
ABSTRACT
The control of mRNA turnover is essential in bacteria to allow rapid adaptation, especially in opportunistic pathogen like Enterococcus faecalis. This mechanism involves RNase and DEAD-box helicases that are key elements in RNA processing and their associations form the degradosome with accessory proteins. In this study, we investigated the function of four RNases (J1, J2, Y and III) and three DEAD-box helicases (CshA, CshB, CshC) present in most Enterococci. The interactions of all these RNA metabolism actors were investigated in vitro, and the results are in accordance with a degradosome structure close to the one of Bacillus subtilis. At the physiological level, we showed that RNase J1 is essential, whereas RNases J2 and III have a role in cold, oxidative and bile salts stress response, and RNase Y in general fitness. Furthermore, RNases J2, Y and III mutants are affected in virulence in the Galleria mellonella infection model. Concerning DEAD-box helicases, all of them are involved in cold shock response. Since the ΔcshA mutant was the most stress impacted strain, we studied this DEAD-box helicase CshA in more detail. This showed that CshA autoregulates its own expression by binding to its mRNA 5ʹUnstranslated Region. Interestingly, CshC is also involved in the expression control of CshA by a hitherto unprecedented mechanism.
Introduction
Enterococcus faecalis is a gram-positive, facultative anaerobic and ubiquitous bacterium commonly found in mammals’ gastrointestinal tract. This bacterium is also a major opportunistic pathogen responsible for diverse hospital-acquired infections such as urinary tract infections bacteraemia and endocarditis [Citation1,Citation2]. Although several virulence factors, such as the haemolysin Cyl, the gelatinase GelE, or the general stress protein Gls24 [Citation3,Citation4] have been described, the ability of E. faecalis to adapt to a wide range of stressing environments is also considered important for the infection process. For example, this bacterium is able to survive to bile salts exposure in the intestinal tract [Citation5], as well as to stresses encountered within macrophages, like acid pH or exposure to reactive oxygen species [Citation6]. Fast and efficient adaptation needs a tight control of gene expression. Thus, mRNA turnover is crucial for bacteria, and this role is performed by a multi-protein complex called the degradosome [Citation7]. In Bacillus subtilis and Staphylococcus aureus, the degradosome is centred on RNase Y, and the other main proteins are RNases J1 and J2, polynucleotide phosphorylase (PNPase), the two glycolytic enzymes phosphofructokinase PfkA and enolase, and the DEAD-box helicase CshA [Citation8,Citation9]. In B. subtilis, it was demonstrated that the complex formation is initiated by RNase Y that binds successively to the PNPase, RNase J1 and J2, and then to the other proteins of the complex, although in a transitional manner [Citation10,Citation11]. RNases J1 and J2 are paralogous enzymes that exert endoribonuclease activity, as well as exoribonuclease activity on 5ʹmonophosphate RNA ends. In B. subtilis, the deletion of J1 has the most impact on bacteria fitness and the enzyme was shown to be involved in many biological processes, while J2 has a weaker activity [Citation7,Citation12,Citation13]. RNase Y can act as endoribonuclease on 5ʹ triphosphate RNA ends but prefers 5ʹ monophosphate ends [Citation7].
In E. faecalis, only the RNase J2 has been studied: this protein plays a role in virulence, being required for expression regulation of the Ebp protein of the pili, and the virulence factor encoded by gls24 [Citation14].
Other RNases play critical roles in bacterial RNA processing and decay, like the RNase III, which is involved in maturation of tRNA and rRNAs, or degradation of regulatory RNA-mRNA duplexes [Citation15–Citation17].
In contrast, among the degradosome interactions probably the most pertinent ones are with helicases, which initiate the major RNA decay with RNases. DEAD-box RNA helicases are also important for RNA processing by unwinding secondary structures of target RNA [Citation10]. They are indeed involved in almost every RNA metabolism steps from their biosynthesis to their degradation [Citation18] and are able to modify RNA-RNA or short DNA-RNA interactions [Citation19]. In B. subtilis, the DEAD-box helicase CshA is composed of two RecA-like domains, and a C-terminal domain essential for its interaction with the other degradosome proteins [Citation8].
Recently, the involvement of DEAD-box helicases in gene regulation, stress resistance or virulence, in Helicobacter pylori [Citation20] and Pseudomonas aeruginosa pathogens [Citation21] has been reported.
In this work, we studied the involvement of the main RNases (J1, J2, III and Y) and three DEAD-box helicases (CshA, B and C) in E. faecalis physiology and virulence. Since CshA appears to play a critical role, we were particularly interested in studying the expression regulation of this DEAD-box helicase and its interactome.
Results
Phylogeny of RNases and DEAD-box helicases in enterococci
We initiated the study of the four key RNases (J1, J2, Y and III) and three DEAD-box helicases in E. faecalis V583 strain ( and Fig. S1). For most enzymes, we used the protein sequences annotated by NCBI. However, in the case of the initiation codon of CshA, we used the TTG as the start codon at position 805,171 of the V583 genome because of the presence of a ribosome binding site (RBS, AGGAGG) absent upstream the initiation codon proposed by NCBI. Of note, the TTG codon and the RBS are conserved in genomes of Enterococci. Operon structures were verified by RT-PCR and potential transcription terminators were identified using the FindTerm software [Citation22] (Fig. S1).
Table 1. Features of the RNases J1, J2, Y and III, and the DEAD-box helicases CshA, B, C of E. faecalis.
The Uniprot database was used to compare the sequence of these 7 proteins with those of 133 entries for Enterococci, including 47 strains of E. faecalis. Each protein was found in 97% to 100% of enterococcal strains (Table S1). To date and based on available data, we observed that the RnjB (RNase J2) and CshA are present and conserved in every Enterococci indexed in the Uniprot database. Using the NCBI genome database and the tBLASTn tool, we identified genes encoding proteins which share more than 70% identity with the different studied helicases and RNases in Enterococci (database of 96 complete genomes), except for CshC. CshC is indeed less conserved within the Enterococcus genus since the corresponding gene encodes a protein having more than 70% identity with CshC only in the E. faecalis species.
Among other genera, the 7 proteins were also found in Carnobacterium, Clostridioides, Tetragenococcus, Vagococcus genus and in Listeria monocytogenes, with more than 50% of identity (Table S1).
In E. faecalis V583, the three DEAD-box helicases have similar sequences and specific conserved domains. However, CshC presents a DEVD motif, and CshA has a C-terminal extension of 65 amino-acids ().
RnjA is essential for E. faecalis
The first step to investigate the role of the different selected actors was to construct deletion mutants for the RNases J1, J2, III and Y, as well as for the three DEAD-box helicases CshA, CshB and CshC in the V19 strain (WT). The genes were deleted as described in Materials and Methods. We obtained a mutant for rnjB, rnc, rny, cshA, cshB and cshC, demonstrating that these enzymes are independently dispensable in E. faecalis.
Different attempts to construct the rnjA mutant in different E. faecalis strains (V19 or JH2-2) were unsuccessful suggesting that this gene is essential for growth. To confirm this hypothesis, E. faecalis strain V19 was first transformed with the stably replicating plasmid pAGEnt [Citation23] harbouring the rnjA gene under the control of an agmatine-inducible promoter. In parallel, we cloned an internal fragment of the rnjA gene into the suicide plasmid pUCB300 [Citation24] (pUCB300-ΔrnjA). The empty vector pUCB300 or the recombinant pUCB300-ΔrnjA were then transformed into E. faecalis V19 containing pAGEnt-rnjA. No erythromycin resistant clones were obtained with the empty plasmid pUCB300 in the presence or absence of agmatine showing absence of non-homologous recombination with the chromosome. When the WT strain carrying pAGEnt-rnjA was transformed with pUCB300-ΔrnjA, we obtained between 200 and 300 clones per µg of DNA, but only in the presence of agmatine. This demonstrated that rnjA is indeed essential in E. faecalis.
Physiological characterization of the RNases and DEAD-box helicases mutants
Growth of the mutants was not significantly different from that of the wild-type strain, with the exception of ΔcshA, which had a doubling time of 50.3 min vs. 42.3 min for the WT strain (). We then tested resistance of the 6 mutants to acid pH (pH 5.7), bile salts (0.06%), or oxidative stress (1 mM H2O2), and low temperature (15°C) (). Compared to the WT strain, significant differences in doubling times were observed for ΔrnjB, Δrnc and Δrny exposed to low temperature, oxidative stress, and bile salts or acid pH exposure, respectively. Growth of the ΔcshB and ΔcshC mutants was decreased at 15°C, and the ΔcshC strain was more sensitive to oxidative stress and ΔcshA was affected under all stress conditions studied. We concluded that all proteins studied participate in resistance to stressful environments, with CshA playing the largest role.
Table 2. Doubling time (in min) of WT, ΔrnjB, Δrnc, Δrny, ΔcshA, ΔcshB and ΔcshC E. faecalis strains in BHI or exposed to different stress.
To determine if these stress conditions induce the expression of the studied genes, RT-qPCR assays were performed (). The cold exposure led to an induction of all seven genes. Moreover, rny and cshA are induced under all tested conditions, suggesting a central role of the RNase Y and CshA in cell adaption to stressing environments, while RNase J2, RNase III, CshB and CshC seem to have a more stress-specific role.
Table 3. Significant fold changes in the expression of rnjA, rnjB, rnc, rny, cshA, cshB, cshC, in E. faecalis WT strain exposed to 30 min of stress or during mice infection and compared to exponential growth cells at 37°C in BHI.
Roles of RNases and DEAD-box helicases in virulence
Results from a previous RNA-Seq analysis suggested that RNases and DEAD-box helicases are induced (2 to 4 fold) during mouse peritonitis [Citation25]. We controlled these preliminary data by RT-pPCR using RNA extracted from bacterial cells recovered after 24 h mice peritonitis. This analysis confirmed induction of the rnjA, rnjB, rny, rnc and cshA genes () and thus suggests a role of the encoded proteins in the E. faecalis survival or colonization inside the host.
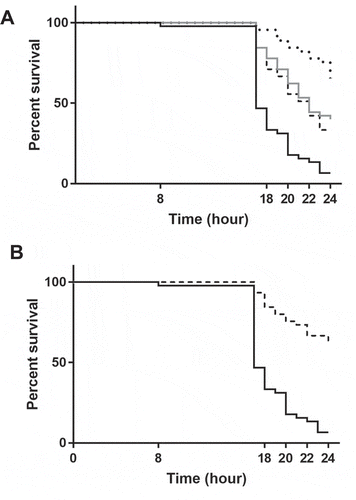
The ability of the different mutants to kill the Galleria mellonella larvae () was also examined. This showed that virulence of the Δrny and ΔcshA mutants was highly attenuated compared to the parental strain (p < 0.0001) (). Also, the ΔrnjB and Δrnc demonstrated decreased virulence also to a lesser extent than the two former mutants (p < 0.0001) (). The ΔcshC and ΔcshB mutants showed phenotypes of virulence similar to that of the WT strain (data not shown).
Figure 1. Survival of G. mellonella inoculated with WT, ΔrnjB, Δrnc, Δrny or ΔcshA, E. faecalis strains. Larvae were inoculated with 2 × 108 CFU of bacteria grown in BHI at 37°C. Survival was monitored between 8 and 24 h post-infection at 37°C using 15 larvae per strain. (A) WT (black line), ΔrnjB (grey line), Δrnc (dashed line), Δrny (dotted line), (B) WT (black line), ΔcshA (dashed line). The assays were performed in triplicates, and log-rank tests were used to evaluate results significance (p < 0.0001).
Identification of the 5ʹUTR of cshA
In a previous study [Citation25], we highlighted the existence of a putative regulatory RNA (named SRC43) located between EF0845 (murF) and EF0846 (cshA) genes. We therefore carried out Northern blot assays to confirm the presence of this sRNA (Fig. S2A): RNA hybridization with a probe specific of this sRNA showed a transcript whose size is identical to that of the mRNA hybridized with a cshA specific probe (approximately 1.5 kb).
In addition, in silico analyses using the Findterm software [Citation22] did not reveal any transcription terminator upstream of cshA. A 5ʹRACE-PCR assay confirmed that the 5ʹend of the putative sRNA (Fig. S2B) and cshA (data not shown) are identical. Considering these results, we concluded that this putative sRNA is actually a 5ʹUTR (Untranslated Region) of cshA, henceforth referred to as 5ʹcshA (from the +1 of transcription to the nucleotide right before the start codon of cshA coding region – coordinates 804,919 to 805,170 of the E. faecalis V583 strain genome).
The transcriptional start site of cshB and of the ef1008-cshC operon were also identified by 5ʹRACE-PCR, and positioned 36 and 25 bp before the ATG start codon of each transcription unit, respectively (data not shown), indicating the absence of 5ʹUTR for these helicase genes.
CshA regulates its own expression
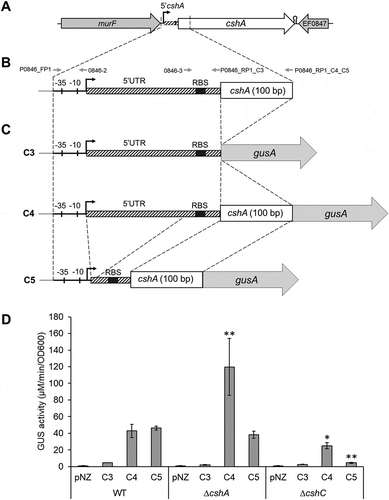
To understand the role of this 5ʹUTR in cshA regulation, translational reporter constructions between different parts of cshA gene and the gusA gene were used ( and detailed in Fig. S3). These constructions were introduced into E. faecalis WT and ΔcshA strains. The empty pNZ273 plasmid was used as the negative control in these experiments. C3 produced a very weak signal (4.6 mol/ml/min) compared to C4 that produced a GUS activity of 42.7 mol/ml/min in the WT strain (). This result suggests that the first hundred base pairs of the cshA gene play a role in gene expression. We also detected a very strong activity of 119.8 mol/mL/min with C4 for ΔcshA strain (). Thus in the absence of CshA, 5ʹcshA has an inducing effect on CshA expression, suggesting a transcriptional or post-transcriptional auto-regulation of cshA via the corresponding sequence. The stability of the cshA mRNA was checked in a strain deleted for the 5ʹUTR 5ʹcshA (described in supplementary material), where we detected the same quantity of cshA mRNA compared to the WT strain, showing that the 5ʹUTR has no influence on mRNA degradation (Fig. S4). Moreover, C3 construction had the same activity in ΔcshA strain than the WT cells (), indicating that the first hundred base pairs of cshA are important for the CshA dependent regulation.
Figure 2. (A) Genetic environment of the gene encoding the CshA DEAD-box helicase, (B) Representation of 5ʹcshA (striped, coordinates 804,919 to 805,170 of the E. faecalis V583 strain genome), including −35 and −10 boxes, +1 of transcription and the Ribosome Binding Site (RBS) (for more details, see Fig. S3). Oligonucleotides location for synthesis of sequences for insertion into the pNZ273 plasmid are indicated. (C) C3, C4 and C5 constructions inserted into the pNZ273 plasmid. C3 contains the promoter of cshA, including the 5ʹcshA (containing the RBS) and the start codon, in frame with the gusA gene of the pNZ273 plasmid (grey). C4 contains 5ʹcshA and the first one hundred base pairs of the cshA ORF (white). The C5 construction is the same as C4, but with a deletion in the 5ʹUTR. (D) β-glucuronidase (GUS) assay with the C3, C4 and C5 constructions in the WT, ΔcshA and ΔcshC strains. pNZ: empty plasmid (negative control). Data are the mean of three independent assays. Statistically significant results in comparison to the same construction in the WT strain are indicated: *p < 0.05, ** p < 0.005 (Kruskal–Wallis test).
To ensure that the effect observed on GUS expression is actually due to ΔcshA mutation, we complemented the deletion by providing the cshA gene ectopically as described in Supplementary Material. The GUS assays with the complemented strain containing the C4 or C5 reporter construction showed the same enzyme activity than the WT strain (Fig. S5).
CshA binds to its 5ʹUTR
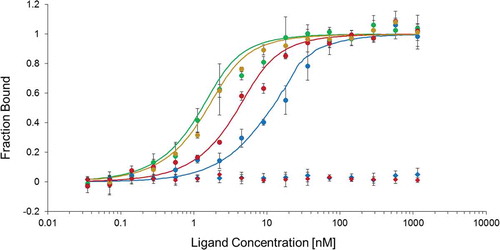
According to our hypothesis, CshA would be able to bind the 5ʹcshA RNA. In order to prove it, we used the MicroScale Thermophoresis (MST) technology. Recombinant CshA (CshA-6xHis) was purified using pQE30 plasmid (Fig. S6), and labelled. RNA corresponding to the region covering the 5ʹcshA ( and Fig. S3) was synthesized using in vitro transcription (see Materials and Methods). The previously identified SRC65 sRNA [Citation26,Citation27] which does not share any sequence homology with 5ʹcshA, was also synthesized and used as negative control. MST experiment revealed an increase of the CshA-6xHis bound fraction following the concentration of the 5ʹcshA RNA, in contrast to the negative control (). This dose–response interaction between CshA and 5ʹcshA RNA allowed us to determine a dissociation constant (KD) of 5.2 ± 2.2 nM, showing a high and specific affinity of CshA for its own RNA. Therefore, we concluded that CshA auto-regulates its own gene expression by binding on 5ʹcshA.
Figure 3. Dose–response curve of MicroScale Thermophoresis (MST) showing the binding of CshA (in red) or CshC (in blue) to the ligand 5ʹcshA RNA (coordinates 804,919 to 805,170 of the E. faecalis V583 strain genome) (circles) or to the negative control SRC65 sRNA control (coordinates 1,170,546 to 1,170,380) (diamonds). Binding of CshA in the presence of saturating concentration of CshC (1 µM, green circles) or CshC in the presence of saturating concentration of CshA (1 µM, orange circles) promotes the interaction of helicases with 5ʹcshA RNA. Solid curves are the fits of the data points. The error bars represent the standard error in triplicate measurements.
Interaction analysis of RNA metabolism proteins
To deepen our understanding of the E. faecalis degradosome, the proteins encoded by the other genes mentioned in this study, i.e. CshB, CshC and RNases J1, J2, Y and III were purified (Fig. S6) and interactions with each other were monitored by MST approach. The KD values were determined () indicating tight and highly specific interactions between RNases Y, J1 and CshA. We also observed that RNases J1, J2 and III and CshA can form homodimers. In addition, MST revealed that RNases J1 and J2 on the one hand and helicases CshA and CshC on the other hand formed heterodimers.
Table 4. Interactions of proteins involved in RNA metabolism. The KD (nM) were revealed by microscale thermophoresis. NI: No interaction monitored.
In order to identify CshA complexing proteins in vivo, a fusion of the cshA gene with the TAP tag (Tandem Affinity Purification) [Citation28] was constructed as described in Supplementary Materials. Thus, CshA-TAP served as a bait protein to identify its interacting partners in E. faecalis, after purification with two successive affinity columns and analysis by mass spectrometry. The first assays showed that the interactome of CshA is composed of 116 proteins (Table S2), including 44 ribosomal proteins of the 50S and 30S subunits. We identified the most important proteins found in the degradosome, i.e. the ribonucleases J1 and J2 (EF2924 and EF1185, respectively), the 6-phosphofructokinase PfkA (EF1045), the enolase Eno (EF1961), and the polynucleotide phosphorylase PnpA (EF3064). RNase Y was not detected in the CshA interactome which may be due to its potentially exclusive presence in the cell membrane fraction, as shown in B. subtilis [Citation8,Citation11]. We also identified several enzymes of cell metabolism, especially from glycolysis (6-phospho-β-glucosidase EF0291, pyruvate kinase EF1046, fructose-biphosphate aldolase EF1167, glucose-6-phosphate isomerase EF1416, glyceraldehyde-3-phosphate dehydrogenase EF1964 – Table S2), likely due to the high quantity of Eno and PfkA. This result prompted us to use more stringent conditions during TAP purification. Given the RNA binding property of CshA, the same experiment was conducted with supplementary steps of incubation with RNase A after cell lysis and crosslinking. The interactome analysis showed that only the enolase was still bound to CshA (Mascot score: 275.1). These data provide evidence that CshA interaction with ribosomal subunits is dependent on RNA encountered in the cell, and that the enolase is the privileged partner of the helicase in vivo.
Expression of CshA is also regulated by the DEAD-box helicase CshC
To understand the role of CshA and CshC interaction, the C4 and C5 constructions were introduced into the ΔcshC mutant and GUS activity was measured. As shown in , gusA in the C5 construction has a 10-fold decreased expression in the ΔcshC background compared to the WT background (4 and 46 µM/min/OD600, respectively). Consequently, CshC is involved in the control of expression of CshA. This effect was also observed with the C4 construction containing the entire 5ʹcshA region. The complementation of ΔcshC mutation was successfully performed as described in Supplementary Material (Fig. S5), showing that the gene deletion does not lead to secondary effect. In contrast, no effect was observed in a cshB mutant (data not shown), providing the evidence of a specific relation between CshA and CshC.
Therefore, we undertook binding studies of CshC on 5ʹcshA RNA. MST approach has allowed us to successfully measure an affinity of 11.9 ± 1.4 nM of the protein for this specific sequence, whereas no binding was observed with SRC65 RNA used as the negative control (coordinates 1,170,546 to 1,170,380 of the E. faecalis V583 strain genome) (). To assess the impact of CshA on CshC binding and conversely, one of the RED-NHS labelled helicase was pre-incubated with a saturating concentration of the other non-labelled helicase (1 µM) and added to the 5ʹcshA RNA (). A positive cooperativity was observed for both assays, since the interaction of CshC with this target was improved 9.0 fold in the presence of CshA (1.52 ± 0.3 nM), and that of CshA 3.4 fold in the presence of CshC (1.32 ± 0.7 nM). Thus, both helicases significantly promote their binding on 5ʹcshA RNA.
Discussion
In this study, we explored expression and functions of key enzymes of RNA metabolism: RNases J1, J2, III, Y and DEAD-box RNA helicases CshA, CshB and CshC. The conservation of these seven enzymes in Enterococci and other Gram-positive bacteria suggests that these proteins play a prominent role among bacteria in the Enterococcus genus, except CshC that is less conserved.
While RNase J1 is essential in E. faecalis, each RNase studied appears to be important for stress response or general fitness, especially the RNase Y. This protein is involved in virulence of E. faecalis, which is in agreement with previous data reported for Streptococcus pyogenes. The RNase Y in this latter bacterium is implicated in the expression control of virulence factors [Citation29], as RNase J2, in E. faecalis [Citation14,Citation30], or RNase III in S. aureus [Citation31].
CshA DEAD-box helicase deletion was shown to affect general bacterial fitness [Citation20], that might affect efficient colonization of G. mellonella responsible for the observed delayed killing of the larvae. However, an important role of CshA in the infection process is supported by overexpression of cshA during mice peritonitis. In contrast, the two other helicases (CshB and CshC) have a more specific role in cold stress response, as shown in numerous studies [Citation32–Citation36], but not in virulence.
We demonstrated that CshA acts as a repressor and autoregulates its own expression. We can therefore hypothesize that CshA operates via its helicase activity, by releasing secondary structures in the 5ʹUTR RNA, which could have an impact on the RNA folding downstream the 5ʹUTR (including the first 100 nucleotides of the coding region) and therefore on the translation efficiency (but does not affect RNA stability). In fact, secondary structures were found in the 5ʹcshA RNA sequence (with RNAfold server [Citation37] for example), that could support our hypothesis. To the best of our knowledge, this is the first report demonstrating auto-regulation of expression of a DEAD-box helicase. Despite the description of RNA helicase autoregulation in cyanobacteria, it occurs indirectly through yet unknown transcriptional and post-transcriptional mechanisms [Citation35]. Furthermore, Caulobacter crescentus might regulate expression of the DEAD-box helicase RhlE through its 5ʹUTR, but as a translation inducer [Citation32]. Of note, the 5ʹUTR of this species does not share any sequence identity with the 5ʹcshA of E. faecalis. No 5ʹUTR were evidenced for cshB and cshC, indicating that regulation of expression of these helicases is different.
We also studied the role of CshA as part of the degradosome through interactome assays. As expected from results of B. subtilis [Citation8] and supported by the KD value measured in this work, RNase Y of E. faecalis is the protein that initiates the degradosome formation and interacts tightly with J1 and CshA in vitro. RNases J1 and J2 can form homo- and heterodimers, and RNase III would dimerizes [Citation38]. Our results also suggest that CshA oligomerizes as shown in B. subtilis [Citation8]. Moreover, we confirmed that CshB and CshC do not participate in any interactions studied here, except the unexpected binding of CshC with CshA with high affinity (KD of 156.9 nM). Thus, we evidenced that CshC and CshA both play a role in cshA expression that is an unprecedented mechanism. In the absence of CshC, CshA even represses its own expression more efficiently. CshC might specifically and alone stimulate cshA expression with or without involving the 5ʹcshA, but also thanks to its direct interaction with CshA. Thus, these two DEAD-box helicases act synergically to regulate cshA expression, as they interact together to bind 5ʹcshA RNA with higher affinity than alone. However, more investigations have to be undertaken to decipher the molecular mechanism of this regulation that might depend on the assistance of other partners.
In the presence of RNA, CshA binds with proteins homologous to those known to belong to the degradosome in B. subtilis [Citation8,Citation11]. It has to be noted that interaction of the enzymes involved in RNA metabolism in B. subtilis was shown to be transitory [Citation11], that could explain that we identified a large number of proteins interacting with CshA, and that the degradosome composition remains quite uncertain. Moreover, more than half of the interactome proteins correspond to ribosomal proteins or proteins involved in translation. This result is consistent with the studies of DEAD-box helicases SrmB and YxiN in E. coli and B. subtilis, respectively, where it was demonstrated that these proteins are involved in rRNA maturation and ribosome assembly [Citation39–Citation44]. We found that CshA shares 38% and 44% identity with these proteins, respectively, and these low homologies are principally due to the absence of the C-terminal extension found in CshA. Interestingly, CshC was also shown to be part of the CshA interactome, confirming the interaction of CshA and CshC shown by MST. In the absence of RNA, we showed that the enolase was the privileged partner of CshA in vivo. This interaction has already been demonstrated before in B. subtilis [Citation8], but to our knowledge not in E. faecalis. No other protein was identified in these conditions that can be explained by the fact that the TAP tag was added at the C-terminus of the protein to keep expression of CshA under the control of its 5ʹUTR. It was previously suggested that this C-terminus end is involved in the degradosome interaction [Citation45], justifying that only few proteins were detected in this interactome study. This also suggests that enolase is interacting with CshA through another part of the helicase.
In summary, our data demonstrate that RNA metabolism proteins have important implications for E. faecalis adaptation and virulence. This study presents new observations regarding the complexity of pathways regulating the DEAD-box helicase CshA expression associated with bacterial stress response. These findings provide new perspectives on the molecular mechanism that lead to the opportunistic behaviour of this bacterium.
Materials and methods
Bacterial strains and growth conditions
In this study, we used the E. faecalis V19 [Citation46] as reference strain WT, which corresponds to the V583 strain [Citation47] cured of its plasmids, that allows more opportunities to use molecular tools since it does not present antibiotic resistance. Cultures were achieved in BHI medium, supplemented or not with 0.06% bile salts, or 1 mM H2O2, or with pH adjusted at 5.7, and incubated at 37°C or 15°C. Chloramphenicol (15 µg/ml) was added for E. faecalis strains containing the pNZ273 or the pMAD plasmids or derivatives. The E. coli strains TOP10, M15 pRep4 and DH5α were used, respectively, for in vitro RNA production, recombinant protein production and translational fusion. These E. coli strains were grown in LB medium at 37°C with agitation, supplemented with kanamycin (25 µg/ml) for the M15 pRep4 strain and pTOPO-containing strains, ampicillin (100 µg/ml) for strains containing pQE30 or pMAD derivatives, or chloramphenicol (10 µg/ml) for strains containing the pNZ273 plasmid.
Molecular techniques
5ʹRACE-PCR were performed using the 5ʹ/3ʹ RACE kit, 2nd generation (Roche, Bâle, Switzerland), with poly-A or poly-C tailing and SP1 to SP3 primers (Table S3). PCR were performed using Phusion High-Fidelity DNA Polymerase (ThermoFisher, Waltham, Massachusetts, USA). PCR products were purified when necessary using the NucleoSpin Gel and PCR Clean-up kit (Macherey-Nagel, Düren, Germany) and plasmid extractions were achieved with the NucleoSpin Plasmid kit (Macherey-Nagel) according to the manufacturer’s recommendations.
Construction of E. faecalis strains
E. faecalis V19 was used for the construction of all mutant strains (ΔrnjA, ΔrnjB, Δrnc, Δrny, ΔcshA, ΔcshB, ΔcshC) and E. coli DH5α for all cloning purposes. E. faecalis competent cells were produced as previously described [Citation48], with 10 µg/ml lysozyme and addition of 10 U mutanolysin. All deletion strains were generated with the same approach: the flanking regions of the gene were PCR amplified with primer pairs 1/2 (upstream fragment) and 3/4 (downstream fragment) that include restriction endonuclease sites (Table S3). The fragments were digested with the corresponding endonucleases and cloned in tandem into the pMAD vector [Citation49]. The resulting plasmids were inserted into the genome of the E. faecalis V19 strain by double crossing-over, as previously described [Citation50], and checked by PCR (with primers pairs 5/6 of each gene) and sequencing.
To prove that rnjA is essential in E. faecalis, the entire gene or an internal fragment were amplified using pAgent2924F/pAgent2924R and pUCB2924F/pUCB2924R primer pairs and were cloned into the plasmids pAGEnt [Citation23] (displaying chloramphenicol resistance) and pUCB300 [Citation24] (displaying erythromycin resistance), respectively, using the NcoI and PstI cloning sites. Once pAGEnt-rnjA was introduced into E. faecalis V19 strain, cells were cultivated in M17 supplemented with ribose 0.5%, to avoid catabolic repression on the PaguB agmatine-inducible promoter, and chloramphenicol (15 µg/ml). The insertion event of the pUCB300-ΔrnjA plasmid in the chromosomal copy of rnjA was selected in presence of erythromycin (50 µg/ml) and chloramphenicol (15 µg/ml) (to maintain pAGEnt-rnjA in the cell), and with or without agmatine.
Constructions in the pNZ273 reporter plasmid
For translational fusions, the pNZ273 plasmid was amplified by PCR with P0846_FP2 and P0846_RP2 for C3, and with P0846_FP2_C4_C5 and P0846_RP2 for C4 and C5. The inserts were amplified with P0846_FP1 and P0846_RP1_C3 for C3, and P0846_FP1 and P0846_RP1_C4_C5 for C4 and C5, giving overlapping tails compatibles with the plasmid. For C5, amplification was performed on Δ5ʹcshA strain. The assembly was performed in vitro using the NEBuilder HiFi DNA Assembly Cloning Kit (New England BioLabs, Ipswich, Massachusetts, USA), and the resulting DNA was used to transform E. coli NEB-5α. After replication of the plasmids, the constructions were extracted and introduced into E. faecalis V19 (wild type) and ΔcshA strains. All steps were controlled by PCR with pNZ273_MCS_for and pNZ273_MCS_rev primers.
β-glucuronidase (GUS) activity assay
The pNZ273 plasmid used for translational fusions includes the reporter gene gusA, encoding for the β-glucuronidase [Citation51]. Strains were cultured at 37°C in GM17 medium until OD600 0.7, and then centrifuged. The pellets were resuspended in an equivalent volume of phosphate buffer, and OD600 was measured. Five hundred µL (v) of the suspension were then mixed with 37.5 µL of lysis buffer (Chloroform 66.7%, SDS 0.03%) and incubated at 37°C for 5 min. One hundred µL of p-nitrophenyl-β-D-glucuronide (4 mg/mL) were added, and the mix was incubated at 37°C for 10 min (t). Two hundred and fifty µL of Na2CO3 1M were next added to stop the reaction, the solution was centrifuged 5 min, and supernatant was used to measure the OD405 and OD550. The GUS activity was expressed in µM/min/OD600 and was determined using the following formula: GUS = (522× OD405)/(t × v× OD600) [Citation51].
Total RNA extraction
Total RNA extractions were performed from 10 mL of culture for RT-qPCR, and 100 mL for Northern blot assay, of bacteria grown at 37°C until OD600 0.5. For stressing conditions, the cultures were grown in BHI medium until OD600 0.3 and centrifuged before being resuspended in stress-inducing medium (BHI supplemented with 0.06% bile salts, or with pH adjusted at 5.7) and incubated 30 min at 37°C. For RNA extraction after mouse infection, cells were prepared as previously described [Citation25]. Briefly, E. faecalis V19 strain was grown overnight in BHI and then resuspended in phosphate-buffered saline (PBS) before intraperitoneal injection of BALB/c mice (109 cells). Peritoneal cells were harvested after 24 h of infection and washed with PBS and then used for RNA extraction. Cells were lysed with the FastPrep apparatus (MP Biomedicals, Illkirch Graffenstaden, France). Extraction and purification were performed using TRIzol Reagent and chloroform/isoamyl alcohol separation followed by Direct-Zol RNA Miniprep kit (Zymo-Research, Irvine, Californie, USA). RNAs were quantified using Nanodrop™ 2000 (ThermoFisher) and their quality was checked by electrophoresis.
RT-qPCR assay
The QuantiTect® Reverse Transcription kit (Qiagen) was used for RT reactions, using 1 µg of RNA and random primers, and qPCR assays were performed with the GoTaq qPCR Master Mix (Promega). Reactions were loaded into a C1000™ Thermal Cycler (Bio-Rad, Hercules, Californie, USA) detection system (Bio-Rad) and the qPCR were performed with the following conditions: 95°C for 3 min, followed by 40 cycles at 95°C for 15 sec, 60°C for 1 min. The transcription level of each gene was normalized to that of the gyrA reference gene. Genomic DNA from WT cells was used to determine PCR efficiency.
Northern blotting
Northern blot assays were performed on 10 µg RNA as previously described [Citation26], with the SRC43 probe (Table S3). Hybridized membranes were exposed for one night to a storage phosphor screen (Packard Instrument Company, Canberra, Australia) and were read with a Cyclone apparatus (Packard Instrument Company).
Production and purification of recombinant proteins
The gene encoding J1, J2, Y and III RNases and CshA, CshB, CshC helicases was amplified with pQE-F and pQE-R primers (Table S3). To allow the purification of the RNase Y, only the DNA sequence corresponding to amino acids 23 to 518 was cloned to avoid the membrane domain. The PCR products and the pQE30 plasmid (Qiagen, Venlo, Netherlands) were digested by restriction endonucleases indicated in Table S3, ligated and used to transform E. coli M15 pRep4. Once the constructions were checked by sequencing, the strain was grown at 37°C in Terrific Broth medium [Citation52] containing 25 µg/mL kanamycin and 100 µg/mL ampicillin until OD600 0.5, and the protein production was induced with 0.5 mM IPTG for 4 h. Recombinant proteins were purified using the Protino Ni-NTA Agarose kit according to the manufacturer’s instructions (Macherey-Nagel), and desalted with PD10 columns (GE Healthcare, Chicago, Illinois, USA). Proteins were quantified with the Bradford Method [Citation53], analysed by SDS-PAGE (12.5%) and their purity was verified using mass-spectrometry: after nLC-ESI-TIMS-TOF analysis of protein samples, MS data (mgf files) were processed using Mascot software and an E. coli M15 Uniprot database (including 4734 entries) to ensure that no RNA binding protein like Hfq were co-purified.
In vitro production of RNA
DNA regions coding the RNA of interest were amplified using primers topo0846_FP1/topo0846_RP1 and topo65_FP1/topo65_RP1 for 5ʹcshA and SRC65, respectively, and the pTOPO plasmid (Zero Blunt TOPO™, Invitrogen, Carlsbad, California, USA) using primers topo_FP2 and topo_RP2. Inserts were independently associated with the plasmid amplimer using the NEBuilder HiFi DNA Assembly Cloning Kit (New England BioLabs) and used to transform E. coli TOP10. After plasmid extraction and linearization with XhoI or SpeI for 5ʹcshA and SRC65, respectively, corresponding RNA were produced using the MAXIScript™ T7 in vitro Transcription Kit (Invitrogen).
Microscale thermophoresis
For RNA–protein interaction studies, recombinant proteins were firstly labelled by using the Protein Labelling Kit RED-NHS 2nd Generation as recommended (Nanotemper Technologies, Munich, Germany), and diluted at a final concentration of 1 nM in Reaction Buffer (20 mM Tris HCl pH 7.4, 150 nM, 5 mM KCl, 1mM CaCl2, 1 mM MgCl2, 0.01% Tween-80). Test and control RNAs were firstly heated for 5 min at 95°C and cooled down at room temperature for approximately 15 min to allow a proper formation of secondary structures. They were then diluted in Reaction Buffer (range of dilutions from 1.15 µM to 0.035 nM). Labelled recombinant CshA or CshC and test or control RNA were then mixed together before transfer in standard capillaries (Monolith NT.115 Capillary), incubated 5 min at room temperature, and subsequently subjected to MST analysis in Monolith NT.115 (Nanotemper Technologies). Binding cooperative analysis was conducted as previously described [Citation54] with RED-NHS CshA or CshC, and a saturating concentration of 1 µM for the non-labelled CshA or CshC. This mixture was incubated 15 min at room temperature, and interaction with RNA was performed as explained below.
To study the degradosome proteins interaction between them, proteins were used as follows: 30 nM of labelled protein and 40 μM as the highest to 0.6 nM as the lowest concentration of the unlabelled protein. Protein samples were incubated at room temperature during 15 min and transferred into capillaries before analysis in Monolith NT.115.
Experiments were conducted in at least three independent replicates and the standard errors were presented as the standard error of the triplicates. The temperature was set to 20°C and measurements were performed at 20% MST power. Data analyses were performed with NTAnalysis software V2.3 (Nanotemper Technologies).
Virulence study on the Galleria mellonella model
G. mellonella larval infections were performed as previously described [Citation55], with an inoculum of 2.108 bacteria per larvae. Fifteen caterpillars were used by analysis and the experiments were repeated at least 3 times.
Authors’ contributions
MS, CM and AR developed the project. MS and CM performed experiments, and BB, TC and OL performed biochemical analysis. MS, CM, BB, OL, TC and AR analyzed the data. MS, CM, BB, TC, OL and AR wrote the paper.
Supplemental Material
Download Zip (1 MB)Acknowledgments
We are grateful to Hilde de Reuse for providing the pILL825C plasmid, and Harald Putzer for his sound advice. The expert technical assistance of Julien Pontin from the Proteogen platform, staff of the Imogere platform, Aurélie Budin-Verneuil and Isabelle Rincé was greatly appreciated.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here
Additional information
Funding
References
- García-Solache M, Rice LB. The Enterococcus: a model of adaptability to its environment. Clin Microbiol Rev. 2019;32:e00058-18.
- Babady NE. Hospital-associated infections. Microbiol Spectr. 2016;4:DMIH2-0003-2015.
- Arias CA, Murray BE. The rise of the Enterococcus: beyond vancomycin resistance. Nat Rev Microbiol. 2012;10:266–278.
- Madsen KT, Skov MN, Gill S, et al. Virulence factors associated with Enterococcus faecalis infective endocarditis: a mini review. Open Microbiol J. 2017;11:1–11.
- Rincé A, Le Breton Y, Verneuil N, et al. Physiological and molecular aspects of bile salt response in Enterococcus faecalis. Int J Food Microbiol. 2003;88:207–213.
- Flannagan RS, Cosío G, Grinstein S. Antimicrobial mechanisms of phagocytes and bacterial evasion strategies. Nat Rev Microbiol. 2009;7:355–366.
- Laalami S, Zig L, Putzer H. Initiation of mRNA decay in bacteria. Cell Mol Life Sci. 2014;71:1799–1828.
- Lehnik‐Habrink M, Pförtner H, Leonie R, et al. The RNA degradosome in Bacillus subtilis: identification of CshA as the major RNA helicase in the multiprotein complex. Mol Microbiol. 2010;77:958–971.
- Roux CM, DeMuth JP, Dunman PM. Characterization of components of the Staphylococcus aureus mRNA degradosome holoenzyme-like complex. J Bacteriol. 2011;193:5520–5526.
- Lehnik-Habrink M, Lewis RJ, Mäder U, et al. RNA degradation in Bacillus subtilis: an interplay of essential endo- and exoribonucleases. Mol Microbiol. 2012;84:1005–1017.
- Cascante-Estepa N, Gunka K, Stülke J. Localization of components of the RNA-degrading machine in Bacillus subtilis. Front Microbiol. 2016;7:1492.
- Jamalli A, Hébert A, Zig L, et al. Control of expression of the RNases J1 and J2 in Bacillus subtilis. J Bacteriol. 2014;196:318–324.
- Laalami S, Putzer H. mRNA degradation and maturation in prokaryotes: the global players. Biomol Concepts. 2011;2:491–506.
- Gao P, Pinkston KL, Bourgogne A, et al. Functional studies of E. faecalis RNase J2 and its role in virulence and fitness. PLoS One. 2017;12:e0175212.
- Arraiano CM, Andrade JM, Domingues S, et al. The critical role of RNA processing and degradation in the control of gene expression. FEMS Microbiol Rev. 2010;34:883–923.
- Johnson CM, Haemig HHA, Chatterjee A, et al. RNA-mediated reciprocal regulation between two bacterial operons is RNase III dependent. MBio. 2011;2:e00189-11.
- Iost I, Chabas S, Darfeuille F. Maturation of atypical ribosomal RNA precursors in Helicobacter pylori. Nucleic Acids Res. 2019;47:5906–5921.
- Cordin O, Banroques J, Tanner NK, et al. The DEAD-box protein family of RNA helicases. Gene. 2006;367:17–37.
- Khemici V, Linder P. RNA helicases in bacteria. Curr Opin Microbiol. 2016;30:58–66.
- El Mortaji L, Aubert S, Galtier E, et al. The sole DEAD-box RNA helicase of the gastric pathogen Helicobacter pylori is essential for colonization. MBio. 2018;9:e02071-17.
- Intile PJ, Balzer GJ, Wolfgang MC, et al. The RNA helicase DeaD stimulates ExsA translation to promote expression of the Pseudomonas aeruginosa type III secretion system. J Bacteriol. 2015;197:2664–2674.
- Solovyev V, Salamov A. Automatic annotation of microbial genomes and metagenomic sequences. In: Li RW, editor. Metagenomics and its applications in agriculture, biomedicine and environmental studies. New York: Nova Science Publishers; 2011; p. 61–78.
- Linares DM, Perez M, Ladero V, et al. An agmatine-inducible system for the expression of recombinant proteins in Enterococcus faecalis. Microb Cell Fact. 2014;13:1–9.
- Frère J, Novel M, Novel G. Molecular analysis of the Lactococcus lactis subspecies lactis CNRZ270 bidirectional theta replicating lactose plasmid pUCL22. Mol Microbiol. 1993;10:1113–1124.
- Muller C, Cacaci M, Sauvageot N, et al. The intraperitoneal transcriptome of the opportunistic pathogen Enterococcus faecalis in mice. Plos One. 2015;10:e0126143.
- Shioya K, Michaux C, Kuenne C, et al. Genome-wide identification of small RNAs in the opportunistic pathogen Enterococcus faecalis V583. Plos One. 2011;6:e23948.
- Salze M, Giard J-C, Riboulet-Bisson E, et al. Identification of the general stress stimulon related to colonization in Enterococcus faecalis. Arch Microbiol. 2019. DOI:10.1007/s00203-019-01735-8
- Stingl K, Schauer K, Ecobichon C, et al. In vivo interactome of Helicobacter pylori urease revealed by tandem affinity purification. Mol Cell Proteomics. 2008;7:2429–2441.
- Broglia L, Materne S, Lécrivain A-L, et al. RNase Y-mediated regulation of the streptococcal pyrogenic exotoxin B. RNA Biol. 2018;15:1336–1347.
- Gao P, Pinkston KL, Nallapareddy SR, et al. Enterococcus faecalis rnjB is required for pilin gene expression and biofilm formation. J Bacteriol. 2010;192:5489–5498.
- Lioliou E, Sharma CM, Caldelari I, et al. Global regulatory functions of the Staphylococcus aureus endoribonuclease III in gene expression. Plos Genet. 2012;8:e1002782.
- Aguirre AA, Vicente AM, Hardwick SW, et al. Association of the cold-shock DEAD-box RNA helicase RhlE to the RNA degradosome in Caulobacter crescentus. J Bacteriol. 2017;199:e00135-17.
- Jiang X, Keto-Timonen R, Skurnik M, et al. Role of DEAD-box RNA helicase genes in the growth of Yersinia pseudotuberculosis IP32953 under cold, pH, osmotic, ethanol and oxidative stresses. Plos One. 2019;14:e0219422.
- Pandiani F, Chamot S, Brillard J, et al. Role of the five RNA helicases in the adaptive response of Bacillus cereus ATCC 14579 cells to temperature, pH, and oxidative stresses. Appl Environ Microbiol. 2011;77:5604–5609.
- Rosana ARR, Chamot D, Owttrim GW. Autoregulation of RNA helicase expression in response to temperature stress in Synechocystis sp. PCC 6803. Plos One. 2012;7:e48683.
- Owttrim GW. RNA helicases. RNA Biol. 2013;10:96–110.
- Mathews DH, Disney MD, Childs JL, et al. Incorporating chemical modification constraints into a dynamic programming algorithm for prediction of RNA secondary structure. Proc Natl Acad Sci USA. 2004;101:7287–7292.
- Nicholson AW. Ribonuclease III mechanisms of double-stranded RNA cleavage. Wiley Interdiscip Rev RNA. 2014;5:31–48.
- Charollais J, Pflieger D, Vinh J, et al. The DEAD-box RNA helicase SrmB is involved in the assembly of 50S ribosomal subunits in Escherichia coli. Mol Microbiol. 2003;48:1253–1265.
- Gentry RC, Childs JJ, Gevorkyan J, et al. Time course of large ribosomal subunit assembly in E. coli cells overexpressing a helicase inactive DbpA protein. RNA. 2016;22:1055–1064.
- Iost I, Dreyfus M. DEAD-box RNA helicases in Escherichia coli. Nucleic Acids Res. 2006;34:4189–4197.
- Trubetskoy D, Proux F, Allemand F, et al. SrmB, a DEAD-box helicase involved in Escherichia coli ribosome assembly, is specifically targeted to 23S rRNA in vivo. Nucleic Acids Res. 2009;37:6540–6549.
- Bizebard T, Dreyfus M. A FRET-based, continuous assay for the helicase activity of DEAD-box proteins. In: Boudvillain M, editor. RNA Remodeling Proteins. New York: Springer New York; 2015. p. 199–209. Available from: http://link.springer.com/10.1007/978-1-4939-2214-7_13
- Kossen K, Uhlenbeck OC. Cloning and biochemical characterization of Bacillus subtilis YxiN, a DEAD protein specifically activated by 23S rRNA: delineation of a novel sub-family of bacterial DEAD proteins. Nucleic Acids Res. 1999;27:3811–3820.
- Giraud C, Hausmann S, Lemeille S, et al. The C-terminal region of the RNA helicase CshA is required for the interaction with the degradosome and turnover of bulk RNA in the opportunistic pathogen Staphylococcus aureus. RNA Biol. 2015;12:658–674.
- Zhao C, Hartke A, La Sorda M, et al. Role of methionine sulfoxide reductases A and B of Enterococcus faecalis in oxidative stress and virulence. Infect Immun. 2010;78:3889–3897.
- Paulsen IT, Banerjei L, Myers GSA, et al. Role of mobile DNA in the evolution of vancomycin-resistant Enterococcus faecalis. Science. 2003;299:2071–2074.
- Bae T, Kozlowicz B, Dunny GM. Two targets in pCF10 DNA for PrgX binding: their role in production of Qa and prgX mRNA and in regulation of pheromone-inducible conjugation11. J Mol Biol. 2002;315:995–1007.
- Arnaud M, Chastanet A, Débarbouillé M. New vector for efficient allelic replacement in naturally nontransformable, low-GC-content, gram-positive bacteria. Appl Environ Microbiol. 2004;70:6887–6891.
- FMD A, Joyet P, Deutscher J, et al. Mutational analysis of glucose transport regulation and glucose-mediated virulence gene repression in Listeria monocytogenes. Mol Microbiol. 2011;81:274–293.
- Platteeuw C, Simons G, de Vos WM. Use of the Escherichia coli beta-glucuronidase (gusA) gene as a reporter gene for analyzing promoters in lactic acid bacteria. Appl Environ Microbiol. 1994;60:587–593.
- Kram KE, Finkel SE, Kelly RM. Rich medium composition affects Escherichia coli survival, glycation, and mutation frequency during long-term batch culture. Appl Environ Microbiol. 2015;81:4442–4450.
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254.
- Gaffarogullari EC, Krause A, Balbo J, et al. Microscale thermophoresis provides insights into mechanism and thermodynamics of ribozyme catalysis. RNA Biol. 2013;10:1815–1821.
- Benachour A, Ladjouzi R, Le Jeune A, et al. The lysozyme-induced peptidoglycan N-acetylglucosamine deacetylase PgdA (EF1843) is required for Enterococcus faecalis virulence. J Bacteriol. 2012;194:6066–6073.