
ABSTRACT
Breast cancer is one of the most prevalent cancers in women worldwide. Through the regulation of many coding and non-coding target genes, oestrogen (E2 or 17β-oestradiol) and its nuclear receptor ERα play important roles in breast cancer development and progression. Despite the astounding advances in our understanding of oestrogen-regulated coding genes over the past decades, our knowledge on oestrogen-regulated non-coding targets has just begun to expand. Here we leverage epigenomic approaches to systematically analyse oestrogen-regulated long non-coding RNAs (lncRNAs). Similar to the coding targets of ERα, the transcription of oestrogen-regulated lncRNAs correlates with the activation status of ERα enhancers, measured by eRNA production, chromatin accessibility, and the occupancy of the enhancer regulatory components including P300, MED1, and ARID1B. Our 3D chromatin architecture analyses suggest that lncRNAs and their neighbouring E2-resonsive coding genes, exemplified by LINC00160 and RUNX1, might be regulated as a 3D structural unit resulted from enhancer-promoter interactions. Finally, we evaluated the expression levels of LINC00160 and RUNX1 in various types of breast cancer and found that their expression positively correlated with the survival rate in ER+ breast cancer patients, implying that the oestrogen-regulated LINC00160 and its neighbouring RUNX1 might represent potential biomarkers for ER+ breast cancers.
Introduction
Breast cancer is the most common malignancy and the second most common cause of death of cancer in women [Citation1]. Up to 70% of total breast cancer cases are oestrogen-receptor α (ERα) positive, making ERα the most important target for breast cancer endocrine therapy [Citation2]. Oestrogen and ERα play a crucial role in regulating the cell proliferation, survival and apoptosis in ERα+ breast cancer [Citation3,Citation4]. Tamoxifen, a selective oestrogen-receptor modulator (SERM), is commonly administered in the adjuvant therapy for ERα+ breast cancers. However, up to 50% of breast cancer patients with metastatic disease do not respond to adjuvant tamoxifen treatment, and many initial responders acquire resistance [Citation5,Citation6]. In order to develop novel therapeutic strategies, new therapeutic targets and specific biomarkers that predict therapeutic response to different therapies need to be identified.
Upon binding to 17β-oestradiol (E2), ERα translocates from the cytoplasm to the nucleus and binds predominantly to distal enhancer regions containing oestrogen response elements (EREs) to regulate a variety of ERα-dependent genes [Citation7,Citation8]. Enhancers are essential distal DNA regulatory elements that control temporal- or spatial-specific gene expression patterns during development and other biological processes [Citation9]. Like other enhancers, ERα enhancers (enhancers bound by ERα)exhibit several common genomic features/signatures, such as open chromatin architecture, a common set of histone modifications (e.g. H3K4me1/2 and H3K27ac), and the binding of chromatin remodelling complexes (e.g. ARID1A/B) and co-activators (e.g. p300/CBP and the mediator complex) [Citation10]. More recently, it’s found that many enhancers are bound by RNA polymerase II and are actively transcribed, generating noncoding enhancer RNAs (eRNA) [Citation11–13]. eRNAs are short transcripts of approximately 50 to 2000 nucleotides in size and often transcribed bi-directionally transcription, and can serve as a robust marker of active enhancers and are widely used to indicate enhancer activity and target gene induction [Citation14–16]. The binding of ERα onto enhancers requires cis-binding pioneer TFs (e.g. FOXA1) that bind to their own DNA binding motifs in close proximity to ERE and open chromatin for ERα to localize onto ERE, followed by the recruitment of epigenetic cofactors [Citation17–21]. We have previously identified a new category of ERα TF ‘co-activators’, MegaTrans TFs, which are recruited by ERα through protein-protein interactions (trans-binding) to active ERα enhancers containing oestrogen response element (ERE) to control the transcription of downstream target genes [Citation8,Citation22]. In addition to protein-coding targets, oestrogen signalling regulates various classes of non-coding RNAs [Citation13,Citation23,Citation24].
Over 90% of the human genome is transcribed into non-protein coding RNAs (ncRNAs) [Citation25], which can be divided into two major groups: small non-coding RNAs (sncRNAs) with size < 200 nt (e.g. miRNA, piRNA, snRNA and snoRNA) and long non-coding RNAs (lncRNAs) with size ≥ 200 nt [Citation26,Citation27]. According to the latest release of Gencode project, there are 19,957 protein coding genes, 7,576 small non-coding RNA (SncRNA) genes, and 17,952 long non-coding RNA (lncRNA) genes in a total of 60,662 genes in the human genome [Citation28]. The large number of lncRNA genes indicates the biological importance of lncRNAs. Over the past years, a wealth of lncRNAs have been found to play a pivotal role in transcriptional regulation [Citation29], and abnormal expression of lncRNAs is involved in the invasion, metastasis and chemoresistance of multiple cancer types [Citation30–32]. Multiple lncRNAs, such as UCA1, DSCAM-AS1 and HOTAIR, are associated with tamoxifen sensitivity in ER+ breast cancers [Citation23,Citation33,Citation34]. lncRNAs are also found to be involved in E2-induced rapid transcriptional regulation in an ER+ breast cancer cell line [Citation13].
Building on previous studies that have begun to understand the role of lncRNAs in breast cancers [Citation13,Citation23,Citation24,Citation35], we set out to perform a systematic analysis at the epigenomic level on oestrogen-regulated lncRNAs. We employed genome-wide epigenomic strategies including GRO-seq and ATAC-seq and identified lncRNAs that are either up- or down-regulated in response to oestrogen treatment in breast cancer cells. The transcription of these oestrogen-regulated lncRNAs correlates with the open chromatin state and the presence of enhancer complex on their neighbouring ERα enhancers. Our findings in both MCF7 and T47D ER+ breast cancer cell lines suggest that some of these oestrogen-regulated lncRNAs and their neighbouring E2-resonsive coding genes might be coordinately regulated as a 3D structural unit through the enhancer-promoter interactions inside this unit. Furthermore, we revealed a positive correlation between the survival rate in ER+ breast cancer patients and the levels of an oestrogen-regulated lncRNA LINC00160 and its neighbouring E2-responsive coding gene RUNX1, indicating that estrogen-regulated lncRNAs might be applied as prognosis biomarkers for ER+ breast cancers.
Results
Identification of lncRNAs transcriptionally regulated by oestrogen signalling in ER+ breast cancer cells
In order to identify lncRNAs that respond to E2 stimulus, we analysed our RNA-seq data [Citation8]to capture differentially expressed lncRNAs in MCF7 cells with or without E2 treatment. We used DESeq2 1.14.1 to call differentially expressed genes and set the threshold as fold expression change ≥ 1.4 and padj ≤ 0.01 [Citation36]. We identified 90 and 160 lncRNAs that were up and down-regulated respectively upon E2 stimulus ( and Supplementary Table 1). As RNA-seq measures changes in the steady-state level of a given RNA, it reflects the combined effect of RNA transcription, processing and degradation. To evaluate changes specifically at transcriptional level, we analysed our global run-on sequencing (GRO-seq) datasets [Citation8] to measure nascent RNAs that are transcribed from chromatin-associated RNA polymerase II. Our GRO-seq identified 253 E2-activated lncRNAs and 96 E2-repressed lncRNAs that are transcriptionally regulated by the oestrogen signalling ( and Supplementary Table 1). To narrow down the E2-responsive candidate lncRNAs, we compared the lncRNAs identified from RNA-seq and GRO-seq by plotting the GRO-seq-identified genes to the RNA-seq data and vice versa (). A total of 47 E2-regulated lncRNAs, including 33 E2-activated and 14 E2-repressed lncRNA genes, were found overlapped between the RNA-seq and GRO-seq datasets (Supplementary Table 1), as demonstrated by Venn diagrams and heatmap ().
Figure 1. Identification of lncRNAs transcriptionally induced/repressed by oestrogen signalling in ER+ breast cancer cells.
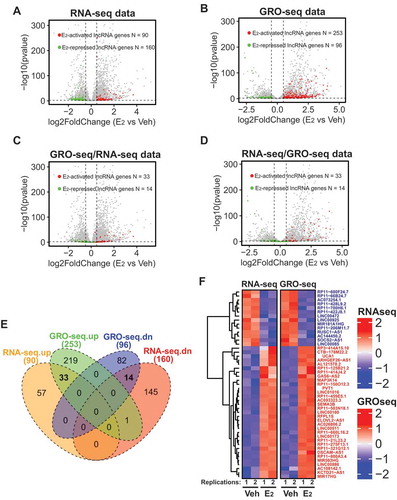
Among the 47 E2-responsive lncRNA genes, LINC01016 and LINC00160 have been previously shown to respond to E2 stimulus in MCF7 and T47D ER+ breast cancer cells [Citation26]. PVT1 is a well-studied oncogenic lncRNA in breast cancer and it was found to regulate the level of its neighbouring oncogene MYC by either directly stabilizing MYC protein [Citation37] or competing for the engagement of enhancers [Citation38]. Interestingly, lncRNA SOCS2-AS1, whose expression was reported to be activated by androgen in androgen receptor (AR) positive LNCaP prostate cancer cells [Citation39], was repressed upon E2 treatment ( and Supplementary Table 1). In addition, other disease-related lncRNAs, such as MIR17HG, a lncRNA that promotes tumorigenesis and metastasis in colorectal cancer [Citation40], were also subjected to E2 regulation ( and Supplementary Table 1). Overall, combining RNA-seq and GRO-seq approaches, we have identified lncRNAs that are transcriptionally regulated by oestrogen signalling in breast cancer cells.
Expression of oestrogen-regulated lncRNAs correlates with eRNA production of neighbouring ERα enhancers
The biological effects of oestrogen are mediated by its binding to oestrogen receptors, which then bind to ERE-containing enhancers to regulate gene expression [Citation41]. We therefore sought to understand the relationship between ERα enhancers and these E2-regulated lncRNAs in ERα+ breast cancer. We retrieved published H3K27ac ChIP-seq data [Citation42] and searched for ERα enhancers located within the region ±40kb from each of the 47 E2-responsive lncRNA genes based on the chromatin occupancy of ERα and H3K27ac profile. We identified 151 and 54 ERα enhancers associated with the 33 E2-activated and the 14 E2-repressed lncRNAs respectively (Supplementary Table 2), implying that the transcription of these lncRNAs might be controlled by ERα enhancers. The presence of ERα enhancers approximate to E2-regulated lncRNA genes is exemplified by ERα and H3K27ac ChIP-seq peaks on the genomic loci of E2-activated LINC00160 and E2-repressed SOCS2-AS1 (), as well as PVT1 and MIR9-3HG (Fig. S1A-B).
Figure 2. Expression of oestrogen-regulated lncRNAs correlates with eRNA production of neighbouring ERα enhancers.
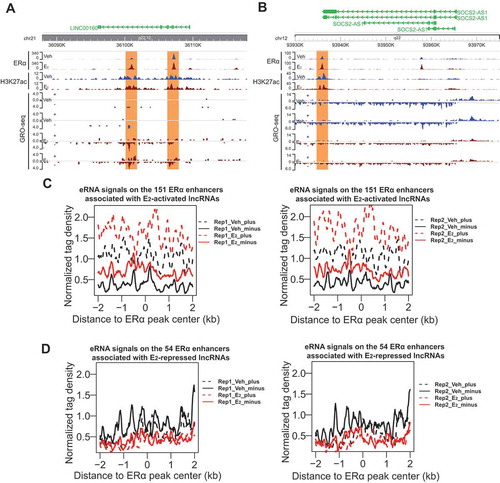
We next asked if the ERα enhancers associated with E2-responsive lncRNAs are active enhancers and how they respond to oestrogen signalling. As GRO-seq captures all RNAs that are being produced by active RNA polymerase, it can be used to study the transcription of RNAs both from the gene bodies and from enhancers simultaneously. eRNAs are often bi-directionally transcribed and have been used as a robust marker for active enhancers [Citation14–Citation16]. We found that the 151 ERα enhancers associated with the 33 E2-activated lncRNAs had strongly increased eRNA production upon E2 treatment (see GRO-seq data from two replications in ), whereas the 54 enhancers related to the 14 E2-repressed lncRNAs showed a slight reduction in eRNA transcription (see GRO-seq data from two replications in ). These E2-induced changes in eRNA generation can also be exemplified by the neighbouring enhancers of representative lncRNAs, including LINC00160, PVT1, SOCS2-AS1 and MIR9-3HG ( and S1A-B). These data suggest that in response to E2 stimulus, ERα binds to and activates enhancers containing ERE, which subsequently activate the lncRNAs that are up-regulated by E2. However, for the E2-repressed lncRNAs, their neighbouring enhancers become less active in response to E2. This is consistent with a recent study showing that binding of ERα to basally active oestrogen-repressed (BAER) enhancers recruits KDM2A, which then brings NEDD4 to BAER enhancers to ubiquitylate and dismiss RNA Pol II from the enhancers to repress target gene expression [Citation43]. Therefore, similar to the protein-coding targets of oestrogen signalling pathway, the lncRNA targets might be also subject to the regulation of ERα enhancers.
Transcription of oestrogen-regulated lncRNAs is associated with the presence of enhancer complex
Having shown the correlation between the expression of E2-responsive lncRNAs and the eRNA level of their neighbouring ERα enhancers, we next explored the components of enhancer complex on these enhancers. We first analysed ERα ChIP-seq data [Citation44] and found that the binding of ERα on both groups of enhancers associated with either E2-activated or E2-repressed lncRNAs was dramatically increased upon E2 treatment (). We also checked the ChIP-seq data of H3K27ac, a histone modification that labels active enhancers. Notably, H3K27ac was present on these ERα enhancers prior to E2 stimulation and its level remained unchanged upon E2 treatment (). This is consistent with the previous report that H3K27ac signals on ERα enhancers remain unchanged upon E2 treatment [Citation42]. As H3K27ac is a well-recognized marker for active enhancers, these data suggest that ERα enhancers might be prepared before the presence of oestrogen, allowing rapid responses upon oestrogen stimulation. We next used our previous ChIP-seq data [Citation8] to examine the occupancy of enhancer co-activators, including the histone acetyltransferase p300/CBP and mediator MED1. For the 151 enhancers associated with E2-activated lncRNAs, the binding of p300 was greatly enhanced in E2-treated breast cancer cells. In contrast, the binding of p300 on the 54 enhancers associated with E2-repressed lncRNAs was reduced upon E2 treatment (). MED1 is known to promote the looping between enhancer and promoter and is involved in the transcription of nearly all RNA polymerase II-dependent genes [Citation45]. We found that the occupancy of MED1 on the 151 enhancers, but not on the 54 enhancers, was enhanced in E2 treated cells ().
Figure 3. Transcription of oestrogen-regulated lncRNAs is associated with the presence of enhancer complex.
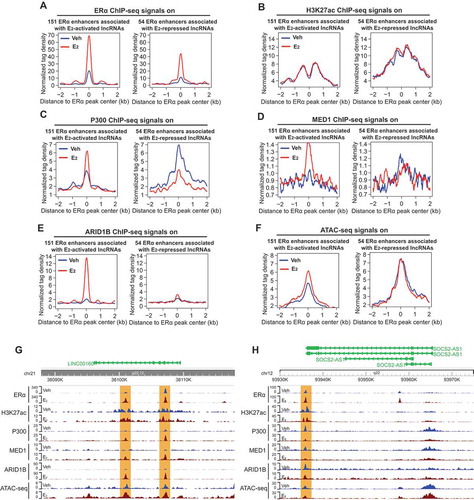
To further investigate the ERα enhancers associated with E2-regulated lncRNAs, we performed ChIP-seq for ARID1B and investigated itsbinding on these enhancers. ARID1A and ARID1B are mutually exclusive subunits of the BAF sub-family of SWI/SNF chromatin remodelling complex that controls accessibility at most promoters and enhancers [Citation46,Citation47]. E2 treatment greatly induced the binding of ARID1B onto the 151 enhancers, but did not affect its binding on the 54 enhancers (). Finally, we carried out ATAC-seq to examine the chromatin accessibility in the genomic regions of the enhancers associated with E2-responsive lncRNAs. We detected enhanced chromatin accessibility on the 151 enhancers in response to E2 treatment, whereas the accessibility on the 54 enhancers remained unchanged (). The E2 effects on the enhancer status described above are also demonstrated by the representative enhancers associated with LINC00160, PVT1, SOCS2-AS1 andMIR9-3HG ( and S2A-B). The differences between the two groups of enhancers (151 enhancers associated with E2-activated lncRNAs vs the 54 enhancers associated with E2-repressed lncRNAs) in their responses to oestrogen signalling might account for the different effects (activation or repression) on their lncRNA target genes. Overall, these data show that the transcription of E2-regulated lncRNAs is tightly linked to the status of their neighbouring ERα enhancers, further supporting the notion that both protein-coding and non-coding targets of oestrogen signalling pathway are regulated by ERα enhancers.
LINC00160 and RUNX1 are co-regulated by E2 and are located within one TAD
As stated above, we have shown that the transcription of oestrogen-regulated lncRNAs correlate with the open chromatin state and the presence of enhancer complex on their neighbouring ERα enhancers (, S1, 3, and S2). This is similar to our previous findings on the regulation of E2-responsive protein-coding genes by ERα enhancers [Citation8,Citation22], and prompted us to ask whether these lncRNAs and the protein-coding targets are co-regulated. Through genome-wide searching, we found that E2-responsive lncRNAs often locate in a close proximity to an E2-responsive coding gene and both lncRNAs and coding genes were activated coordinately by oestrogen, exemplified by the LINC00160-RUNX1 pair and the PVT1-MYC pair ( and S3A). This coordinate activation of LINC00160 and RUNX1 by E2 was also demonstrated by our qPCR data for LINC00160 and RUNX1 in both MCF7 and T47DER+ breast cancer cell lines (Fig. S3B-C). These results suggest that oestrogen may co-regulate a lncRNA and its neighbouring coding gene within a 3D structural unit on the chromatin.
Figure 4. LINC00160 and RUNX1 are co-regulated by E2 and are located within one TAD.
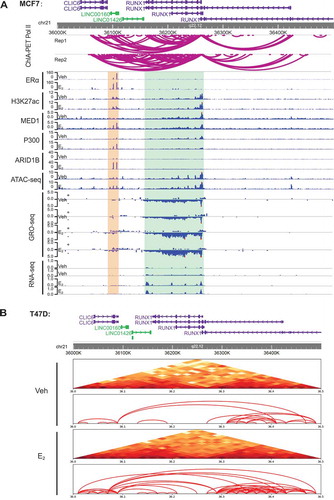
RUNX1 is a transcription factor involved in luminal development [Citation48] and frequently found to be involved in ER+ breast cancer progression [Citation49].Thus, we next sought to study the chromatin architecture regulation mechanism underlying the coordinate induction of LINC00160 and RUNX1 by oestrogen. The ChIP-seq and ATAC-seq epigenomic data revealed that there were at least two ERα enhancers within the LINC00160 gene body (). We employed the published ChIA-PET Pol II data [Citation50] from MCF7 cell line to investigate the 3D chromatin interactions between LINC00160 and RUNX1. ChIA-PET Pol II data revealed frequent looping events between the enhancers on LINC00160 and the RUNX1 promoter (). To further confirm the spatial relationship between LINC00160 and RUNX1, we performed Hi-C to measure any potential interactions near the LINC00160-RUNX1 loci in T47D ER+ breast cancer cell line. We found that the interaction between LINC00160 and RUNX1 was enhanced by the treatment of E2 (). Our Hi-C data also demonstrated that LINC00160 and RUNX1 were organized in a 3D structural unit, as they were located within a topologically associating domain (TAD) revealed by Hi-C (). Altogether, our findings suggest that some of these oestrogen-regulated lncRNAs might be transcribed together as a whole unit with their neighbouring E2-resonsive protein-coding genes through 3D spatial chromatin looping.
The levels of LINC00160 and RUNX1 correlate with the disease outcome in ER+ breast cancer patients
Given the transcriptional regulation of LINC00160 and RUNX1 by oestrogen signalling, we asked if their expression has a prognostic potential in breast cancer patients. We first performed the bodymap analyses using GEPIA website [Citation51]. We found that LINC00160 was predominantly expressed in mammary gland, and the expression level of LINC00160 was significantly elevated in breast tumours compared to paired normal tissues (). To further confirm bodymap results, we took advantage of the RNA-seq data from TCGA and found a notable upregulation of LINC00160 in breast tumors when compared to normal tissues (), suggesting the biological relevance of LINC00160 in breast cancer oncogenesis.
Figure 5. The levels of LINC00160 and RUNX1 correlate with the disease outcome in ER+ breast cancer patients.
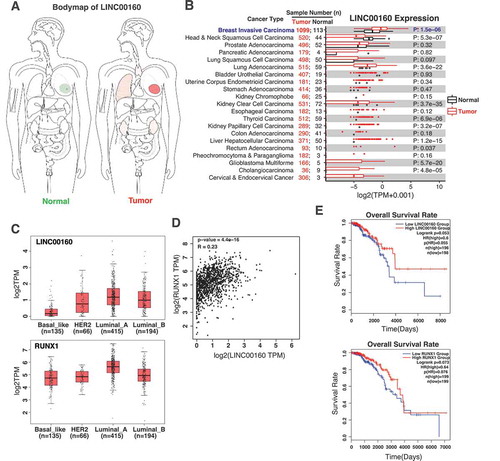
As LINC00160 and its neighbouring coding gene RUNX1 are co-regulated by oestrogen and they reside within a 3D structural unit on chromatin ( and S3), we next examined the relationship between both of their expression and their different roles in ER+ vs ER- breast cancer subtypes. We analysed the expression levels of LINC00160 and RUNX1 in the four subtype of breast cancer using TCGA datasets. Comparing with ER- breast cancer subtypes, including basal and HER2+ subtypes, the expression of both LINC00160 and RUNX1 was significantly higher in ER+ luminal subtypes, particularly in luminal A subtype (). Consistently, the expression correlation analyses demonstrated significant positive correlation of LINC00160 and RUNX1 with each other in ER+ breast cancer patients (p = 4.4e-16, R = 0.23) (), suggesting these two E2-regulated genes may both be involved in the oncogenesis of ER+ breast cancer.
Finally, we analysed the relationship between the expression levels of LINC00160-RUNX1 and the survival rate of ER+ breast cancer patients. When we analysed TCGA data from annotated and transcriptionally profiled ER+ breast cancer samples, we found that the group with high LINC00160 level had a better overall survival rate than the group with low LINC00160 level. Similarly, the group with high RUNX1 expression displayed an improved survival rate when compared to the group with low RUNX1 level (). This correlation between the overall survival rate and the expression levels of LINC00160 and RUNX1 is in agreement with the notion that higher levels of E2-responsive LINC00160 and RUNX1 may indicatea higher sensitivity to endocrine therapies and a better prognostic outcome.
Overall, consistent with our finding that LINC00160 and RUNX1 are coordinately regulated by oestrogen signalling in ER+ breast cancer cell lines, these analyses on clinical datasets suggest that LINC00160 and RUNX1, a pair of oestrogen-regulated non-coding and coding targets, might represent potential prognostic biomarkers in ER+ breast cancer.
Discussion
In this study, we identified oestrogen-activated and -repressed lncRNAs in breast cancer cells through genome-wide epigenomic strategies. Similar to protein-coding targets, the transcription of these lncRNA targets correlates with the open chromatin state and the occupancy of enhancer complex on their neighbouring ERα enhancers. We have then focused on one of the lncRNA targets delineated the concomitant regulation of LINC00160 and RUNX1 by oestrogen signalling. Moreover, by performing ChIA-PET Pol II and Hi-C, we demonstrated that LINC00160 and its neighbouring RUNX1 coding gene were organized within one TAD domain and the several ERα enhancers locating on LINC00160 had a strong chromatin spatial interaction with the promoter of RUNX1, and such interaction was augmented by E2. Finally, we revealed a correlation between disease outcome and the levels of LINC00160 and RUNX1, indicating that they might be applied as prognosis biomarkers for ER+ breast cancers.
E2-responsive lncRNAs are regulated by ERα enhancers
Oestrogen/ERα signalling pathway plays a crucial role in breast cancer tumorigenesis and progression. Upon ligand stimulation, ERα regulates coding target gene expression by binding to distal enhancers. On ERα enhancers, cis-binding pioneer TFs (such as FOXA1) open chromatin for ERα to localize onto ERE, followed by the recruitment of epigenetic cofactors including histone acetyltransferase p300 and mediator MED1 [Citation17–Citation21], resulting in a global increase in eRNA transcription on enhancers adjacent to E2-upregulated coding genes [Citation42]. Additionally, we have previously demonstrated that a new category of ERα TF ‘co-activators’, termed as MegaTrans TFs, was selectively recruited to the most active ERα enhancers to regulate coding target gene expression [Citation22]. Despite the advance in our understanding on the protein-coding target genes of this pathway, the regulation and function of non-coding targets remain largely unknown. To understand how these non-coding targets are regulated, we searched for ERα enhancers associated with the E2-activated and -repressed lncRNAs and investigated how they respond to oestrogen signalling. Like ERα enhancers that regulate coding genes, these lncRNA-associated ERα enhancers recruit ERα upon E2 stimulus. For the enhancers associated with E2-activated lncRNAs, we detected increased ATAC-seq signals, elevated occupancy of enhancer complex such as p300, MED1 and ARID1B, as well as enhanced eRNA transcription in cells treated with E2 ( and ). However, the enhancers associated with E2-repressed lncRNAs showed different responses to E2: unchanged occupancy of chromatin remodelling factor ARID1B accompanied with unaltered chromatin accessibility, reduced binding of epigenetic co-activators p300 and MED1, and slightly reduced eRNA production. The difference between the two groups of enhancers in the responses to oestrogen might account for the opposite effects (activation or repression) on the target lncRNAs. Notably, a recent study has shown that binding of ERα to oestrogen-repressed enhancers recruits KDM2A and NEDD4 to modify and dismiss RNA Pol II from the enhancers to repress target coding gene expression [Citation43]. It’s not clear whether the same KDM2A-mediated repression strategy is utilized to repress non-coding target gene expression. Regardless, our data has suggested that, similar to the protein-coding targets of oestrogen signalling pathway, lncRNA targets are regulated by ERα enhancers, and that the regulation of the lncRNA targets also involves enhancer chromatin opening, enhancer complex recruitment and eRNA production.
Co-regulation of lncRNAs and adjacent coding genes within a TAD
The chromosomes of mammalian cells are hierarchically organized into hundreds of megabase-sized topologically associated domains (TADs) and the cell-type-specific enhancer-promoter contacts take place within the TAD scaffold, leading to signal-regulated gene expression pattern [Citation52]. Although LINC00160 and RUNX1 have been individually implied in breast cancers [Citation24,Citation49], they had not been linked to each other previously. For the first time, we revealed that LINC00160 and its neighbouring RUNX1 were co-activated by oestrogen in several different ER+ breast cancer cell lines ( and S3). Interestingly, LINC00160 and RUNX1 appear to reside within the same TAD chromatin 3D domain, and two ERα enhancers are located within LINC00160 gene (). We therefore speculate that these ERα enhancers regulate both LINC00160 and RUNX1 through looping to both LINC00160 and RUNX1 promoters. Our ChIA-PET Pol II and Hi-C data demonstrated strong chromatin spatial interactions between these ERα enhancers and the promoter of RUNX1 and showed that such interactions could be enhanced by E2 treatment (). Unfortunately, current Hi-C resolution is not sufficient to detect the interactions between these enhancers and LINC00160 promoter due to the small spatial distance. Future studies are needed to functional test the regulatory role of each individual ERα enhancer located in LINC00160 gene by deleting each enhancer with CRISPR/Cas9 and measuring the expression levels of LINC00160 and RUNX1. As many lncRNAs are known to function as regulatory elements gene expression regulation, it will be interesting to knockdown LINC00160 or to delete its promoter with CRISPR/Cas9 and test if LINC00160 lncRNA itself is involved in enhancer-promoter looping and gene regulation in this TAD domain.
LINC00160 and RUNX1 as potential prognostic biomarkers
Our analyses on clinical data showed that LINC00160 is primarily expressed in breast tissue and is expressed at a higher level in breast tumours than the paired normal breast tissues (). Among all different ER+ or ER- breast cancer subtypes, LINC00160 and its neighbouring coding gene RUNX1 were significantly up regulated in ER+ luminal breast cancer, especially luminal A. The higher level of both LINC00160 and RUNX1 in luminal A subtype is probably due to the higher expression of ERα in Luminal A subtype [Citation53]. These clinical data are in agreement with the co-regulation of LINC00160 and RUNX1 by oestrogen in ER+ breast cancer cell lines, and suggest that as E2/ERα-regulated downstream target genes, LINC00160 and RUNX1 may mediate oestrogen signalling to promote the early stage oncogenesis of ER+ breast cancers. Interestingly, our prognosis analyses showed that high expression of both LINC00160 and RUNX1 was associated with high overall survival rate in ER+ breast cancer patients. This might be because that higher levels of E2-responsive LINC00160 and RUNX1 in ER+ breast cancer patients could render higher sensitivity to endocrine therapies, and subsequently a better prognostic outcome. Similarly, in a previous study, another E2/ERα-regulated target GREB1 was found to mediate oestrogen signalling to promote breast cancer oncogenesis, but high level of GREB1 was found to correlate with better prognostic outcome [Citation54]. This finding is also consistent with a previous report that overexpression of RUNX1 inhibited the proliferation and growth of breast cancer stem cells and thus resulting in a better survival rate in breast cancer [Citation55]. Therefore, the oestrogen-regulated LINC00160 and its neighbouring RUNX1 might represent potential biomarkers for ER+ breast cancers. Future studies to dissect the in vivo function of LINC00160 and RUNX1 will help to evaluate their roles as prognostic biomarkers and therapeutic targets.
Materials and methods
Cell culture studies
MCF7 and T47D cell lines were obtained from ATCC. MCF7 cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% FBS and penicillin/streptomycin. T47D cells were cultured in Roswell Park Memorial Institute (RPMI) 1640 Medium supplemented with 10% FBS and penicillin/streptomycin. Both cell lines were grown in a humidified incubator with 5% CO2. For E2 stimulation, cells were hormone stripped for 3 days in phenol-free media with 5% charcoal-stripped FBS before receiving 100 nM 17β-oestradiol (E2) (Sigma) or ethanol vehicle control treatment for 1 hour or 6 hours for oestrogen signalling induction.
ChIP-seq
ARID1B ChIP-seq was performed as previously described [Citation22] with slight modification. Briefly, cells were cross-linked with 1% formaldehyde for 10 min at room temperature.For selected ARID1B ChIP experiments, cells were double cross-linked with 2 mM DSG (CovaChem) for 45 min followed by secondary fixation with 1% formaldehyde for 10 min. Cross-linking was quenched with 0.125 M glycine for 5 min. Cells were successively lysed in lysis buffer LB1 (50 mM HEPES-KOH, pH 7.5, 140 mM NaCl, 1 mM EDTA, 10% glycerol, 0.5% NP-40, 0.25% Triton X-100, 1x PI), LB2 (10 mM Tris-HCl, pH 8.0, 200 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, 1x PI), LB3 (10 mM Tris-HCl, pH 8.0, 100 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, 0.1% Na-Deoxycholate, 0.5% N-lauroylsarcosine, 1x PI) (Note: after passing through buffers LB1 and LB2, the pellet becomes nuclear fraction and will be lysed in LB3 lysis buffer for sonication). Chromatin was sonicated to an average size of ~200-500 bp using QSonica’s Q800 R sonicator system (20% amplitude, 10 s on and 20 s off for 10 min). A total of 3–6 μg of antibody was added to the sonicated chromatin and incubated overnight at 4°C. Subsequently, 50 μl of Dynabeads Protein G (Invitrogen) were added to each ChIP reaction and incubated for 4 hours at 4°C. Dynabeads were washed with RIPA buffer (50 mM HEPES pH 7.6, 1 mM EDTA, 0.7% Na-Deoxycholate, 1% NP-40, 0.5 M LiCl) 6 times, and once with TE. The chromatin was eluted, followed by reverse cross-linking and DNA purification. ChIP DNA was resuspended in 10 mM Tris-HCl pH 8.5. The purified DNA was subjected to qPCR to confirm target region enrichment before moving on to deep sequencing library preparation. For sequencing, the extracted DNA was used to construct the ChIP-seq library using KAPA Hyper Prep kit (KK8504), followed by deep sequencing with the Illumina’s HiSeq 3000 system according to the manufacturer’s instructions.
ATAC-seq
ATAC-seq library prep was performed as previously described [Citation56]. Briefly, 20,000 cells were washed three times with cold PBS, collected by centrifugation then lysed in lysis buffer (10 mM Tris-HCl, pH 7.4, 10 mM NaCl, 3 mM MgCl2, 0.1% NP-40). After purification of nuclei, transposition was performed with Tn5 transposase from Nextera DNA Library Prep Kit (Illumina, catalogue # FC-121-1030). Purified DNA was then ligated with adapters, amplified and size selected for sequencing. Libraries were sequenced with Mid 75 bp PE on Illumina NextSeq 500.
RNA isolation and quantitative RT-PCR
Total RNA was isolated with RNeasy Mini Kit (Qiagen) according to the manufacture’s protocol and 1 μg RNA was used to convert to cDNA using iScript Select cDNA Synthesis Kit (Bio-Rad) in the presence of both oligo (dT) and random primers. qPCR was conducted with SsoAdvanced Universal SYBR Green Supermix (Bio-Rad) using CFX384 Real-Time PCR Detection System (Bio-Rad) according to the manufacturer’s instructions. Relative expression of RNAs was determined by the ΔΔCT method using GAPDH as an internal control for quantification analyses of gene targets. The primers used for qPCR are listed. Primer sets used in qPCR are as follows:
GAPDH: F: ACATCATCCCTGCCTCTACTGG, R: GTTTTTCTAGACGGCAGGTCAGG; LINC00160: F: CCCAACCTCAGCCATTCTTG, R: GTGGCCCAGGAGTGACTTTA;RUNX1: F:CAGGTTTGTCGGTCGAAGTG, R:TGATGGCTCTGTGGTAGGTG.
RNA-seq data analysis
The sequencing reads were aligned to human genome (hg19) with STAR 2.5.2b. Gencode v19 was used as the transcriptome annotation model, and read counts for each gene were conducted by featureCounts package with default parameter [Citation57,Citation58]. DESeq2 1.14.1 was used to call differentially expressed genes with fold expression change ≥ 1.4 and padj ≤ 0.01 as the cut-off[Citation36].
GRO-seq data analysis
GRO-seq reads were aligned to human genome (hg19) using bowtie with ‘–best – strata – m 1 – v 2’ parameters. Duplicated reads were eliminated for subsequent analysis. To balance the clonal amplification bias and total useful reads, only three reads at most were allowed for each unique genomic position. When measuring the expression level of genes, mapped reads from the first 30 kb of gene body were counted, excluding promoter-proximal region (transcription start site (TSS) to 1000 bp downstream of TSS; if the length of a gene is shorter than 10 kb, then the reads mapped to the first 10%*length regions were excluded). If the length of a gene is shorter than 30 kb, then the mapped reads from the whole gene were counted, excluding promoter-proximal region and gene end (500 bp upstream of transcription termination site (TTS) to TTS). Differential expression analysis was then performed by DESeq2 with a fold change threshold of 1.4 and FDR≤0.01.
ChIP-seq data analysis
Reads were aligned to human genome (hg19) using bowtie with ‘–best – strata – m 1’ parameters [Citation59]. Only uniquely mapped and non-duplicated reads were selected for subsequent analysis. MACS2 was employed to call peaks with default parameters and a q-value cut-off of 1e-5 [Citation60]. For H3K27ac, the broad mode of MACS2 was switched on. The peaks overlapping with the blacklist regions from UCSC were removed. All the ERα peaks within 40kb from the gene body of the E2-regulated lncRNAs were designated as the potential regulatory elements.
ATAC-seq data analysis
Cutadapt 1.11 was used to trim adapters in ATAC-seq reads, which were then aligned to human genome (hg19) using bowtie with parameters ‘–best – strata – m 1 – v 2’[Citation61]. Aligned reads with the same genomic position and orientation were collapsed to a single read. The reads were extended to 200bp and normalized to a sequencing depth of 10 million reads for each library.
Clinical data analysis
The bodymap of LINC00160 expression level in normal and cancer tissues, the correlation of LINC00160 with RUNX1, and the relationship between expression of LINC00160-RUNX1 with survival rate were all performed and visualized in GEPIA website [Citation51].
Deep sequencing data visualization
All the genome browser viewers were visualized in WashU Epigenome Browser [Citation62]. The two saturated replicates of Pol II ChIA-PET interaction data were retrieved from the browser website. ChIP-seq and GRO-seq samples were normalized to 10 million mapped reads per experiment, while RNA-seq samples were normalized to 1 million reads.
Hi-C experiment and data analysis
TCC (tethered chromatin conformation, a modified Hi-C protocol) data for T47D (vehicle treatment vs 1-hour E2 treatment) have been deposited in GEO under accession number GSE119890 [Citation63]. The data were mapped to hg19 human genome with bowtie2 [Citation64], filtered and normalized by HiC-Pro [Citation65]. Then the loops were called by Fit-Hi-C [Citation66] with the cut-off of FDR < 0.1.
Deep sequencing data availability
The ATAC-seq data and ChIP-seq data of ARID1B generated for this paper have been deposited in the Gene Expression Omnibus (GEO) under accession code GSE144927. The RNA-seq and GRO-seq data were retrieved from published GSE125606 and GSE125607, respectively. The ChIP-seq data of H3K27ac, P300, MED1 and ERαwere retrieved from published GSE45822, GSE60270, GSE125594, and ERR011973 respectively. Deep sequencing datasets are summarized in Supplementary Table 3.
Statistical analysis
The RT-qPCR experiments were performed with at least three independent biological replicates and three technical replicates for each reaction. Results are reported as mean ± SEM of three independent experiments. Data were analysed and statistics were performed using unpaired two-tailed Student’s t-tests or one-way ANOVA (Prism 5 GraphPad). Significant differences between two groups were noted by asterisks (* P < 0.05, ** P < 0.01, *** P < 0.001).
Supplemental Material
Download Zip (1.6 MB)Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
References
- Miller KD, Nogueira L, Mariotto AB, et al. Cancer treatment and survivorship statistics, 2019. CA Cancer J Clin. 2019;69(5):363–385.
- Burstein HJ, Temin S, Anderson H, et al. Adjuvant endocrine therapy for women with hormone receptor-positive breast cancer: american society of clinical oncology clinical practice guideline focused update. J Clin Oncol. 2014;32:2255–2269.
- Moggs JG, Orphanides G. Estrogen receptors: orchestrators of pleiotropic cellular responses. EMBO Rep. 2001;2:775–781.
- Pink JJ, Jordan VC. Models of estrogen receptor regulation by estrogens and antiestrogens in breast cancer cell lines. Cancer Res. 1996;56:2321–2330.
- Murata T, Jinno H, Takahashi M, et al. Clinicopathologic features of hormone-receptor-positive breast cancer patients with late recurrence. Breast J. 2019;25(1):9–15.
- Ring A, Dowsett M. Mechanisms of tamoxifen resistance. Endocr Relat Cancer. 2004;11:643–658.
- Carroll JS, Meyer CA, Song J, et al. Genome-wide analysis of estrogen receptor binding sites. Nat Genet. 2006;38(11):1289–1297.
- Zhu C, Li L, Zhang Z, et al. A Non-canonical Role of YAP/TEAD Is Required for Activation of Estrogen-Regulated Enhancers in Breast Cancer. Mol Cell. 2019;75(4):791–806 e8.
- Visel A, Rubin EM, Pennacchio LA. Genomic views of distant-acting enhancers. Nature. 2009;461(7261):199–205.
- Heinz S, Romanoski CE, Benner C, et al. The selection and function of cell type-specific enhancers. Nat Rev Mol Cell Biol. 2015;16(3):144–154.
- De Santa F, Barozzi I, Mietton F, et al. A large fraction of extragenic RNA pol II transcription sites overlap enhancers. PLoS Biol. 2010;8(5):e1000384.
- Kim TK, Hemberg M, Gray JM, et al. Widespread transcription at neuronal activity-regulated enhancers. Nature. 2010;465:182–187.
- Hah N, Danko CG, Core L, et al. A rapid, extensive, and transient transcriptional response to estrogen signaling in breast cancer cells. Cell. 2011;145(4):622–634.
- Wang D, Garcia-Bassets I, Benner C, et al. Reprogramming transcription by distinct classes of enhancers functionally defined by eRNA. Nature. 2011;474(7351):390–394.
- Hah N, Murakami S, Nagari A, et al. Enhancer transcripts mark active estrogen receptor binding sites. Genome Res. 2013;23(8):1210–1223.
- Kim T-K, Shiekhattar R. Architectural and Functional Commonalities between Enhancers and Promoters. Cell. 2015;162(5):948–959.
- Carroll JS, Liu XS, Brodsky AS, et al. Chromosome-wide mapping of estrogen receptor binding reveals long-range regulation requiring the forkhead protein FoxA1. Cell. 2005;122(1):33–43.
- Hurtado A, Holmes KA, Ross-Innes CS, et al. FOXA1 is a key determinant of estrogen receptor function and endocrine response. Nat Genet. 2011;43(1):27–33.
- Jozwik KM, Carroll JS. Pioneer factors in hormone-dependent cancers. Nat Rev Cancer. 2012;12(6):381–385.
- Kraus WL, Kadonaga JT. p300 and estrogen receptor cooperatively activate transcription via differential enhancement of initiation and reinitiation. Genes Dev. 1998;12(3):331–342.
- Zhang X, Krutchinsky A, Fukuda A, et al. MED1/TRAP220 exists predominantly in a TRAP/Mediator subpopulation enriched in RNA polymerase II and is required for ER-mediated transcription. Mol Cell. 2005;19:89–100.
- Liu Z, Merkurjev D, Yang F, et al. Enhancer activation requires trans-recruitment of a mega transcription factor complex. Cell. 2014;159(2):358–373.
- Niknafs YS, Han S, Ma T, et al. The lncRNA landscape of breast cancer reveals a role for DSCAM-AS1 in breast cancer progression. Nat Commun. 2016;7(1):12791.
- Jonsson P, Coarfa C, Mesmar F, et al. Single-Molecule Sequencing Reveals Estrogen-Regulated Clinically Relevant lncRNAs in Breast Cancer. Mol Endocrinol. 2015;29(11):1634–1645.
- Mattick JS, Makunin IV, Non-coding RNA Human molecular genetics 2006; 15 Spec No 1:R17–29.
- Palazzo AF, Lee ES. Non-coding RNA: what is functional and what is junk? Front Genet. 2015;6:2.
- Derrien T, Johnson R, Bussotti G, et al. The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res. 2012;22(9):1775–1789.
- GENCODE. Statistics about the current GENCODE Release (version 33). 2019, Aug.
- Agliano F, Rathinam VA, Medvedev AE, et al. Long Noncoding RNAs in Host-Pathogen Interactions. Trends Immunol. 2019;40:492–510.
- Deng SJ, Chen HY, Ye Z, et al. Hypoxia-induced LncRNA-BX111 promotes metastasis and progression of pancreatic cancer through regulating ZEB1 transcription. Oncogene. 2018;37:5811–5828.
- Li W, Li H, Zhang L, et al. Long non-coding RNA LINC00672 contributes to p53 protein-mediated gene suppression and promotes endometrial cancer chemosensitivity. J Biol Chem. 2017;292(14):5801–5813.
- Chen X, Xie R, Gu P, et al. Long Noncoding RNA LBCS Inhibits Self-Renewal and Chemoresistance of Bladder Cancer Stem Cells through Epigenetic Silencing of SOX2. Clin Cancer Res. 2019;25(4):1389–1403.
- Wang H, Guan Z, He K, et al. LncRNA UCA1 in anti-cancer drug resistance. Oncotarget. 2017;8(38):64638–64650.
- Xue X, Yang YA, Zhang A, et al. LncRNA HOTAIR enhances ER signaling and confers tamoxifen resistance in breast cancer. Oncogene. 2016;35:2746–2755.
- Sun M, Gadad SS, Kim DS, et al. Discovery, Annotation, and Functional Analysis of Long Noncoding RNAs Controlling Cell-Cycle Gene Expression and Proliferation in Breast Cancer Cells. Mol Cell. 2015;59:698–711.
- Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550.
- Tseng YY, Moriarity BS, Gong W, et al. PVT1 dependence in cancer with MYC copy-number increase. Nature. 2014;512:82–86.
- Cho SW, Xu J, Sun R, et al. Promoter of lncRNA Gene PVT1 Is a Tumor-Suppressor DNA Boundary Element. Cell. 2018;173(6):1398–412 e22.
- Misawa A, Takayama K, Urano T, et al. Androgen-induced Long Noncoding RNA (lncRNA) SOCS2-AS1 Promotes Cell Growth and Inhibits Apoptosis in Prostate Cancer Cells. J Biol Chem. 2016;291(34):17861–17880.
- Xu J, Meng Q, Li X, et al. Long Noncoding RNA MIR17HG Promotes Colorectal Cancer Progression via miR-17-5p. Cancer Res. 2019;79(19):4882–4895.
- Thomas C, Gustafsson JA. The different roles of ER subtypes in cancer biology and therapy. Nat Rev Cancer. 2011;11:597–608.
- Li W, Notani D, Ma Q, et al. Functional roles of enhancer RNAs for oestrogen-dependent transcriptional activation. Nature. 2013;498(7455):516–520.
- Tan Y, Jin C, Ma W, et al. Dismissal of RNA Polymerase II Underlies a Large Ligand-Induced Enhancer Decommissioning Program. Mol Cell. 2018;71(4):526–39 e8.
- Schmidt D, Schwalie PC, Ross-Innes CS, et al. A CTCF-independent role for cohesin in tissue-specific transcription. Genome Res. 2010;20(5):578–588.
- Thomas-Claudepierre AS, Robert I, Rocha PP, et al. Mediator facilitates transcriptional activation and dynamic long-range contacts at the IgH locus during class switch recombination. J Exp Med. 2016;213(3):303–312.
- Raab JR, Resnick S, Magnuson T. Genome-Wide Transcriptional Regulation Mediated by Biochemically Distinct SWI/SNF Complexes. PLoS Genet. 2015;11(12):e1005748.
- Bao X, Rubin AJ, Qu K, et al. A novel ATAC-seq approach reveals lineage-specific reinforcement of the open chromatin landscape via cooperation between BAF and p63. Genome Biol. 2015;16(1):284.
- Chimge NO, Little GH, Baniwal SK, et al. RUNX1 prevents oestrogen-mediated AXIN1 suppression and beta-catenin activation in ER-positive breast cancer. Nat Commun. 2016;7(1):10751.
- Mercado-Matos J, Matthew-Onabanjo AN, Shaw LM. RUNX1 and breast cancer. Oncotarget. 2017;8(23):36934–36935.
- Li G, Ruan X, Auerbach RK, et al. Extensive promoter-centered chromatin interactions provide a topological basis for transcription regulation. Cell. 2012;148(1–2):84–98.
- Tang Z, Li C, Kang B, et al. GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017;45(W1):W98–W102.
- Dixon JR, Selvaraj S, Yue F, et al. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 2012;485(7398):376–380.
- Yu NY, Iftimi A, Yau C, et al. Assessment of Long-term Distant Recurrence-Free Survival Associated With Tamoxifen Therapy in Postmenopausal Patients With Luminal A or Luminal B Breast Cancer. JAMA Oncol. 2019;5(9):1304.
- Wu Y, Zhang Z, Cenciarini ME, et al. Tamoxifen Resistance in Breast Cancer Is Regulated by the EZH2-ERalpha-GREB1 Transcriptional Axis. Cancer Res. 2018;78(3):671–684.
- Hong D, Fritz AJ, Finstad KH, et al. Suppression of Breast Cancer Stem Cells and Tumor Growth by the RUNX1 Transcription Factor. Mol Cancer Res. 2018;16(12):1952–1964.
- Buenrostro JD, Wu B, Chang HY, et al. ATAC-seq: A Method for Assaying Chromatin Accessibility Genome-Wide. Curr Protoc Mol Biol. 2015;109(1)::21 9 1–9.
- Dobin A, Davis CA, Schlesinger F, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29(1):15–21.
- Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30(7):923–930.
- Langmead B, Trapnell C, Pop M, et al. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10(3):R25.
- Zhang Y, Liu T, Meyer CA, et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008;9(9):R137.
- Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnetjournal. 2011;17:10–12.
- Zhou X, Maricque B, Xie M, et al. The Human Epigenome Browser at Washington University. Nat Methods. 2011;8(12):989–990.
- Zhou Y, Gerrard DL, Wang J, et al. Temporal dynamic reorganization of 3D chromatin architecture in hormone-induced breast cancer and endocrine resistance. Nat Commun. 2019;10(1):1522.
- Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9(4):357–359.
- Servant N, Varoquaux N, Lajoie BR, et al. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol. 2015;16(1):259.
- Ay F, Bailey TL, Noble WS. Statistical confidence estimation for Hi-C data reveals regulatory chromatin contacts. Genome Res. 2014;24(6):999–1011.