
ABSTRACT
RNase J is a prokaryotic 5′-3′ exo/endoribonuclease that functions in mRNA decay and rRNA maturation. Here, we report a novel duplex unwinding activity of mpy-RNase J, an archaeal RNase J from Methanolobus psychrophilus, which enables it to degrade duplex RNAs with hairpins up to 40 bp when linking a 5′ single-stranded overhangs of ≥ 7 nt, corresponding to the RNA channel length. A 6-nt RNA-mpy-RNase J-S247A structure reveals the RNA-interacting residues and a steric barrier at the RNA channel entrance comprising two archaeal loops and two helices. Mutagenesis of the residues key to either exoribonucleolysis or RNA translocation diminished the duplex unwinding activity. Substitution of the residues in the steric barrier yielded stalled degradation intermediates at the duplex RNA regions. Thus, an exoribonucleolysis-driven and steric occlusion-based duplex unwinding mechanism was identified. The duplex unwinding activity confers mpy-RNase J the capability of degrading highly structured RNAs, including the bacterial REP RNA, and archaeal mRNAs, rRNAs, tRNAs, SRPs, RNase P and CD-box RNAs, providing an indicative of the potential key roles of mpy-RNase J in pleiotropic RNA metabolisms. Hydrolysis-coupled duplex unwinding activity was also detected in a bacterial RNase J, which may use a shared but slightly different unwinding mechanism from archaeal RNase Js, indicating that duplex unwinding is a common property of the prokaryotic RNase Js.
Introduction
RNA decay plays essential roles in the control of gene expression and RNA metabolism, which are critical biological processes for the survival and optimal physiology of both eukaryotes and prokaryotes. In particular, the RNA decay process at the post-transcriptional stage controls the RNA lifespan and translation efficiency in response to the changing requirement for protein synthesis of cells [Citation1–Citation5]. This fundamental biological process involves the synergistic action of a suite of endonucleases and/or 5′-3′ or 3′-5′ exonucleases [Citation6]. In eukaryotes, RNA decay is primarily implemented by either 3′-5′ exonuclease (i.e., exosome) on the de-adenylated 3′-end or the 5′-3′ exonuclease Xrn1 on the 5′-cap removed transcript [Citation2,Citation4,Citation7–Citation10]. In bacteria, mRNA decay is generally initiated by endoribonucleolytic cleavage [Citation11–Citation15], and followed by digestions of 3′-5′ exonucleases, such as polynucleotide phosphorylase (PNPase), RNase II, and RNase R [Citation6,Citation15–Citation18]. PNPase, RNase II, and also the archaeal RNA exosome are capable of exoribonucleolytically degrading single-stranded RNAs with a high processivity, but stall when encountering stable stem-loop structures [Citation18,Citation19] unless otherwise aided by associated RNA helicases. For example, bacterial PNPase degrades structured RNAs by complexing with an ATP-dependent RNA helicase RhlB, a member of the DEAD-box family [Citation20–Citation22]. Similarly, the eukaryotic RNA exosome complex contains the RNA helicase Mtr4 and degrades structured RNAs and specific substrates [Citation23]. However, some exoribonucleases, like the bacterial RNase R [Citation24], and the eukaryotic cytoplasmic Xrn1, nuclear Xrn2 [Citation25] and 3′–5′ exoribonuclease Rrp44 (Dis3) [Citation26], have intrinsic ATP-independent helicase-like activities, and thus are capable of degrading extensive secondary structured RNAs by themselves without requiring other proteins. Consequently, these characteristics confer them the primary roles in RNA metabolisms, such as mRNA decay, rRNA and tRNA quality control, as well as non-functional rRNA and tRNA degradation [Citation24,Citation25,Citation27–Citation30]. Thus, helicase-like activity, also referred to as duplex unwinding activity, is essential for exoribonucleases to degrade structural RNAs independently and allows them to play fundamental roles in pleiotropic RNA metabolism.
RNA degradation through the 5′-to-3′ exoribonucleolytic pathway is conventionally believed to be absent in bacteria until the discovery of the 5′-3′ exonuclease RNase J [Citation31–Citation33]. RNase J plays an important role in the 5′-end maturation of pre-16S rRNA and mRNA degradation from 5′-termini [Citation31,Citation32,Citation34]. The B. subtilis RNase J1 as well as its paralog RNase J2 are both involved in governing global mRNA stability and the cellular abundance of bulk transcripts, and hence are regarded as global post-transcriptional controllers for cell growth, morphology, and sporulation [Citation35–Citation37]. RNase J has also been reported to function as a pleiotropic post-transcriptional regulator involved in growth, morphology, acid tolerance, natural competence, biofilm formation, and virulence of Staphylococcus and Streptococcus species [Citation38–Citation40].
However, mRNA decay and ribonucleases are far less thoroughly understood in Archaea than that in Eukarya and Bacteria [Citation4,Citation6,Citation41]. The recent discoveries of the archaeal β-CASP family proteins, with which the archaeal RNase Js affiliate, would shed light on such knowledge [Citation41–Citation43]. In-depth phylogenomic analysis has revealed that bacterial RNase J orthologs are highly represented in Archaea [Citation42–Citation44]. So far, highly processive 5′-3′ exoribonuclease activities have been identified in four archaeal RNase Js, namely, Pab-RNase J of Pyrococcus abyssi[Citation44], Tk-RNase J of Thermococcus kodakaraensis[Citation44], Mja-RNase J of Methanocaldococcus jannashii (RNase J3 in [Citation45]), and mpy-RNase J of Methanolobus psychrophilus R15 [Citation46]. Given the wide distribution and the analogous 5′-3′ exoribonuclease activity to bacterial RNase Js, archaeal RNase Js are predicted to play similar roles in RNA maturation, degradation, and/or other types of RNA metabolism in Archaea [Citation41]. The solved RNA-free and -bound structures of mpy-RNase J further reveal that archaeal RNase J lacks the bacterial C-terminal domain and only possesses a metallo-β-lactamase domain (MβL) with the insertion of a β-CASP domain, and RNA is bound in a single-stranded RNA (ssRNA) channel composed of the MβL and β-CASP domains [Citation46]. Previously, we determined that a hydrogen bond network facilitates a two-zinc ion catalytic mechanism in mpy-RNase J, and the specific 5′-monophosphate (5′P) pocket and conserved sandwich pocket are critical to its high 5′-3′ exoribonucleolytic catalysis and processivity [Citation46]. Moreover, two unique structural archaeal loops (loops I and II) were predicted involving in interaction with RNA [Citation44,Citation46].
Here, we report the novel duplex unwinding activity of mpy-RNase J, and by solving a 6-nt RNA-bound structure in combination with site mutagenesis and enzymatic assays on various duplex RNAs, we have determined that the duplex unwinding activity is achieved by an exoribonucleolysis-driven and steric occlusion-coupled mechanism. Using 5′-3′ exoribonucleolysis released energy, mpy-RNase J is capable of unwinding duplex RNA via a steric barrier, which is composed of the two unique archaeal loops I and II, and helices α5 and α9 at the RNA channel entrance. Furthermore, we found that the duplex RNA unwinding activity facilitates mpy-RNase J to efficiently degrade cellular structured RNAs, including rRNAs, tRNAs, functional RNAs, and highly structured mRNA 5′ untranslated regions (5′UTRs). A similar duplex unwinding activity was also detected in a bacterial RNase J homolog, sco-RNase J from Streptomyces coelicolor, suggesting that this is a shared property among prokaryotic 5′-3′ exonucleases.
Materials and methods
Microbial strains and culture conditions
Methanolobus psychrophilus R15 was grown at 30°C in a defined mineral medium containing 20 mM trimethylamine and under a gas phase of N2/CO2 (80: 20, v/v; 0.1 MPa) as previously described [Citation5,Citation47]. Escherichia coli strains DH5α, JM109, and BL21(DE)3plyS were grown in Luria-Bertani (LB) medium supplemented with 100 μg/mL of ampicillin or 50 μg/mL of kanamycin at 37°C with shaking.
Extraction of genomic DNA and total RNA
The genomic DNA of M. psychrophilus R15 was extracted and purified from mid-exponential phase 37°C-grown cultures when the OD600 was at 0.5. DNA was extracted using the TIANamp bacteria DNA kit (TIANGEN Biotech, Beijing, China) following the manufacturer’s instructions. Total RNA was extracted and purified from the mid-exponential phase cells cultured at 37°C using the TRIzolTM reagent (Invitrogen, Waltham, MA, USA). Purified RNA was quantified using a NanoPhotometer spectrophotometer (IMPLEN, CA, USA), and RNA quality was confirmed using 2% agarose gel electrophoresis.
Protein expression and purification
The expression plasmid pET28a-Mpsy_0886 (gene encoding the mpy-RNase J) and mutants with mutations at the catalytic centre (H84A, H84A/H87A, H388A), in the adjacent hydrogen-bond network (S247A, S153A), and in the 5′P pocket (H384A, S386A and H384A/S386A) were previously constructed [Citation46]. Mutants with mutations at archaeal loop I (D38A, Q41A, T48A, and D49A), loop II (R298A, R299A), helix α5 (H114A, S118A), helix α9 (R275A), and at the conserved residue Phe245 (F245A) were constructed via site-directed mutation using the pET28a-Mpsy_0886 plasmid as a template and a site-directed mutagenesis kit (Stratagene, La Jolla, CA, USA). The primers (Sangon, Beijing, China) used for cloning and mutagenesis are listed in Table S1. The PCR products of all constructs were verified by DNA sequencing.
Mpy-RNase J and its various mutants were expressed in the E. coli BL21(DE)3plyS strain carrying the plasmid pET28a-Mpsy_0886 and its mutant variants. Cells were cultured in LB medium containing 50 μg/mL of kanamycin at 37°C until OD600 reaching at 0.6–0.8, and then subjected to 16 h of induction with 0.5 mM isopropyl-β-D-thiogalactoside (IPTG) at 22°C. Cells were then harvested by centrifugation at 5,000 g for 30 min at 4°C and stored at −80°C. Mpy-RNase J and its mutants were purified using Ni2+-nitrilotriacetatic acid-agarose columns (Novagen), HiTrap Q HP anion exchange columns, and a gel filtration chromatography column according to the manufacturer’s protocols (GE Healthcare) as previously described [Citation46]. The purified protein was concentrated using Amicon Ultrafra-30 concentrators (Millipore), flash frozen, and then stored at −80°C prior to crystallization and nuclease assays. Protein concentrations were determined using a Pierce BCA protein assay kit (Thermo Scientific).
Synthesis of single-stranded and hairpin RNAs in addition to hybrid RNA-DNA duplex substrates
Hairpin Gx, Dx RNAs, and single-stranded S20 and S10 RNA molecules of varying lengths (Table S2) were synthesized with 3′ or 5′ ends labelled with 6-carboxfluorescein (6-FAM) at Genscript (Nanjing, China). The 3′ end radiolabeled Gx or Dx RNAs used in were generated by ligating the [32P]pCp at the 3′ hydroxy ends of the synthesized RNAs using T4 RNA ligase (Ambion). Double-stranded duplex substrates were prepared by mixing the 3′ end 6-FAM labelled 5′ monophosphorylated single-stranded S20 RNA with the 3′-end complemented single-stranded oligonucleotides of size 20, 17, 15, 13, 12, 10, and 8 nt in order to form the Hx duplex RNA substrates (x: 5′ overhang length) with 0, 3, 5, 7, 8, 10, and 12 nt 5′ overhangs, respectively (Table S2). The single-stranded S20 RNA molecule and each complementary oligonucleotide were incubated and denatured at 90°C for 5 min and then slowly cooled to 25°C to generate the duplex RNA-DNA substrates. The long hybrid RNA-DNA duplex substrates of dx (x: duplex length) (Table S2) were generated similarly as Hx.
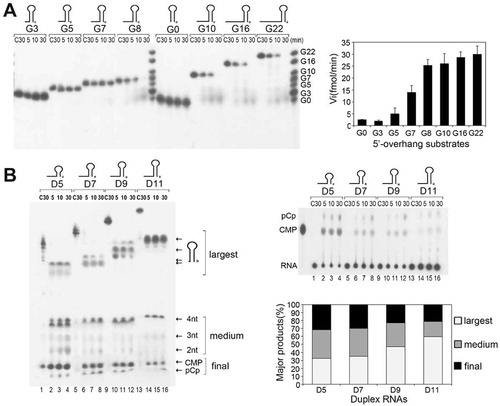
Figure 1. The duplex unwinding activity of mpy-RNase J on hairpin RNAs. (A) Kinetic analysis of mpy-RNase J degrading 3′-radiolabeled hairpin RNAs (Gx) that have a common 9-bp duplex but linking variable nt (suffixed numbers) of 5′ single-stranded overhangs. Nuclease assays were conducted as described in the Materials and Methods, and the radiolabeled Gx substrates with indicated sizes were used as the migration ladders of cleavage products. The initial reaction rates of plotting the decrease of RNA substrates are shown as histograms at right. (B) Kinetic analysis of degrading the 3′-radiolabeled 8-nt 5′ ss overhang RNAs (Dx) carrying duplex lengths of 5, 7, 9, and 11 bp indicated as suffix of D. Nuclease assays were conducted as in (A) except using five-fold higher protein concentrations. Reaction products were analysed on 10% PAGE gels (left panel). Arrows indicate the three types of major products noted as ‘largest, medium, and final’ according to the migrations. The final products, CMP and pCp, were also determined with thin layer chromatography (TLC) plates in parallel (right upper panels). Percentages of the three major products released at 30 min are shown as a histogram in the right lower panel.
In vitro transcription of the highly structured RNAs
The highly structured RNAs, including the M. psychrophilus tRNAMet, sR3CD-box snoRNA, SRP RNA, RNase P RNA, the 5′ UTRs of Mpsy_1580, Mpsy_1767, Mpsy_2714, and Mpsy_3111, and E. coli malEF REP RNA (Table S2) were produced by in vitro transcription reactions as previously described [Citation46]. The DNA templates for in vitro transcription of the structured RNAs were amplified by PCR using the primers listed in Table S1. The amplicons were purified using the Wizard Gel and PCR Clean-UP System (Promega). In vitro transcriptions were then conducted using the MEGA shortscript T7 Kit (Ambion) according to manufacturer’s instructions. The RNA products were further purified using 10% denatured PAGE gels containing 8 M urea by ethidium bromide staining and the ZR small-RNATMPAGE Recovery Kit (ZYMO), followed by quantification with a NanoPhotometer spectrophotometer (IMPLEN, CA, USA).
Nuclease activity assays
The nuclease activities of mpy-RNase J and its mutants were assayed similarly as previously described [Citation46,Citation48]. A routine ribonuclease assay included 2 μM RNA and 10 μM (or otherwise indicated concentrations) of purified mpy-RNase J, or its variant mutants. The two were mixed in a reaction buffer containing 20 mM HEPES (pH 7.5), 150 mM NaCl, 0.1 mg/mL BSA, 1 mM DTT, and 1 mM MnCl2. Reactions were initiated by addition of the enzyme, incubated at 37°C for 5–45 min, and then stopped by incubation with 10 μg/mL of proteinase K (Ambion) at 55°C for 15 min. After incubation, the reaction solutions were mixed with a formamide-containing dye and analysed on 10% or 20% urea-PAGE for substrates in lengths <20 nt. The synthesized oligoribonucleotides with 3′ end radiolabeling or 6-FAM labelling were used as molecular ladders to indicate the migration positions of RNA substrates and products. For imaging non-radioactive or 6-FAM labelled RNAs used in , S3 and S4, a SYBR Gold (Invitrogen) staining was performed after electrophoresis. RNAs in urea-PAGE gels were analysed by fluorescence imaging or phosphorimaging with a Storm Phosphor-Imager and the Image-Quant software programme (GE Healthcare), and/or autoradiography with X-ray film.
Crystallization and structure determination
Crystallization and structure determination of mpy-RNase-J S247A mutants complexed with a 6-nt polyA RNA were conducted as previously described [Citation45]. Briefly, the co-crystal structure was obtained using the hanging-drop vapour diffusion method with a recipe containing 0.01 M MgSO4, 0.05 M (CH3)2AsO2Na (pH 6.5), and 2 M (NH4)2SO4. The diffraction data were collected at beamline BL19U of the Shanghai Synchrotron Radiation Facility. The data were processed with XDS and Aimless [Citation49,Citation50]. The initial model was obtained and then refined with PHENIX [Citation51] using the molecular replacement method with the previously determined mpy-RNase-J S247A structure as the search model. The data collection and refinement statistics are provided in Table S3. A total of 96.30%, 3.70%, and 0% of the residues were present in the favoured, allowed, and disallowed regions of the Ramachandran plot, respectively. All of the structure figures were prepared with PyMOL (http://www.pymol.org).
Data availability
The structural factors and coordinated files were deposited in Protein Data Bank with accession ID of 6LLB.
Results
Mpy-RNase J exhibits a duplex RNA unwinding activity
In the previously solved apo and 5-nt RNA-bound mpy-RNase J structures [Citation46], we invariably observed a sulphate ion (SO42-) positioned over the N-terminus of helix α10 at the RNA channel entrance (Fig. S1). The sulphate ion was located upstream the phosphate group of the fifth nucleotide (nt +4, the fourth nucleotide downstream the cleavage site) at a distance of 8.4 Å. This is longer than the average distance of 5.7–6.2 Å between two adjacent phosphate groups, implying a phosphate binding site of nt +6, with two additional nucleotides binding and distortion. This suggests that the ssRNA channel of mpy-RNase J can accommodate an ssRNA molecule of up to 7 nt in length. Considering that duplex structures would be blocked at the ssRNA channel entrance, we first evaluated the actual length of the channel via assaying the kinetics of mpy-RNase J on degrading an array of Gx substrates, which carry varying lengths (x) of 5′ single-stranded (ss) overhang RNAs but linked to an invariant 9 base-paired duplex structure, and 3′-end radiolabeled for detection. Kinetic assays revealed that duplex RNAs linking short 5′ overhangs ≤7 nt, G0, G3, G5, and G7 were nearly not degraded; however, those linked with >7 nt 5′ overhangs, G8, G10, G16, and G22, were efficiently degraded (). This indicates that to initiate an efficient exoribonucleolytic hydrolysis of Gx substrates, mpy-RNase J requires a 5′ ss overhang with no less than 7 nt in length to reach the catalytic site at the channel end. This verifies that the ssRNA channel of mpy-RNase J is in a length of 7 nt.
However, the major degradation products from duplex RNAs of G8, G10, G16, and G22 were G1 and G4, but not G7, i.e., the 5′ ss overhangs of the products were shorter than the channel length of 7 nt. This inspired a hypothesis that mpy-RNase J may unwind part of the duplex. To evaluate whether mpy-RNase J possesses duplex unwinding activity, we analysed the ribonucleolytic kinetics on an array of 8 nt 5′ ss overhang RNAs that link with varying duplex lengths of 5, 7, 9, and 11 bps (). At a five-fold higher protein concentration, mpy-RNase J degraded all tested duplex RNAs to the last monoribonucleotide, the radiolabeled pCp and CMP, which were verified by thin layer chromatography (TLC). Thus, mpy-RNase J indeed exhibited duplex unwinding ability. However, lower release rates of pCp and CMP were detected on the longer RNA duplexes and also the concomitant accumulation of degradation intermediates (), presumably due to the likely stalling of mpy-RNase J at the duplex regions. Consistently, degradation intermediates in length ≤4 nt were accumulated in parallel with pCp and CMP from the four duplex RNAs (), reminiscent of the same degradation pattern of ssRNA substrates [Citation46], verifying again that mpy-RNase J loses exoribonucleolytic processivity on RNAs shorter than 5 nt. In contrast, no degradation of the D9 RNA was found in the parallelly purified catalytic inactive mutants, H84A, H84AH87A, H388A (Fig. S2A) [Citation46], excluding the activity from a possible contamination of the E. coli enzymes but verifying the intrinsic duplex unwinding activity of mpy-RNase J.
Mpy-RNase J unwinds duplexes in length up to 40 bp
To further evaluate the duplex unwinding activity of mpy-RNase J, we then assayed its activity on the commonly used RNA-DNA hybrid duplexes [Citation24,Citation25,Citation52,Citation53] that carry varying lengths of 5ʹ ss overhangs. The hybrid duplex substrates, noted as Hx (x: length of 5′ overhangs), share a 5ʹ monophosphorylated 20 nt RNA strand with 3ʹ end 6-FAM labelling for a convenient fluorescence detection, but hybridized with varying lengths of DNA strands carrying 5ʹ-hydroxyl ends (). Enzymatic assays indicated that, similar to that observed on Gx RNAs, mpy-RNase J hardly degraded the Hx duplex RNAs without a 5′ overhang, H0, or with 5′ overhangs of ≤7 nt, H3, H5, and H7, whereas those with 5′ overhangs of >7 nt, H8, H10, and H12, were degraded at efficiencies similar to that of the single-stranded S20 RNA (). This indicates that regardless of the duplex RNA types, mpy-RNase J requires a 5′ ss overhang of at least 7 nt that is long enough to reach the catalytic site at the channel end to initiate duplex RNA degradation ( and ), i.e., the 5′ ss overhang exoribonucleolysis is likely essential for mpy-RNase J to unwind the RNA duplex.
Figure 2. The duplex unwinding activity of mpy-RNase J on hybrid RNA-DNA substrates (Hx). (A) Kinetic analysis was assayed using the 3′-end 6-FAM labelled Hx substrates that share a common 20-nt 3′-labelled ssRNA and hybridized with varying-length complementary DNA strands. Suffixed numbers refer to the 5′ ss overhang lengths. 3′-labelled ssRNAs with indicated sizes were used as ladders. A ssRNA S20 was included in the assays. (B) Degradation of 3′-labelled 10 nt 5′ ss overhang RNAs (dx). Suffixed numbers refer to the duplex (d) lengths of 20, 30, and 40 bp. Nuclease assays were conducted as in (A), but with gradient concentrations of mpy-RNase J as indicated. The cleavage products were analysed on 10% PAGE gels (left panel). The final products, UMP-6FAM and the bottom 6FAM are indicated.
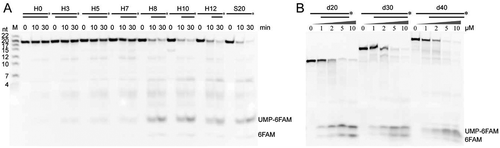
Next, we assayed the capability of mpy-RNase J of degrading long duplexes using RNA-DNA hybrids that carry a 10 nt 5ʹ ss overhang and complementary duplexes in lengths of 20, 30, or 40 bps (). Unexpectedly, mpy-RNase J efficiently degraded all long duplex substrates, again indicating that as long as the 5ʹ ss overhang is long enough for initiating the 5ʹ-3ʹ exoribonucleolytic activity, mpy-RNase J is able to unwind up to 40 bp RNA duplex. Moreover, consistent with that the 3′ end radiolabeled pCp can be finally degraded to the radiolabeled CMP ( and [Citation46]), the 6-FAM band can also be cleaved from the final product of the 3′ end 6-FAM labelled ribonucleotide (NMP-6FAM) by mpy-RNase J when degrading both the hairpin and single-stranded RNAs ( and S2B).
A 6-nt RNA-mpy-RNase J co-crystal structure reveals a potential hydrolysis-coupled duplex unwinding mechanism based on steric occlusion
To gain insights into the duplex unwinding mechanism employed by mpy-RNase J, we first examined its RNA-free and -bound structures [Citation46]. None of the ATP-binding sites or characteristic helicase domains or motifs, such as Walker A and Walker B motifs, were identified, thus precluding the possibility that mpy-RNase J possesses a classical helicase unwinding mechanism. In contrast, the duplex unwinding initiated by exoribonucleolytic hydrolysis suggests a possible mechanism in which hydrolysis driven substrate translocation past a steric barrier excludes the duplex regions. To investigate this hypothetical mechanism and explore the presence of a possible steric barrier, we attempted to resolve the structures of mpy-RNase J complexed with both duplex RNAs and ssRNAs longer than 5 nt, and eventually obtained a co-crystal structure of mpy-RNase J-S247A, a catalytic inactive mutant of mpy-RNase J, complexed with a 6-nt poly(A) RNA, i.e., rA6-mpy-RNase J-S247A complex (2.7 Å, Table S3).
In the rA6-mpy-RNase J-S247A structure, the poly(A)6 RNA is located at the interface of the β-CASP and MβL domains with its phosphate-ribose backbone grasped by the β-CASP domain, and the bases of nt +1 to +5 situated in the direction of the MβL domain or solvent (nt −1) (). Notably, the phosphate group of nt +4 is fixed over the N-terminus of the alpha-helix α9 via an interaction of negatively charged group and the α-helix dipole. The nt +2 to +4 bases that stack against each other are sandwiched by residues Leu37 and Arg271 and situate approximately 5 Å to archaeal loop I (residues 39–51). The nt +4 base is situated proximal (4.8 Å) to helix α5 (residue S118). The nt +5 base stacks against the planar guanine group of Arg271, exposes to helix α5 at approximately 6 Å to His114, and forms a hydrogen bond with Arg275 (2.9 Å) (). These observations support that the RNA binding channel only accommodates ssRNAs.
Figure 3. The rA6-mpy-RNase J-S247A structure suggests a hydrolysis-coupled and steric occlusion-based duplex unwinding mechanism. (A) Cartoon overall structure of rA6-mpy-RNase J-S247A. A 6 nt poly(A) RNA within the channel is shown in sticks and surrounded by a ligand-omitted 1Fo-Fc map contoured at 3o’. (B) Interactions of rA6 RNA with mpy-RNase J. (C) A proposed steric barrier comprising two unique archaeal loops in addition to helices α5 and α9. Carbon, oxygen, and nitrogen atoms are indicated in red, grey, and blue, respectively. Archaeal loop I, loop II, helix α5, and helix α9 are shown in green, cyan, yellow, and red, respectively. Key residues in the predicted elements that constitute the steric barrier and RNA interactions are depicted as sticks.
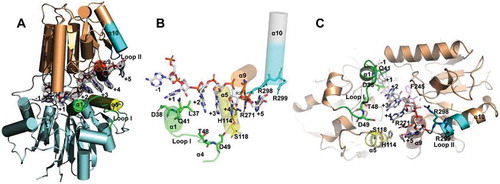
In contrast to the previously resolved rA5-mpy-RNase J-S247A structure [Citation46], the rA6-mpy-RNase J-S247A structure does not exhibit obvious conformational changes and clearly reveals the position of nt +5 along with the structural motifs involved in the +5 base binding and the formation of the ssRNA channel entrance. It showed that three alpha-helices, namely, helix α5 (His114, Ser118), α9 (Arg271, Arg275), and α10 (Arg298, Arg299), constitute the entrance of the ssRNA channel entrance (), and archaeal loop II (residues 297–305) extends α10 as part of the channel entrance. The archaeal loop I is assumed to involve stabilization of the channel entrance conformation and the channel-bound RNA. Moreover, the highly conserved residue Phe245 located at the other side of the bound RNA () could also play a role in RNA conformation stabilization. Thus, we propose that the archaeal loops I and II, and helices α5, α9, and α10 form a narrow entrance of 15 Å width (distance between His114 and helix α10), possibly serving as a steric barrier that could exclude and unzip duplexes at the channel entrance along with the forward RNA translocation in the channel.
The 5ʹ-3ʹ exoribonuclease activity drives the duplex unwinding of mpy-RNase J
To investigate whether the duplex unwinding of mpy-RNase J is fuelled by the exoribonucleolytic hydrolysis, we assayed the mutation effects on the activity of mpy-RNase J using a series of mpy-RNase J mutants that carry substitutions in residues that are key to the exoribonucleolytic activity and processivity [Citation46]. Enzymatic assays found that H84A, H84A/H87A, H388A, and S247A mutants that carry mutations in the catalytic centre and the adjacent hydrogen-bond-network, all lost 5ʹ-3ʹ exoribonucleolytic activity on 5ʹ monophosphorylated single-stranded S20 RNA (). Simultaneously, these mutants did not show the duplex unwinding activity on RNA-DNA duplex H8 (). These findings indicate that the duplex unwinding activity of mpy-RNase J is driven by its 5ʹ-3ʹ exoribonucleolytic activity.
Figure 4. The coupled duplex unwinding and exoribonuclease activities of mpy-RNase J. Residue substitution mutants were constructed in the key elements involving in the exoribonucleolytic cleavage and processivity, which include the catalytic active site, hydrogen-bond network flanking the catalytic centre, and the 5ʹ monophosphorylated (5ʹP) pocket. The 5ʹ end exoribonucleolytic cleavage activity (A), the 5ʹ to 3ʹ exoribonucleolytic processivity (B), and the duplex unwinding activity (C) of mpy-RNase J and its variants were compared on the 5ʹ end-labelled S10, 3ʹ end-labelled S20, and 3ʹ end-labelled H8 RNA substrates. 10 μM of mpy-RNase J and its mutants were used in the nuclease assays. Cleavage products were analysed using PAGE gel, and migrations of ssRNA markers with indicated lengths are labelled on the left of the gels.
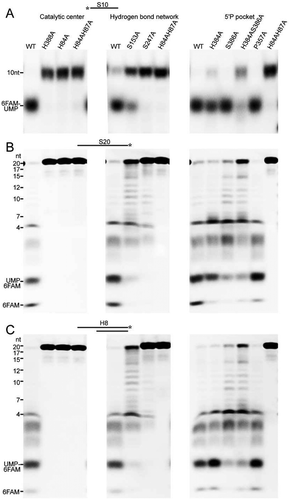
Next, we determined the role of RNA translocation in the duplex unwinding of mpy-RNase J using its mutants carrying mutations in the 5ʹ monophosphorylate (5ʹP) pocket. The 5ʹP pocket specifically recognizes the 5ʹP end of RNA and contributes to the RNA 5ʹ terminal coordination for efficient catalysis and exoribonucleolytic processivity (i.e., RNA translocation) [Citation46]. Enzymatic assays showed that, consistent with the cleavage pattern on single-stranded S20 RNA (), the mutants of H384A, S386A, and also S153A produced similar stalled degradation intermediates of ≥ 4 nt from the duplex RNA-DNA H8 (). However, the H384A/S386A double mutation exhibited similar significant reduced hydrolytic activity and produced similar stalled degradation intermediates of ≥ 4 nt on both of S20 ssRNA and the duplex H8 (–C). These results experimentally verify that the duplex unwinding activity of mpy-RNase J is fuelled and driven by its 5ʹ-3ʹ exoribonucleolytic hydrolysis and the forward RNA translocation within the ssRNA channel.
Stacking interaction of Arg271 with bases +2 to +5 contributes to the processive RNA translocation of mpy-RNase J
As described above, the nt +5 base stacked against Arg271 results in continuous π-π stacking interactions ordering as base +2, base +3, base +4, R271, and base +5 in the rA6-S247A structure. This suggests a possible role of the stacking in efficient RNA translocation, which has been determined to be essential to the duplex unwinding of mpy-RNase J. To evaluate the role of such base stackings in RNA translocation, we compared the degradation patterns of mpy-RNase J on the synthetic ssRNAs S19 and S19-M4 that possesses four consecutive bases deleted in the middle nucleotides of 9–12 (). Compared to the cleavage products of S19, five additional degradation intermediates were produced from S19-M4. Referenced to the migration of S11-M4, a synthetic RNA marker carrying the last 11 nucleotides of S19-M4 and lacking the same four consecutive bases, the smallest intermediate was identified as S11-M4, and the other intermediates by each 1 nt larger were identified as S12-M4 to S15-M4 (). Thus, S15-M4 could be an intermediate with the first deleted base (of nt 9) that was translocated at the position of nt +5 base and lost a base stacking against Arg271. The S11-M4 band should be an intermediate with the first and last deleted bases (of nts 9 and 12) that were translocated at positions of nt +2 and nt +5 base and lost the base stacking against Leu37 and Arg271, respectively. Thus, S12-M4, S13-M4, and S14-M4 should be the intermediates that had lost base stacking interactions with Arg271 from both sides. These results indicate that the stacking interactions among base +4, R271, and base +5 are important to efficient RNA translocation. Consequently, an exoribonucleolytic product that lost such stackings would not be translocated properly. Consistently, the mutant R271A accumulated several intermediates when degrading S19 as well, but the mutant R271 K did not (Fig. S3). This could be because, similar as arginine, lysine has a flexible sidechain, which could insert into the space sandwiched by the two bases +4 and +5, and therefore form a Van der Waals interaction between the bases and the flexible sidechain, which may mimic or partially replace the pi-pi stacking.
Figure 5. Roles of base stacking at Arg271 and the proposed steric barrier involved in RNA translocation and duplex unwinding of mpy-RNase J. (A) Contribution of stacking interactions between bases +2 to +5 and Arg271 to RNA translocation in the processive 5ʹ to 3ʹ exoribonucleolysis. The 5ʹ to 3ʹ exoribonucleolytic processivity of mpy-RNase J was compared using 3ʹ-labelled S19 and its mutant S19-M4 that carries four consecutive base lacking nucleotides. A mixture of ssRNAs with indicated lengths was used as size markers at the gel left (lane M). A synthetic 3ʹ-labelled S11-M4 shown at right was also used as size marker for the smallest degradation intermediate. The schematic in the right panel shows the five degradation intermediates from S19-M4. (B, C) Effects of the proposed steric barrier mutations on duplex unwinding activity of mpy-RNase J. Ten key residues in the archaeal loops I and II and the helices α5 and α9 that constitute the proposed steric barrier were mutated, and the duplex unwinding activities of mpy-RNase J and the mutants were determined by using the hairpin G8 (B) and hybrid H8 RNA (C) substrates. Stalled degradation intermediates at duplex structures are indicated by arrows. 10 μM of mpy-RNase J and its mutants were used in the nuclease assays, and CK indicates the assay without enzyme. The cleavage products were analysed on PAGE gel, and migrations of ssRNA markers with the indicated sizes are shown on the left.
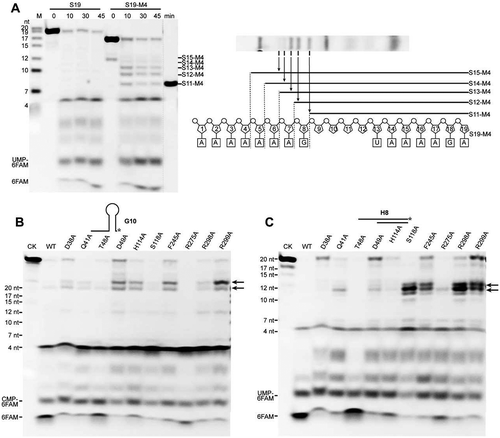
Key residues of the proposed steric barrier contribute to duplex RNA unwinding
Next, we evaluated contributions of the proposed steric barrier that is composed of the archaeal loops I/II and helices α5/α9, to the duplex RNA unwinding activity of mpy-RNase J. Key residues of these elements were mutated by residue substitutions, including His114 and Ser118 of helix α5, Arg275 of α9, Arg298 and Arg299 of archaeal loop II, and Asp38, Gln41, Thr48, and Asp49 of archaeal loop I. In addition, Phe245 was also mutated because it is highly conserved across bacterial and archaeal RNase Js and presumably important in the preassembly and conformation stabilization of bound RNA. A total of 10 mutants were obtained, by each carrying a single-site mutation. The 5ʹ-3ʹ exoribonucleolytic activities and processivities were first evaluated using a 5ʹ end-labelled 10 nt RNA and a 3ʹ end-labelled S20 ssRNA, respectively. Compared with the wild-type mpy-RNase J, no significant reduction in exoribonucleolytic activity was observed for the mutants, except for R299A that exhibited a < 40% activity reduction (Fig. S4A). Moreover, no additional stalled degradation intermediates > 4 nt were accumulated in all mutants, indicating that these mutations do not affect the exoribonucleolytic processivity (Fig. S4B). Duplex unwinding activities were then evaluated for the 10 mutants using a G10 RNA but with a 3ʹ-end 6-FAM labelling. Compared with the wild-type mpy-RNase J that efficiently degraded RNA into the hairpin region to generate a final product of ≤ 4 nt, mutants D38A, Q41A, D49A, H114A, S118A, F245A, R298A, and R299A accumulated degradation intermediates of 22 nt (). Thus, mutation of any of these residues in the proposed steric barrier presumably causes mpy-RNase J to stall at the stem-loop hairpin structure. Similarly, when evaluating degradation of the RNA-DNA duplex H8, mutants Q41A, S118A, F245A, R298A, and R299A clearly generated 12-nt degradation intermediates, and the D49A, H114A, and R275A mutants accumulated lower amounts of the intermediates (). These suggest that mutation of any of these residues also triggers mpy-RNase J stalling at the duplex structure of H8, further verifying the contribution of the steric barrier residues to RNA duplex unwinding. Taken together, the residues of archaeal loop I and loop II (α10), in addition to helices α5 and α9, together constitute a steric barrier at the ssRNA channel entrance and play a role in excluding and unwinding duplex RNA structures for their degradation. In addition, the conformation stabilizing residue Phe245 also plays a role in duplex unwinding.
Bacterial RNase J also exhibits duplex unwinding activity
To determine whether the widely distributed bacterial RNase Js [Citation33] possess a similar duplex unwinding activity, we selected Streptomyces coelicolor sco-RNase J [Citation54] for investigation. Enzymatic assays showed that similar to the archaeal RNase J, sco-RNase J did not hydrolyse the Hx RNA without a 5′ overhang (H0). However, it efficiently degraded those with ≥3 nt 5′ overhangs (H3, H5, H7, H8, and H10) to the last nucleotide (). These findings indicate that the bacterial sco-RNase J possesses the same duplex RNA unwinding activity, but requires a shorter 5′ ss overhang of only 3 nt rather than 7 nt required by the archaeal mpy-RNase J.
Figure 6. Duplex unwinding activity of bacterial sco-RNase J determined using hybrid RNA substrates (Hx). (A) Using the same Hx RNAs as in , degradation kinetics of sco-RNase J were assayed in the same way as those of mpy-RNase J. (B) Degradation of the 3′-labelled 10 nt 5′ ss overhang dx RNAs by sco-RNase J. The same dx RNAs in and 10 μM of sco-RNase J protein were used for nuclease assays. The reaction products were analysed by 10% PAGE gels, and the final products, UMP-6FAM and the bottom 6FAM are indicated.
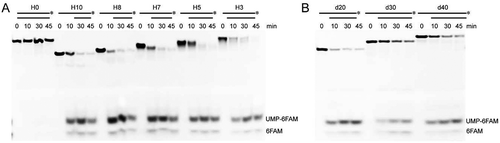
Further assays found that sco-RNase J was capable of degrading d20, d30, and d40 RNAs efficiently (), similar to that was observed in mpy-RNase J, thereby indicating that the bacterial RNase J can unwind and degrade RNAs with duplexes up to 40 bp as well. Taken together, these results indicate that the bacterial and archaeal RNase Js exhibit shared duplex RNA unwinding activities, but appear employing slightly different mechanisms because of the different RNA channel lengths and the entrance elements.
The archaeal mpy-RNase J can degrade highly structured RNAs
Duplex RNA unwinding activity could facilitate ribonucleases to degrade highly structured RNAs independent of RNA helicases [Citation24–Citation26]. To evaluate the capability of mpy-RNase J in degrading the cellular structured RNAs, we first assayed its activity on rRNAs, the known structured RNAs, by using the total RNA extracts of M. psychrophilus as substrates. Enzymatic assays showed that mpy-RNase J at a concentration of 0.4 μM could efficiently degrade 23S and 16S rRNAs in 1 μg of total RNA (Figs. S5A and B), and the maximal activity was observed at above 37°C (Fig. S5C). In contrast, the parallel purified catalytic inactive mutant, H84A, did not degrade the rRNAs, excluding a possible contamination of E. coli enzymes during expression and purification of the recombinant mpy-RNase J. Similarly, bacterial sco-RNase J was also capable of degrading the cellular rRNAs although with a weaker activity than mpy-RNase J on tested M. psychrophilus rRNAs (Fig. S5D).
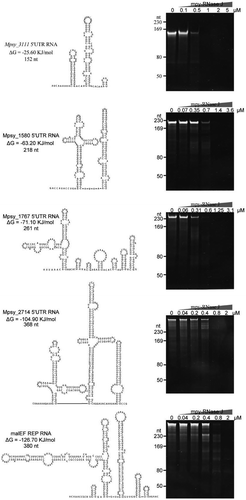
Based on the prevalence of long 5ʹ UTRs in M. psychrophilus mRNAs [Citation5,Citation55], we used those possessing predicted highly folded secondary structures and 5ʹ ss overhangs > 7 nts as native structured mRNA substrates of mpy-RNase J. Four structured 5ʹ UTR RNAs were selected, which are derived from Mpsy_3111, Mspy_1580, Mspy_1767, and Mpsy_2714 and have predicted folded structures with ΔG values of −25.6 KJ/mol to −104.9 KJ/mol (). In addition, a well-known highly structured RNA of 375 nt transcribed from the E. coli malEF intergenic region was also used, which contains a repetitive extragenic palindrome (REP) stem-loop structure [Citation20,Citation21]. After the addition of gradient concentrations of mpy-RNase J (), all five highly structured RNAs were efficiently degraded, thus demonstrating the capability of mpy-RNase J in degrading highly structured cellular mRNAs independent of an RNA helicase, which is most likely facilitated by its structured RNA unwinding activity.
Figure 7. The degradation activity of mpy-RNase J on highly structured mRNAs. Five highly structured mRNAs were used as substrates, including four 5ʹ UTR RNAs from Mpsy_3111, Mpsy_1580, Mspy_1767, and Mpsy_2714 of M. psychrophilus, and a REP-structured RNA from the E. coli malEF intergenic region. RNA lengths, secondary structures, and folding ΔG values are shown in the left panels. Nuclease assays were conducted using the indicated concentrations of mpy-RNase J and the reaction conditions described in the Materials and Methods. Cleavage products were analysed by 10% PAGE gels. Migrations of the ssRNA markers with indicated length are shown at the left of the gels.
Subsequently, we evaluated the potential role of mpy-RNase J in the quality control and clearance of the non-functional highly structured intermediates during functional RNAs maturation. Functional RNAs including RNase P, tRNAs, CD-box snoRNA, and SRP RNAs of M. psychrophilus [Citation55] were selected and each was fused with a 15 nt 5ʹ ss overhang to facilitate mpy-RNase J loading at the RNA 5ʹ ends (Fig. S6). After being exposed to gradient concentrations of mpy-RNase J, these functional RNAs were all efficiently degraded including the highly structured regions (Fig. S6), indicating that mpy-RNase J may play a role in the supervision of the functional RNAs and clearance of their non-functional intermediates.
Discussion
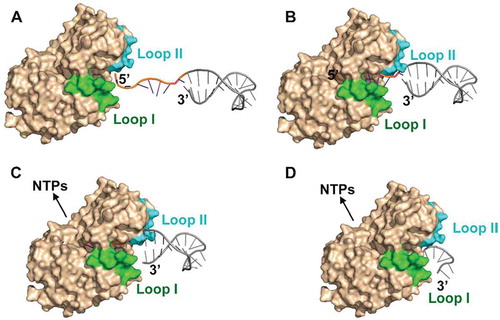
Here, we report that an archaeal RNase J exhibits a novel helicase-independent duplex unwinding activity. Based on structural analysis, mutagenesis experiments, and biochemical assays, an exoribonucleolysis-coupled and steric occlusion-based duplex unwinding mechanism is proposed. As depicted in , an unstructured 5′ ssRNA overhang with a minimal length of 7 nt is allowed to access the deeply buried catalytic centre at the end of ssRNA channel to initiate exoribonucleolytic hydrolysis at the 5′ end by 1 nt per cycle. RNA hydrolysis-released energy could power translocation of the 5′p (n-1) RNA forward to the catalytic site. The unique archaeal loops, I and II, in synergy with helices α5 and α9 serve as the entrance barrier that only permits the entry of single-stranded RNA but excludes double-stranded regions. Due to the processive exoribonucleolytic nature of the enzyme and the single-stranded restriction of the narrow entrance barrier, the translocation of an RNA substrate 1 nt at one time between consecutive catalytic cycles could mechanically tear 1 bp duplex at the entrance barrier. Thus, with repeated cleavage at the 5′ end of an RNA chain, duplex RNAs are unzipped and degraded in concert with the exoribonucleolysis-fuelled RNA translocation. Importantly, the duplex unwinding activity can equip archaeal RNase J to degrade the cellular highly structured RNAs, therefore functioning as a key player in archaeal pleiotropic RNA metabolisms. Moreover, the bacterial RNase J exhibits a shared duplex unwinding activity, although may employ a slightly different mechanism.
Figure 8. The proposed exoribonucleolysis-coupled and steric occlusion-based duplex unwinding mechanism of archaeal RNase J. (A) To initiate duplex RNA degradation, mpy-RNase J requires a ≥ 7 nt 5′ single-stranded overhang, which is long enough to reach the catalytic site at the RNA channel end for initiating exoribonucleolysis. (B) Following each 5′ end cleavage, the 5′p (n-1) RNA is translocated by advancing 1 nt in each exoribonucleolytic cycle using the hydrolysis released energy. (C) Along with the forward RNA translocation, a steric barrier at the RNA channel entrance, which is constituted by the archaeal loops I and II and helices α5 and α9, only allows the passage of single-stranded RNAs but sterically restricts and unwinds double-stranded RNAs. (D) The synergistic action of the forward RNA translocation within the channel and the steric occlusion at the channel entrance unwinds the duplex regions, thereby enabling mpy-RNase J to degrade the structured RNAs.
Duplex unwinding activity equips archaeal RNase J as an important player in cellular RNA metabolism
Exoribonucleases are essential to the fundamental RNA metabolisms in life, such as RNA surveillance and quality control, turnover and processing for maturation etc. To accomplish these important and multifarious biological processes, both the highly processive exoribonucleolysis and structured RNA degradation activities of exoribonucleases are required. In particular for degrading extensive RNA secondary structures, the intrinsic ATP-independent helicase-like duplex unwinding activities of exoribonucleases are usually essential for them to play fundamental roles in pleiotropic RNA metabolism. Such as the duplex unwinding ability enables the eukaryotic 5′–3′ exoribonucleases Xrn1 and Xrn2 to degrade structured RNAs and function in many fundamental physiological processes, i.e., Xrn1 functions in mRNA decay, and 18S non-functional rRNA decay, and tRNA quality control in yeast [Citation25,Citation27,Citation28], and Xrn2 specifically functions in transcriptional termination [Citation56]. The duplex unwinding activity of Rrp44 (Dis3) assists RNA exosome to digest structured RNAs including hypomethylated tRNAs [Citation29]. This ability also facilitates the bacterial RNase R to degrade repetitive extragenic palindromic sequences in mRNAs and highly structured regions of rRNAs and tRNAs [Citation24,Citation30,Citation57,Citation58].
Here, we report a newly identified duplex-unwinding activity and the mechanism of an archaeal RNase J, which also degrades the highly structured RNAs in vitro, including the highly structured 5ʹ UTRs and REP regions (), functional RNAs of RNase P, tRNAs, CD-box snoRNA, and SRP RNAs (Fig. S6), and rRNAs (Fig. S5). Therefore, archaeal RNase J most likely functions as a housekeeping ribonuclease in the control of cellular diverse RNA metabolisms of Archaea, such as mRNA decay for regular cellular RNA turnover in addition to rRNA and tRNA quality control by clearing non-functional or incorrectly processed structured intermediates. Additionally, this enzyme may also operate in the supervision of stable and highly structured functional RNAs including SRPs, RNase P, and CD-box snoRNAs as well, and its potential role in rRNA and tRNA maturation processes still cannot be excluded. Although the in vivo functions of the archaeal RNase J need to be exploited further, the gene encoding mmp-RNase J, an archaeal RNase J ortholog, has been established as an essential gene in Methanococcus maripaludis[Citation59], which could provide a circumstantial evidence for its essential cellular functions, presumably in archaeal RNA metabolism. Our preliminary experiment shows that depletion of mmp-RNase J expression results in growth retardation and prolonged half-life of bulk mRNA of M. maripaludis, further verifying the essential role of archaeal RNase J in RNA metabolism and physiology of Archaea.
Shared duplex unwinding activity of bacterial RNase J but with a slightly different mechanism
B. subtilis RNase J1 also exhibits enzymatic activity on structured RNAs including polyG sequences and 16S rRNA bodies, leading to the proposal of intrinsic ‘helicase-like’ activity [Citation32]. Here, using sco-RNase J from S. coelicolor[Citation54], a similar duplex unwinding activity as archaeal RNase J was observed in the bacterial sco-RNase J, including the unwinding of long duplexes up to 40 bp and the degradation of highly structured rRNAs, suggesting a common duplex unwinding property among the RNase J family proteins. Although the two archaeal loops are absent in bacterial RNase J, an additional long C-terminal domain is present and responsible for its dimerization [Citation46]. Thus, bacterial RNase J is likely to use a slightly different duplex unwinding strategy or steric barrier. Moreover, sco-RNase J apparently requires a shorter 5′ overhang of only 3 nt in duplex unwinding () than the minimal 7 nt 5′ overhang required by mpy-RNase J ( and ). Superposition of the rA6-mpy-RNase J and the rCGCCU-sco-RNase J structures revealed that residues 42–52 and 442–446 (within the C-terminal domain) of Sco-RNase J situate equivalently as archaeal loop I in mpy-RNase J, whereas no archaeal loop II corresponding structural motifs were observed (Fig. S7). Therefore, an mpy-RNase J equivalent steric barrier is not present in sco-RNase J. Moreover, the RNA channel width of sco-RNase J at the fourth nucleotide, nt +3, is approximately 20 Å, i.e., the distances of His111 to Met305 and of Lys107 to Ala306, whereas that in mpy-RNase J is only 15 Å, i.e., the distances of Glu326 to Asp49 and Pro327 to Asp49 (Fig. S7). Thus, the wider RNA channel of bacterial RNase J could accommodate the duplex structure, and a short 5′ overhang of 3 nt is presumably long enough to reach the catalytic site and initiate duplex RNA degradation.
The hydrolysis-driven and steric occlusion-based duplex unwinding mechanism is widely shared by various exoribonucleases
In this study, an exoribonucleolysis-coupled and steric occlusion-based duplex unwinding mechanism is determined in an archaeal RNase J (). This mechanism also draws unexpected mechanistic parallels with some other exonucleases from eukaryotes and bacteria that work in either 5′-3′ or 3′-5′ directions. Such examples include the eukaryotic 5′-3′ exoribonuclease Xrn1 [Citation25] and the RNase II family of processive 3′-5 exoribonucleases, like the bacterial RNase R [Citation52,Citation53,Citation57], and the eukaryotic Rrp44/Dis3 [Citation26,Citation60]. These proteins all also apparently require an initial loading on a single-stranded terminal fragment to initiate exoribonucleolysis, and are driven by the forward RNA translocation of 1 nt per cleavage cycle and coordinated with a steric exclusion at the ssRNA channel entrance, resulting in the unwinding of duplex RNAs and the degradation of structured RNAs. These parallels suggest an evolutionary conserved strategy shared by the exoribonucleases among the three domains of life. The hydrolysis-coupled structured RNA degradation activity without a requirement of energy input or helicase assistance enables the exoribonucleases to be the major players in RNA metabolism and involved in various biological processes. Moreover, the unexpected mechanistic parallels among divergent exoribonucleases in the three domains of life provide evidence for the evolutionary principle of ‘disparate enzymes but convergent strategies’.
Author contributions
J.L., D.F.L., and X.Z.D. proposed the study. J.L. and X.N.G. designed and performed the biochemical experiments. Y.J.H and D.F.L. designed and performed the crystallographic experiments. All of the researchers interpreted the experimental data and assisted with the preparation of the manuscript. J.L., D.F.L., and X.Z.D. wrote the manuscript. All authors approved the final manuscript.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Material
Download Zip (5.2 MB)Acknowledgments
We thank Shanghai National Synchrotron Radiation Centre for the use of Synchrotron Radiation Facility. The National Natural Science Foundation of China under grant nos. 31670049, 91751203, and 31430001 supported this study. We thank LetPub (www.letpub.com) for providing linguistic assistance during the preparation of this manuscript.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
References
- Merritt J, Chen Z, Liu N, et al. Posttranscriptional regulation of oral bacterial adaptive responses. Curr Oral Health Rep. 2014;1(1):50–58.
- Garneau NL, Wilusz J, Wilusz CJ. The highways and byways of mRNA decay. Nat Rev Mol Cell Biol. 2007;8(2):113–126.
- Arraiano CM, Andrade JM, Domingues S, et al. The critical role of RNA processing and degradation in the control of gene expression. FEMS Microbiol Rev. 2010;34(5):883–923. .
- Belasco JG. All things must pass: contrasts and commonalities in eukaryotic and bacterial mRNA decay. Nat Rev Mol Cell Bio. 2010;11(7):467–478.
- Qi L, Yue L, Feng D, et al. Genome-wide mRNA processing in methanogenic archaea reveals post-transcriptional regulation of ribosomal protein synthesis. Nucleic Acids Res. 2017;45(12):7285–7298.
- Hui MP, Foley PL, Belasco JG. Messenger RNA degradation in bacterial cells. Annu Rev Genet. 2014;48(1):537–559.
- Nagarajan VK, Jones CI, Newbury SF, et al. XRN 5ʹ–>3ʹ exoribonucleases: structure, mechanisms and functions. Biochim Biophys Acta. 2013;1829(6–7):590–603.
- Makino DL, Baumgartner M, Conti E. Crystal structure of an RNA-bound 11-subunit eukaryotic exosome complex. Nature. 2013;495(7439):70–75.
- Liu Q, Greimann JC, Lima CD. Reconstitution, activities, and structure of the eukaryotic RNA exosome. Cell. 2006;127(6):1223–1237.
- Chlebowski A, Lubas M, Jensen TH, et al. RNA decay machines: the exosome. Biochim Biophys Acta. 2013;1829(6–7):552–560.
- Shahbabian K, Jamalli A, Zig L, et al. RNase Y, a novel endoribonuclease, initiates riboswitch turnover in Bacillus subtilis. EMBO J. 2009;28(22):3523–3533.
- Yao S, Bechhofer DH. Initiation of decay of Bacillus subtilis rpsO mRNA by endoribonuclease RNase Y. J Bacteriol. 2010;192(13):3279–3286.
- Callaghan AJ, Marcaida MJ, Stead JA, et al. Structure of Escherichia coli RNase E catalytic domain and implications for RNA turnover. Nature. 2005;437(7062):1187–1191.
- Koslover DJ, Callaghan AJ, Marcaida MJ, et al. The crystal structure of the Escherichia coli RNase E apoprotein and a mechanism for RNA degradation. Structure. 2008;16(8):1238–1244. .
- Carpousis AJ. The RNA Degradosome of Escherichia coli: an mRNA-degrading machine assembled on RNase E. Annu Rev Microbiol. 2007;61(1):71–87.
- Oussenko IA, Abe T, Ujiie H, et al. Participation of 3ʹ-to-5ʹ exoribonucleases in the turnover of Bacillus subtilis mRNA. J Bacteriol. 2005;187(8):2758–2767.
- McLaren RS, Newbury SF, Dance GS, et al. mRNA degradation by processive 3ʹ-5ʹ exoribonucleases in vitro and the implications for prokaryotic mRNA decay in vivo. J Mol Biol. 1991;221(1):81–95.
- Spickler C, Mackie GA. Action of RNase II and polynucleotide phosphorylase against RNAs containing stem-loops of defined structure. J Bacteriol. 2000;182(9):2422–2427.
- Lorentzen E, Conti E. Structural basis of 3ʹ end RNA recognition and exoribonucleolytic cleavage by an exosome RNase PH core. Mol Cell. 2005;20(3):473–481.
- Py B, Higgins CF, Krisch HM, et al. A DEAD-box RNA helicase in the Escherichia coli RNA degradosome. Nature. 1996;381(6578):169–172.
- Coburn GA, Miao X, Briant DJ, et al. Reconstitution of a minimal RNA degradosome demonstrates functional coordination between a 3ʹ exonuclease and a DEAD-box RNA helicase. Gene Dev. 1999;13(19):2594–2603.
- Liou GG, Chang HY, Lin CS, et al. DEAD box RhlB RNA helicase physically associates with exoribonuclease PNPase to degrade double-stranded RNA independent of the degradosome-assembling region of RNase E. J Biol Chem. 2002;277(43):41157–41162.
- Weick EM, Puno MR, Januszyk K, et al. Helicase-dependent RNA decay illuminated by a Cryo-EM structure of a human nuclear RNA exosome-MTR4 Complex. Cell. 2018;173(7):1663–77 e21.
- Cheng ZF, Deutscher MP. An important role for RNase R in mRNA decay. Mol Cell. 2005;17(2):313–318.
- Jinek M, Coyle SM, Doudna JA. Coupled 5ʹ nucleotide recognition and processivity in Xrn1-mediated mRNA decay. Mol Cell. 2011;41(5):600–608.
- Lorentzen E, Basquin J, Tomecki R, et al. Structure of the active subunit of the yeast exosome core, Rrp44: diverse modes of substrate recruitment in the RNase II nuclease family. Mol Cell. 2008;29(6):717–728.
- Chernyakov I, Whipple JM, Kotelawala L, et al. Degradation of several hypomodified mature tRNA species in Saccharomyces cerevisiae is mediated by Met22 and the 5’-3’ exonucleases Rat1 and Xrn1. Gene Dev. 2008;22(10):1369–1380.
- Cole SE, LaRiviere FJ, Merrikh CN, et al. A convergence of rRNA and mRNA quality control pathways revealed by mechanistic analysis of nonfunctional rRNA decay. Mol Cell. 2009;34(4):440–450.
- Schneider C, Anderson JT, Tollervey D. The exosome subunit Rrp44 plays a direct role in RNA substrate recognition. Mol Cell. 2007;27(2):324–331.
- Hossain ST, Deutscher MP. Helicase activity plays a crucial role for RNase R Function in vivo and for RNA metabolism. J Biol Chem. 2016;291(18):9438–9443.
- Condon C. What is the role of RNase J in mRNA turnover? RNA Biol. 2010;7(3):316–321.
- Mathy N, Benard L, Pellegrini O, et al. 5ʹ-to-3ʹ exoribonuclease activity in Bacteria: role of RNase J1 in rRNA maturation and 5ʹ stability of mRNA. Cell. 2007;129(4):681–692.
- Even S, et al. Ribonucleases J1 and J2: two novel endoribonucleases in B.subtilis with functional homology to E. coli RNase E. Nucleic Acids Res. 2005;33(7):2141–2152. .
- Britton RA, Wen T, Schaefer L, et al. Maturation of the 5’ end of Bacillus subtilis 16S rRNA by the essential ribonuclease YkqC/RNase J1. Mol Microbiol. 2007;63(1):127–138. .
- Mader U, Zig L, Kretschmer J, et al. mRNA processing by RNases J1 and J2 affects Bacillus subtilis gene expression on a global scale. Mol Microbiol. 2008;70(1):183–196.
- Durand S, Gilet L, Bessieres P, et al. Three Essential Ribonucleases—RNase Y, J1, and III—Control the abundance of a majority of Bacillus subtilis mRNAs. Plos Genet. 2012;8(3):e1002520.
- Figaro S, Durand S, Gilet L, et al. Bacillus subtilis mutants with knockouts of the genes encoding ribonucleases RNase Y and RNase J1 are viable, with major defects in cell morphology, sporulation, and competence. J Bacteriol. 2013;195(10):2340–2348.
- Linder P, Lemeille S, Redder P. Transcriptome-wide analyses of 5ʹ-ends in RNase J mutants of a gram-positive pathogen reveal a role in RNA maturation, regulation and degradation. Plos Genet. 2014;10(2):e1004207.
- Bugrysheva JV, Scott JR. Regulation of virulence gene expression in Streptococcus pyogenes. RNA Biol. 2010;7(5):569–572.
- Merritt X, Liu N, Khajotia S, et al. RNases J1 and J2 are critical pleiotropic regulators in Streptococcus mutans. Microbiology. 2015;161(4):797–806.
- Dominski Z, Carpousis AJ, Clouet-d’Orval B. Emergence of the beta-CASP ribonucleases: highly conserved and ubiquitous metallo-enzymes involved in messenger RNA maturation and degradation. Biochim Biophys Acta. 2013;1829(6–7):532–551.
- Clouet-d’Orval B, Phung DK, Langendijk-Genevaux PS, et al. Universal RNA-degrading enzymes in Archaea: prevalence, activities and functions of beta-CASP ribonucleases. Biochimie. 2015;118:278–285.
- Phung DK, Rinaldi D, Langendijk-Genevaux PS, et al. Archaeal beta-CASP ribonucleases of the aCPSF1 family are orthologs of the eukaryal CPSF-73 factor. Nucleic Acids Res. 2013;41:1091–1103.
- Clouet-d’Orval B, Rinaldi D, Quentin Y, et al. Euryarchaeal beta-CASP proteins with homology to bacterial RNase J have 5ʹ- to 3ʹ-exoribonuclease activity. J Biol Chem. 2010;285(23):17574–17583.
- Hasenohrl D, Konrat R, Blasi U. Identification of an RNase J ortholog in Sulfolobus solfataricus: implications for 5’-to-3’ directional decay and 5’-end protection of mRNA in Crenarchaeota. RNA. 2011;17(1):99–107.
- Zheng X, Feng N, Li DF, et al. New molecular insights into an archaeal RNase J reveal a conserved processive exoribonucleolysis mechanism of the RNase J family. Mol Microbiol. 2017;106(3):351–366.
- Li LY, Ren MF, Xu YQ, et al. Enhanced glycosylation of an S-layer protein enables a psychrophilic methanogenic archaeon to adapt to elevated temperatures in abundant substrates. FEBS Lett. 2020;594(4):665–677.
- Zhao Y, Lu MH, Zhang H, et al. Structural insights into catalysis and dimerization enhanced exonuclease activity of RNase J. Nucleic Acids Res. 2015;43(11):5550–5559. .
- Winn MD, Ballard CC, Cowtan KD, et al. Overview of the CCP 4 suite and current developments. Acta Crystallogr D. 2011;67(4):235–242.
- Kabsch W. Xds. Acta Crystallogra D. 2010;66(2):125–132.
- Adams PD, Afonine PV, Bunkoczi G, et al. PHENIX : a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D. 2010;66(2):213–221. .
- Hossain ST, Malhotra A, Deutscher MP. How RNase R Degrades Structured RNA: role of the helicase activity and the s1 domain. J Biol Chem. 2016;291(15):7877–7887.
- Vincent HA, Deutscher MP. Substrate recognition and catalysis by the exoribonuclease RNase R. J Biol Chem. 2006;281(40):29769–29775.
- Pei XY, Bralley P, Jones GH, et al. Linkage of catalysis and 5ʹ end recognition in ribonuclease RNase J. Nucleic Acids Res. 2015;43(16):8066–8076.
- Li J, Qi L, Guo Y, et al. Global mapping transcriptional start sites revealed both transcriptional and post-transcriptional regulation of cold adaptation in the methanogenic archaeon Methanolobus psychrophilus. Sci Rep. 2015;5(1):9209. .
- Eaton JD, Davidson L, Bauer DLV, et al. Xrn2 accelerates termination by RNA polymerase II, which is underpinned by CPSF73 activity. Gene Dev. 2018;32(2):127–139.
- Chu LY, Hsieh TJ, Golzarroshan B, et al. Structural insights into RNA unwinding and degradation by RNase R. Nucleic Acids Res. 2017;45(20):12015–12024.
- Vincent HA, Deutscher MP. The roles of individual domains of RNase R in substrate binding and exoribonuclease activity. The nuclease domain is sufficient for digestion of structured RNA. J Biol Chem. 2009;284(1):486–494.
- Sarmiento F, Mrazek J, Whitman WB. Genome-scale analysis of gene function in the hydrogenotrophic methanogenic archaeon Methanococcus maripaludis. Proc Natl Acad Sci USA. 2013;110(12):4726–4731.
- Lee G, Bratkowski MA, Ding F, et al. Elastic coupling between RNA degradation and unwinding by an exoribonuclease. Science. 2012;336(6089):1726–1729.