. The fold change was estimated as the ratio of the average of NSCs in the 3D group to that in the control group. Because all proteins in all replicates could not be identified, we purposely assigned the SCs of unidentified proteins or missing values in a certain sample as the number of one to avoid overestimating the fold changes and dividing by zero.
The biological pathways and processes involved with the EV-A71 3D-interacting proteins were revealed by the KEGG database and the DAVID (version 6.8), respectively [Citation61]. The known and predicted associations between the 3D-interacting partners were analysed with STRING software (version 10.5). A combined confidence score of ≥0.9 was used as the cut-off criterion [Citation62].
Immunoprecipitation for EV-A71-infected cells
For analysis of the interaction of EV-A71 3D and RPS6, RD cells were transfected with the 3× FLAG-tagged RPS6-expressing plasmid for 12 h and then infected with the EV-A71 4643 strain at MOI of 40 for 8 h. The cells were lysed and subjected to immunoprecipitation with anti-FLAG-M2 affinity resin following the manufacturer’s instructions. For analysis of the interaction of the EV-A71 3D and endogenous RPS6 proteins, RD cells infected with the EV-A71 4643 strain at MOI of 40 for 8 h were lysed and subjected to immunoprecipitation with the anti-EV-A71 3D antibody (GeneTex, USA) mixed with protein A and protein G agarose beads (Blossom Biotechnologies, Taiwan). The mixtures were rotated at 4°C for 12 h and then centrifuged to precipitate the agarose beads and the associating antibody and proteins. After the precipitates were washed, the associated proteins were eluted with 1x sample buffer (Bionovas, Canada).
Immunoblotting and immunofluorescence
Cell lysates, immunoprecipitates, or proteins in the fractions of polysome profiling were separated by SDS-PAGE and then transferred to PVDF membranes. The membranes were reacted individually with anti-FLAG (Sigma-Aldrich), anti-EV71 3D (GeneTex), anti-EV-A71 3A (provided by Jim-Tong Horng in Chang Gung University), anti-RPS6 (Cell Signaling), anti-RPL0 (GeneTex), anti-eIF4G (Santa Cruz, USA) and anti-β-actin (Sigma-Aldrich) primary antibodies, incubated with species-specific secondary antibodies conjugated with HRP (GE Healthcare Life Sciences), and then detected by a chemiluminescent HRP substrate (Millipore, USA).
HeLa cells seeded on coverslips were infected with the EV-A71 strain 4643 at the indicated MOI for 6 h. The infected cells were fixed with 4% paraformaldehyde and subsequently treated with 0.5% Triton X-100. After the fixation solution was removed, the cells were treated with a blocking buffer containing 2% bovine serum albumin, 1% gelatin, and 0.02% saponin, incubated with mouse anti-3D (GeneTex) and rabbit anti-RPS6 (Cell Signaling, USA) antibodies, and then reacted with anti-species-specific IgG conjugated with Alexa Fluor® 488 dye or with Alexa Fluor® 594 dye (Thermo Fisher, USA). The cell nuclei were stained with Hoechst 33258 (Sigma-Aldrich). After mounting, the cells were observed under a confocal fluorescence microscope.
GST-pulldown assay
GST and GST-RPS6 fusion proteins were overproduced in Escherichia coli strain BL21 and purified by using Glutathione Sepharose 4B resin (GE Healthcare Life Sciences) according to the manufacturer’s instructions. Purified recombinant proteins were then dialysed against S10 dialysis buffer [Citation63]. The recombinant EV-A71 3D protein was prepared as previously described [Citation7]. To pull down EV-A71 3D in vitro, 10 μg of GST or GST-RPS6 was incubated with 20 μg of 3D recombinant protein in a 200 μl mixture containing S10 dialysis buffer at 30°C for 30 min. Proteins selected by glutathione-Sepharose were detected by immunoblotting with anti-3D, anti-RPS6 and anti-GST (Santa Cruz Biotechnology).
EV-A71 virus replication analysis
RD cells were first transfected with control siRNA and siRPS6 at final concentration 60 nM, respectively. At 24-h post-transfection, the transfected cells were infected by EV-A71 strain of 4643 at MOI of 0.001 in triplicate in medium with 2% FBS. One hour later, cells were thoroughly washed by PBS and refreshed in 2% serum-containing media. Virus was harvested at different time-points post-infection and plaque assay was performed to quantify the virus titre [Citation63].
Polysome profiling
Lysates of infected or transfected cells were subjected to 10–50% sucrose gradient sedimentation to separate monosome and polysome fractions. RD cells grown in a 15-cm dish at 90% confluency (∼1.8 x 107 cells) were either mock-infected or infected by MP4 at MOI of 10 and incubated at 37°C for 6 h. Transfection was performed in HEK293T cells by calcium phosphate [Citation64], and ∼1.2 × 107 cells grown in a 15-cm dish were transiently transfected with empty vector (10 μg) or an expression vector encoding the FLAG-tagged 3D of the EV-A71 C2 (10 μg) and C4 subgenotype (20 μg). Infected cells or transfectants were lysed in 1 ml lysis buffer containing 20 mM Tris-HCl pH 8.0, 140 mM KCl, 5 mM MgCl2, 1% (v/v) Triton-X100, 100 μg/ml cycloheximide, and 1× EDTA-free protease inhibitor (Roche) at 4°C for 10 min [Citation65]. Later, the lysates were subjected to 5–7 passages through a 26-gauge syringe followed by centrifugation at 4°C for 10,000 xg for 20 min, and the clarified lysates were used for gradient sedimentation analysis. Clarified lysates (~1.5 mg proteins) were then layered onto the top of a 10–50% gradient, which was prepared by diffusion using a gradient buffer composed of 20 mM Tris-HCl pH 8.0, 140 mM KCl, 5 mM MgCl2, and 100 μg/ml cycloheximide [Citation65]. Centrifugation was carried out by using the SW41 rotor in an Optima XE Ultracentrifuge (Beckman coulter) at 39,000 rpm at 4°C for 2.5 h. The OD254nm detection and collection of gradient fractions were performed using Density Gradient Fractionation Systems (Teledyne ISCO). RNA recovery from the fraction was performed using LS TRIzol (Thermo Fisher) according to the manufacturer’s instructions, and proteins were precipitated by using 10% trichloroacetic acid.
Determination of the translation efficiency in vivo and in vitro
A dual luciferase assay was carried out to monitor the translation activity in vivo [Citation22]. First, 0.1 μg reporter plasmid was cotransfected into 4 × 105 HEK293T cells with empty vector (2 μg) or an expression vector encoding FLAG-3D derived from the EV-A71 C2 (1 μg) and C4 strain (2 μg). Twenty-four-hour post-transfection, cells were lysed in 200 μl 1× passive buffer (Promega) at room temperature for 15 min followed by centrifugation at 12,000 rpm at 4°C for 2 min. The Renilla luciferase and firefly luciferase activities were measured using the Dual-Luciferase Reporter Assay System (Promega) in the GloMax 20/20 Luminometer (Promega) according to the manufacturer’s instructions. For RPS6 knockdown experiment, siRNA (at final concentration 100 nM) was cotransfected into HEK293T cells with reporter and effector plasmids described above. Transfectants were incubated for 48 h to increase the knockdown efficiency.
Cell-free translation using S10 extracts of HEK293T cells was carried out essentially as described by Lee et al. [Citation63]. To investigate the cap-dependent translation, micrococcal nuclease (New England Biolabs) was used; 20 units of nuclease were added in the presence of 2.5 mM CaCl2 per 100 μl S10 extract at room temperature for 15 min. The reaction was stopped by the addition of 3.75 μl 0.2 M EGTA (at a final concentration of 7.5 mM). For the cap-depletion experiment, nuclease-treated lysates were further incubated with 1 mM m7G-cap (New England Biolabs) at 4°C for 10 min. RNA encoding firefly luciferase with or without the IRES sequence at the 5ʹ terminus was synthesized by a MEGAscript T7 Transcription Kit (Thermo Fisher) according to the manufacturer’s instructions. The firefly luciferase activity was measured using the Luciferase Assay System (Promega) according to the manufacturer’s instructions.
Disclosure of potential conflicts of interest
No potential conflict of interest was reported by the authors.
Supplemental Material
Download MS Excel (49.2 KB)Supplemental Material
Download MS Excel (1.3 MB)Acknowledgments
We thank Jen-Ren Wang for providing the viruses and Kai-Yin Lo and Bo-Shiun Chen for the helpful discussions. This research was supported by grants from the Ministry of Science and Technology, Taiwan (106-2320-B-182-024-MY3, and 108-2320-B-182-030-MY3) and the Chang Gung Memorial Hospital (CMRPD1G0571~2 and BMRPC09). The work was also supported by grants to Research Center for Emerging Viral Infections, Chang Gung University from Featured Areas Research Center Program within the Framework of Higher Education Sprout Project by Ministry of Education and Ministry of Science and Technology, Taiwan (MOST 108-3017-F-182-001).
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
References
- Solomon T, Lewthwaite P, Perera D, et al. Virology, epidemiology, pathogenesis, and control of enterovirus 71. Lancet Infect Dis. 2010;10:778–790.
- Suresh S, Forgie S, Robinson J. Non-polio Enterovirus detection with acute flaccid paralysis: A systematic review. J Med Virol. 2018;90:3–7.
- Ooi MH, Wong SC, Lewthwaite P, et al. Clinical features, diagnosis, and management of enterovirus 71. Lancet Neurol. 2010;9:1097–1105.
- Chang LY, Lin HY, Gau SS, et al. Enterovirus A71 neurologic complications and long-term sequelae. J Biomed Sci. 2019;26:57.
- Owino CO, Chu JJH. Recent advances on the role of host factors during non-poliovirus enteroviral infections. J Biomed Sci. 2019;26:47.
- Shih SR, Stollar V, Li ML. Host factors in enterovirus 71 replication. J Virol. 2011;85:9658–9666.
- Liu YC, Kuo RL, Lin JY, et al. Cytoplasmic viral RNA-dependent RNA polymerase disrupts the intracellular splicing machinery by entering the nucleus and interfering with Prp8. PLoS Pathog. 2014;10:e1004199.
- Huang PN, Jheng JR, Arnold JJ, et al. UGGT1 enhances enterovirus 71 pathogenicity by promoting viral RNA synthesis and viral replication. PLoS Pathog. 2017;13:e1006375.
- Wang W, Xiao F, Wan P, et al. EV71 3D protein binds with NLRP3 and enhances the assembly of inflammasome complex. PLoS Pathog. 2017;13:e1006123.
- Wang LC, Chen SO, Chang SP, et al. Enterovirus 71 proteins 2A and 3D antagonize the antiviral activity of gamma interferon via signaling attenuation. J Virol. 2015;89:7028–7037.
- Kuo RL, Chen CJ, Wang RYL, et al. Role of enteroviral RNA-dependent RNA polymerase in regulation of MDA5-mediated beta interferon activation. J Virol. 2019;93 :e00132-19.
- Ben-Shem A, Garreau de Loubresse N, Melnikov S, et al. The structure of the eukaryotic ribosome at 3.0 A resolution. Science. 2011;334:1524–1529.
- Rabl J, Leibundgut M, Ataide SF, et al. Crystal structure of the eukaryotic 40S ribosomal subunit in complex with initiation factor 1. Science. 2011;331:730–736.
- Klinge S, Voigts-Hoffmann F, Leibundgut M, et al. Crystal structure of the eukaryotic 60S ribosomal subunit in complex with initiation factor 6. Science. 2011;334:941–948.
- Etchison D, Milburn SC, Edery I, et al. Inhibition of HeLa cell protein synthesis following poliovirus infection correlates with the proteolysis of a 220,000-dalton polypeptide associated with eucaryotic initiation factor 3 and a cap binding protein complex. J Biol Chem. 1982;257:14806–14810.
- Etchison D, Fout S. Human rhinovirus 14 infection of HeLa cells results in the proteolytic cleavage of the p220 cap-binding complex subunit and inactivates globin mRNA translation in vitro. J Virol. 1985;54:634–638.
- Lloyd RE, Jense HG, Ehrenfeld E. Restriction of translation of capped mRNA in vitro as a model for poliovirus-induced inhibition of host cell protein synthesis: relationship to p220 cleavage. J Virol. 1987;61:2480–2488.
- Belsham GJ, Sonenberg N. RNA-protein interactions in regulation of picornavirus RNA translation. Microbiol Rev. 1996;60:499–511.
- Lee YF, Nomoto A, Detjen BM, et al. A protein covalently linked to poliovirus genome RNA. Proc Natl Acad Sci U S A. 1977;74:59–63.
- Jackson RJ, Hunt SL, Gibbs CL, et al. Internal initiation of translation of picornavirus RNAs. Mol Biol Rep. 1994;19:147–159.
- Lin JY, Shih SR, Pan M, et al. hnRNP A1 interacts with the 5ʹ untranslated regions of enterovirus 71 and Sindbis virus RNA and is required for viral replication. J Virol. 2009;83:6106–6114.
- Lin JY, Li ML, Shih SR. Far upstream element binding protein 2 interacts with enterovirus 71 internal ribosomal entry site and negatively regulates viral translation. Nucleic Acids Res. 2009;37:47–59.
- Huang PN, Lin JY, Locker N, et al. Far upstream element binding protein 1 binds the internal ribosomal entry site of enterovirus 71 and enhances viral translation and viral growth. Nucleic Acids Res. 2011;39:9633–9648.
- Teterina NL, Levenson E, Rinaudo MS, et al. Evidence for functional protein interactions required for poliovirus RNA replication. J Virol. 2006;80:5327–5337.
- Yin J, Liu Y, Wimmer E, et al. Complete protein linkage map between the P2 and P3 non-structural proteins of poliovirus. J Gen Virol. 2007;88:2259–2267.
- Kuo RL, Chen CJ, Tam EH, et al. Interactome analysis of ns1 protein encoded by influenza A H7N9 virus reveals an inhibitory role of NS1 in host mRNA maturation. J Proteome Res. 2018;17:1474–1484.
- Flather D, Semler BL. Picornaviruses and nuclear functions: targeting a cellular compartment distinct from the replication site of a positive-strand RNA virus. Front Microbiol. 2015;6:594.
- Wang J, Du J, Jin Q. Class I ADP-ribosylation factors are involved in enterovirus 71 replication. PLoS One. 2014;9:e99768.
- Belov GA, Sztul E. Rewiring of cellular membrane homeostasis by picornaviruses. J Virol. 2014;88:9478–9489.
- Lee KM, Gong YN, Hsieh TH, et al. Discovery of enterovirus A71-like nonstructural genomes in recent circulating viruses of the enterovirus A species. Emerg Microbes Infect. 2018;7:111.
- Cherry S, Doukas T, Armknecht S, et al. Genome-wide RNAi screen reveals a specific sensitivity of IRES-containing RNA viruses to host translation inhibition. Genes Dev. 2005;19:445–452.
- Yang C, Zhang C, Dittman JD, et al. Differential requirement of ribosomal protein S6 by plant RNA viruses with different translation initiation strategies. Virology. 2009;390:163–173.
- Huang JY, Su WC, Jeng KS, et al. Attenuation of 40S ribosomal subunit abundance differentially affects host and HCV translation and suppresses HCV replication. PLoS Pathog. 2012;8:e1002766.
- Fitzgerald KD, Semler BL. Re-localization of cellular protein SRp20 during poliovirus infection: bridging a viral IRES to the host cell translation apparatus. PLoS Pathog. 2011;7:e1002127.
- Merz C, Urlaub H, Will CL, et al. Protein composition of human mRNPs spliced in vitro and differential requirements for mRNP protein recruitment. RNA. 2007;13:116–128.
- Rakotondrafara AM, Hentze MW. An efficient factor-depleted mammalian in vitro translation system. Nat Protoc. 2011;6:563–571.
- Krausslich HG, Nicklin MJ, Toyoda H, et al. Poliovirus proteinase 2A induces cleavage of eucaryotic initiation factor 4F polypeptide p220. J Virol. 1987;61:2711–2718.
- Kuo RL, Kung SH, Hsu YY, et al. Infection with enterovirus 71 or expression of its 2A protease induces apoptotic cell death. J Gen Virol. 2002;83:1367–1376.
- Sweeney TR, Abaeva IS, Pestova TV, et al. The mechanism of translation initiation on Type 1 picornavirus IRESs. Embo J. 2014;33:76–92.
- Kuo RL, Lin YH, Wang RYL, et al. Proteomics analysis of EV71-infected cells reveals the involvement of host protein NEDD4L in EV71 replication. J Proteome Res. 2015;14:1818–1830.
- Paul AV, Wimmer E. Initiation of protein-primed picornavirus RNA synthesis. Virus Res. 2015;206:12–26.
- Paul AV, van Boom JH, Filippov D, et al. Protein-primed RNA synthesis by purified poliovirus RNA polymerase. Nature. 1998;393:280–284.
- Hsu NY, Ilnytska O, Belov G, et al. Viral reorganization of the secretory pathway generates distinct organelles for RNA replication. Cell. 2010;141:799–811.
- Saxena P, Lomonossoff GP. Virus infection cycle events coupled to RNA replication. Annu Rev Phytopathol. 2014;52:197–212.
- Novak JE, Kirkegaard K. Coupling between genome translation and replication in an RNA virus. Genes Dev. 1994;8:1726–1737.
- Liu HM, Aizaki H, Machida K, et al. Hepatitis C virus translation preferentially depends on active RNA replication. PLoS One. 2012;7:e43600.
- Shulla A, Randall G. Spatiotemporal analysis of hepatitis C virus infection. PLoS Pathog. 2015;11:e1004758.
- Bentley DL. Coupling mRNA processing with transcription in time and space. Nat Rev Genet. 2014;15:163–175.
- Simms CL, Thomas EN, Zaher HS. Ribosome-based quality control of mRNA and nascent peptides. Wiley Interdiscip Rev RNA. 2017;8:e1366.
- Melchior F, Paschal B, Evans J, et al. Inhibition of nuclear-protein import by nonhydrolyzable analogs of Gtp and identification of the small Gtpase Ran/Tc4 as an essential transport factor. J Cell Biol. 1993;123:1649–1659.
- Gorlich D, Pante N, Kutay U, et al. Identification of different roles for RanGDP and RanGTP in nuclear protein import. Embo J. 1996;15:5584–5594.
- Porter FW, Bochkov YA, Albee AJ, et al. A picornavirus protein interacts with Ran-GTPase and disrupts nucleocytoplasmic transport. Proc Natl Acad Sci U S A. 2006;103:12417–12422.
- Tang WF, Huang RT, Chien KY, et al. Host microRNA miR-197 plays a negative regulatory role in the enterovirus 71 infectious cycle by targeting the RAN protein. J Virol. 2016;90:1424–1438.
- Han L, Li K, Jin C, et al. Human enterovirus 71 protein interaction network prompts antiviral drug repositioning. Sci Rep. 2017;7:43143.
- Huang HI, Chang YY, Lin JY, et al. Interactome analysis of the EV71 5ʹ untranslated region in differentiated neuronal cells SH-SY5Y and regulatory role of FBP3 in viral replication. Proteomics. 2016;16:2351–2362.
- Kuo RL, Li ZH, Li LH, et al. Interactome analysis of the NS1 protein encoded by influenza A H1N1 virus reveals a positive regulatory role of host protein PRP19 in viral replication. J Proteome Res. 2016;15:1639–1648.
- Li HP, Peng CC, Wu CC, et al. Inactivation of the tight junction gene CLDN11 by aberrant hypermethylation modulates tubulins polymerization and promotes cell migration in nasopharyngeal carcinoma. J Exp Clin Cancer Res. 2018;37:102.
- Hsu CW, Chang KP, Huang Y, et al. Proteomic profiling of paired interstitial fluids reveals dysregulated pathways and salivary NID1 as a biomarker of oral cavity squamous cell carcinoma. Mol Cell Proteomics. 2019;18:1939–1949. In press.
- Keller A, Nesvizhskii AI, Kolker E, et al. Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal Chem. 2002;74:5383–5392.
- Nesvizhskii AI, Keller A, Kolker E, et al. A statistical model for identifying proteins by tandem mass spectrometry. Anal Chem. 2003;75:4646–4658.
- Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57.
- Szklarczyk D, Franceschini A, Wyder S, et al. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015;43:D447–452.
- Lee KM, Gong YN, Shih SR. Methods for detection and study of virus-derived small RNAs produced from the intramolecular base-pairing region of the picornavirus genome. Methods. 2019. DOI:10.1016/j.ymeth.2019.08.011
- Lee KM, Ia W H, Tarn WY. TRAP150 activates pre-mRNA splicing and promotes nuclear mRNA degradation. Nucleic Acids Res. 2010;38:3340–3350.
- Heyer EE, Moore MJ. Redefining the Translational Status of 80S Monosomes. Cell. 2016;164:757–769.