
ABSTRACT
In eukaryotes, the beak structure of 40S subunits is formed by the protrusion of the 18S rRNA helix 33 and three ribosomal proteins: eS10, eS12 and eS31. The exact role of these proteins in ribosome biogenesis is not well understood. While eS10 is an essential protein encoded by two paralogous genes in Saccharomyces cerevisiae, eS12 and eS31 are not essential proteins encoded by the single-copy genes RPS12 and UBI3, respectively. Here, we have analysed the contribution of yeast eS12 to ribosome biogenesis and compared it with that of eS31. Polysome analysis reveals that deletion of either RPS12 or UBI3 results in equivalent 40S deficits. Analysis of pre-rRNA processing indicates that eS12, akin to eS31, is required for efficient processing of 20S pre-rRNA to mature 18S rRNA. Moreover, we show that the 20S pre-rRNA accumulates within cytoplasmic pre-40S particles, as deduced from FISH experiments and the lack of nuclear retention of 40S subunit reporter proteins, in rps12∆ and ubi3∆ cells. However, these particles containing 20S pre-rRNA are not efficiently incorporated into polyribosomes. We also provide evidence for a genetic interaction between eS12 or eS31 and the late-acting 40S assembly factors Enp1 and Ltv1, which appears not to be linked to the dynamics of their association with or release from pre-40S particles in the absence of either eS12 or eS31. Finally, we show that eS12- and eS31-deficient ribosomes exhibit increased levels of translational misreading. Altogether, our data highlight distinct important roles of the beak region during ribosome assembly and function.
Introduction
In eukaryotes, ribosome biogenesis is a complex process that, in addition to the ribosomal RNAs (rRNAs) and ribosomal proteins (r-proteins), requires a plethora of RNA and protein trans-acting factors [Citation1]. Most of our current knowledge concerning this process comes from studies in the yeast Saccharomyces cerevisiae, where about 80 small nucleolar RNAs (snoRNAs) and about 300 protein trans-acting factors have been characterized [Citation2]. However, the precise function of many of these factors, also known as ribosome assembly factors, is only beginning to emerge owing to the recent advances in cryogenic electron microscopy (cryo-EM), which has enabled the structural characterization of distinct pre-ribosomal particles at near-atomic resolution (for a review, see ref. [Citation3]). Different studies in yeast and humans indicate that r-proteins also play active roles in the process of ribosome assembly (for a review, see ref. [Citation4]). Understanding the exact function of r-proteins in ribosome assembly is a very pertinent issue as some of the human diseases classified as ribosomopathies are linked to ribosome defects caused by loss-of-function mutations in r-protein genes (for reviews, see ref. [Citation5–Citation7]). The most common of these pathologies is Diamond-Blackfan anemia (DBA), an inherited and congenital disease linked to recessive mutations in r-protein genes; it has been established that approximately 60% of the DBA patients bear a mutated allele of one of the following genes RPS19, RPS24, RPS17, RPS7, RPS10 and RPS26 encoding eS19, eS24, eS17, eS7, eS10 and eS26 r-proteins, respectively, from the small (40S) r-subunit (e.g. refs [Citation6,Citation7].).
The structure of ribosomes has been resolved at atomic resolution (see [Citation8] and references therein). The structure of 40S r-subunits is less complex than that of large (60S) r-subunits. The 18S rRNA and its equivalent prokaryotic 16S rRNA can be divided into four, practically independently folding domains, which upon the interaction of specific r-proteins form the typical morphological features of the 40S r-subunit [Citation8,Citation9]: the 5ʹ domain of 18S rRNA forms the shoulder and the foot and, together with the 3ʹ minor domain (helices h44 and h45), constitutes the body of the 40S r-subunit; the central domain of 18S rRNA forms the platform and the 3ʹ major domain forms the head. Protruding from the head, the universally conserved region referred to as the beak is special in eukaryotes since it has been transformed during evolution from the all-rRNA structure (helix h33) found in bacterial 30S r-subunits to a protuberance of essentially the same structure but containing a reduced h33 and three r-proteins (eS10, eS12 and eS31) [Citation9,Citation10] (see below).
The role during ribosome biogenesis of most, but not all, r-proteins from the 40S r-subunit has been examined by either specific studies or systematic analyses (e.g. refs. [Citation11–Citation17]). These analyses revealed that 40S r-subunit assembly occurs co-transcriptionally and hierarchically in the 5ʹ-to-3ʹ direction with respect to the nascent rRNA; thus, in general, r-proteins that bind to the body of the 40S r-subunit appear to bind early during transcription of the 35S precursor rRNA (pre-rRNA) while most of those from the platform assemble slightly later but before many of those from the head, which are the latest r-proteins that bind, still in the nucleus, to pre-40S r-particles. Few 40S r-proteins seem to assemble in the cytoplasm, among them Asc1, eS10 and perhaps eS26, which belong to the head of the 40S r-subunit (see refs. [Citation11–Citation13]). Remarkably, the order in which r-proteins assemble with yeast and mammalian 40S r-subunits seems to parallel the incorporation of their bacterial counterparts in vivo and importantly in vitro (see, ref. [Citation18]); thus, the essential information for the assembly of small r-subunits seems to intrinsically reside in the biophysical properties of the different interactions that the r-proteins could undergo with the 16S/18S rRNA (see ref. [Citation19]). Functional analyses have nicely demonstrated that, in general, the timing of action in pre-rRNA processing of the r-proteins from the 40S r-subunit correlates with their specific location within the subunits and, hence, with their specific course of assembly [Citation11–Citation13]. In this sense, loss-of-function of the r-proteins located in the body region of 40S r-subunits (as examples, eS6, eS8, eS24, uS4, uS17) strongly impairs processing of early pre-rRNAs (35S pre-rRNA in yeast, 47S pre-rRNA in human cells; for a scheme of pre-rRNA processing in yeast see Figure S1), whereas loss-of-function of r-proteins located in the head (as examples, eS31, uS2, uS3, uS5, uS10, uS19) impairs downstream maturation steps, such as the nucleo-cytoplasmic export of pre-40S r-particles and processing of late pre-rRNAs (20S pre-rRNA in yeast, 21S and 18S-E pre-rRNA in human cells) [Citation4,Citation11–Citation13]. Importantly, the recent publications of diverse cryo-EM structures of yeast and human 90S and pre-40S r-particles is practically fully in agreement with this hierarchical assembly pathway of early versus late 40S r-proteins following 18S rRNA maturation in a 5ʹ-to 3ʹ direction (see [Citation3,Citation20–Citation22] and references therein). However, surprisingly, the beak region of the head seems to be practically but not totally moulded in the early 90S pre-ribosomal particles as deduced from cryo-EM studies; thus, the apical structure formed by eS12, eS31 and helix h33 is already shaped whereas its base, built in mature 40S r-subunits by eS10, is occupied by the trans-acting factor Enp1 in these particles (see e.g. ref. [Citation23]). The subsequent nuclear maturation of 90S into pre-40S r-particles comprises the release of most trans-acting factors, with few exceptions such as Dim1, Enp1 and Pno1, and also the association of others factors, among them Tsr1, Nob1, Rio2 or Ltv1 (e.g. refs. [Citation24–Citation27]). The release of Ltv1 and Enp1, which directly interact [Citation24,Citation28–Citation30], from cytoplasmic pre-40S r-particles then remodels the head region of 40S r-subunits to relocate the r-protein uS3, thus permitting the assembly of eS10 and Asc1 (e.g. refs. [Citation24,Citation31–Citation33]).
We are interested in understanding the contribution of the r-proteins of the beak to ribosome biogenesis and function. We and others have previously shown that deletion of UBI3, the gene encoding eS31 as a precursor protein fused to a single ubiquitin moiety, strongly impairs cytoplasmic 20S pre-rRNA processing in S. cerevisiae [Citation11,Citation17,Citation34]. Here, we have undertaken the functional characterization of the yeast r-protein eS12 in the biogenesis and function of ribosomes. Our results indicate that eS12 is a quasi-essential protein for cell growth required for the normal accumulation of 40S r-subunits. Deletion of RPS12 leads to a similar cytoplasmic accumulation of 20S pre-rRNA as the deletion of UBI3; this accumulation is not enhanced by the simultaneous deletion of both genes. Moreover, we reveal a genetic interaction between the absence of either eS12 or eS31 and that of Ltv1 or a mutant allele of ENP1, which is not apparently linked to the dynamics of incorporation into or release from the pre-40S r-particles of these trans-acting factors. Finally, we also show that the eS12-deprived ribosomes are more error-prone than wild-type ones.
Results
Loss of eS12 results in a severe growth defect
The r-protein eS12 is a eukaryote-specific constituent of 40S r-subunits [Citation35]. Unlike most yeast r-proteins [Citation1], eS12 is encoded by a non-essential single-copy gene (RPS12, YOR369C) [Citation36]. This gene produces a small globular protein of ca. 16 kDa located in the beak of the 40S r-subunit, which interacts with helix h33 of the 18S rRNA, the eukaryote-specific r-protein eS10 and the archaea- and eukaryote-specific r-protein eS31 [Citation9]. The role of yeast r-proteins eS10 and eS31 in ribosome biogenesis has been previously studied [Citation11,Citation17,Citation34,Citation37]. Seemingly, the role of yeast eS12 in ribosome biogenesis has also been earlier explored, with the authors concluding that it was not required for the formation of 40S r-subunits [Citation11]. As we discuss below, this conclusion was unfortunately erroneous as, most likely, the authors inadvertently used a non-appropriate strain.
To study the role of yeast eS12 in ribosome biogenesis and function, we first analysed the phenotypic consequences of deleting the RPS12 gene. In contrast to the rps12∆ strain employed by Ferreira-Cerca et al., which grew identically to the wild-type strain at 30°C on YPD plates [Citation11], the haploid rps12∆ strains we generated either in the W303 or the BY4743 genetic background displayed a severe slow-growth (sg) defect at 30°C on YPD plates that was enhanced at 22 and 37°C ( and Figure S2). In liquid YPD at 30°C, the rps12∆ strains doubled each ca. 7 h while their respective isogenic wild-type counterparts did each ca. 1.8 h. Our results were, however, consistent with previous ones by Steffen et al., who also observed a severe sg phenotype for haploid rps12∆ segregants upon sporulation of a heterozygous RPS12/rps12∆ diploid in the BY4743 background [Citation38]. Thus, we conclude that Ferreira-Cerca et al. have likely analysed a spontaneous fast-growing bypass suppressor of the rps12∆ mutant (see Discussion).
Figure 1. Deletion of RPS12 results in a severe slow-growth phenotype. Growth analysis of the rps12 null mutant in the W303 background. Cells of the strains W303-1A (Wild type), SMY315 (rps12∆), TLY14.3C (ubi3∆) and SMY372 (rps12∆ ubi3∆) were spotted in 10-fold serial dilutions onto YPD plates and incubated at 22°C for 5 days and at 30 and 37°C for 3 days.

The r-protein eS12 is a close neighbour of eS31 in the 40S r-subunit [Citation9]. We and others have shown that a mutant strain lacking eS31 displayed a strong sg defect and a cold-sensitive (cs) phenotype [Citation17,Citation34]. To check whether the simultaneous absence of eS12 and eS31 have an enhanced growth defect, we constructed a rps12∆ ubi3∆ double deletion strain in the W303 background. As shown in , this double mutant displayed growth defects very similar to that of the single rps12∆ mutant. Thus, the rps12∆ mutation seem to be epistatic to the ubi3∆ allele.
Loss of eS12 leads to a strong 40S r-subunit shortage
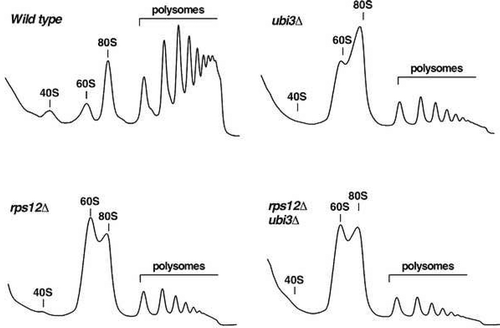
To study the contribution of yeast eS12 to ribosome biogenesis, we first analysed the polysome profile of the rps12∆ mutant and compared it to those of isogenic ubi3∆ and wild-type strains. As shown in , polysome profile analysis using cell extracts of the rps12∆ mutant revealed a clear reduction in free 40S r-subunits and a huge increase in free 60S r-subunits; the polysomal content was also drastically reduced. The profile was similar to those of the isogenic ubi3∆ single and the ubi3∆ rps12∆ double mutants (). Thus, the rps12∆ deletion alone or in combination with the ubi3∆ mutation leads to a shortage of 40S r-subunits. In agreement, quantification of total r-subunits by low-Mg2+ sucrose gradients showed that deletion of either eS12 or eS31 leads to a ca. 55% specific reduction of 40S with respect to 60S r-subunits compared to the wild-type control. This reduction was of about 65% for the double rps12∆ ubi3∆ mutant (Figure S3). We conclude that the absence of eS12 r-protein results in a severe deficiency in 40S relative to 60S r-subunits, which is slightly more pronounced when eS31 r-protein is simultaneously lacking.
Figure 2. Deletion of RPS12 results in a deficit in 40S r-subunits. The strains W303-1A (Wild type), SMY315 (rps12∆), TLY14.3C (ubi3∆) and SMY372 (rps12∆ ubi3∆) were grown in YPD medium at 30°C to an OD600 of around 0.8 and cell extracts were prepared. Ten A260 units of each extract were subjected to polysome analysis in 7–50% sucrose gradients. After centrifugation, the A254 values along each gradient were continuously recorded by an on-line spectrophotometer. Sedimentation is from left to right. The peaks of free 40S and 60S r-subunits, 80S free couples/monosomes and polysomes are indicated.
Deletion of RPS12 impairs rRNA processing
To determine whether the rps12∆ mutant was defective in the formation rather than the stability of 40S r-subunits, we examined the kinetics of rRNA production by pulse-chase analyses with the rps12∆ strain and its isogenic wild-type counterpart. Both strains were first transformed with an empty YCplac33 plasmid (CEN, URA3, Table S2) to make them prototrophic for uracil. Then, they were grown in liquid SD-Ura medium to mid-log phase at 30°C. The cells were pulse-labelled for 2 min with [5,6-3H]uracil, then chased for 5, 15, 30 and 60 min with a large excess of non-radioactive uracil. Total RNA was extracted from each sample and analysed by agarose and acrylamide gel electrophoresis, followed by transfer to nylon membranes and exposition to tritium sensitive phosphor screens. As shown in , in wild-type cells, the early pre-rRNA precursors were converted rapidly into 27S and 20S species, which were further processed into mature 25S and 18S rRNA, respectively. After 15 min of chase, practically all label was in the mature rRNAs. Compared to the wild-type control, processing of 27S pre-rRNAs was delayed in the rps12∆ mutant since these precursors could still be detected after the 30 min chase time point; consistently, practically no labelled mature 25S rRNA was detected at early 5 min or 15 min chase time points (). Similarly, a delayed conversion of 7S pre-rRNAs into mature 5.8S rRNAs was detected (). More significantly, synthesis of 18S rRNA was strongly inhibited since the levels of the 20S pre-rRNA remained unchanged up to the 30 min time point of chase and almost no mature 18S rRNA was produced. Only after 60 min of chase, the amounts of the 20S pre-rRNA were clearly reduced and some mature 18S rRNA could be detected, suggesting that either the 40S particles containing the immature 20S pre-rRNA or the produced mature 40S r-subunits are instable and turned over.
Figure 3. Deletion of RPS12 leads to delayed conversion of the 20S pre-rRNA to mature 18S rRNA. The strains W303-1A (Wild type) and SMY315 (rps12∆) were transformed with the YCplac33 plasmid and then grown in SD-Ura to an OD600 of around 0.8 at 30°C. Cells were pulse-labelled for 2 min with [5,6-3H]uracil and then chased for 5, 15, 30 and 60 min with an excess of unlabelled uracil. Total RNA was extracted and 3,000 cpm per sample were loaded and separated on (A) a 1.2% agarose-formaldehyde gel or (B) a 7% polyacrylamide-8 M urea gel, transferred to nylon membranes, exposed to a tritium sensitive phosphor screen and analysed by phosphorimager scanning. The position of the different pre-rRNAs and mature rRNAs is indicated.
![Figure 3. Deletion of RPS12 leads to delayed conversion of the 20S pre-rRNA to mature 18S rRNA. The strains W303-1A (Wild type) and SMY315 (rps12∆) were transformed with the YCplac33 plasmid and then grown in SD-Ura to an OD600 of around 0.8 at 30°C. Cells were pulse-labelled for 2 min with [5,6-3H]uracil and then chased for 5, 15, 30 and 60 min with an excess of unlabelled uracil. Total RNA was extracted and 3,000 cpm per sample were loaded and separated on (A) a 1.2% agarose-formaldehyde gel or (B) a 7% polyacrylamide-8 M urea gel, transferred to nylon membranes, exposed to a tritium sensitive phosphor screen and analysed by phosphorimager scanning. The position of the different pre-rRNAs and mature rRNAs is indicated.](/cms/asset/b8e48f79-e69f-4364-9368-21b330bc099f/krnb_a_1767951_f0003_b.gif)
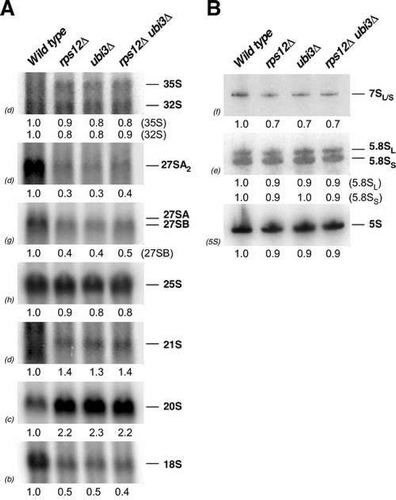
To investigate in more detail the pre-rRNA processing defects occurring in the absence of eS12, we analysed the steady-state levels of pre- and mature rRNAs by northern hybridization in the rps12∆ mutant and compared them to those detected in ubi3∆ and double rps12∆ ubi3∆ strains. As shown in , the single rps12∆ and ubi3∆ strains and the double rps12∆ ubi3∆ mutant exhibited practically identical pre-rRNA processing defects. Thus, none of these strains showed significant differences in the levels of 35S and 32S pre-rRNAs compared to the wild-type strain. However, some delay in the early pre-rRNA processing reactions might occur as an aberrant 21S pre-rRNA, which extends from site A1 to A3, slightly accumulated in the three mutants. Moreover, the 27S and 7S pre-rRNAs clearly decreased, however, levels of mature 25S and 5.8S rRNAs only slightly decreased. In agreement with the delayed conversion of 20S pre-rRNA to 18S rRNA seen in pulse-chase analyses, steady-state levels of 20S pre-rRNA visibly accumulated in the rps12∆ strain, in a similar manner as in the ubi3∆ and rps12∆ ubi3∆ mutants (); as a consequence, levels of mature 18S rRNA significantly decreased. Altogether, our results indicate that the absence of eS12 leads to a general delay in pre-rRNA maturation that is much more pronounced for the 20S pre-rRNA conversion into mature 18S rRNA, which likely accounts for the deficit in 40S r-subunits detected in rps12∆ cells. These pre-RNA processing defects are quite similar to those observed in the ubi3∆ strain and apparently no further enhanced by the simultaneous absence of eS31.
Figure 4. Absence of eS12 leads to robust accumulation of 20S pre-rRNA and concomitant reduction in the steady-state levels of mature 18S rRNA. The strains W303-1A (Wild type), SMY315 (rps12∆), TLY14.3C (ubi3∆) and SMY372 (rps12∆ ubi3∆) were grown in YPD medium at 30°C to an OD600 of around 0.8. Total RNA was extracted from each strain; equal amounts of RNA (5 µg) were analysed by northern blot. (A) Northern hybridisation of high-molecular-mass pre- and mature rRNAs. (B) Northern blot analysis of low-molecular-mass pre- and mature rRNAs. Signal intensities were measured by phosphorimager scanning (indicated below each panel) and normalised to those obtained for the wild-type, arbitrarily set to 1.0. Probes, between parentheses, are described in Figure S1 and Table S3.
The rps12∆ deletion mutant accumulates the 20S pre-rRNA in the cytoplasm
Nuclear pre-40S r-particles are rapidly released through the nuclear pore complexes to the cytoplasm, where processing of 20S pre-rRNA to mature 18S rRNA occurs [Citation39,Citation40]. Therefore, a defect in 20S pre-rRNA processing might result from either reduced nuclear export of pre-40S r-particles or impaired cleavage of cytoplasmic 20S pre-rRNA. To assess this issue, we analysed the subcellular localization of a GFP-tagged version of the uS3 r-protein, which assembles in the nucleus [Citation32,Citation41], in wild-type, rps12∆ and ubi3∆ cells. As shown in Figure S4, both uS3-GFP and the 60S r-subunit reporter uL23-GFP, which also assembles in the nucleus (e.g. ref. [Citation42]), were cytoplasmic in the wild-type and mutant rps12∆ and ubi3∆ strains. We also analysed the localisation of GFP-tagged versions of eS10 and eS31, which are close neighbours of eS12 at the beak [Citation9]. While eS31 has been shown to assemble in the nucle(ol)us [Citation23,Citation37], eS10 has been suggested to predominantly assemble late in the cytoplasm [Citation26,Citation31]. As shown in Figure S5, no nuclear accumulation of these two reporters could either be observed in the rps12∆ strain.
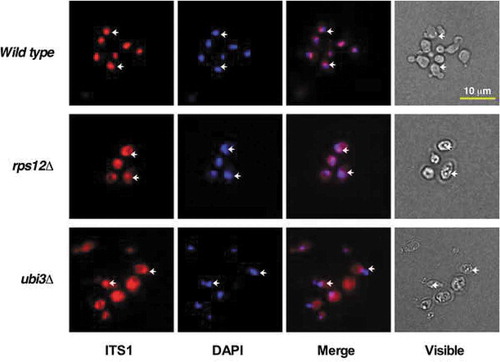
We also visualised the 20S pre-rRNA and its precursors (35S, 33S and 32S pre-rRNAs) by fluorescent in situ hybridisation (FISH) in the rps12∆ and ubi3∆ strains using a probe complementary to positions within the D-A2 region of ITS1. As shown in , a mainly nucleolar FISH signal was detected in wild-type cells, as expected since 35S, 33S and 32S pre-rRNAs are nucleolar and cytoplasmic 20S pre-rRNA is very rapidly processed to mature 18S rRNA in the cytoplasm. However, the FISH signal turned out to be also abundant in the cytoplasm in the rps12∆ and ubi3∆ strains; thus, suggesting that 20S pre-rRNA is exported in these mutants but inefficiently processed. Altogether, these results reveal that export of pre-40S r-particles from the nucleus is neither blocked in the rps12∆ nor ubi3∆ mutant. Therefore, the increased levels of 20S pre-rRNA detected in rps12∆ and ubi3∆ mutant cells are the result of an impairment of cytoplasmic 20S pre-rRNA processing.
Figure 5. The 20S pre-rRNA accumulates in the cytoplasm of rps12∆ and ubi3∆ cells. The strains W303-1A (Wild type), SMY315 (rps12∆) and TLY14.3C (ubi3∆) were grown to mid-log phase in YPD medium at 30°C. Cells were fixed with formaldehyde, spheroblasted and subjected to FISH using a Cy3-labelled probe complementary to a sequence within the D-A2 segment of ITS1, which hybridises to the 35S, 33S, 32S and 20S pre-rRNAs (Table S3). DAPI staining visualises mostly the nucleoplasm. Arrows point to the nucleolus revealed by the partial overlapping with the DAPI staining. The visible pictures (bright field microscopy) are also shown. Scale bar, 10 µm.
The accumulated 20S pre-rRNA is not efficiently incorporated into translating ribosomes in rps12∆ cells
It has been previously reported that, in some mutants, pre-40S r-particles containing the 20S pre-rRNA could be efficiently incorporated into translating ribosomes (e.g. refs [Citation17,Citation43,Citation44]). In contrast, in wild-type cells and most mutants accumulating 20S pre-rRNA, pre-40S r-particles were only found to peak together with free 40S r-subunits or 80S ribosomes (e.g. refs. [Citation14,Citation45–Citation48]).
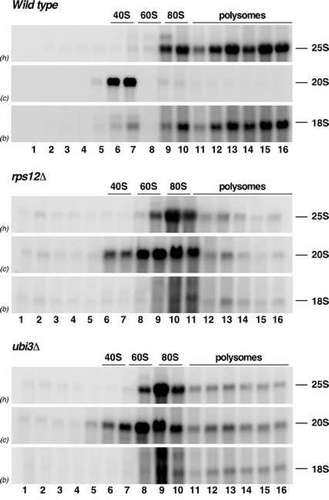
To examine whether the pre-40S r-particles containing the 20S pre-rRNA that accumulate in rps12∆ cells could engage in translation, we subjected cell extracts of the rps12∆ strain to sucrose gradient centrifugation and subsequently analysed the distribution of the 20S pre-rRNA in the different fractions of the gradients by northern blotting. The results were compared to those obtained with wild-type and ubi3∆ cells. As expected, the bulk of 20S pre-rRNA co-migrated basically with the free 40S peak in wild-type cells ( and Figure S6). However, in rps12∆ and ubi3∆ cells, 20S pre-rRNA was predominantly found in the 80S peak, although its maximum was shifted by one fraction to the left of the maximum of 25S/18S in this peak ( and Figure S6). Some portion of 20S pre-rRNA was also contained in the polysomal fractions of the gradients in rps12∆ and ubi3∆ cells. Given that there is a reduced polysome content in these two mutants, the 20S pre-rRNA only accumulated at similarly low levels as the mature 25S and 18S rRNAs in these fractions (see ). Indeed, quantification of the signal intensities of 20S pre-rRNA and the mature 25S and 18S rRNAs confirmed the similar distribution of these three rRNA species in the gradients corresponding to rps12∆ and ubi3∆ cells, but not in those of isogenic wild-type cells (Figure S6). These data suggest that pre-40S r-particles lacking either eS12 or eS31 can form 80S ribosomes but only inefficiently engage in translation.
Figure 6. The absence of either eS12 or eS31 alters the sedimentation behaviour of pre-ribosomal particles containing the 20S pre-rRNA. The strains W303-1A (Wild type), SMY315 (rps12∆) and TLY14.3C (ubi3∆) were grown to mid-log phase in YPD medium at 30°C. Cell extracts were prepared and 10 A260 units of each extract were resolved in 7–50% sucrose gradients and fractionated. RNA was extracted from each fraction and analysed by northern blotting using the indicated probes (between parentheses; see Figure S1 and Table S3). The position of free 40S and 60S r-subunits, 80S ribosomes and polysomes are shown.
Functional interaction between trans-acting factors Enp1 and Ltv1 and the eS12 and eS31 r-proteins
Cryo-EM analyses have shown that the trans-acting factors Enp1 and Ltv1 are present in cytoplasmic pre-40S r-particles at a position connecting the beak to the head structure [Citation26,Citation29,Citation30,Citation49]. Moreover, the release of these two factors (at least that of Ltv1) from cytoplasmic pre-40S r-particles upon their phosphorylation by the kinase Hrr25 [Citation24] seems to induce a conformational rearrangement of the beak/head area, which allows the stable positioning of uS3 [Citation32,Citation33] and the efficient recruitment of eS10, uS10, uS14 and RACK1 to the 40S head [Citation31]. Given that Enp1 already associates with 90S pre-ribosomal particles, which apparently also contain eS12 and eS31 [Citation23], and that Ltv1 only binds later to nucleoplasmic pre-40S r-particles [Citation25,Citation27,Citation50], we found it particularly pertinent to examine the functional relationship between the r-proteins eS12 and eS31 and the trans-acting factors Enp1 and Ltv1.
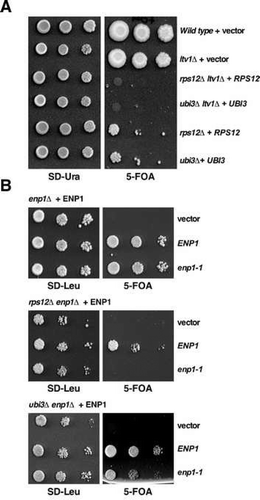
We first evaluated whether the growth defects caused by the absence of eS12 or eS31 could be enhanced by the absence of Ltv1 or the partial loss of function of Enp1. It has been reported that the ltv1∆ null allele leads to a sg phenotype for growth at 30°C (see ) and significantly impairs 40S r-subunit synthesis due mainly to defects in 20S pre-rRNA maturation but also to a slight export defect of pre-40S r-particles from the nucleus to the cytoplasm [Citation44,Citation50,Citation51]. In turn, the enp1-1 mutation confers a temperature sensitive (ts) growth phenotype at 37°C, which is linked to a severe 40S r-subunit deficiency due to both the inhibition of pre-rRNA processing at the early A0-A2 cleavage sites and the block of nucleocytoplasmic 40S r-subunit export [Citation25,Citation52]; however, at 30°C, the growth of enp1-1 cells was apparently comparable to that of an isogenic wild-type strain (see ). As shown in , the ltv1∆ mutation confers synthetic lethality when combined with either the rps12∆ or the ubi3∆ allele. Moreover, as shown in , the combination of the enp1-1 allele with the rps12∆ or the ubi3∆ null allele resulted in a synthetically lethal or enhanced phenotype, respectively. As the individual growth defect of rps12∆ and ubi3∆ mutants is very severe, these genetic interactions could be just due to an additive effect of the ltv1∆ and the enp1-1 mutations. Thus, we also evaluated whether mild nob1 alleles could exacerbate the growth defect of the rps12∆ allele. As shown in Figure S7, none of the combinations tested showed synthetic enhancement of the rps12∆ growth phenotype. These findings suggest an intimate functional interaction between the trans-acting factors Ltv1 and Enp1 and the r-proteins eS12 and eS31 during 40S r-subunit biogenesis.
Figure 7. Synthetic lethality or enhancement of the slow-growth phenotype of the rps12∆ and ubi3∆ mutants by the ltv1∆ and the enp1-1 alleles. (A) The strains W303-1A (Wild type) and SMY307 (ltv1∆), harbouring an empty YCplac33 plasmid (+ vector) and the strains SMY352 (rps12∆ ltv1∆), SMY345 (ubi3∆ ltv1∆), SMY315 (rps12∆) and TLY14.3C (ubi3∆) harbouring the rps12 or ubi3 null alleles complemented by a YCplac33-RPS12 (+ RPS12) or YCplac33-UBI3 (+ UBI3), respectively, were grown in complete SD liquid medium and then spotted in 10-fold serial dilutions onto SD-Ura and SD containing 5-FOA plates. Plates were incubated 3 and 10 days at 30°C, respectively. (B) Strains SMY310 (enp1∆), SMY431 (rps12∆ enp1∆) and SMY439 (ubi3∆ enp1∆), which harbour the enp1 null allele complemented by a pRS316-ENP1 (+ ENP1) were transformed with an empty pRS415 vector (vector) or pRS315 plasmids that carry either the wild-type ENP1 (ENP1) or the mutant enp1-1 (enp1-1) allele. Then, cells were grown in SD-Leu liquid medium and spotted in 10-fold serial dilution steps onto SD-Leu or SD containing 5-FOA plates. Plates were incubated 3 and 5 days at 30°C, respectively.
Second, we studied whether the dynamics of Enp1 and Ltv1 association with or release from pre-ribosomal particles changed in the absence of either eS12 or eS31. To do so, we analysed the localization of GFP-tagged reporters of Enp1 and Ltv1, expressed from genomic loci, in the rps12∆ and ubi3∆ mutants and compared this to their localization in an isogenic wild-type control. Consistent with previous reports, we found that while Enp1-GFP has a nucleolar steady-state localization in wild-type cells (Figure S8; see ref. [Citation25,Citation27,Citation52]), Ltv1-GFP had a cytoplasmic localization (Figure S8; see refs. [Citation27,Citation32,Citation50]). We also made use of a Ltv1∆NES-GFP reporter, which, in agreement with previous findings [Citation32,Citation53], localized to the nucleus of wild-type cells (Figure S8). As clearly also shown in Figure S8, absence of eS12 or eS31 did not noticeably modify the subcellular localization of Enp1 or Ltv1. Altogether, these results indicate a functional interaction between the trans-acting factors Enp1 and Ltv1 and the r-proteins eS12 and eS31 in the process of 40S r-subunit biogenesis that apparently does not involve changes in the dynamics of association and release of either Enp1 or Ltv1 in the absence of these two r-proteins from the beak structure.
The absence of eS12 or eS31 induces an increase in translational misincorporation
Numerous examples of mutations in yeast r-proteins and translation factors causing a reduction in the accuracy of translation have been previously reported (e.g. [Citation54–Citation60]). To assess whether ribosomes lacking either eS12 or eS31 are defective in the accuracy of translation, we first compared the efficiency of stop-codon recognition in isogenic wild type and rps12∆ and ubi3∆ mutant cells. To do so, we made use of a plasmid-borne tandem Renilla and firefly luciferase reporter system [Citation54]. In this system, the two types of luciferase genes are separated by an in-frame linker sequence in the control plasmid, resulting in the synthesis of both enzymes as a single polypeptide chain. In the readthrough reporter, there is a single in-frame stop codon within the linker sequence (); therefore, in this reporter, the activity of firefly luciferase, encoded by the distal open reading frame (ORF), results from translation beyond the stop-codon as the result of a readthrough event. The activity of Renilla luciferase, encoded by the proximal ORF, serves as an internal control for mRNA abundance and efficiency of translation initiation as the translation of both luciferase enzymes is initiated from the same start codon (for details, see refs. [Citation54,Citation61]). As shown in , no significant differences were observed in the stop-codon suppression when the rps12∆ and ubi3∆ strains were compared to the isogenic counterpart. Next, we assessed the extent of misincorporation by making use of an equivalent misreading reporter system. In this system, the CAC codon coding for a His residue at position 245, which is instrumental for the enzymatic activity of the firefly luciferase, has been substituted by a CGC codon coding for an Arg residue, thus resulting in a protein with only residual luciferase activity [Citation54]. Therefore, any increased activity of firefly luciferase results from a misreading event that allow misincorporation of a non-deleterious amino acid (normally His via His-tRNA(GUG)His) at the mutated CGC codon [Citation54]. Interestingly, as also shown in , a significant increase in misreading was observed in cells lacking eS12; notably, this effect was even more pronounced in the absence of eS31. These results suggest that eS12 and eS31 are involved in the fidelity of translation elongation, thus ribosomes lacking either eS12 or eS31 are error-prone and lead to an increase in the misreading rate.
Figure 8. Effects of RPS12 or UBI3 deletion on readthrough and misreading during translation. (A) Scheme of the dual luciferase readthrough and misincorporation reporter systems. (B) Measurement of the readthrough and misincorporation frequencies in yeast strains lacking eS12 or eS31. Strains W303-1A (Wild type), SMY315 (rps12∆) and TLY14.3C (ubi3∆) were transformed with plasmids pDB720 or pDB721 (UAG stop codon), pDB722 or pDB723 (UAA stop codon), pDB690 or pDB691 (UGA stop codon) to measure readthrough or with pDB688 and pDB868 to measure misreading (Arg245(GGC) to His245(CAC)). Transformants were grown in SD-Ura to mid-log phase at 30°C and then Renilla and firefly luciferase activity was measured. Assays were done in quadruplicate, and the data were expressed as the mean ± the standard deviation. The percentage of readthrough in each strain was expressed as the firefly/Renilla luciferase activity (nonsense) divided by the firefly/Renilla luciferase activity (sense) multiplied by 100. The percentage of misreading was expressed as the firefly (H245R)/Renilla luciferase activity divided by the firefly (wild type)/Renilla luciferase activity multiplied by 100. For further details see [Citation54,Citation61]. Significance levels were determined by Student’s t test (*, p < 0.05).
![Figure 8. Effects of RPS12 or UBI3 deletion on readthrough and misreading during translation. (A) Scheme of the dual luciferase readthrough and misincorporation reporter systems. (B) Measurement of the readthrough and misincorporation frequencies in yeast strains lacking eS12 or eS31. Strains W303-1A (Wild type), SMY315 (rps12∆) and TLY14.3C (ubi3∆) were transformed with plasmids pDB720 or pDB721 (UAG stop codon), pDB722 or pDB723 (UAA stop codon), pDB690 or pDB691 (UGA stop codon) to measure readthrough or with pDB688 and pDB868 to measure misreading (Arg245(GGC) to His245(CAC)). Transformants were grown in SD-Ura to mid-log phase at 30°C and then Renilla and firefly luciferase activity was measured. Assays were done in quadruplicate, and the data were expressed as the mean ± the standard deviation. The percentage of readthrough in each strain was expressed as the firefly/Renilla luciferase activity (nonsense) divided by the firefly/Renilla luciferase activity (sense) multiplied by 100. The percentage of misreading was expressed as the firefly (H245R)/Renilla luciferase activity divided by the firefly (wild type)/Renilla luciferase activity multiplied by 100. For further details see [Citation54,Citation61]. Significance levels were determined by Student’s t test (*, p < 0.05).](/cms/asset/7b7b70ce-4dea-477e-b8ed-c68985f7a44e/krnb_a_1767951_f0008_b.gif)
Discussion
In this work, we have dissected the contribution of beak r-proteins to ribosome biogenesis and function and demonstrated that the yeast eS12 r-protein has major roles in cell growth, 40S r-subunit biogenesis and translation. Our findings were independent of the yeast genetic background used (Figures S2 and S9). However, in an outstanding publication, Ferreira-Cerca et al. had previously described that eS12, although needed for efficient production of both 18S and 25S rRNAs, did not significantly participate in the formation of cytoplasmic ribosomes [Citation11]; the rps12∆ strain used in that study, which was directly obtained from the Euroscarf collection, appeared to grow just as well as the wild-type counterpart and showed only a significant growth reduction when cultivated in the presence of a sublethal dose of cycloheximide [Citation11]. In contrast, the rps12∆ strains used in our study, as well as the one reported by Steffen et al. [Citation38], exhibited a strongly impaired growth, which is fully restored upon complementation with a wild-type RPS12 allele from a centromeric plasmid. This discrepancy is likely due to the appearance of an undesired spontaneous suppressor of the growth defect in the clone of the Euroscarf collection, a phenomenon that is very common among haploid r-protein gene deletion strains [Citation38,Citation62] (see also ref. [Citation63]). The future identification of such suppressor mutation(s) might provide further insights into the functional environment of eS12, however only if being a true extragenic mutation, since often the suppression event is not informative as it arises from a duplication of the gene to be disrupted in a different locus by some unknown mechanism (our unpublished results, see also ref. [Citation64]).
Polysome profile analysis and examination of pre-rRNA processing by pulse-chase and northern blotting experiments in the rps12∆ strain showed unequivocally that eS12 is required for efficient maturation of 40S r-subunits. Specifically, the absence of eS12 caused a modest delay in pre-rRNA processing at site A2, thereby affecting the levels of 27SA2 pre-rRNA and its further products as well as leading to the appearance of an aberrant 21S pre-rRNA. More importantly, the rps12 null mutation resulted in a strong delay in pre-rRNA processing at site D, a late step in the maturation of 40S r-subunits [Citation39,Citation65]. Thus, the 20S pre-rRNA accumulated and was not efficiently converted into mature 18S rRNA. This accumulation was apparently cytoplasmic, indicating that the rps12∆ mutation does therefore not impair significantly nuclear export of late pre-40S r-particles. While we believe that the cytoplasmic 20S pre-rRNA to 18S rRNA maturation defect is likely the direct consequence of the lack of eS12, the modest delay in pre-rRNA processing at site A2, which occurs in the nucleus (see [Citation65] for a review) and is likely the cause of the accumulation of the aberrant 21S precursor in rps12∆ cells, is probably due to some failure during recycling of certain shuttling factors from cytoplasmic pre-40S r-particles that this way become limiting for function. Interestingly, these phenotypes are very similar to those previously observed for the ubi3∆ mutant (; see also [Citation17,Citation34,Citation37]). Moreover, comparable results were obtained upon knocking down the expression levels of human RPS12, which leads to cytoplasmic accumulation of the 18S-E pre-rRNA, the equivalent form of yeast 20S pre-rRNA [Citation13,Citation66,Citation67]
In yeast, cytoplasmic accumulation of the 20S pre-rRNA has previously been reported following the depletion of or the mutation in other r-proteins and trans-acting factors. Among others, in addition to eS31, these r-proteins include the 40S r-subunit proteins uS2 [Citation11,Citation15,Citation68], uS5 [Citation11], uS3 [Citation11,Citation41], uS7 [Citation69], eS10 [Citation11], uS11 [Citation11,Citation14], uS19 [Citation11], uS10 [Citation11,Citation70], eS26 [Citation11,Citation71,Citation72], eS28 [Citation11] and likely eS21 [Citation68] (Figure S10) as well as the 60S r-subunit protein uL3 [Citation43,Citation73]. Regarding trans-acting factors, these comprise, in addition to the D-site endonuclease Nob1 [Citation74,Citation75] and its partner Pno1/Dim2/Rrp20 [Citation76], other proteins such as the DEAH-box RNA helicase Prp43 [Citation44,Citation77,Citation78] and its co-factor Pfa1 [Citation44,Citation77], Ltv1 [Citation32,Citation33,Citation44,Citation50,Citation51], the kinase Hrr25 [Citation24,Citation33], the ATPases Rio1/Rrp10 [Citation79–Citation81] and Rio2 [Citation79,Citation82], the GTPase-like Tsr1 [Citation81,Citation83], the aminocarboxypropyl transferase Tsr3 [Citation84,Citation85], the dedicated uS2-chaperone Tsr4 [Citation84,Citation86,Citation87], the dedicated uS3-chaperone Yar1 [Citation41], the uS11-specific chaperone Fap7 [Citation46,Citation48,Citation81], the eS26-escortin Tsr2 [Citation81] and the translation initiation factor eIF5B/Fun12 [Citation47,Citation84,Citation88].
Along the cytoplasmic 40S r-subunit maturation pathway, it has been found that r-proteins are required either for association, release, activity or repositioning of the above-mentioned factors as exemplified by the reported roles of uS3, uS10, eS10, uS11 and eS26. Thus, the final assembly of uS3, which initially depends on Ltv1, and uS10 coordinates Hrr25’s kinase activity and the Ltv1 and Rio2 release [Citation32,Citation33,Citation70]. Moreover, eS10 appears to assemble only upon release of Ltv1 and Enp1 [Citation26,Citation33]. Stable assembly of uS11 and eS26 requires the functional interplay with Fap7; the stable assembly of these two r-proteins is linked to Fap7 and Pno1 release [Citation26,Citation72,Citation89,Citation90]. Further work is required to understand the specific role of eS12 and eS31 in efficient cytoplasmic maturation of 40S r-subunits, but the epistatic relationship of rps12∆ over the ubi3∆ mutation suggests that both mutations impair 40S r-subunit maturation in the same pathway. Interestingly, our data suggest that, although both eS12 and eS31 functionally interact with Ltv1 and Enp1, their roles in 40S r-subunit maturation seem not to be strictly dependent on the release dynamics of these factors, which both bind at the base of the beak (e.g. [Citation26,Citation30]). In contrast, mutations in uS3 and uS10 showed faulty pre-40S release of Ltv1 (e.g. cytoplasmic mislocalisation of the Ltv1∆NES-GFP reporter) and Enp1, among other phenotypes [Citation32,Citation70]. In agreement with a non-impaired release of Ltv1 in rps12∆ and ubi3∆ cells, eS12- or eS31-lacking pre-40S r-particles can enter into the translation-like cycle by forming 80S-like r-particles; previously published results have demonstrated that inhibition of Ltv1 release blocked joining of pre-40S to 60S r-particles [Citation33].
Our data indicate that in rps12∆ and ubi3∆ cells, most pre-40S r-particles containing 20S pre-rRNA accumulated in 80S-like r-particles. We cannot currently discern whether these particles correspond to truly blocked 80S-like r-particles or vacant ribosomes formed non-specifically between 60S r-subunits and the pre-40S r-particles that accumulate in these mutants under Mg2 +-containing buffer conditions. Indeed, our experiments suggest that, in contrast to the situation found in other 40S-deficient mutants (e.g. ubi3∆ub, rpl3[W255C] and ltv1∆ pfa1∆ mutants, or Rio1-depleted cells [Citation17,Citation43–Citation45,Citation91]), the pre-40S r-particles containing 20S pre-rRNA that accumulated in rps12∆ and ubi3∆ cells are only inefficiently engaging in translation, thus accumulating as 80S-like r-particles. Although hardly detected in wild-type conditions, 80S-like r-particles have been described upon depletion of Fap7 [Citation47,Citation48] and Rio1 [Citation45,Citation91] or catalytic inactivation of Nob1 [Citation30]. Whether or not the formation of 80S-like r-particles in the absence of eS12 or eS31 is linked to the loss-of-function of any of these above factors needs further experiments. Strikingly, the 20S pre-rRNA containing particles we observed in rps12∆ and ubi3∆ cells sedimented slightly lighter than the 80S peak (see and S6). It is likely that 80S-like r-particles accumulating in the absence of eS12 or eS31 could contain some still non-stably associated r-proteins and/or trans-acting factors that fall off during polysome extraction or centrifugation. Further experiments are required to test this hypothesis.
Finally, we show that the perturbation of the beak structure by the deletion of RPS12 or UBI3 promoted a significant increase in the frequency of translational misincorporation events. Owing to the universal presence of the beak structure in all ribosomes and the fact that this region is located at the mRNA entry site [Citation10], it is surprising that no previous studies, except for a twenty-year-old work showing that eS31 is required for efficient translation of the GCN4 mRNA [Citation92], have apparently addressed the effects of eS12- or eS31-lacking 40S subunits on translation. Herein, we revealed that the absence of eS12 or eS31 induce amino acid misincorporation during decoding. More specifically, the misincorporation event that we observed with Bedwell’s reporter constructs involves the recognition of a CGC codon (Arg245) by, very likely, a near-cognated His-charged tRNA(GUG)His [Citation54]. This improper codon-anticodon recognition could occur due to structural alterations of the region around the A-site of ribosomes lacking these important r-proteins, a possibility that we have previously suggested when studying the functional consequences of the truncation of the eukaryote-specific N-terminal extension of eS31, which extends towards the A-site [Citation37]. As the polysome profiles of rps12∆ and ubi3∆ cells are significantly perturbed, we cannot formally exclude the possibility that the increased misreading rate of these cells could be the indirect consequence of having impaired the function of other relevant extrinsic protein such as a translation factor or of just having reduced the number of functional ribosomes (for further discussion, see ref. [Citation93]). Further studies are definitively required to clarify this issue.
In summary, we have started tackling the functional characterization of the eS12 and eS31 r-proteins. Our results envisage significant and related roles for these two r-proteins during cytoplasmic maturation of 40S r-subunits and translation. These results also establish the basis for specific forthcoming genetic, biochemical and structural studies that will be instrumental to precisely understand the molecular consequences of the defects caused by their absence in these processes. Finally, mutations in RPS10, the gene coding for human beak r-protein eS10 have been reported in about 3% of DBA patients [Citation94,Citation95]. It is so far unclear whether mutations in the RPS27A and RPS12 genes, encoding human eS31 and eS12, respectively, are linked to ribosomopathies [Citation94]. However, the fact that, in human cells, siRNA-dependent knockdown of RPS10 expression entails pre-rRNA processing defects (e.g. 18S-E pre-rRNA accumulation) similar to those observed upon knockdown of RPS26 (coding for eS26) – note that mutations in this gene are commonly found in DBA patients (ca. 6%) [Citation94] –, RPS12 or RPS27A [Citation13,Citation94] opens the possibility that mutant variants of eS12 or eS31 could also cause DBA in a rare subset of patients. In any circumstances, as at least eS10 is clearly linked to DBA, studying in detail the formation of the beak structure and its translation functions could help to better understand the pathogenesis and eventually provide new insights for the treatment of this disease.
Materials and methods
Strains and microbiological methods
All yeast strains used in this work are listed in Table S1. Unless otherwise indicated, experiments were conducted in the W303 genetic background. Strains SMY227 and SMY231 are meiotic rps12∆ and ubi3∆ deletion-disruption segregants of strains Y21666 and Y24116 (Euroscarf), respectively. Strain SMY315 was generated by PCR-based gene disruption of the RPS12 gene in the diploid W303 strain. To this end, we amplified the rps12::kanMX4 disruption cassette using genomic DNA of the SMY227 strain as a template and the oligonucleotide pair EcoRI_S12up and S12_check_down (see Table S3). Transformants were selected on YPD plates containing 200 µg/ml G418, some candidates were chosen and the correctness of the integration verified by PCR. Positive heterozygous candidates were transformed with the YCplac33-RPS12 plasmid, then sporulated, and tetrads were dissected. SMY315 was selected as a representative rps12∆ null haploid mutant.
To generate SMY372, TLY14.3C was crossed to SMY315 [YCplac33-RPS12]; the resulting diploid was sporulated, and tetrads were dissected. Spore clones from different tetratype tetrads were obtained and restreaked on 5-FOA-containing plates. Other strains, listed in Table S1, harbouring combinations of different alleles were obtained by a similar approach. Information on the respective crosses and analyses of the progenies are available on request.
Growth and handling of yeast as well as yeast media were done following established procedures [Citation96]. Strains were grown at the indicated temperatures either in rich YPD medium (1% yeast extract, 2% peptone and 2% glucose) or synthetic minimal medium (0.15% yeast nitrogen base, 0.5% ammonium sulphate) supplemented with the appropriate amino acids and bases as nutritional requirements, and containing 2% glucose (SD) as carbon source. To prepare plates, 2% agar was added to the media before sterilization. Yeast cells were transformed by a lithium acetate method [Citation97]. Tetrads were dissected with a MSM200 micromanipulator (Singer Instruments, UK). Escherichia coli strain DH5α was used for cloning and propagation of plasmids. All recombinant DNA techniques were done according to established procedures [Citation98].
Plasmids
Plasmids used in this work are listed in Table S2. To generate YCplac33-RPS12, a PCR was performing using yeast genomic DNA as template and the oligonucleotides BamHI_S12up and S12_SacIdown (Table S3), placed plus-minus 1 kb upstream and downstream from the start-stop codon of the RPS12 ORF. The ca. 2.4 kb PCR product was restricted with BamHI and SacI and cloned into YCplac33, which was also digested with the same enzymes. The rest of plasmids have been previously reported elsewhere (see Table S2).
Sucrose gradient centrifugation
Polysome and ribosomal subunit analyses were performed as previously described [Citation99]. Ten A260 units of total cell extract were load onto 7–50% sucrose gradients. These gradients were centrifuged at 39,000 rpm in a Beckman Coulter SW 41 Ti rotor for 2 h 45 min (for polysome profile analyses) or for 4 h 30 min (for ribosomal subunit analyses) at 4°C; then, the A254 was continuously monitor using an ISCO UA-6 system.
When required, fractions of 0.5 ml were collected from the gradients. RNA was extracted from each fraction and an equal volume of RNA from individual fractions was subjected to northern analyses (see below) as previously described [Citation100].
RNA extractions and northern hybridization analyses
Total RNA was extracted from samples corresponding to 10 OD600 units of exponentially grown cells by the acid-phenol method [Citation101]. Equal amounts of total RNA were loaded on 1.2% agarose-6% formaldehyde or on 7% polyacrylamide-8 M urea gels as previously described [Citation102]. Specific antisense-oligonucleotides (Table S3) were used to detect distinct pre- and mature rRNAs. Oligonucleotides were 5ʹ-end labelled with [γ-32P]ATP (6000 Ci/mmol; Perkin Elmer) as previously described [Citation102]. Phosphorimager analysis was performed with a Typhoon™ FLA-9000 imaging system (GE Healthcare) and signals intensities were quantified using the GelQuant.NET software (biochemlabsolutions.com).
Pulse-chase labelling of pre-RNA
For [5,6-3H]uracil pulse-chase labelling analysis, strains W303-1A and SMY315 were first transformed with an empty YCplac33 plasmid to make them prototrophic for uracil. Transformants were grown in 50 ml of liquid SD-Ura medium to mid-log phase at 30°C. Then, the cells were concentrated in 1 ml of SD-Ura medium and pulse-labelled using 100 µCi of [5,6-3H]uracil (45 to 50 Ci/mmol; Perkin Elmer) per 40 OD600 units of yeast cells as exactly described in Kressler et al. [Citation103]. Cells were chased for 5, 15, 30 and 60 min after diluting 200 µl aliquots of pulse-labelled cells in 4 ml of SD medium containing an excess (1 mg/ml) of non-radioactive uracil. Total RNA was extracted by the acid-phenol method as above.
Uracil incorporation was measured by scintillation counting and 3,000 cpm per RNA sample was loaded and resolved on 1.2% agarose–6% formaldehyde and 7% polyacrylamide-8 M urea gels. RNA was then transferred and crosslinked to Hybond-N nylon membranes (GE Healthcare), and exposed to a tritium screen (BAS-IP TR2040E; GE Healthcare). Visualization was performed with a Typhoon™ FLA-9000 imaging system (GE Healthcare).
Detection of fluorescent proteins by microscopy
To test pre-ribosomal particle export, the appropriate strains were transformed with pRS315-RPL25-yEGFP-mRFP-NOP1, pRS315-RPS3-yEGFP-mRFP-NOP1 (gifts from J. Bassler and E. Hurt), YCplac111-RPS10-yEGFP or YCplac111-UBI3-yEGFP plasmids (see Table S2). Transformants were grown in selective SD medium. Cells were washed and resuspended in PBS buffer (140 mM NaCl, 8 mM Na2HPO4, 1.5 mM KH2PO4, 2.75 mM KCl, pH 7.3) before their microscopy inspection. When required, Hoechst was used to stain nuclear DNA. Cells were examined with an Olympus BX61 fluorescence microscope equipped with a digital camera; images were analysed using the CellSens software (Olympus).
To study the subcellular location of GFP-tagged Enp1, Ltv1 and Ltv1∆NES, the appropriate strains (see Table S1) were grown in YPD medium. Hoechst was used to stain DNA in the nucleus. Fluorescently labelled cells were inspected as described above.
Fluorescence in situ hybridization microscopy
To examine the localization of the 20S pre-rRNA, fluorescence in situ hybridisation (FISH) was performed in spheroblasted cells fixed with formaldehyde, as previously described [Citation104,Citation105]. A Cy3-labelled ITS1-specific probe (see Table S3) was used. Cells were also stained with DAPI (4,6-diamidino-2-phenylindole) to visualize DNA. Cells were then examined by fluorescence microscopy as described above.
Quantification of translation accuracy
To measure the efficiency of translation termination and of amino acid misincorporation, the strains W303-1A, SMY315 and TLY14.3C were transformed with the different dual-luciferase reporter plasmids generously provided by D. Bedwell (see Table S2 for plasmids and references). Luciferase activities were measured as previously described [Citation54] using the Dual-Glo® Luciferase Assay System (Promega). Cells from each strain were grown in liquid SD-Ura medium to mid-log phase at 30°C and firefly and Renilla luciferase luminescence levels were measured at room temperature with a CLARIOstar 1.20 microplate reader (BMG Labtech, Germany) adjusted to endpoint read-type and default settings. Assays were repeated four times (biological replicas), each of these was replicated three times (technical replicas) and the data were expressed as the mean ± the standard deviation. Error rates for each strain were calculated as the percentage of the firefly/Renilla luciferase activity (mutant plasmid) divided by the firefly/Renilla luciferase activity (wild-type plasmid).
Supplemental Material
Download MS Word (5.5 MB)Acknowledgments
This paper is dedicated to the memory of Prof J. R. Warner. We thank those colleagues mentioned in the text for supplying material used in this work. We specially thank D. M. Bedwell, J. Bassler and E. Hurt for plasmids, and B. Pertschy and D. Kressler for strains, plasmids and critical reading of the manuscript.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here
Additional information
Funding
References
- Warner JR. The economics of ribosome biosynthesis in yeast. Trends Biochem Sci. 1999;24:437–440.
- Woolford JL Jr. , Baserga SJ . Ribosome biogenesis in the yeast Saccharomyces cerevisiae . Genetics. 2013;195:643–681.
- Bassler J , Hurt E . Eukaryotic ribosome assembly. Annu Rev Biochem. 2019;88:281–306.
- de la Cruz J , Karbstein K , Woolford JL Jr. Functions of ribosomal proteins in assembly of eukaryotic ribosomes in vivo . Annu Rev Biochem. 2015;84:93–129.
- Armistead J , Triggs-Raine B . Diverse diseases from a ubiquitous process: the ribosomopathy paradox. FEBS Lett. 2014;588:1491–1500.
- Teng T , Thomas G , Mercer CA . Growth control and ribosomopathies. Curr Opin Genet Dev. 2013;23:63–71.
- Narla A , Ebert BL . Ribosomopathies: human disorders of ribosome dysfunction. Blood. 2010;115:3196–3205.
- Yusupova G , Yusupov M . High-resolution structure of the eukaryotic 80S ribosome. Annu Rev Biochem. 2014;83:467–486.
- Rabl J , Leibundgut M , Ataide SF , et al. Crystal structure of the eukaryotic 40S ribosomal subunit in complex with initiation factor 1. Science. 2011;331:730–736.
- Wilson DN , Doudna Cate JH . The structure and function of the eukaryotic ribosome. Cold Spring Harb Perspect Biol. 2012;4:a011536.
- Ferreira-Cerca S , Pöll G , Gleizes PE , et al. Roles of eukaryotic ribosomal proteins in maturation and transport of pre-18S rRNA and ribosome function. Mol Cell. 2005;20:263–275.
- Ferreira-Cerca S , Pöll G , Kuhn H , et al. Analysis of the in vivo assembly pathway of eukaryotic 40S ribosomal proteins. Mol Cell. 2007;28:446–457.
- O’Donohue MF , Choesmel V , Faubladier M , et al. Functional dichotomy of ribosomal proteins during the synthesis of mammalian 40S ribosomal subunits. J Cell Biol. 2010;190:853–866.
- Jakovljevic J , de Mayolo PA , Miles TD , Nguyen TM , Léger-Silvestre I , Gas N , et al. The carboxy-terminal extension of yeast ribosomal protein S14 is necessary for maturation of 43S preribosomes. Mol Cell. 2004;14:331–342.
- Ford CL , Randal-Whitis L , Ellis SR . Yeast proteins related to the p40/laminin receptor precursor are required for 20S ribosomal RNA processing and the maturation of 40S ribosomal subunits. Cancer Res. 1999;59:704–710.
- Tabb AL , Utsugi T , Wooten-Kee CR , et al. Genes encoding ribosomal proteins Rps0A/B of Saccharomyces cerevisiae interact with TOM1 mutants defective in ribosome synthesis. Genetics. 2001;157:1107–1116.
- Lacombe T , García-Gómez JJ , de la Cruz J , et al. Linear ubiquitin fusion to Rps31 and its subsequent cleavage are required for the efficient production and functional integrity of 40S ribosomal subunits. Mol Microbiol. 2009;72:69–84.
- Davis JH , Williamson JR . Structure and dynamics of bacterial ribosome biogenesis. Philos Trans R Soc Lond B Biol Sci. 2017; 372:20160181.
- Sykes MT , Williamson JR . A complex assembly landscape for the 30S ribosomal subunit. Annu Rev Biophys. 2009;38:197–215.
- Chaker-Margot M . Assembly of the small ribosomal subunit in yeast: mechanism and regulation. RNA. 2018;24:881–891.
- Kressler D , Hurt E , Bassler J . A puzzle of life: crafting ribosomal subunits. Trends Biochem Sci. 2017;42:640–654.
- Klinge S , Woolford JL Jr. Ribosome assembly coming into focus. Nat Rev Mol Cell Biol. 2019;20:116–131.
- Sun Q , Zhu X , Qi J , et al. Molecular architecture of the 90S small subunit pre-ribosome. eLife. 2017;6:e22086.
- Schäfer T , Maco B , Petfalski E , et al. Hrr25-dependent phosphorylation state regulates organization of the pre-40S subunit. Nature. 2006;441:651–655.
- Schäfer T , Strauss D , Petfalski E , et al. The path from nucleolar 90S to cytoplasmic 40S pre-ribosomes. Embo J. 2003;22:1370–1380.
- Strunk BS, Loucks CR, Su M, Vashisth H, Cheng S, Schilling J, et al . Ribosome assembly factors prevent premature translation initiation by 40S assembly intermediates. Science. 2011;333:1449–1453.
- Moriggi G , Nieto B , Dosil M . Rrp12 and the exportin Crm1 participate in late assembly events in the nucleolus during 40S ribosomal subunit biogenesis. PLoS Genet. 2015;10:e1004836.
- Campbell MG , Karbstein K . Protein-protein interactions within late pre-40S ribosomes. PLoS One. 2011;6:e16194.
- Heuer A , Thomson E , Schmidt C , et al. Cryo-EM structure of a late pre-40S ribosomal subunit from Saccharomyces cerevisiae . eLife. 2017;6: e30189.
- Scaiola A , Pena C , Weisser M , et al. Structure of a eukaryotic cytoplasmic pre-40S ribosomal subunit. Embo J. 2018;37:e98499.
- Collins JC , Ghalei H , Doherty JR , et al. Ribosome biogenesis factor Ltv1 chaperones the assembly of the small subunit head. J Cell Biol. 2018;217:4141–4154.
- Mitterer V , Murat G , Rety S , et al. Sequential domain assembly of ribosomal protein S3 drives 40S subunit maturation. Nat Commun. 2016;7:10336.
- Ghalei H , Schaub FX , Doherty JR , et al. Hrr25/CK1δ-directed release of Ltv1 from pre-40S ribosomes is necessary for ribosome assembly and cell growth. J Cell Biol. 2016;208:745–759.
- Finley D , Bartel B , Varshavsky A . The tails of ubiquitin precursors are ribosomal proteins whose fusion to ubiquitin facilitates ribosome biogenesis. Nature. 1989;338:394–401.
- Lecompte O , Ripp R , Thierry JC , et al. Comparative analysis of ribosomal proteins in complete genomes: an example of reductive evolution at the domain scale. Nucleic Acids Res. 2002;30:5382–5390.
- Giaever G , Chu AM , Ni L , et al. Functional profiling of the Saccharomyces cerevisiae genome. Nature. 2002;418:387–391.
- Fernández-Pevida A , Martín-Villanueva S , Murat G , de la Cruz J et al.The eukaryote-specific N-terminal extension of ribosomal protein S31 contributes to the assembly and function of 40S ribosomal subunits. Nucleic Acids Res. 2016;44:7777–7791.
- Steffen KK , McCormick MA , Pham KM , et al. Ribosome deficiency protects against ER stress in Saccharomyces cerevisiae . Genetics. 2012;191:107–118.
- Udem SA , Warner JR . The cytoplasmic maturation of a ribosomal precursor ribonucleic acid in yeast. J Biol Chem. 1973;248:1412–1416.
- Trapman J , Planta RJ . Maturation of ribosomes in yeast. I. Kinetic analysis by labelling of high molecular weight rRNA species. Biochim Biophys Acta. 1976;442:265–274.
- Koch B , Mitterer V , Niederhauser J , et al. Yar1 protects the ribosomal protein Rps3 from aggregation. J Biol Chem. 2012;287:21806–21815.
- Hurt E , Hannus S , Schmelzl B , et al. A novel in vivo assay reveals inhibition of ribosomal nuclear export in Ran-cycle and nucleoporin mutants. J Cell Biol. 1999;144:389–401.
- García-Gómez JJ , Fernández-Pevida A , Lebaron S , et al. Final pre-40S maturation depends on the functional integrity of the 60S subunit ribosomal protein L3. PLoS Genet. 2014;10:e1004205.
- Pertschy B , Schneider C , Gnadig M , et al. RNA helicase Prp43 and its co-factor Pfa1 promote 20 to 18S rRNA processing catalyzed by the endonuclease Nob1. J Biol Chem. 2009;284:35079–35091.
- Soudet J , Gelugne JP , Belhabich-Baumas K , et al. Immature small ribosomal subunits can engage in translation initiation in Saccharomyces cerevisiae . Embo J. 2010;29:80–92.
- Ghalei H , Trepreau J , Collins JC , et al. The ATPase Fap7 tests the ability to carry out translocation-like conformational changes and releases Dim1 during 40S ribosome maturation. Mol Cell. 2017;67:990–1000.e3. DOI:10.1016/j.molcel.2017.08.007
- Strunk BS , Novak MN , Young CL , et al. A translation-like cycle is a quality control checkpoint for maturing 40S ribosome subunits. Cell. 2012;150:111–121.
- Granneman S , Nandineni MR , Baserga SJ . The putative NTPase Fap7 mediates cytoplasmic 20S pre-rRNA processing through a direct interaction with Rps14. Mol Cell Biol. 2005;25:10352–10364.
- Johnson MC , Ghalei H , Doxtader KA , et al. Structural heterogeneity in pre-40S ribosomes. Structure. 2017;25:329–340.
- Seiser RM , Sundberg AE , Wollam BJ , et al. Ltv1 is required for efficient nuclear export of the ribosomal small subunit in Saccharomyces cerevisiae . Genetics. 2006;174:679–691.
- Fassio CA , Schofield BJ , Seiser RM , et al. Dominant mutations in the late 40S biogenesis factor Ltv1 affect cytoplasmic maturation of the small ribosomal subunit in Saccharomyces cerevisiae . Genetics. 2010;185:199–209.
- Chen W , Bucaria J , Band DA , et al. Enp1, a yeast protein associated with U3 and U14 snoRNAs, is required for pre-rRNA processing and 40S subunit synthesis. Nucleic Acids Res. 2003;31:690–699.
- Merwin JR , Bogar LB , Poggi SB , et al. Genetic analysis of the ribosome biogenesis factor Ltv1 of Saccharomyces cerevisiae . Genetics. 2014;198:1071–1085.
- Salas-Marco J , Bedwell DM . Discrimination between defects in elongation fidelity and termination efficiency provides mechanistic insights into translational readthrough. J Mol Biol. 2005;348:801–815.
- Dresios J , Derkatch IL , Liebman SW , et al. Yeast ribosomal protein L24 affects the kinetics of protein synthesis and ribosomal protein L39 improves translational accuracy, while mutants lacking both remain viable. Biochemistry. 2000;39:7236–7244.
- Synetos D , Frantziou CP , Alksne LE . Mutations in yeast ribosomal proteins S28 and S4 affect the accuracy of translation and alter the sensitivity of the ribosomes to paromomycin. Biochim Biophys Acta. 1996;1309:156–166.
- Anthony RA , Liebman SW . Alterations in ribosomal protein RPS28 can diversely affect translational accuracy in Saccharomyces cerevisiae . Genetics. 1995;140:1247–1258.
- Alksne LE , Anthony RA , Liebman SW , et al. An accuracy center in the ribosome conserved over 2 billion years. Proc Natl Acad Sci USA. 1993;90:9538–9541.
- Cui Y , Dinman JD , Kinzy TG , et al. The Mof2/Sui1 protein is a general monitor of translational accuracy. Mol Cell Biol. 1998;18:1506–1516.
- Wawiórka L , Molestak E , Szajwaj M , et al. Multiplication of ribosomal P-stalk proteins contributes to the fidelity of translation. Mol Cell Biol. 2017;37:e00060–17.
- Keeling KM, Lanier J, Du M, Salas-Marco J, Gao L, Kaenjak-Angeletti A, et al . Leaky termination at premature stop codons antagonizes nonsense-mediated mRNA decay in S cerevisiae . RNA. 2004;10:691–703.
- Steffen KK , MacKay VL , Kerr EO , et al. Yeast life span extension by depletion of 60S ribosomal subunits is mediated by Gcn4. Cell. 2008;133:292–302.
- Puddu F , Herzog M , Selivanova A , et al. Genome architecture and stability in the Saccharomyces cerevisiae knockout collection. Nature. 2019;573:416–420.
- Santos C , Ballesta JPG . Ribosomal protein P0, contrary to phosphoproteins P1 and P2, is required for ribosome activity and Saccharomyces cerevisiae viability. J Biol Chem. 1994;269:15689–15696.
- Fernández-Pevida A , Kressler D , de la Cruz J . Processing of preribosomal RNA in Saccharomyces cerevisiae . Wiley Interdiscip Rev RNA. 2015;6:191–209.
- Nicolas E , Parisot P , Pinto-Monteiro C , et al. Involvement of human ribosomal proteins in nucleolar structure and p53-dependent nucleolar stress. Nat Commun. 2016;7:11390.
- Wild T , Horvath P , Wyler E , et al. A protein inventory of human ribosome biogenesis reveals an essential function of exportin 5 in 60S subunit export. PLoS Biol. 2010;8:e1000522.
- Tabb-Massey A , Caffrey JM , Logsden P , et al. Ribosomal proteins Rps0 and Rps21 of Saccharomyces cerevisiae have overlapping functions in the maturation of the 3ʹ end of 18S rRNA. Nucleic Acids Res. 2003;31:6798–6805.
- Neueder A , Jakob S , Pöll G , et al. A local role for the small ribosomal subunit primary binder rpS5 in final 18S rRNA processing in yeast. PLoS One. 2010;5:e10194.
- Mitterer V , Shayan R , Ferreira-Cerca S , et al. Conformational proofreading of distant 40S ribosomal subunit maturation events by a long-range communication mechanism. Nat Commun. 2019;10:2754.
- Schütz S , Fischer U , Altvater M , et al. A RanGTP-independent mechanism allows ribosomal protein nuclear import for ribosome assembly. eLife. 2014;3:e03473.
- Peña C , Schütz S , Fischer U , et al. Prefabrication of a ribosomal protein subcomplex essential for eukaryotic ribosome formation. eLife. 2016;5:e21755.
- Mitlin JA , Cannon M . Defective processing of ribosomal precursor RNA in Saccharomyces cerevisiae . Biochem J. 1984;220:461–467.
- Fatica A , Oeffinger M , Dlakic M , et al. Nob1p is required for cleavage of the 3ʹ end of 18S rRNA. Mol Cell Biol. 2003;23:1798–1807.
- Fatica A , Tollervey D , Dlakic M . PIN domain of Nob1p is required for D-site cleavage in 20S pre-rRNA. RNA. 2004;10:1698–1701.
- Woolls HA , Lamanna AC , Karbstein K . Roles of Dim2 in ribosome assembly. J Biol Chem. 2011;286:2578–2586.
- Lebaron S , Papin C , Capeyrou R , et al. The ATPase and helicase activities of Prp43p are stimulated by the G-patch protein Pfa1p during yeast ribosome biogenesis. Embo J. 2009;28:3808–3819.
- Combs DJ , Nagel RJ , Ares M Jr. , et al. Prp43p is a DEAH-box spliceosome disassembly factor essential for ribosome biogenesis. Mol Cell Biol. 2006;26:523–534.
- Vanrobays E , Gelugne JP , Gleizes PE , et al. Late cytoplasmic maturation of the small ribosomal subunit requires RIO proteins in Saccharomyces cerevisiae . Mol Cell Biol. 2003;23:2083–2095.
- Vanrobays E , Gleizes PE , Bousquet-Antonelli C , et al. Processing of 20S pre-rRNA to 18S ribosomal RNA in yeast requires Rrp10p, an essential non-ribosomal cytoplasmic protein. Embo J. 2001;20:4204–4213.
- Peng WT , Robinson MD , Mnaimneh S , et al. A panoramic view of yeast noncoding RNA processing. Cell. 2003;113:919–933.
- Geerlings TH , Faber AW , Bister MD , et al. Rio2p, an evolutionarily conserved, low abundant protein kinase essential for processing of 20S Pre-rRNA in Saccharomyces cerevisiae . J Biol Chem. 2003;278:22537–22545.
- Gelperin D , Horton L , Beckman J , et al. Bms1p, a novel GTP-binding protein, and the related Tsr1p are required for distinct steps of 40S ribosome biogenesis in yeast. RNA. 2001;7:1268–1283.
- Li Z , Lee I , Moradi E , et al. Rational extension of the ribosome biogenesis pathway using network-guided genetics. PLoS Biol. 2009;7:e1000213.
- Meyer B , Wurm JP , Sharma S , et al. Ribosome biogenesis factor Tsr3 is the aminocarboxypropyl transferase responsible for 18S rRNA hypermodification in yeast and humans. Nucleic Acids Res. 2016;44:4304–4316.
- Black JJ , Musalgaonkar S , Johnson AW . Tsr4 is a cytoplasmic chaperone for the ribosomal protein Rps2 in Saccharomyces cerevisiae . Mol Cell Biol. 2019;39:e00094–19.
- Rossler I , Embacher J , Pillet B , et al. Tsr4 and Nap1, two novel members of the ribosomal protein chaperOME. Nucleic Acids Res. 2019;47:6984–7002.
- Lebaron S , Schneider C , van Nues RW , et al. Proofreading of pre-40S ribosome maturation by a translation initiation factor and 60S subunits. Nat Struct Mol Biol. 2012;19:744–753.
- Loc’h J , Blaud M , Rety S , et al. RNA mimicry by the fap7 adenylate kinase in ribosome biogenesis. PLoS Biol. 2014;12:e1001860.
- Hellmich UA , Weis BL , Lioutikov A , et al. Essential ribosome assembly factor Fap7 regulates a hierarchy of RNA-protein interactions during small ribosomal subunit biogenesis. Proc Natl Acad Sci USA. 2013;110:15253–15258.
- Belhabich-Baumas K , Joret C , Jady BE , et al. The Rio1p ATPase hinders premature entry into translation of late pre-40S pre-ribosomal particles. Nucleic Acids Res. 2017;45:10824–10836.
- Mueller PP , Grueter P , Hinnebusch AG , et al. A ribosomal protein is required for translational regulation of GCN4 mRNA. Evidence for involvement of the ribosome in eIF2 recycling. J Biol Chem. 1998;273:32870–32877.
- Mills EW , Green R . Ribosomopathies: there’s strength in numbers. Science. 2017;358:eaan2755.
- Doherty L , Sheen MR , Vlachos A , et al. Ribosomal protein genes RPS10 and RPS26 are commonly mutated in diamond-blackfan anemia. Am J Hum Genet. 2010;86:222–228.
- Boria I , Garelli E , Gazda HT , et al. The ribosomal basis of diamond-blackfan anemia: mutation and database update. Hum Mutat. 2010;31:1269–1279.
- Kaiser C , Michaelis S , Mitchell A . Methods in yeast genetics: a cold spring harbor laboratory course manual. Cold Spring Harbor, N. Y: Cold Spring Harbor Laboratory Press; 1994.
- Gietz D , St. Jean A , Woods RA , et al. Improved method for high efficiency transformation of intact yeast cells. Nucleic Acids Res. 1992;20:1425.
- Sambrook J , Fritsch EF , Maniatis T . Molecular cloning: a laboratory manual. Cold Spring Harbor, N. Y.: Cold Spring Harbor Laboratory Press; 1989.
- Kressler D , de la Cruz J , Rojo M , et al. Fal1p is an essential DEAD-box protein involved in 40S-ribosomal-subunit biogenesis in Saccharomyces cerevisiae . Mol Cell Biol. 1997;17:7283–7294.
- de la Cruz J , Kressler D , Rojo M , et al. Spb4p, an essential putative RNA helicase, is required for a late step in the assembly of 60S ribosomal subunits in Saccharomyces cerevisiae . RNA. 1998;4:1268–1281.
- Ausubel FM , Brent R , Kingston RE , et al. Saccharomyces cerevisiae. Current protocols in molecular biology. New York, N. Y.: John Wiley & Sons, Inc.; 1994. p. 13.0.1-.4.7.
- Venema J , Planta RJ , Raué HA . In vivo mutational analysis of ribosomal RNA in Saccharomyces cerevisiae . In: Martin R , editor. Protein synthesis: methods and Protocols. Totowa, N. J.: Humana Press; 1998. p. 257–270.
- Kressler D , de la Cruz J , Rojo M , et al. Dbp6p is an essential putative ATP-dependent RNA helicase required for 60S-ribosomal-subunit assembly in Saccharomyces cerevisiae . Mol Cell Biol. 1998;18:1855–1865.
- Grosshans H , Hurt E , Simos G . An aminoacylation-dependent nuclear tRNA export pathway in yeast. Genes Dev. 2000;14:830–840.
- Rodríguez-Galán O , García-Gómez JJ , Kressler D , de la Cruz J . Immature large ribosomal subunits containing the 7S pre-rRNA can engage in translation in Saccharomyces cerevisiae . RNA Biol. 2015;12:838–846.