
ABSTRACT
Many bacteria and archaea possess an RNA-guided adaptive and inheritable immune system that consists of clustered regularly interspaced short palindromic repeats (CRISPRs) and CRISPR-associated (Cas) proteins. In most CRISPR-Cas systems, the maturation of CRISPR-derived small RNAs (crRNAs) is essential for functionality. Cas6 endonucleases function as the most frequent CRISPR RNA maturation enzymes. In the cyanobacterium Anabaena sp. PCC 7120, ten CRISPR loci are present, but only two cas gene cassettes plus a Tn7-associated eleventh array. In this study, we deleted the two cas6 genes alr1482 (Type III-D) or alr1566 (Type I-D) and tested the specificities of the two corresponding enzymes in the resulting mutant strains, as recombinant proteins and in a cell-free transcription-translation system. The results assign the direct repeats (DRs) to three different groups. While Alr1566 is specific for one group, Alr1482 has a higher preference for the DRs of the second group but can also cleave those of the first group. We found that this cross-recognition limits crRNA accumulation for the Type I-D system in vivo.
We also show that the DR of the cas gene-free CRISPR array of cyanophage N-1 is processed by these enzymes, suggesting that it is fully competent in association with host-encoded Cas proteins. The data support the functionality of CRISPR arrays that frequently appear fragmented to multiple genomic loci in multicellular cyanobacteria and disfavour other possibilities, such as the nonfunctionality of these orphan repeat-spacer arrays. Our results show the functional coordination of Cas6 endonucleases with both neighbouring and remote repeat-spacer arrays in the CRISPR-Cas system of cyanobacteria.
Introduction
Clustered regularly interspaced short palindromic repeats (CRISPR) and CRISPR-associated (Cas) proteins provide an adaptive immune system directed against invading nucleic acids in many bacteria and most archaea [Citation1–Citation3]. Immunity relies on the transcription of the CRISPR repeat-spacer array, ultimately leading to short CRISPR RNAs (crRNAs) that guide the CRISPR complexes in the recognition and destruction of invading nucleic acids. In most systems, first, a long precursor transcript (pre-crRNA) originates from the repeat-spacer array (Brouns et al., 2008; Hale et al., 2008) that is cleaved into short crRNAs (Hochstrasser and Doudna, 2015). The maturation of crRNAs is essential for the functionality of CRISPR-Cas systems and facilitates the recognition and destruction of invading nucleic acids upon base pairing through the interference complex [Citation4,Citation5]. In Type I, III, V and VI systems, endoribonucleases encoded by specific cas genes recognize their cognate direct repeat (DR) sequences and mediate crRNA maturation [Citation6–Citation10]. In the majority of Type I and III systems, the cognate endoribonucleases belong to the Cas6 protein family [Citation5,Citation7,Citation11]. However, these specialized endoribonucleases can also be replaced by host enzymes in some instances [Citation12,Citation13].
Cyanobacteria are of considerable interest in biotechnology and ecology, as they are the only bacteria that perform oxygenic photosynthesis. In addition to the fixation of inorganic carbon through photosynthesis, some species are additionally capable of converting atmospheric nitrogen into organic biomass, thereby sustaining diazotrophic growth. Some of these cyanobacteria are multicellular and feature complex genomes. In these strains, the presence of multiple orphan CRISPR repeat-spacer cassettes not linked to any cas genes is typical [Citation14]. Free-standing repeat-spacer arrays were also observed in up to ~11% of previously investigated CRISPR systems of other phyla, although these findings might include some false-CRISPR elements [Citation15,Citation16]. Among the multicellular, filamentous cyanobacteria, the high frequency of orphan CRISPR arrays is illustrated by such examples as Tolypothrix bouteillei VB521301 with 44 repeat-spacer arrays but only five cas1 genes or the strain Nostoc calcicola FACHB-389, which has 19 instances of repeat-spacer arrays and only two cas1 genes [Citation14].
The strain Anabaena (Nostoc) sp. PCC 7120 (from here: Anabaena 7120) is a model for the group of multicellular, nitrogen-fixing cyanobacteria. A previously performed search for interspaced DR sequences suggested the existence of 11 CRISPR-like repeat-spacer cassettes in Anabaena 7120, which were designated CR_1 to CR_11 [Citation14]. One of these loci, CR_9, belongs to a Tn7-associated CRISPR derivative similar to those described in Scytonema hofmanni UTEX B 2349 and Anabaena cylindrica PCC 7122, two other multicellular cyanobacteria [Citation17]. In this article, below we will differentiate between ‘DR’ which stands for ‘direct repeat’ and ‘CR’, which stands for ‘CRISPR-like repeat-spacer cassette’.
All 11 arrays are transcribed and showed the crRNA-typical accumulation patterns of precursors and matured crRNAs, which is in contrast to the fact that only two instances of cas gene cassettes were found in this organism plus one additional isolated CRISPR-Cas integration cassette containing a cas1, cas2 and a csx18 gene [Citation14,Citation18]. According to the specific cas gene content found in these cassettes, the existence of a Type I-D and a III-D system in Anabaena 7120 was predicted [Citation14,Citation15]. These cas gene cassettes are linked to CR_4 (the I-D system) or CR_2 and CR_3 in the case of the III-D system. Hence, together with the Tn7-linked CR_9 array, four arrays appear connected to a particular system, while the other seven (CR_1, CR_5 to CR_8 and CR_10 and CR_11) qualify as orphan arrays. In addition, a short CRISPR array was described in cyanophage N-1 [Citation19], for which Anabaena 7120 is a suitable host [Citation20,Citation21]. This CRISPR array was demonstrated to be transcribed in Anabaena 7120 during infection [Citation19].
In this study, we deleted the two cas6 genes in Anabaena 7120 and tested the specificities of the two corresponding enzymes in the resulting mutant strains as recombinant proteins and in a cell-free transcription-translation (TXTL) system.
The results assign five different arrays each to one or to the other of the two Cas6 riboendonucleases and clearly show that processing of the Tn7-associated CR_9 is independent from either of the two enzymes. Hence, the data support the functionality of repeat-spacer arrays that frequently appear fragmented to multiple genomic loci in multicellular cyanobacteria [Citation14]. In addition, we observed a striking in vivo cross-specificity between the two Cas6 enzymes in which the presence of one protein lowers the processing and crRNA accumulation of maturation products produced by the other enzyme.
Materials and methods
Cultures and construction of mutant strains
Cultures of Anabaena 7120 were grown photoautotrophically in BG11 liquid medium or on agar plates [Citation22] under white light illumination of 30–50 µmol photons m−2 s−1 at 30°C. The medium was supplemented with 30 μg mL−1 neomycin if needed.
The CRISPR-Cas12a (Cpf1) genome editing tool together with the pSL2680 plasmid (Addgene No. 85581) was used for the construction of Anabaena mutants, as previously reported [Citation23]. First, a pair of complementary oligonucleotides that target the genes to be knocked out were annealed, treated with T4 polynucleotide kinase (Thermo Fisher), and cloned into the AarI (Thermo Fisher) digested plasmid pSL2680 by T4 DNA ligase (NEB), resulting in the gRNA-cassette editing plasmids. Second, the repairing templates (1 kb upstream and downstream flanked regions, respectively) were amplified from genomic DNA of Anabaena 7120 and inserted into the KpnI (Thermo Fisher) sites of the editing plasmids by seamless assembly, resulting in the final gRNA and repairing cassette editing plasmids. Third, the final editing plasmids were introduced into wild-type Anabaena 7120 by triparental mating conjugal transfer. Finally, genotypes of exconjugants were confirmed by PCR (Figure S1), and the editing plasmids were diluted and finally removed by subculture in BG11 medium without antibiotics. Primer pairs of alr1482gRNA-1/2 and alr1566gRNA-1/2 were used to prepare the gRNA-cassette editing plasmids, and primer pairs of alr1482KO-1/2 and alr1566KO-1/2 were used to prepare the gRNA and repairing cassette editing plasmids for constructing Δalr1482 and Δalr1566, respectively. Conjugal transfer was performed as previously reported [Citation24]. The plasmids pRL443 and pRL623 and the final CRISPR editing plasmids generated in this study were used as conjugal, helper and cargo plasmids, respectively. The sequences of all oligonucleotides are listed in . All PCR fragments, plasmids generated in this study, and gene mutation regions in the mutants were verified by Sanger sequencing.
Table 1. Oligonucleotide primers used in this work.
Isolation and analysis of RNA
Total RNA was isolated from 50 mL exponentially grown cultures. Cells were harvested by centrifugation or filtration on Pall Supor 800 0.8 µm membranes. Cells were transferred to screw cap tubes together with 250 µL glass beads (0.1–0.25 µm in diameter) and 1 mL PGTX [Citation25] and were frozen in liquid nitrogen. Cell disruption (3 cycles of 3 x 6,500 rpm for 15 s with 10 s breaks in between) was performed using the Precellys 24 Dual homogenizer (Bertin) under nitrogen cooling. The supernatant was transferred to a new tube. Samples were incubated for 10 min at 65°C in a water bath, one volume chloroform:isoamyl alcohol (24:1) was added, and samples were incubated for 10 min at room temperature with several vortexing cycles. After centrifugation for 3 min at 6,500 rpm in a swing-out rotor, the supernatant was transferred to a fresh tube, and one volume of chloroform:isoamyl alcohol was added. This step was repeated twice. Finally, RNA was precipitated by the addition of one volume of isopropanol. Alternatively, for the analyses of cas6 deletion mutants, we also used the Retsch MM400 system for cell disruption. Nitrogen frozen samples were thawed and mixed with one volume of glass beads (0.1–0.25 µm and 0.5 µM in diameter, mixed in equal amounts), and cell lysis was performed in three cycles for 10 min at a frequency of 10 Hz. Between each cycle, it was checked that the samples did not become warm and were cooled if necessary. The supernatant was transferred to a fresh tube and mixed with one volume of PGTX. Then, extraction was preceded as described above.
CRISPR-related transcript accumulation was analysed by Northern hybridization using single-stranded radioactively labelled RNA probes transcribed in vitro from PCR-generated templates (see for primers), as described [Citation26].
Transcriptome analysis of the small RNA fraction of Anabaena 7120
The small RNA fraction (< 200 nt) was isolated from a single sample of total RNA from cultures grown under standard conditions using the RNeasy MinElute Cleanup kit (Qiagen). The preparation of cDNA libraries and sequencing were performed by vertis AG, Freising, Germany. The small RNA fraction sample was split into two parts. One half was first treated with Antarctic Phosphatase and rephosphorylated with T4 Polynucleotide Kinase (+ PNK), and the other half was left untreated (- PNK). Then, oligonucleotide adapters were ligated to the 5ʹ and 3ʹ ends of the RNA samples. First-strand cDNA synthesis was performed using M-MLV reverse transcriptase and the 3ʹ adapter as the primer. The resulting cDNAs were amplified by 12 PCR cycles using a high-fidelity DNA polymerase. The cDNA was purified using the Agencourt AMPure XP kit (Beckman Coulter) and analysed by capillary electrophoresis. The cDNA libraries were single-end sequenced with a NextSeq 500 system using 150 bp read-length yielding 294,333,852 (+ PNK) and 36,509,792 (- PNK) reads. RNA-seq data analysis was performed with the tools installed on usegalaxy.eu. The single-end reads were trimmed, adapters and reads shorter than 14 nt were filtered out using cutadapt 1.16 [Citation27]. Mapping was performed on the chromosome and plasmids of Anabaena 7120 using bowtie2 version 2.3.4.3 with the parameters for single-end reads [Citation28]. The data have been uploaded to NCBI’s short reads archive and were assigned the accession number PRJNA624132.
TXTL system
For testing the Cas6 enzymes Alr1566 and Alr1482 in a cell-free transcription-translation system, the E. coli-based TXTL assay was used [Citation29,Citation30]. The myTXTL Sigma 70 Cell-Free Master Mix was purchased from Arbor Biosciences. The included p70a plasmid was used as a template for cloning of the CRISPR repeat-spacer sequences. All PCRs were performed using PCRBio HiFi polymerase (PCR Biosystems). The p70a plasmid was linearized by PCR omitting the open reading frame for the destabilized enhanced GFP (deGFP) and its 5ʹUTR. CRISPR sequences were PCR-amplified from genomic DNA of Anabaena 7120, and overlaps with p70a were added. The repeat-spacer sequences were thus fused directly downstream of the bacteriophage λ p70a promoter and upstream of the T500 terminator.
E. coli K12 codon-optimized sequences of alr1566 and alr1482 were purchased from IDT as gBlocks and subsequently PCR-amplified with overlaps to a linearized pet28a(+) plasmid. The Cas6 proteins were thus expressed from an IPTG-inducible T7 promoter with a C-terminal 6xHis tag. Fragment assembly was performed at room temperature for 30 min upon transformation into chemically competent E. coli DH5alpha cells for cloning. Assembled plasmids were isolated, and regions of interest were sequenced (Eurofins Genomics).
For the expression of proteins encoded on pet28a(+) in the TXTL assay, T7 RNA polymerase, expressed in this instance from p70a, and IPTG are necessary. Reactions were performed for 16 h at 29°C in a total volume of 12 µL with 9 µL TXTL master mix, 1 mM IPTG, 0.5 nM p70a_T7RNAP, 2 nM pet28a(+)_alr1482/alr1566, and 10 nM p70a_CR_1-CR_11_S1-S3. For Western blot analysis, 2 µL was loaded on a 10% polyacrylamide SDS gel. Western blot analysis was performed using the Penta His HRP conjugate kit (Qiagen). To check for equal loading, the membrane was stained with Ponceau S (0.1% (w/v) in 5% acetic acid). For RNA analysis, 90 µL H2O and 100 µL phenol:chloroform:isoamyl alcohol (25:24:1) were added to 10 µL of the reactions. Phases were separated using 5Prime Phase Lock Gel heavy 2 (Quantabio). The supernatant was purified with RNA Clean and Concentrator 25 columns (Zymo Research). Ten micrograms of each sample was loaded onto a 10% polyacrylamide gel containing 8 M urea. Northern hybridizations were performed as described [Citation26] (see for probe template PCR primers).
Expression and purification of Alr1482 and Alr1566 and cleavage assays
E. coli K12 codon-optimized alr1566 and alr1482 were expressed from pet28a(+) upon 1 mM IPTG induction in E. coli BL21(DE3) in a 1 L culture volume overnight at room temperature. Cell disruption (3 cycles of 3 x 6,000 rpm for 10 s interrupted by 5 s breaks) was performed with the Precellys 24 Dual homogenizer (Bertin) under nitrogen cooling. Filtered lysate was used for protein purification with an Äkta start system (GE Healthcare). Lysis buffer containing 20 mM imidazole and wash buffer with 40 mM imidazole was used. Elution was performed gradually from 40 to 500 mM imidazole. Fractions containing the purified protein were pooled and desalted with PD midi Trap G-25 (GE Healthcare) using the spin protocol, and buffer was thereby exchanged with PBS (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4 1.8 mM KH2PO4; pH 7.4). Protein concentrations were determined with a direct detection spectrometer (Merck), and purified proteins were stored at −80°C.
Enzymatic reactions were performed in a total reaction volume of 10 µL at 37°C for 1 h in a reaction buffer with 20 mM Tris-HCl, pH 7.8 and 400 mM KCl. As a substrate, 1 µM synthetic RNA oligonucleotides were used. The proteins Alr1482 or Alr1566 were added to a concentration of 3 µM. For experiments with 32P isotope-labelled probes, 30 pmol RNA was 5′-end-labelled using 40 µCi γ-ATP (3000 Ci/mmol, 10 mCi/mL, Hartmann Analytic) and 30 U of T4 polynucleotide kinase (Thermo Fisher) for 30 min at 37°C in a volume of 30 μL. Reactions were stopped by the addition of 1.5 μL 0.5 M EDTA (pH 8.0) and incubation for 10 min at 80°C. Unincorporated nucleotides were removed by using MicroSpin G-25 Columns (GE Healthcare). For the enzymatic reactions, the labelled RNA was incubated with 0.3 µM Cas6 at a final concentration of 0.1 µM. A total of 150 ng of the ZR small RNA ladder (Zymo Research) and 120 pmol of a homemade mix of synthetic RNA (MX with 31, 29, 26, P-22 nt) were labelled analogously. To stop the reaction, RNA loading dye was added, and the fragments were separated on an 8 M urea 10% polyacrylamide sequencing gel upon staining with SYBR gold [Citation32]. Radioactive signals were detected after drying the gel and exposing it to a storage phosphor screen (Kodak) with a GE Typhoon FLA 9500 imaging system. Synthetic CRISPR repeat oligonucleotides () were purchased from Biomers.
Results
Two Cas6 proteins facing eight major types of repeats
Multiple sequence alignments of the ten CRISPR DRs (with the exclusion of the Tn7-associated CR_9 array) showed considerable differences between them (). As in many CRISPR-Cas systems, minor sequence variants occur (Figure S2). For instance, three variants exist for the CR_2 and CR_3 arrays, which belong to the same subtype III-D system but are split due to the insertion of a gene encoding Cas1-reverse transcriptase fusion protein [Citation14]. Some DRs, such as DR1 and DR7, have a sequence identity of <50% with each other ().
Figure 1. CRISPR repeats and Cas6 proteins in Anabaena 7120.
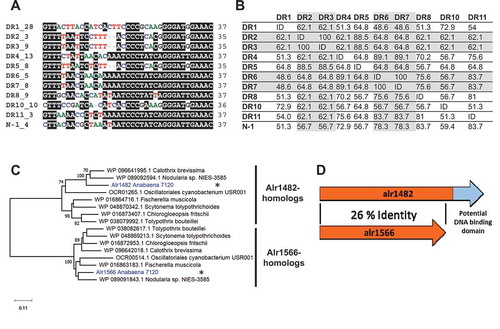
On the other hand, some DRs are identical, such as the major variants of CR_6 and CR_7, which fits the observation that CR_6 and CR_7 belong to one and the same array and probably only recently became separated by the insertion of a 134 nt miniature inverted repeat element (MITE) [Citation14]. Hence, some of the array DRs were joined (all variants of CR_2 and CR_3 as well as CR_6 and CR_7), leaving 8 variants of clearly distinct sequences (plus the even more divergent CR_9). In addition, we included the DR of the array in phage N-1, which differed in six positions from the most closely related host-located sequence, DR8 ().
These differences raised the question of whether all these different types of DRs were substrates for one of the two identified Cas6 proteins, Alr1482 and Alr1566, encoded in Anabaena 7120, or both. These two Cas6 proteins exhibit a sequence identity of only 26% with each other and differ considerably in length from either 290 aa (Alr1566) or 377 aa (Alr1482). The extension of Alr1482 relative to Alr1566 includes an additional domain of 70 amino acids. According to the structural prediction by HHpred [Citation31], this domain possesses similarity to helix-turn-helix domains, as well as similarity to domain 4 of σ-factor 70 and replication initiator proteins (TrpR- and DnaA- domains) (Figure S11). The possible functionality of this motif is currently elusive, but these similarities suggest a double-strand DNA-binding role.
To safeguard that no additional Cas6 proteins exist in Anabaena 7120, a library of 9891 annotated Cas6, Csf5 and GSU0054 (a subtype I-U Cas5/6 fusion protein [Citation33] recently re-classified as belonging to subtype I-G [Citation2]) proteins from NCBI was created and compared by blastP against the entire protein set of Anabaena 7120. This search yielded in addition to Alr1482 and Alr1566 four further proteins with a sequence overlap ≥ 150 amino acids (E-value ≤ 0.01). Three of these proteins were only aligned to the reverse transcriptase-domain fused to some Cas6 and showed no similarity to Cas6 itself. The other protein, All0207, possesses only weak sequence similarity in its respective Cas6 alignment (identity: 26.14 %). Furthermore, an HHpred analysis and a search against NCBI's Conserved Domain Database [Citation34] indicated the presence of a phosphodiesterase/alkaline phosphatase domain in All0207. Together with the fact that All0207 was detected in the Anabaena 7120 exoproteome [Citation35], a function as an extracellular protein involved in extracellular DNA or RNA degradation appears likely, which would explain the weak similarity to Cas6. We conclude that Alr1482 and Alr1566 are the only two Cas6 proteins in Anabaena 7120.
Interestingly, there are close homologs to both enzymes in a large number of other cyanobacteria, and in many instances, homologs of the two proteins coexist, similar to the situation in Anabaena 7120. Phylogenetic analysis showed that the two Cas6 variants belong to a clearly distinct clade and a partition of all found homologs to one or the other clade ( and S3). We identified 38 species of Cyanobacteria with at least one homolog of both types of cas6 genes (Figure S3). All of these species belong to the order Nostocales, except for Oscillatoriales cyanobacterium USR001. Despite the general relatedness of these homologs, they varied up to 44% compared to their respective Cas6 variants in Anabaena 7120. The extra C-terminal domain was consistently present in all homologs in the Alr1482 clade and showed high sequence conservation.
Analyses in a cell-free transcription-translation system
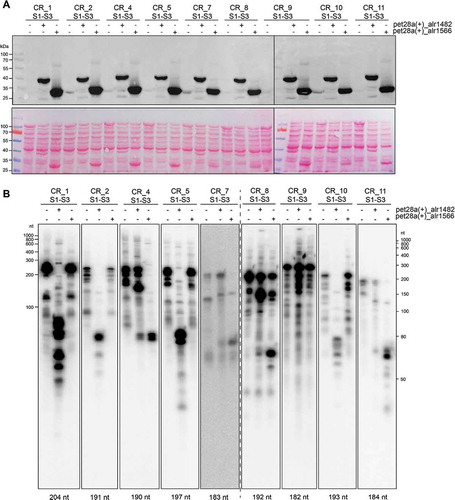
The two Anabaena 7120 Cas6 proteins Alr1482 and Alr1566 were expressed with a C-terminal 6xHis tag in a cell-free E. coli-based transcription-translation system (TXTL [Citation29,Citation30]). The expression from vector pet28a(+) was driven by T7 RNA polymerase and IPTG inducible. Both proteins were detected by Western blot analysis with respective sizes of 43 kDa for Alr1482 and 33.6 kDa for Alr1566 (). In parallel, crRNA precursors matching the sequences of CR_1, 2, 4, 5, 7, 8, 9, 10 and 11 were constitutively expressed from vector p70a. Each of these precursors consisted of the first three spacers and two DR instances (S1-R-S2-R-S3). The full-length transcripts of these different pre-crRNAs were detected by Northern hybridization (). The length of all transcripts appeared slightly higher compared to the calculated lengths, although transcription should have been terminated by the p70a terminator T500. In addition to the full-length transcripts, all reactions showed additional bands, which can often be seen for in vitro transcripts and is due to premature transcription termination. Nevertheless, one can clearly see that the addition of one or the other Cas6 protein changed the pattern of the pre-crRNA to shorter products with less than 100 nt. Alr1566 was able to process pre-crRNA derived from CR_4, 7, 8 and 11. Alr1482 was able to process not only CR_1, 2, 5 and 10 precursors but also CR_4, 7, 8 and 11, albeit less effectively than Alr1566. The CR_9 precursor was processed by neither Alr1482 nor Alr1566. The results of these analyses are summarized in .
Table 2. Activities of the two Cas6 enzymes in the TXTL assay (cf. ).
Figure 2. TXTL assay of Cas6 specificities.
Cleavage assays of CR_1-11 synthetic RNA repeats with recombinant Alr1482 or Alr1566
Both Cas6 proteins were purified as recombinant proteins and tested against all nine major DR types. shows that the DRs of CR_4, 6/7, 8 and 11 were mainly cleaved by Alr1566 but to a lesser extent by Alr1482. In contrast, the DRs of CR_1, 2/3, 5 and 10 were only cleaved by Alr1482. The DR of CR_9 was not cleaved by either of the two Cas6 proteins. For CR_1, 4, 5, 6/7 and 11, two cleavage products were observed irrespective of which of the two enzymes was responsible for the cleavage. To address the origin of the cleavage products, we performed cleavage reactions with 5ʹ radioactively labelled RNA, which is shown as an example for DR_1 and DR_4 in . The shorter of the two cleavage products for both repeats that were visible with SYBR Gold staining was not labelled radioactively. This finding shows that the second cleavage site was not located near the other cleavage site at the 3ʹ repeat end but was located close, between 2 and 4 nucleotides, to the 5ʹ end of the repeat.
Figure 3. Assays using purified Cas6 proteins.
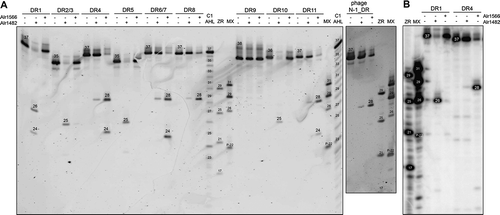
The genome of cyanophage N-1 contains a CRISPR repeat-spacer array [Citation19]. The corresponding DR was included in the in vitro assay with the Cas6 proteins of Anabaena 7120. The N-1 DR was preferentially cleaved by Alr1566 and, to a lesser extent, by Alr1482 (). Thus, the phage repeat behaved similar to the repeats of CR_4, 6/7, 8 and 11 of Anabaena 7120. This finding is consistent with the fact that these DR sequences shared 72.9 to 83.7% sequence identity with the N-1 DR, while all other DRs showed <60% identity ().
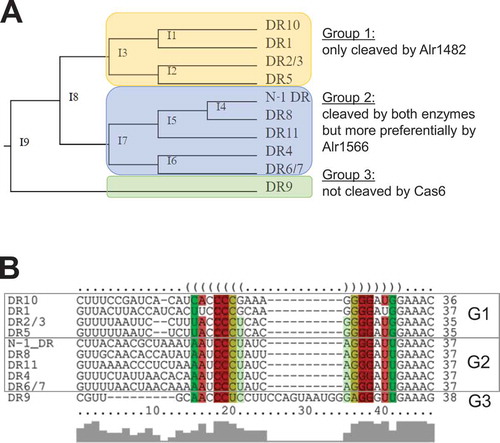
According to the in vitro cleavage results, the DRs could be classified into three groups that precisely matched the classification based on a sequence-structure comparison (). Analyses using the LocARNA algorithm that aligns sequences based on sequence and structure comparison [Citation36,Citation37] grouped the DRs from CR_1, 2/3, 5 and 10 into one group (group 1, G1). All these DRs were cleaved by Alr1482 and not by Alr1566. The DRs of CR_4, 6/7, 8, 11 and of the N-1 phage also grouped together (group 2, G2). The DRs of G2 have in common that they were cleaved by both enzymes but more preferentially by Alr1566. In contrast to all other DRs, those of CR_9 could not be assigned to either G1 or G2 and therefore belonged to separate group 3 (G3). This classification is consistent with the TXTL results, as well. The secondary structure prediction shows a single stem-loop formed by the palindromic sequence within the DRs. The stem consists of all DRs of six to eight paired bases. An apparent difference is the nonmatching C:A in the middle of the predicted stem in all group 1 DRs (). The longer single–stranded loop region of 13 nt in DR_9 sets the only group 3 secondary structure apart from all other DRs that have loops of only three or four nucleotides ().
Figure 4. Classification of crRNA maturation in Anabaena 7120 according to sequence-structure alignments and Cas6 cleavage specificities.
Analysis of cas6 deletion mutants provides in vivo evidence for crosstalk
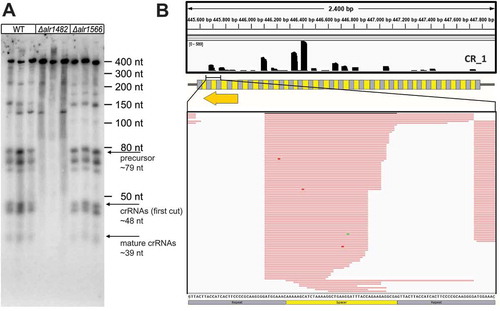
Cas6 enzymes generate the crRNA 5ʹ tags, in many systems by a single cleavage eight nucleotides from the 3ʹ end of the respective repeat (for review see Behler and Hess (2019). The results of the in vitro assays shown in seem to deviate from this rule because in several instances, two bands were generated. Moreover, the longer cleavage fragments differed in lengths, seemingly one or two nucleotides too short to leave 8 nucleotide-long tags. Hence, to compare the results of the in vitro and in silico analyses with the in vivo situation, an sRNA transcriptome analysis of WT cultures was performed and deletion mutants of the two cas6 genes (Δalr1482 and Δalr1566) were constructed and analysed. Consistent with the in vitro data, the crRNAs of all group 1 arrays were expressed and processed in the WT and Δalr1566 but not in Δalr1482, as shown in , for this group using the CR_1 array as an example. Moreover, the data from the sRNA transcriptome analysis of WT cultures showed that in vivo, the mature crRNAs started precisely with the 8 nt 5ʹ tags that are needed for self/nonself discrimination and loading of the mature crRNA into the effector complexes (). Similar results were obtained for the other group 1 DRs (Figures S5 to S7).
Figure 5. Analysis of cas6 deletion mutants for the CR_1 array representing group 1 arrays in vivo.
In contrast, the CRISPR arrays of group 2 were processed just in the WT and in Δalr1482, while in Δalr1566, only some precursors >200 nt in length were detected, exemplified for this group by means of the CR_8 array in . Again, the comparison to sRNA transcriptome data from WT cultures showed that in vivo, the mature crRNAs started precisely with the 8 nt 5ʹ tags in vivo ().
Figure 6. Analysis of cas6 deletion mutants for the CR_8 array representing group 2 arrays in vivo.
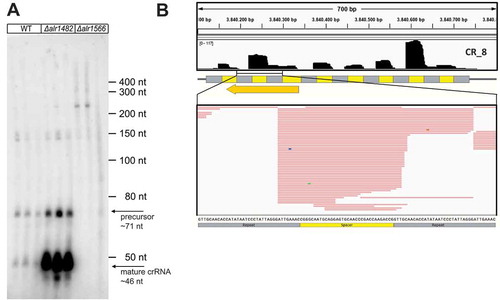
These data suggest that Alr1482 is not able to properly cleave the group 2 repeats in vivo. However, a striking observation was the highly increased accumulation of mature CR_8 crRNAs in Δalr1482 compared to the WT. This overaccumulation is also visible for the other group 2 crRNAs (Figures S8 to S10).
Discussion
While the coexistence of multiple CRISPR-Cas systems in one organism is frequent, there are notably few studies in which the role of co-occurring Cas6 was investigated. Reimann et al. (2017) found evidence for weak promiscuity of Cas6-1 and Cas6-2a in the cyanobacterium Synechocystis sp. PCC 6803 in vitro but not in vivo. In the archaeon Pyrococcus furiosus, three CRISPR-Cas systems exist (Types I-A, I-G and III-B) and seven repeat-spacer arrays, while only a single cas6 gene is present [Citation38]. In another Euryarchaeon, Methanosarcina mazei strain Gö1, a Type I-B and a Type III-C system coexist, with each being associated with its own CRISPR array and a single cas6 gene [Citation39]. Assays in vitro unexpectedly indicated that both Cas6 were able to process the precursor RNA from both CRISPR arrays, while in vivo, only one of the enzymes, that of the I-B system, mediated the maturation of crRNAs from both systems [Citation40]. In contrast to observations made in Anabaena 7120, the DR sequences in Methanosarcina mazei Gö1 are highly similar and differ by a maximum of two positions.
In our specificity assays, we included the major DR sequence of the CRISPR array in cyanophage N-1 (). This sequence was cleaved by both enzymes but more preferentially by Alr1566, consistent with its assignment to group 2 according to the classification based on sequence and secondary structure (). These data strongly suggest that the cas gene-free system of phage N-1 is fully competent to interact with host-encoded Cas proteins. While there are no reliable targets predicted for the four spacers present in the N-1 array, we identified a single spacer in Anabaena 7120 to target the N-1 genome (Figure S4). Spacer 12 of the CR_1 system is identical in 40 of its 45 nt positions to the region from nucleotides 5,214 to 5,258 on the reverse strand of the N-1 genome (GenBank accession KU234532), in which an unknown protein is encoded that in database searches revealed a single homolog in cyanophage A-1. These findings further support the tight interaction between this phage and Anabaena 7120. However, the five mismatches between CR_1 spacer 12 and the phage N-1 genome likely render the immunity inactive because we observed a high efficiency of plaque formation (not shown).
Our data provide further evidence that all the distributed CRISPR arrays in Anabaena 7120 are likely functional. The results clearly assign all 11 identified repeat spacer arrays to three systems:
group 1 crRNAs, which result in vivo and in vitro only from processing by Alr1482, belong to the Type III-D system,
group 2 crRNAs, which are primarily processed by Alr1566, belong to the Type I-D system
the group 3 array belongs to the Tn7-associated system and is independent of either Cas6.
The most striking finding was the group 2 arrays that are primarily processed by Alr1566 and overaccumulated in the Δalr1482 mutant compared to the WT (, Figures S8 to S10). The fully matured crRNAs are central for the assembly of functionally active defence complexes. Therefore, it is likely that more functional CRISPR-Cas complexes that carry group 2 crRNAs exist in the Δalr1482 mutant. This property probably is biologically meaningful. The enhanced number of CRISPR-Cas complexes with group 2 crRNAs could make up for a deficiency in the other type of complexes. Such deficiency could resulting, for instance, from mutations or neutralization by an anti-CRISPR factor. Anti-CRISPR proteins have been characterized for several different types of CRISPR-Cas systems and have also been predicted for Type I-D [Citation41] and Type III-D systems [Citation42].
The main conclusion from these observations is that the parallel presence of Alr1482 in the WT normally has a dampening effect on the expression of the Type I-D system carrying group 2 crRNAs. This effect could be mediated at the posttranscriptional level. In this scenario, the observed weak cross-reactivity of Alr1482 for group 2 crRNA maturation () might lead to an association of the Alr1482 Cas6 endonuclease with the respective products of this maturation. The resulting group 2 crRNA-Alr1482 complexes are likely incompatible with the assembly of the I-D interference complexes and therefore become rapidly degraded. In an alternative scenario, this crosstalk could be mediated directly via the potential DNA binding domain in Alr1482 (). Clarifying the functional relevance of this domain as well as identifying the evolutionary forces and genetic mechanisms driving the previously observed frequent fragmentation of CRISPR-Cas repeat-spacer arrays in filamentous cyanobacteria [Citation14], warrant further research.
Disclosure of potential conflicts of interest
The authors have no conflicts of interest to declare.
Author contributions
VR, TZ and MZ performed the experimental analyses, and bioinformatics analyses were performed by MZ and WRH. HL contributed to the strain construction, and JB contributed to the small RNA analysis. WRH, VR, TZ and XL drafted the manuscript with contributions from all authors.
Supplemental Material
Download PDF (2.6 MB)Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
References
- Hille F, Richter H, Wong SP, et al. The biology of CRISPR-Cas: backward and forward. Cell. 2018;172:1239–1259.
- Makarova KS, Wolf YI, Iranzo J, et al. Evolutionary classification of CRISPR-Cas systems: a burst of class 2 and derived variants. Nat Rev Microbiol. 2020;18:67–83.
- Westra ER, Dowling AJ, Broniewski JM, et al. Evolution and ecology of CRISPR. Annu Rev Ecol Evol Syst. 2016;47:307–331.
- Charpentier E, Richter H, van der Oost J, et al. Biogenesis pathways of RNA guides in archaeal and bacterial CRISPR-Cas adaptive immunity. FEMS Microbiol Rev. 2015;39:428–441.
- Hochstrasser ML, Doudna JA. Cutting it close: CRISPR-associated endoribonuclease structure and function. Trends Biochem Sci. 2015;40:58–66.
- Behler J, Hess WR. Approaches to study CRISPR RNA biogenesis and the key players involved. Methods. 2019;172:12–26.
- Hale C, Kleppe K, Terns RM, et al. Prokaryotic silencing (psi)RNAs in Pyrococcus furiosus. RNA. 2008;14:2572–2579.
- Hale CR, Zhao P, Olson S, et al. RNA-guided RNA cleavage by a CRISPR RNA-Cas protein complex. Cell. 2009;139:945–956.
- Karginov FV, Hannon GJ. The CRISPR system: small RNA-guided defense in Bacteria and Archaea. Mol Cell. 2010;37:7–19.
- Przybilski R, Richter C, Gristwood T, et al. Csy4 is responsible for CRISPR RNA processing in Pectobacterium atrosepticum. RNA Biol. 2011;8:517–528.
- Carte J, Wang R, Li H, et al. Cas6 is an endoribonuclease that generates guide RNAs for invader defense in prokaryotes. Genes Dev. 2008;22:3489–3496.
- Behler J, Sharma K, Reimann V, et al. The host-encoded RNase E endonuclease as the crRNA maturation enzyme in a CRISPR–Cas subtype III-Bv system. Nat Microbiol. 2018;3:367–377.
- Chou-Zheng L, Hatoum-Aslan A. A type III-A CRISPR-Cas system employs degradosome nucleases to ensure robust immunity. Elife. 2019;8:e45393.
- Hou S, Brenes-Álvarez M, Reimann V, et al. CRISPR-Cas systems in multicellular cyanobacteria. RNA Biol. 2019;16:518–529.
- Makarova KS, Wolf YI, Alkhnbashi OS, et al. An updated evolutionary classification of CRISPR-Cas systems. Nat Rev Microbiol. 2015;13:722–736.
- Zhang Q, Ye Y. Not all predicted CRISPR–Cas systems are equal: isolated cas genes and classes of CRISPR like elements. BMC Bioinformatics. 2017;18:92.
- Strecker J, Ladha A, Gardner Z, et al. RNA-guided DNA insertion with CRISPR-associated transposases. Science. 2019;365:48–53.
- Shah SA, Alkhnbashi OS, Behler J, et al. Comprehensive search for accessory proteins encoded with archaeal and bacterial type III CRISPR-cas gene cassettes reveals 39 new cas gene families. RNA Biol. 2019;16:530–542.
- Chénard C, Wirth JF, Suttle CA. Viruses infecting a freshwater filamentous Cyanobacterium (Nostoc sp.) encode a functional CRISPR array and a proteobacterial DNA polymerase B. MBio. 2016;7:e00667–16.
- Adolph KW, Haselkorn R. Isolation and characterization of a virus infecting the blue-green alga Nostoc muscorum. Virology. 1971;46:200–208.
- Adolph KW, Haselkorn R. Blue-green algal virus N-1: physical properties and disassembly into structural parts. Virology. 1973;53:427–440.
- Rippka R, Deruelles J, Waterbury JB, et al. Generic assignments, strain histories and properties of pure cultures of cyanobacteria. Microbiology. 1979;111:1–61.
- Ungerer J, Pakrasi HB. Cpf1 is a versatile tool for CRISPR genome editing across diverse species of Cyanobacteria. Sci Rep. 2016;6:39681.
- Elhai J, Wolk CP. Conjugal transfer of DNA to cyanobacteria. Meth Enzymol. 1988;167:747–754.
- Pinto FL, Thapper A, Sontheim W, et al. Analysis of current and alternative phenol based RNA extraction methodologies for cyanobacteria. BMC Mol Biol. 2009;10:79.
- Steglich C, Futschik ME, Lindell D, et al. The challenge of rRegulation in a minimal photoautotroph: non-coding RNAs in Prochlorococcus. PLoS Genet. 2008;4:e1000173.
- Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal. 2011;17:10.
- Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–359.
- Maxwell CS, Jacobsen T, Marshall R, et al. A detailed cell-free transcription-translation-based assay to decipher CRISPR protospacer-adjacent motifs. Methods. 2018;143:48–57.
- Shin J, Noireaux V. An E. coli cell-free expression toolbox: application to synthetic gene circuits and artificial cells. ACS Synth Biol. 2012;1:29–41.
- Zimmermann L, Stephens A, Nam S-Z, et al. A completely reimplemented MPI bioinformatics toolkit with a new HHpred server at its core. J Mol Biol. 2018;430:2237–2243.
- Reimann V, Alkhnbashi OS, Saunders SJ, et al. Structural constraints and enzymatic promiscuity in the Cas6-dependent generation of crRNAs. Nucleic Acids Res. 2017;45:915–925.
- Almendros C, Nobrega FL, McKenzie RE, et al. Cas4-Cas1 fusions drive efficient PAM selection and control CRISPR adaptation. Nucleic Acids Res. 2019;47:5223–5230.
- Marchler-Bauer A, Bo Y, Han L, et al. CDD/SPARCLE: functional classification of proteins via subfamily domain architectures. Nucleic Acids Res. 2017;45:D200–3.
- Oliveira P, Martins NM, Santos M, et al. The Anabaena sp. PCC 7120 exoproteome: taking a peek outside the box. Life (Basel). 2015;5:130–163.
- Raden M, Ali SM, Alkhnbashi OS, et al. Freiburg RNA tools: a central online resource for RNA-focused research and teaching. Nucleic Acids Res. 2018;46:W25–9.
- Will S, Joshi T, Hofacker IL, et al. LocARNA-P: accurate boundary prediction and improved detection of structural RNAs. RNA. 2012;18:900–914.
- Majumdar S, Zhao P, Pfister NT, et al. Three CRISPR-Cas immune effector complexes coexist in Pyrococcus furiosus. RNA. 2015;21:1147–1158.
- Nickel L, Weidenbach K, Jäger D, et al. Two CRISPR-Cas systems in Methanosarcina mazei strain Gö1 display common processing features despite belonging to different types I and III. RNA Biol. 2013;10:779–791.
- Nickel L, Ulbricht A, Alkhnbashi OS, et al. Cross-cleavage activity of Cas6b in crRNA processing of two different CRISPR-Cas systems in Methanosarcina mazei Gö1. RNA Biol. 2019;16:492–503.
- He F, Bhoobalan-Chitty Y, Van LB, et al. Anti-CRISPR proteins encoded by archaeal lytic viruses inhibit subtype I-D immunity. Nat Microbiol. 2018;3:461–469.
- Gussow AB, Shmakov SA, Makarova KS, et al. Vast diversity of anti-CRISPR proteins predicted with a machine-learning approach. bioRxiv. 2020. DOI:2020.01.23.916767.
- Kumar S, Stecher G, Li M, et al. Genetics Analysis across computing platforms. Mol Biol Evol. 2018;35:1547–1549.
- Rzhetsky A, Nei M. A simple method for estimating and testing minimum evolution trees. Mol Biol Evol. 1992;9:945–967.
- Felsenstein J. Confidence limits on phylogenies: an approach using the bootstrap. Evolution. 1985;39:783–791.
- Zuckerkandl E, Pauling L. Evolutionary divergence and convergence in proteins. In: Bryson V, Vogel HJ, editors. Evolving Genes and Proteins. New York, NY: Academic Press; 1965. p. 97–166.
- Nei M, Kumar S. Molecular Evolution and Phylogenetics. New York, NY: Oxford University Press; 2000.
- Saitou N, Nei M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4:406–425.