
ABSTRACT
Here, we describe SR7, a dual-function antisense RNA encoded on the Bacillus subtilis chromosome. This RNA was earlier described as SigB-dependent regulatory RNA S1136 and reported to reduce the amount of the small ribosomal subunit under ethanol stress. We found that the 5ʹ portion of SR7 encodes a small protein composed of 39 amino acids which we designated SR7P. It is translated from a 185 nt SigB-dependent mRNA under five different stress conditions and a longer SigB-independent RNA constitutively. About three-fold higher amounts of SR7P were detected in B. subtilis cells exposed to salt, ethanol, acid or heat stress. Co-elution experiments with SR7PC-FLAG and Far-Western blotting demonstrated that SR7P interacts with the glycolytic enzyme enolase. Enolase is a scaffolding component of the B. subtilis degradosome where it interacts with RNase Y and phosphofructokinase PfkA. We found that SR7P increases the amount of RNase Y bound to enolase without affecting PfkA. RNA does not bridge the SR7P-enolase-RNase Y interaction. In vitro-degradation assays with the known RNase Y substrates yitJ and rpsO mRNA revealed enhanced enzymatic activity of enolase-bound RNase Y in the presence of SR7P. Northern blots showed a major effect of enolase and a minor effect of SR7P on the half-life of rpsO mRNA indicating a fine-tuning role of SR7P in RNA degradation.
Introduction
Although short open reading frames (sORFs) are present in all genomes, they have often been missed in annotations. Therefore, small proteins comprising less than 50 amino acids (aa) are understudied. Nevertheless, a number of small proteins involved in different pathways have been identified serendipitously and investigated in more detail (rev. in [Citation1–3]). Only recently, some focused efforts were made to study whole peptidomes (rev. in [Citation4]). Among the few peptides studied so far are type I toxins that are often integrated into the membrane (rev. in [Citation5]), chaperones of nucleic acids and metals, membrane components, factors stabilizing or disrupting larger protein complexes and regulatory peptides.
Type I toxins usually encompass <50 aa and either induce pores in bacterial membranes, act as RNase or DNase or interfere with cell envelope biosynthesis without affecting the membrane potential (rev. in [Citation5]), like B. subtilis BsrG [Citation6]. To be inserted into the membrane, these peptides carry trans-membrane domains.
RNA chaperones like Hfq (60–100 aa) and CsrA (≈60 aa) belong to small proteins and have been studied in much detail [Citation7–10]. In addition, in B. subtilis, the small proteins FbpA (59 aa), FbpB (53 aa) and FbpC (29 aa) were proposed to be potential RNA chaperones [Citation11]. Among them, FbpB has been shown to be required for the action of the sRNA FsrA on certain target mRNAs [Citation12], although no biochemical analyses have been performed so far to demonstrate its RNA binding activity. By contrast, other peptides bind metal ions, as e.g. E. coli MntS (42 aa) that binds manganese and delivers it to other proteins [Citation13].
SpoVM (26 aa) from B. subtilis [Citation14] whose NMR structure has been solved recently [Citation15] is an example for a small protein as membrane component. It recognizes – most probably as multimer – convex membrane curvature, acts as a cue for the deposition of the endospore coat and tethers the endospore coat to the developing forespore [Citation16].
An example for a small protein interacting with a regulatory protein in B. subtilis is Sda (46 aa) that inhibits KinA, the first kinase needed for activation of the key regulator of sporulation Spo0A [Citation17]. A small protein that disrupts a large complex is the 40 aa comprising B. subtilis MciZ [Citation18]. It binds to the C-terminal polymerization interface of FtsZ, where it causes shortening of protofilaments and blocks the assembly of higher order FtsZ structures [Citation19].
Small regulatory RNAs (sRNAs), the most important posttranscriptional regulators in all three kingdoms of life, can act by base-pairing or by protein-binding [Citation20–23]. Chromosome-encoded sRNAs inhibit or activate translation or affect RNA stability, although the detailed mechanisms vary. The majority of base-pairing sRNAs do not comprise an ORF and are, therefore, not translated. However, a handful of trans-encoded sRNAs contain an ORF and have two functions, a base-pairing and a peptide-encoding. They have been designated dual-function sRNAs [Citation24]. To date, only one dual-function sRNA from Gram-negative bacteria, SgrS [Citation25], is known and has been intensively studied, whereas the other dual-function sRNAs are encoded in the genomes of Gram-positive bacteria: RNAIII (rev. in [Citation26]) and psm-mec RNA from Staphylococcus aureus [Citation27], Pel RNA from Streptococcus pyogenes [Citation28] and SR1 from Bacillus subtilis [Citation29–32]. SR1 encodes a 39 aa protein, SR1P, which interacts with the glyceraldehyde-3P-dehydrogenase A (GapA), thereby promoting the interaction of GapA with RNase J1 and increasing RNase J1 activity [Citation33]. On a few other sRNAs, small ORFs have only been detected, but it has not been elucidated so far whether or not they are translated and what their biological functions are [Citation24].
We chose ncr2360 RNA that was originally found in 2010 [Citation34] among 54 newly identified sRNAs from B. subtilis to investigate the function of its encoded peptide. In preliminary experiments, we could detect the RNA in Northern blots and show – using a translational ncr2360-lacZ fusion under control of the heterologous promoter pIII [Citation35] – that the putative ncr2360 SD sequence is recognized in B. subtilis. Here, we rename ncr2360 as sr7 and report that sr7 is transcribed from a SigB-dependent promoter under five stress conditions. Its encoded peptide SR7P (39 aa) is synthesized in B. subtilis under different stress conditions and additionally produced to a low extent from a longer RNA originating at a SigA-dependent promoter located 1.6 kb upstream. We demonstrate that SR7P directly interacts with the glycolytic enzyme enolase which moonlights as scaffolding component in the putative B. subtilis degradosome [Citation36]. The SR7P-enolase interaction promotes binding of RNase Y to enolase. SR7P does not seem to interact with the other scaffolding component, phosphofructokinase PfkA. In vitro RNA degradation assays and Northern blotting using two known RNase Y substrates reveal a contribution of SR7P to RNase Y-dependent RNA degradation. Interestingly, in 2015, SR7 was described as SigB-dependent antisense RNA S1136, which is convergently transcribed to rpsD RNA and affects the amount of the small ribosomal subunit under ethanol stress [Citation37]. However, it escaped the authors’ attention that S1136 contains an ORF which might code for a small protein. Based on their and our data, SR7 is the second dual-function regulatory sRNA found in Bacillus subtilis. In contrast to the trans-encoded sRNA SR1 [Citation30,Citation38], SR7 is a cis-encoded bona-fide antisense RNA.
Results
Two independent RNA species are detectable in the intergenic region between tyrS and rpsD
The sr7 gene is located on the B. subtilis chromosome between the tyrS and rpsD genes and transcribed convergently to the rpsD gene (, B). As Mars et al. had previously reported that S1136 – here renamed as SR7 – is transcribed under control of SigB and induced under ethanol stress [Citation37], we mapped the transcriptional start site of SR7 under non-stress and five stress conditions. To this end, B. subtilis DB104 was cultivated at 37°C in TY medium, either induced with 4% ethanol, 0.5 M NaCl, 1 mM Mn2+, HCl (pH 5.0) or heat shock (transfer to 48°C) for 15 min, RNA prepared, treated with DNase and subjected to primer extension. For comparison, both an isogenic ΔsigB strain and a strain where the SigB-dependent promoter upstream of sr7 was deleted [DB104(Δpsr7)] grown under nonstress and ethanol stress conditions were used (). Without treatment, we observed an extremely weak transcription start signal at the expected position 14 bp downstream of the −10 box of the SigB-dependent promoter and a second signal far upstream, which most probably originated at an upstream promoter (). Under all stress conditions, we detected strong SigB-dependent signals, the strongest of them under ethanol stress. These signals were missing in the RNA from the ΔsigB strain as well as from DB104(Δpsr7) confirming that they originate at the SigB-dependent promoter psr7.
Figure 1. Location of the sr7 gene and determination of transcription start and processing sites
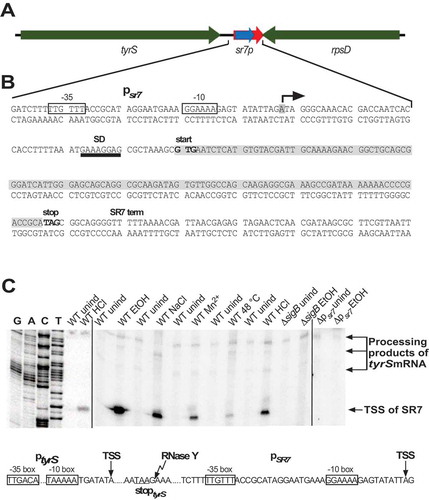
To find out if the bands upstream of the SR7 transcription start site in the primer extension experiment were processing products or derived from an additional upstream promoter, we employed RNA from a Δrny and a Δrnc strain in comparison to the wild-type (Fig. S1). In case of the Δrny strain, the signal was strongly reduced and instead a new band appeared 74 bp upstream, indicating that this band was a processing product from a longer RNA originating far upstream. As the only SigA-dependent promoter located upstream of psr7 is the tyrS promoter, these signals most probably originated there. We mapped the processing site of the longer SR7 species directly 3ʹ of the tyrS stop codon (see Fig. S1A). In the case of the Δrnc strain, the original signal disappeared and no upstream signal was detectable. This suggests that RNase III might be either involved in cleavage of a duplex formed between transcripts originating at the rpsD promoter and those originating at the tyrS promoter in the absence of stress or, alternatively, in cleavage of a longer double-strand formed within the tyrS transcript.
In summary, we conclude that two independent transcripts are observable in the intergenic tyrS-rpsD region, one – SR7 – originating at the SigB-dependent promoter psr7 and a processing product of a transcript from the SigA-dependent tyrS promoter.
The small protein SR7P is synthesized in B. subtilis, and its amount increases under NaCl, ethanol, acid and heat stress
In preliminary experiments, we employed a translational sr7p-lacZ fusion expressed under control of the constitutive strong promoter pIII [Citation35] to investigate if the putative SD sequence of sr7p is recognized in B. subtilis. The results of β-galactosidase measurements indicated that this is the case (Fig. S2). To determine under which conditions the 39 aa protein SR7P is synthesized in Bacillus subtilis, strain DBSR7PF encoding a C-terminally FLAG-tagged peptide (SR7PC-FLAG) in its native locus was constructed as described in Materials and Methods. The SR7 transcriptional terminator was replaced by the heterologous BsrF terminator [Citation39]. To confirm the synthesis of SR7PC-FLAG, 100 ml of strain DBSR7PF were grown in complex TY medium, not induced or subjected to different stresses for 15 min (NaCl, ethanol, low pH and heat-shock) and crude extracts prepared by sonication. After equilibration to the same protein amounts, they were passed through M2 anti-FLAG columns, elution fractions pooled, precipitated, separated in a 15% SDS-PAA gel and subjected to Western blotting using M2 anti-FLAG antibodies. In non-stressed DB104, a weak protein band could be visualized at ≈ 6.5 kDa () corroborating synthesis of SR7PC-FLAG in B. subtilis. Without the column step, a protein of nearly the same size that unspecifically interacted with the FLAG antibodies masked the SR7P signal. 3.5-fold higher amounts of SR7PC-FLAG were detected under ethanol stress, and 2.5-fold higher amounts under salt stress and heat shock (). This increase was lower than expected from the SR7 signals in the primer extension experiment, but most probably due to the synthesis of certain amounts of SR7P already under non-stress conditions from the processing product of the SigA-dependent tyrS mRNA. The very weak SR7P signal obtained after acid stress was probably due to a reduced binding of SR7PC-FLAG after acid treatment to the anti-FLAG column. Therefore, strain DBSR7PS expressing C-terminally Strep-tagged SR7P was subjected to HCl stress, crude extracts prepared and passed through a Strep-Tactin column. Afterwards, a 3.6-fold increase in the amount of SR7P was detectable ().
Figure 2. SR7P is synthesized in B. subtilis under non-stress and stress conditions
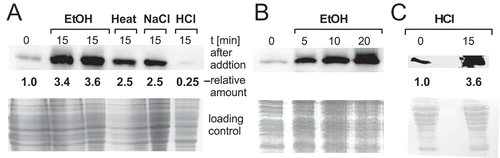
In an additional experiment, cultures were subjected to ethanol stress, time samples taken after 5, 10 and 20 min and analysed as above. Already 10 min after ethanol addition, the highest amount of SR7P was observed (). This is in good correlation with a SigB-dependent response.
Northern blotting confirms the induction of SigB-dependent psr7 under five different stress conditions
To analyse all stress conditions under which sr7 is transcribed from the SigB-dependent promoter, B. subtilis strain DB104 was grown in TY, either 4% ethanol, 0.5 M NaCl, 1 mM manganese or HCl to pH 5.0 added or the culture shifted from 37°C to 48°C (heat shock). After 15 min stress, aliquots were flash-frozen, total RNA prepared and subjected to Northern blotting. Upon stress, a strong SR7 band of ≈185 nt appeared which was not visible under non-stress conditions (Fig. S3). No induction of SR7 was observed under oxygen stress or deficiency, vancomycin or iron stress (not shown). In a rifampicin experiment, a half-life of ≈29 min was determined for the 185 nt SR7 under NaCl stress conditions indicating that it is very stable (). The same stability was observed for SR7 after ethanol stress (Fig. S3B). In Fig. S3E Northern blots are displayed with samples from the isogenic wild-type, Δrny and Δrnc strains after induction with ethanol or NaCl. In all cases, the 185 nt long SR7 is visible under NaCl or ethanol stress. In addition – in contrast to primer extension which only shows 5ʹ ends of RNAs originating from psr7 or the upstream tyrS promoter – Northern blots reveal a number of longer sr7 transcripts differing in their 3ʹ ends that are due to read-through of the bidirectional terminator between sr7 and rpsD.
Figure 3. Determination of the half-life of SR7 under salt stress
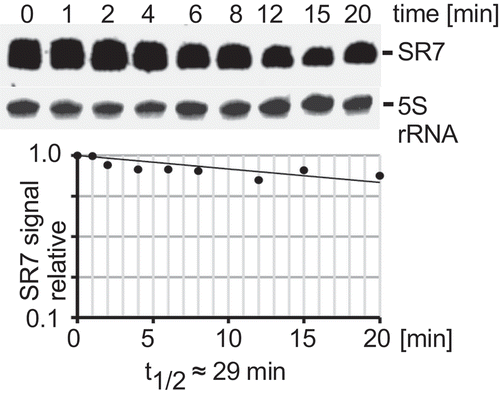
As sr7 is transcribed from the complementary strand to the essential rpsD gene, the entire sr7 gene cannot be simply deleted. Therefore, we added the heterologous BsrF terminator [Citation39] to the 3ʹ end of the rpsD gene and simultaneously replaced the sr7 gene including its promoter psr7 by the chloramphenicol resistance gene. The resulting strain DB104(Δsr7) was grown as above and induced with 0.5 M NaCl. Northern blotting confirmed that the knockout strain does not express the 185 nt SR7 (Fig. S3). Therefore, this strain was employed below to analyse the function of SR7P.
SR7P interacts with enolase
To identify potential interaction partners of SR7P, coelution experiments were performed. To this end, strain DBSR7PF expressing SR7PC-FLAG was grown in 0.8 l complex TY medium. Strain DB104 expressing the untagged SR7P served as negative control. Crude extracts from both cultures were prepared (see Materials and Methods), applied to M2 anti-FLAG columns and elution fractions analysed on 15% SDS-PAA gels. After staining, one band of about 50 kDa was visible in elution fractions two and three from DBSR7PF, whereas no bands were detected in the negative control (). The 50 kDa band was excised, subjected to tryptic in gel-digestion followed by mass spectrometry (see Materials and Methods) and identified to be enolase. To confirm the SR7PC-FLAG -Eno interaction, a reciprocal experiment was performed using strain DBSR7PFE encoding both N-terminally Strep-tagged enolase (EnoN-Strep) and in addition SR7PC-FLAG from their native loci. Cultures were grown under the same conditions as above, crude extracts prepared, applied to Strep-Tactin columns, eluted with desthiobiotin, separated on 15% SDS-PAA gels and stained (). Subsequent Western blotting with M2 anti-FLAG antibodies revealed a direct correlation between the amount of eluted EnoN-Strep and the amount of co-eluted SR7PC-FLAG ().
Figure 4. Enolase co-purifies with SR7P
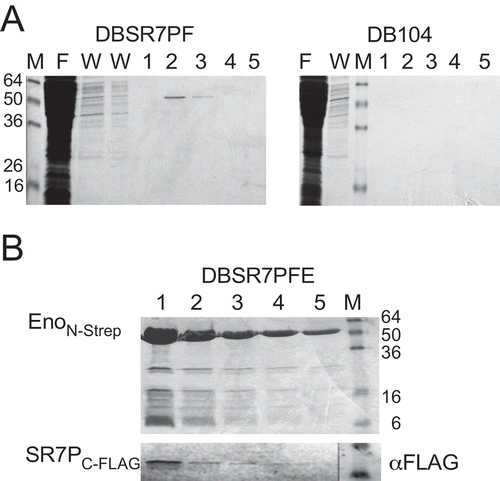
To rule out that enolase might interact with any FLAG-tagged protein, strain DB104 expressing another FLAG-tagged small protein of the same abundance and almost the same size (38 aa), SP2184 C-FLAG in addition with EnoN-Strep from the chromosome was employed in two co-elution experiments. In the first experiment, elution fractions were passed through an anti-FLAG column, where we detected SP2184 C-FLAG in Western blots, but no EnoN-Strep in the Western blot with anti-Strep-tag antibodies (Fig. S4A). In the second experiment, elution fractions were passed through a Strep-Tactin-column and displayed EnoN-Strep on the stained gel, whereas no SP2184 C-FLAG was visible in the Western blot (Fig. S4B). To exclude a hypothetical interaction of the FLAG-tag with the Strep-Tag or the Strep-Tactin column, an additional control experiment was performed: The gapAC-Strep gene [Citation33] was integrated into strain DBSR7PF expressing sr7pC-FLAG. After preparing crude extracts as above and separating elution fractions from the Strep-Tactin column in SDS-PAA gels, GapAC-Strep could be visualized on the stained gel, but no FLAG-tagged protein was detectable on the Western blot (Fig. S4C). In addition, we constructed two strains for the expression of SR7PC-Strep (DBSR7PS) and SR7PC-His10 (DBSR7PH) which could be used for co-elution of enoN-FLAG (Fig. S4E) or native enolase ().
Figure 5. SR7P co-elutes RNase Y bound to enolase, but not phosphofructokinase PfkA
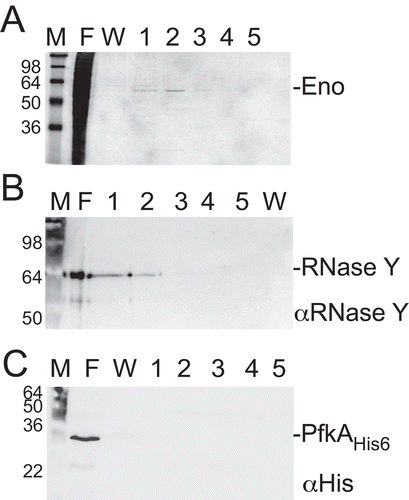
Figure 6. Far-Western blotting
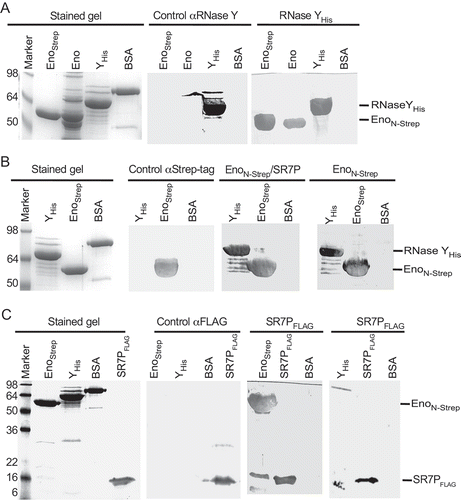
Taken together, these experiments demonstrate that SR7P interacts with B. subtilis enolase.
SR7PC-FLAG co-elutes RNase Y bound to enolase but not phosphofructokinase PfkA
Recently, we have discovered that B. subtilis GapA binds RNase J1 and RNase Y [Citation24,Citation33]. By Western blotting, we detected in GapA preparations from B. subtilis DB104 small amounts of RNase Y (one molecule RNase Y in 100 molecules GapA). Far-Western blotting confirmed that RNase Y can bind GapA and vice versa. As enolase has been reported to interact with both RNase Y and phosphofructokinase PfkA in the B. subtilis degradosome [Citation36,Citation40] we wanted to find out whether SR7PC-FLAG co-elutes RNase Y or PfkA bound to enolase. To enable detection of PfkA in Western blots, strain DBPFC-His encoding PfkAC-His6 and SR7PC-FLAG in their native loci was constructed. This strain was used for a co-elution experiment as above, followed by Western blotting with antibodies against native RNase Y and against the His-tag. As shown in , enolase was co-eluted as before (), and RNase Y (58 kDa) could be identified in the same elution fractions (). By contrast, no co-eluted phosphofructokinase could be detected (). Therefore, we conclude that SR7PC-FLAG co-elutes enolase carrying RNase Y but not phosphofructokinase.
Far-Western blotting confirms the interaction between RNase Y or SR7P with enolase but excludes a direct SR7P-RNase Y interaction
To confirm the SR7P-enolase interaction by an independent method and to exclude a direct SR7P-RNase Y interaction, Far-Western blotting was employed. First, we used EnoN-Strep isolated from the Δsr7 strain and wild-type enolase isolated through SR7PC-His10 as targets and RNase YC-His6 as bait (). Detection was performed with antibodies against native RNase Y, which proved to be specific (control, central panel). RNase Y bound specifically both Eno preparations (right panel), indicating that the Strep-tag does not interfere with binding. In the second Far-Western blot (), we used RNase YC-His6 as target and EnoN-Strep with or without SR7P as bait, and detection was with anti-Strep-tag antibodies, which were also specific (control panel). As expected, EnoN-Strep and EnoN-Strep/SR7P bound to RNase Y (right). To exclude a direct interaction between SR7P and RNase Y we run RNase YC-His6 and – as positive control – EnoN-Strep on a gel and used SR7PC-FLAG purified from IPTG-induced E. coli strain BL21DE3(pDRSR7P), which does not contain RNase Y, as bait (). Whereas a strong signal for SR7PC-FLAG bound to enolase was visible, no signal for an interaction with RNase Y was detectable.
Taken together, Far-Western blotting corroborated the enolase-SR7P and the enolase-RNase Y interactions discovered in co-elution experiments and ruled out a direct SR7P-RNase Y interaction.
Enolase co-eluted with SR7PC-FLAG carries significant amounts of RNase Y
To investigate if SR7P influences the binding of RNase Y to enolase, we performed co-elution experiments with two strains both encoding EnoN-Strep at its native locus. One strain contained in addition the sr7pC-FLAG gene, in the other strain the entire sr7 gene was replaced by the cmR gene. EnoN-Strep co-eluted by SR7PC-FLAG was compared to EnoN-Strep eluted from the Δsr7 strain through a Strep-Tactin column. The elution fractions were subjected to Western blotting. As shown in , in the presence of SR7P, enolase carried about 10 times more RNase Y than in its absence. In fact, an approximately 1:1 ratio of enolase and RNase Y was detected in the presence of SR7P. These data indicate that SR7P increases the amount of RNase Y bound by enolase or promotes the enolase-RNase Y interaction.
Figure 7. Enolase-SR7P co-elutes significant amounts of RNase Y and RNase Y promotes binding of enolase to SR7P
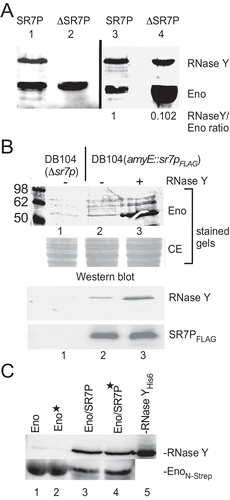
RNase Y promotes the interaction between enolase and SR7P
When we added RNase YC-His6 purified from E. coli to a crude-protein extract obtained from a B. subtilis strain expressing an IPTG-inducible SR7PC-FLAG before loading it onto an anti-FLAG M2 column, we observed in the elution fractions a significant increase in the amount of enolase bound to SR7PC-FLAG (, lane 3 vs. 2) although the amount of SR7PC-FLAG detected in Western blots was identical. The background bands in the stained gels above the enolase band are due to treatment with IPTG. Since we have shown in Far-Western blots () that SR7P cannot directly bind RNase Y, i.e. does not bridge the enolase-RNase Y interaction, this result suggests that RNase Y facilitates the binding of enolase to SR7P. To exclude that RNase YC-His6 can directly interact with the M2-anti-FLAG column (or a Strep-Tactin column), we performed a control experiment shown in Fig. S4D.
RNA does not bridge the interaction between SR7P, enolase and RNase Y
To investigate if RNA is required for the SR7P/enolase and enolase/RNase Y interactions, a co-elution experiment was conducted using two B. subtilis strains, both expressing enoN-Strep from the chromosome and one lacking the sr7 gene, the other expressing sr7pC-His10. Crude protein extracts – 50% treated with RNase A and 50% untreated – were passed through Ni-NTA agarose columns. Afterwards, elution fractions were analysed by Western blotting with anti-RNase Y antibodies and anti-Strep-tag antibodies (). As shown in lanes 3 and 4, identical amounts of enolase and RNase Y were co-eluted with SR7PHis10 in RNase A-treated and in untreated extracts. This confirmed the Far-Western blotting data that SR7P can bind enolase without the help of RNA (). Furthermore, it also corroborated that the enolase/RNaseY/SR7P complex forms without the help of RNA as scaffold. However, when we purified EnoN-Strep from a Δsr7 strain through a Strep-Tactin column and assayed co-eluted RNase Y in Western blots (, lanes 1 and 2), we observed about 20% to 30% less RNase Y in the RNase A-treated samples. This suggests that although enolase interacts with RNase Y in the absence of SR7P (, B, and [Citation40]) the interaction is to a minor degree supported by RNA.
In vitro degradation of RNase Y substrates yitJ 5ʹ UTR and rpsO mRNA
A number of RNAs have been reported to be substrates of RNase Y [Citation41]. Among them is the 5ʹ UTR of the riboswitch yitJ, which had been confirmed both in vivo and in vitro to be an RNase Y substrate [Citation42]. All other substrates have been characterized in vivo in Northern blots, but none of them has been analysed in vitro with purified RNase Y. Therefore, we set up an in vitro degradation assay with 5ʹ 32P-labelled substrate RNA, RNase YC-His6 purified from E. coli and EnoN-Strep purified from B. subtilis sr7pC-FLAG or Δsr7 strains. EnoN-Strep contains the co-eluted RNase Y. When we used enolase carrying SR7PC-FLAG, degradation of yitJ RNA was visible with 0.3 pmol EnoN-Strep/RNase Y/SR7P (, lane 4). By contrast, even 12 pmol of EnoN-Strep/RNase Y purified from the Δsr7 strain were not sufficient for a significant increase in yitJ RNA degradation (, lane 10). This result suggests that SR7P does not only enhance the binding of RNase Y to enolase but, in addition, it increases the enzymatic activity of RNase Y in this triple complex. Previous Northern blots showed that rpsO mRNA is also a substrate of RNase Y [Citation43]; however, no in vitro cleavage had been studied previously and no transcription start site published. Since we observed in Northern blots three rpsO RNA species, the second of which was the main RNase Y target (see below), we determined their 5ʹ ends by primer extension (Fig. S5). For our in vitro degradation studies, we used the in vitro-synthesized 5ʹ labelled 386 nt rpsO mRNA as substrate (). First, we employed different amounts of RNase YC-His purified from E. coli to analyse the degradation pattern of rpsO mRNA (lanes 2–4). Then, we used enolase that does not contain RNase Y as well as SR7P purified from E. coli as negative controls to ensure that they do not have intrinsic RNase activities (lanes 5 and 6). When we compared the activity of RNase Y in the EnoN-Strep/RNase Y/SR7P complex (lanes 12–15) with that in the EnoN-Strep/RNase Y complex purified from the Δsr7 strain (lanes 16–18), we found it to be significantly more active. Like in the case of yitJ RNA, RNase Y bound to 0.3 pmol EnoN-Strep in the triple complex was sufficient for complete disappearance of the full-length rpsO species whereas no substantial degradation was observed with even 3.2 pmol SR7P-free EnoN-Strep/RNase Y. Interestingly, when SR7P purified from E. coli (SR7PE) was added afterwards to 0.8, 1.6 or 3.2 pmol EnoN-Strep/RNase Y (lanes 19–21) at 3.2 pmol, full-length rpsO RNA was degraded almost completely (lane 21), and the same distinct pattern of degradation products was observed as with EnoN-Strep/RNase Y/SR7P at 0.1 pmol enolase (lane 12). This indicates that even the subsequent addition of purified SR7P can improve degradation of rpsO RNA by enolase-bound RNase Y, although the complex formed in vivo seems to be more active. Surprisingly, 1 pmol of RNase Y purified from E. coli (lane 4) was less active in degrading rpsO mRNA than the small amount of RNase Y present in the complex with 0.1 pmol enolase and SR7P (lane 12). This again suggests that SR7P might increase the enzymatic activity of RNase Y bound to enolase.
Figure 8. SR7P affects RNA degradation in vitro.
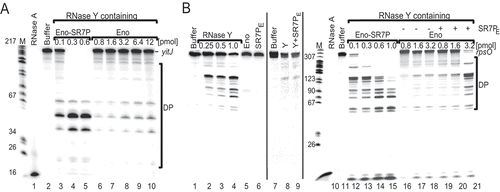
From these in vitro data, we reason that SR7P promotes degradation of yitJ and rpsO RNAs by enolase-bound RNase Y.
Analysis of effects of SR7P, enolase and RNase Y on rpsO mRNA in vivo
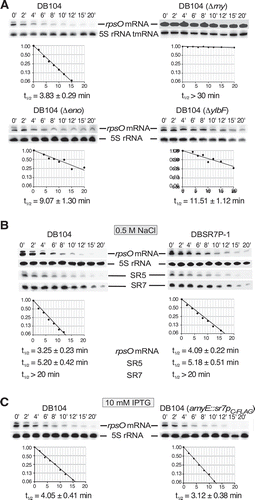
To analyse the effects of SR7P, enolase and RNase Y on the half-life of rpsO mRNA in vivo, we performed Northern blotting experiments with wild-type DB104 and isogenic Δrny, Δeno and ΔylbF strains grown in TY until stationary phase. The ylbF knockout strain was included, because recently the Y-complex composed of YlbF, YmcA and YaaT has been discovered to affect the degradation of many mRNAs by RNase Y [Citation44]. As expected for an RNase Y substrate, the half-life of rpsO mRNA was with ≈29 min at least tenfold longer in the absence of RNase Y (). In the absence of enolase, the half-life increased about 2.5-fold, and in the absence of the Y complex component YlbF about threefold. As we observed 2.5-fold higher amounts of SR7P after salt stress () we determined the half-life of rpsO mRNA in the wild-type and the isogenic sr7p-start-to stop-mutant strain DBSR7P-1 after treatment with 0.5 M NaCl. We did not employ ethanol stress, because ethanol had also an effect on the abundance of some RNase Y targets in vivo [Citation45,Citation46]. With 3.25 vs. 4.09 min we observed a slight, but reproducibly (with four biological replicates) higher half-life in the absence of SR7P (). Reprobing of the filters against SR7 indicates that the abundance and stability of SR7 were not altered by the mutation, so that the effects observed on rpsO mRNA are due to the small protein SR7P and not to the RNA SR7 ( and S6). To corroborate this effect, we constructed strain DB104 (amyE::sr7pC-FLAG specR) for inducible overexpression of sr7p from the chromosomal amyE locus. When we induced this strain with IPTG, the rpsO RNA half-life decreased from 4.05 (DB104) to 3.12 min (). This was again a slight effect, which was, however, also reproducible with four biological replicates. To corroborate that the effect of SR7P is specific for an RNase Y target, we reprobed all filters against SR5, a 163 nt RNA antitoxin that is neither a substrate of RNase Y nor affected by 0.5 M NaCl [46]. As shown in and S6, the half-life of SR5 was not influenced by SR7P.
Figure 9. Effect of SR7P, RNase Y, enolase and YlbF on rpsO mRNA stability in B. subtilis.
Half-life of SR7 in the presence or absence of RNase Y, enolase and YlbF
As it is also not excluded that the stability of SR7 itself is dependent on RNase Y or the Y complex, we determined the stability of SR7 in the isogenic Δrny and ΔylbF strains grown in TY until stationary phase. Fig. S3 C shows that the stability of SR7 is neither affected by RNase Y nor by YlbF.
SR7P is highly conserved among 10 Bacillus species
To ascertain if the 39 aa SR7P is restricted to Bacillus subtilis, we performed a BLAST analysis (both nucleotide and peptide sequence) using the B. subtilis SR7 sequence against all known genomic sequences followed by a refined BLAST search for all found homologues. SR7P homologues were only detected in B. subtilis strains and in nine other species belonging to the genus Bacillus. Neither in the genomes of other firmicutes nor in those of other Gram-positive or Gram-negative bacteria, a small protein with a similar primary sequence was found to be encoded. From all phylogenetic groups of Bacillus species, our hits were restricted to a subdivision of one group. These species are closely related and belong to one side arm of the Bacilli. presents an alignment of the aa sequences of the SR7P homologues. In all cases, an almost identical stretch of 20 aa is present in the N-terminal half of the protein, whereas the C-terminal half reveals more differences. In contrast to B. subtilis SR7P, all other homologues contain three additional hydrophilic aa near their C-termini and are, therefore, 42 aa long. When we assume that the SR7P-enolase interaction is conserved, the interaction surface is located most likely within the 20 conserved aa of SR7P.
Figure 10. Alignment of SR7P homologues

The peptide is predicted to form an α-helix. However, NMR data obtained so far with a purified SR7P (designated SP-12 in [Citation47]) indicated that only 20.5% of free SR7P is structured and 79.4% is disordered. Interestingly, in predictions with the FuzPred algorithm for the bound form, the percentage of structured form increased to 41% compared to 59% disordered. Both CD and NMR structural analysis indicated a rather molten globule for free SR7P [Citation47].
At DNA level, all homologous sr7 genes are preceded by SigB-dependent promoters, and upstream of all peptide sequences SD sequences are located (Fig. S7). Only in B. amyloliquefaciens and B. velezensis, the distance between SD sequence and GTG start codon is with 14 nt rather long, but an alternative AG-rich sequence nearer to the start codon could also serve as RBS. In all cases, the start codon is GTG. In the 5ʹ half of the ORF, nt exchanges are found almost exclusively at wobble positions indicating that this is the most highly conserved region. All sr7 genes carry Rho-independent transcription terminators at their 3ʹ ends. Consequently, the SigB-dependent SR7 homologues are also conserved at DNA level.
Only in B. halotolerans, B. vallismortis and B. cereus, the SR7P encoding genes are located downstream of the tyrS gene and convergently transcribed to the rpsD gene as in B. subtilis (see ). However, not the entire genomes are annotated for the other species so far. Therefore, we cannot exclude a conserved gene arrangement.
Discussion
The B. subtilis degradosome seems to have a more dynamic structure than the E. coli degradosome as it cannot be isolated in the absence of cross-linking reagents [Citation48]. Furthermore, not only Eno and PfkA, but also a third glycolytic enzyme, glyceraldehyde-3P-dehydrogenase A (GapA), affects its composition [Citation33]: GapA interacts with RNase J1, and this interaction is improved by a small protein, SR1P, which is expressed under gluconeogenic conditions when the metabolic function of GapA is not required. Furthermore, the enzymatic activity of RNase J1 is enhanced in the GapA/RNase J1/SR1P complex compared to that of the unbound enzyme.
Here, we report on the discovery and characterization of another small protein in B. subtilis, the 39 aa SR7P. It is encoded on two RNAs, a constitutively transcribed and subsequently processed one of 259 nt and on a stress-induced SigB-dependent 185 nt RNA renamed as SR7 (formerly S1136 [Citation37]). SR7P interacts with the glycolytic enzyme enolase. This interaction promotes the binding of endoribonuclease RNase Y to enolase (see ) and significantly increases the degradation of two known RNase Y substrates, yitJ RNA and rpsO mRNA, in vitro. In addition, rpsO mRNA degradation is also affected in vivo about threefold by the presence of enolase and to a minor, but reproducibly measurable extent, by SR7P (). Under salt stress, the 2.5-fold amount of SR7P is synthesized and bound by enolase, resulting in an increase of the enolase fraction carrying RNase Y and in turn, a slight increase in degradation of RNase Y substrates (). These data support that the Eno/SR7P-RNase Y interaction has a biological function. The small effect of inducible overexpression or deletion of sr7p might be due to the fact that the bulk of its interaction partner enolase is located in the cytosol to fulfil its role in glycolysis, and only a small percentage is present in the degradosome where it interacts with RNase Y and PfkA. Furthermore, the effect of the enolase deletion is higher than that of the sr7p deletion, because enolase moonlights in RNA degradation under a variety of conditions and SR7P only modulates this activity under specific stress conditions.
Figure 11. Working model for the role of SR7P in the proposed B. subtilis degradosome
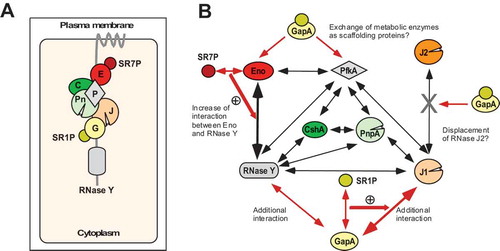
Binding of SR7P to enolase and formation of the SR7P/enolase/RNase Y complex are independent of RNA. Therefore, we can rule out a bridging function of RNA in the trimeric complex (). Unexpectedly, binding of enolase to RNase Y in an sr7 knockout strain was to a low extent supported by RNA (). This is in contrast to what we found for the GapA-RNase Y or the GapA-RNase J1 interactions which were independent of RNA [Citation33]. Furthermore, we could exclude that SR7P binds directly to RNase Y (). We hypothesize that binding of SR7P alters the conformation of enolase and increases its binding affinity to RNase Y. In turn, the addition of RNase Y promoted the enolase-SR7P interaction (). SR7P is predominantly unfolded free in solution, but bioinformatic methods predicted a propensity for structure induction upon interaction with proteins [Citation47]. These theoretical predictions are in line with our experimental data that propose that the formation of a stable trimeric SR7P-enolase-RNase Y-complex in the degradosome is coupled to structure induction of SR7P.
Already in 2011, it was reported that citrate alters the activity of E. coli PNPase by directly binding to the enzyme [Citation49]. When Newman et al. found that citrate also influences the activity of B. subtilis enolase, they argued that glycolytic enzymes may act as sensors of nutritional stress and coordinate this stress with the RNA degrading machinery [Citation40]. This would entail a decrease of global mRNA turnover under energy-limiting conditions. Our previous data on SR1P/GapA supported this idea: SR1P is only synthesized when glucose is exhausted and might also act as a sensor to link RNA degradation to the nutritional state of the cell [Citation33]. Similarly, SR7P might act as a sensor to connect RNA turnover to different stresses: sr7 transcription was induced under five stress conditions, and a 2.5- to 3.5-fold increase of SR7P under stress was found (Fig.s2 and S3). Under salt stress, we observed a small, but reproducible effect of SR7P on the half-life of the RNase Y substrate rpsO mRNA.
The discovery of an impact of two 39 aa proteins, SR1P and SR7P, on components of the putative B. subtilis degradosome suggests that small proteins might also play a broader role in fine-tuning RNA degradation in other bacteria. Like SR1P [Citation32], SR7P is restricted to the Bacillales (, Fig. S7). Sequence homology could not be found to any other small protein in other Gram-positive or in Gram-negative bacteria. However, it is not excluded that in degradosomes of other bacteria that contain enolase as scaffolding component [Citation50], small proteins with a different primary sequence play a comparable role under specific stress conditions.
The SigB-dependent RNA encoding SR7P was formerly reported as S1136, an antisense RNA whose convergent transcription to rpsD mRNA results in reduced amounts of the small ribosomal subunit under ethanol stress [Citation37]. We found that this RNA, which we renamed as SR7, has in addition to its antisense RNA function an mRNA function: It encodes SR7P which – via enolase binding – affects RNA degradation. Therefore, SR7 is after SR1 [Citation29,Citation30,Citation33,Citation38] the second dual-function regulatory sRNA identified in Bacillus subtilis. All dual-function regulatory RNAs reported so far act only in trans on their target mRNAs [Citation24]. By contrast, Mars et al. found that SR7 acts only in cis – most probably due to transcriptional interference [Citation37]. Consequently, the class of dual-function regulatory sRNAs can be extended by dual-function antisense RNAs.
Future investigations will focus on the structure of SR7P, structural alterations of both interacting partners, SR7P and enolase, upon binding, and on mapping of the enolase/SR7P interaction surface, as initiated already [Citation47]. Moreover, transcriptomics will show whether the abundance of other RNase Y substrates is also affected by SR7P.
Materials and methods
Strains, media and growth conditions
E. coli strains DH5α and BL21DE3 and B. subtilis strains DB104 [Citation51] were used. TY medium served as complex medium for E. coli [Citation52]. All B. subtilis strains were grown in TY medium until OD560 = 4.
Enzymes and chemicals
Chemicals used were of the highest purity available. Q5 DNA polymerase, T7 RNA polymerase, CIP and polynucleotide kinase were purchased from NEB, Firepol Taq polymerase from Solis Biodyne, and sequenase from Affymetrix.
Isolation of chromosomal DNA from Bacillus subtilis
0.5 ml of B. subtilis DB104 stationary phase culture was centrifuged, the pellet washed with 1 ml TES (10 mM Tris-HCl pH 8.0, 1 mM EDTA, 100 mM NaCl), re-suspended in 750 µl TES containing 25 µl lysozyme (10 mg/ml) and incubated at 37°C for 5 min. Subsequently, 50 µl pronase E and 50 µl 10% SDS were added and mixed gently, followed by a 30 min incubation at 37°C. Afterwards, one phenol/chloroform extraction and one chloroform extraction were performed using decapitated 1 ml tips, and the final supernatant was added to 2 ml 96% ethanol. After one min of gentle shaking the precipitated DNA was carefully taken out with a yellow tip, dissolved in 100 µl bidest, incubated at 37°C for 30 min followed by 1 h on ice.
Primer extension
Primer extension experiments were carried out as described [Citation29] using total RNA from B. subtilis strain DB104, DB104 (Δrny::specR) and DB104 (Δrnc::cmR) and 5ʹ-labelled primer SB3182 (all primers are listed in Table S1).
In vitro transcription, preparation of total RNA and Northern blotting
In vitro transcription was performed as described [Citation38]. Preparation of total RNA and Northern blotting including the determination of RNA half-lives were carried out as described previously [Citation29] except that 1 ml time samples were taken and directly added to 250 μl RNAprotect Bacteria Reagent (Qiagen) or to 250 μl containing 5% phenol and 95% ethanol, vortexed and incubated for 5 min at room temperature. After centrifugation, the pellets were flash-frozen in liquid nitrogen and stored until use at −20°C.
Strain constructions
All PCRs were performed with Q5 polymerase using 25 cycles with an annealing temperature of 48°C. If not stated otherwise, primers (listed in Table S1) and as template, chromosomal DNA of B. subtilis DB104 were used. All constructed recombinant strains were confirmed by sequencing using the primers listed in Table S1.
sr7pC-FLAG, sr7pC-His10 and sr7pC-Strep knock-in strains
A 340 bp fragment encoding SR7PC-FLAG was obtained in a two-step PCR. In PCR 1 with primer pair SB2963/SB2978, a C-terminal FLAG-tag was added at the end of the sr7 ORF. In the subsequent PCR with primer SB2843, a stop codon was introduced after the FLAG-tag sequence and the native terminator replaced by the heterologous BsrF terminator [Citation39] yielding the sr7pC-FLAG fragment. The chloramphenicol resistance (cmR) cassette was amplified from plasmid pINT12 C [Citation46] with primer pair SB2938/SB2939. This cassette has 20 bp complementarity to the sr7pC-FLAG fragment on its 5ʹ end and 20 bp complementarity to the back cassette on its 3ʹ end. A 1 kb front cassette that has 20 bp complementarity on its 3ʹ end to the sr7pC-FLAG fragment was obtained using primer pair SB2964/SB2966. Similarly, a 1 kb back cassette was generated with primer pair SB2967/SB2968 which has a 5ʹ 20 bp complementarity to the cmR gene. The four PCR fragments were purified and joined by a 10-cycles’ primer-less PCR. Afterwards, the final 3 kb fragment was produced using a 25 cycles’ PCR with primer pair SB2964/SB2968. This fragment was used to transform B. subtilis DB104. The resulting strain was designated DBSR7PF (all strains are listed in ).
Table 1. Bacterial strains used in this study
A 1 kb fragment with 10 histidine residues at the 3ʹ end of the sr7 ORF was obtained using primer pair SB3013/SB3575. This fragment has 20 bp complementarity to the 5ʹ primer region of the cmR (see above) gene. A 1 kb back cassette with 20 bp complementarity to the 3ʹ region of the cmR gene was generated using primer pair SB2967/SB3156. All three fragments were joined, primer pair SB3013/SB3156 added, a final 3 kb fragment was amplified and used to transform B. subtilis DB104 yielding strain DBSR7PH.
The same approach as above was used to construct strain DBSR7PS for the expression of SR7PC-Strep; however, the primer pair SB3013/SB3282 was used to generate the 1 kb FRONT cassette, while the other fragments were as in the case of DBSR7PH.
Construction of the sr7 promoter deletion strain, the Δsr7 strain and the start- to stop-codon mutant strain
A 40 bp deletion of the sr7 promoter including both −35 and −10 boxes, but retaining the transcription start site +1 was constructed as follows: a 1 kb front cassette upstream of the sr7 gene was generated by PCR 1 with primer pair SB3013/SB3014. The 0.8 kb spectinomycin resistance (specR) gene was obtained by PCR 2 on plasmid pMG16 [Citation46] using primer pair SB3039/SB3040. The 3ʹ end of the front cassette has a 20 bp complementarity to the 5ʹ region of the specR gene. The 1.14 kb back cassette comprising the promoterless sr7 gene together with the rpsD gene located downstream on the complementary strand was generated by PCR 3 with primer pair SB3042/SB3020. It displays a 20 bp complementarity to the 3ʹ end of the specR gene. As above, the purified fragments were joined, a 3 kb fragment obtained with primer pair SB3013/SB3020 and used to transform B. subtilis DB104 resulting in promoter deletion strain DBSR7PΔP.
To delete the sr7 ORF, a 1 kb front cassette was generated using primer pair SB3013/SB3014 with 20 bp complementarity to the 5ʹ of the cmR cassette. The 1 kb back cassette was amplified using primer pair SB3243/SB3156. The BsrF terminator [Citation39] was added to the rpsD ORF present on the opposite strand with 20 bp complementarity to the 3ʹ end of the cmR gene. The fragments were joined, a final 3 kb fragment obtained as above using primer pair SB3013/SB3156 and used to transform B. subtilis DB104 resulting in DB104 (Δsr7).
To construct an sr7 start- to stop-codon mutant strain, a 1 kb fragment upstream of the sr7 locus was amplified using primer pair SB3013/SB3014. This fragment has 20 bp complementarity to the 5´ region of the cmR gene. The sr7 start codon was replaced by a stop codon through a PCR with primer pair SB3017/SB3227 introducing simultaneously a 20 bp complementarity to the 3‘ region of the cmR gene. A 1 kb back fragment was generated with primer pair SB3226/SB3156. All four fragments were joined, a final 3.2 kb fragment was obtained with SB3013/SB3156 and used to transform B. subtilis DB104 yielding strain DBSR7P-1.
pfkAC-His6 and pfkAC-FLAG knock-in strains
To add a His or a FLAG tag to the C-terminus of the pfkA ORF, a 1 kb front cassette encoding 6 His residues or 3x FLAG was generated using primer pair SB3269/SB3347 or SB3269/SB3270, respectively. The neomycin resistance (kmR) gene was amplified from plasmid pMG9 using primer pair SB3039/SB3180. In the subsequent PCR with primer SB3267 or SB3167 a stop codon and a 20 bp complementarity to the 5ʹ region of the kmR cassette were introduced. A 1 kb back cassette with 20 bp complementarity to the 3ʹ primer region of the kmR gene was obtained using primer pair SB3271/SB3272. All fragments were purified, joined in a final PCR with SB3269/SB3272 as above to generate a 3 kb fragment which was used to transform B. subtilis DBSR7PF yielding B. subtilis DBPFC-His or DBPFC-FLAG.
Plasmid constructions
For the construction of a translational ncr2360-lacZ fusion (later renamed as sr7p-lacZ fusion) under control of the constitutive heterologous promoter pIII [Citation35] a PCR fragment was generated with primer pair SB2816/SB2817, cleaved with BamHI and EcoRI and inserted into the BamHI/EcoRI pGAB1 vector resulting in pGABP2360. For the construction of a plasmid for IPTG-inducible overexpression of SR7PC-FLAG a PCR fragment lacking the sr7 promoter was obtained on chromosomal DNA using primer pair SB2872/SB2878. In the subsequent PCR, the native stop codon was replaced by a 3 x FLAG tag with primer pair SB2872/SB3439. The resulting fragment was cleaved with Hind III and SphI and inserted into the pDR111 [Citation54] Hind III/SphI vector. Both recombinant vectors pGAB2360 and pDRSR7P were amplified in E. coli DH5α, linearized with ScaI and integrated into the amyE locus of B. subtilis strains DB104 and DB104 (Δsr7), respectively.
Preparation of protein crude extracts for the detection of SR7PC-FLAG, Western Blotting and Far-Western Blotting
Protein crude extracts were prepared by either sonication (see below) or DNase I/lysozyme treatment. In the latter case, pellets were treated with 10 mg/ml lysozyme/1 mg/ml DNase I in PBS for 30 min at 37°C followed by two 5 min centrifugation steps (10 min at RT) to obtain the supernatant.
Western blotting for the detection of SR7PC-FLAG was performed as follows: The pellet from a 100 ml culture grown in TY was resolved in 4 ml TBS and sonicated three times for 5 min. Supernatants obtained by two 10 min centrifugation steps at 4°C were run through an anti-FLAG M2 column (bed volume 200 µl), washed 6 times in 200 µl TBS followed by 5 elution steps with 200 μl of 100 mM glycine HCl pH 3.5. 1 ml of elution fractions was mixed with 1 ml 50% ice-cold TCA, placed on ice for 30 min and centrifuged for 30 min at 13000 rpm. The pellet was washed three times with ice-cold acetone (20 min centrifugation), air-dried for 5 min and dissolved in 50 μl 2x Laemmli buffer. Ten microlitres was loaded onto a 15% SDS-PAA gel run at 35 mA for 75 min and transferred onto PVDF membranes at 12 V for 40 min by semidry blotting in transfer buffer (5.8 g Tris-HCl, 2.9 g glycine, 0.37 g SDS and 200 ml methanol per litre). Membranes were blocked for 1 h in PBST [Citation31] with 0.5% gelatine and incubated for 1 h with M2 anti-FLAG antibodies (1:2500) followed by 5 times 8 min washing in PBST. Subsequently, membranes were incubated with secondary antibodies (horseradish peroxidase conjugated anti-mouse) for 1 h followed by 5 washing steps. For development, membranes were incubated in 20 ml substrate solution (50 mM Tris pH 7.5, 0.9 g/ml diaminobenzidine and 20 µl H2O2) until bands were visible. The reaction was stopped by washing in distilled water. For enolase detection, rabbit against enolase antibodies (1:5000) were used as primary and horseradish peroxidase conjugated anti-rabbit antibodies (1:2500) as secondary antibodies.
Detection by chemiluminescence was used for RNase Y, PfkAC-His6, quantification of SR7P amounts and RNase Y after crude extracts were treated with RNase A: The membrane was washed briefly with deionized water and the pH adjusted with a solution containing 100 mM NaCl and 100 mM Tris-HCl pH 9.5. Afterwards, the membrane was incubated with developing solution (Invitrogen Novex AP Chemiluminescent substrate) for 5 min in the dark and signals were detected with a CCD camera (ImageQuant LAS 4000).
For Far-Western blotting, proteins were separated on 10% or 15% SDS/PAA gels, transferred onto PVDF membranes and afterwards blocked for 2 h. Blots were incubated overnight with 10 ml PBST-gelatine or PBST-gelatine supplemented with either 100 μg EnoN-Strep purified from wild-type or Δsr7 B. subtilis strains or 170 μg RNase YC-His6 purified from E. coli. Binding of EnoN-Strep and RNase YC-His6 was detected by incubation with mouse-anti-Strep-tag antibody (1:1000; IBA Göttingen) or mouse-anti-His-tag antibody (1:2000; IBA Göttingen), respectively, and subsequently, alkaline phosphatase coupled anti-mouse antibody (1:7500; Santa Cruz Biotechnology).
Purification of FLAG-tagged, Strep-tagged and His-tagged proteins from B. subtilis
B. subtilis strain DBSR7PF was grown in 0.8 l TY medium till onset of stationary phase and harvested by centrifugation at 4°C and 8000 rpm. Pellets were frozen overnight. Subsequently, they were disrupted in 2 ml total volume of equilibration buffer (150 mM NaCl, 100 mM Tris-HCl pH 8.0, 1 mM EDTA) for 1.5 min at 2000 rpm in the Mikro-Dismembrator (Sartorius). Resulting crude extracts were subjected to sonication in 30 ml buffer and afterwards centrifuged twice at 13.000 rpm and 4°C. Supernatants were applied to M2 anti-FLAG (strain DBSR7PF) or to Strep-Tactin columns (strain GP1215). Washing, elution and regeneration of the 1 ml columns were performed according to the manufacturers’ instructions. Six 500 μl elution fractions were collected for each column, and 25 μl of each fraction tested on SDS-PAA gels. The purification of SR7PC-His10 was performed as described before [Citation31].
Mass spectrometry
Proteins were identified by LC-ESI-MS/MS. Tryptic in-gel protein digests were analysed with an LC-ESI-MS equipment consisting of an Ettan MDLC™-HPLC (GE Healthcare, Munich) and an LTQÔ mass spectrometer (Thermo Scientific, Waltham, MA). For protein identification, the Thermo Proteome Discoverer 1.0™ software (Thermo Scientific) and the B. subtilis protein database were used.
RNA degradation assay
Two μl in vitro transcribed, [γ-32P]-ATP labelled RNA (10.000 cpm) were incubated with 1 μl 10x reaction buffer (200 mM Tris-HCl pH 8.0; 80 mM MgCl2, 1 M NH4 Cl; 0.5 mM DTT including 1 U RNasin) and 7 μl diluted protein for 30 min at 37°C. The reaction was stopped by addition of 10 μl formamide loading dye [Citation29] and 5 min at 95°C. Samples were separated on 6% denaturing PAA gels. Dried gels were analysed by phosphorImaging using Aida Image Analyzer v.4.5.
Abbreviations
| aa | = | amino acid |
| nt | = | nucleotide |
| bp | = | base pair |
| ORF | = | open reading frame |
| PAA | = | polyacrylamide |
| sRNA | = | small RNA |
| RBS | = | ribosome binding site. |
Supplemental Material
Download PDF (5.7 MB)Acknowledgments
The authors thank Bernhard Schlott (FLI Jena) and Karl-Heinz Gührs for performing the mass spectrometry analysis to identify enolase. We thank Jörg Stülke (Göttingen) for strains GP1215 and GP1214, antibodies against native RNase Y and comments on the manuscript. We are grateful to Harald Putzer (Paris) for E. coli strain SSC420 for overexpression of RNase YC-His6 and to Byoung-Mo Koo (San Francisco) for the Δeno strain. Furthermore, we thank Laura Remmel and Nina Kubatova (AG Schwalbe, Frankfurt/Main) for SR7P purified from E. coli.
Disclosure statement
No potential conflicts of interest were disclosed.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
References
- Hobbs EC, Fontaine F, Yin X, et al. An expanding universe of small proteins. Curr Opin Microbiol. 2011;14:167–173.
- Storz G, Wolf YI, Ramamurthi KS. Small proteins can no longer be ignored. Annu Rev Biochem. 2014;83:753–777.
- Orr MW, Mao Y, Storz G, et al. Alternative ORFS and small ORFs: shedding light on the dark proteome. Nucleic Acids Res. 2020;48:1029–1042.
- Andrews SJ, Rothnagel JA. Emerging evidence for functional peptides encoded by short open reading frames. Nat Rev Genet. 2014;15:193–204.
- Brantl S, Jahn N. sRNAs in bacterial type I and type III toxin-antitoxin systems. FEMS Microbiol Rev. 2015;39:413–427.
- Jahn N, Brantl S, Strahl H. Against the mainstream: the membrane-associated type I toxin BsrG from Bacillus subtilis interferes with cell envelope biosynthesis without increasing membrane permeability. Mol Microbiol. 2015;98:651–666.
- Vogel J, Luisi BF. Hfq and its constellation of RNA. Nat Rev Microbiol. 2011;9:578–589.
- Kavita K, de Mets F, Gottesman S. New aspects of RNA-based regulation by Hfq and its partner sRNAs. Curr Opin Microbiol. 2018;42:53–61.
- Vakulskas CA, Potts AH, Babitzke P, et al. Regulation of bacterial virulence by Csr (Rsm) systems. Microbiol Mol Biol Rev. 2015;79:193–224.
- Müller P, Gimpel M, Wildenhain T, et al. A new role for CsrA: promotion of complex formation between an sRNA and its mRNA target in Bacillus subtilis. RNA Biol. 2019;16:972–987.
- Gaballa A, Antelmann H, Aguilar C, et al. The Bacillus subtilis iron-sparing response is mediated by a Fur-regulated small RNA and three small, basic proteins. Proc Natl Acad Sci USA. 2008;105:11927–11932.
- Smaldone GT, Revelles O, Gaballa A, et al. A global investigation of the Bacillus subtilis iron-sparing response identifies major changes in metabolism. J Bacteriol. 2012;194:2594–2605.
- Martin JE, Waters LS, Storz G. The E. coli small protein MntS and exporter MntP optimize the intracellular concentration of manganese. PLoS Genet. 2015;11:e1004977.
- Cutting S, Anderson M, Lysenko E, et al. SpoVM, a small protein essential to development in Bacillus subtilis, interacts with the ATP-dependent protease FtsH. J Bacteriol. 1997;17:5534–5542.
- Gill RL, Castaing JP, Hsin J, et al. Structural basis for the geometry-driven localization of a small protein. Proc Natl Acad Sci USA. 2015;112:E1908–15.
- Ramamurthi KS, Lecuyer S, Stone HA, et al. Geometric cue for protein localization in a bacterium. Science. 2009;323:1354–1357.
- Cunningham KA, Burkholder WF. The histidine kinase inhibitor Sda binds near the site of autophosphorylation and may sterically hinder autophosphorylation and phosphotransfer to Spo0F. Mol Microbiol. 2009;71:659–677.
- Ray S, Kumar A, Panda D. GTP regulates the interaction between MciZ and FtsZ: A possible role of MciZ in bacterial cell division. Biochemistry. 2013;52:392–401.
- Bisson-Filho AW, Discola KF, Castellen P, et al. FtsZ filament capping by MciZ, a developmental regulator of bacterial division. Proc Natl Acad Sci USA. 2015;12:2130–2138.
- Brantl S. Bacterial chromosome-encoded small regulatory RNAs. Future Microbiol. 2009;4:85–103.
- Brantl S. Acting antisense: plasmid- and chromosome-encoded sRNAs from Gram-positive bacteria. Future Microbiol. 2012;7:853–871.
- Brantl S, Brückner R. Small regulatory RNAs from low-GC Gram-positive bacteria. RNA Biol. 2014;11:443–456.
- Wagner EGH, Romby P. Small RNAs in bacteria and archaea: who they are, what they do, and how they do it. Adv Genet. 2015;90:133–208.
- Gimpel M, Brantl S. Dual function small regulatory RNAs in bacteria. Mol Microbiol. 2017;103:387–397.
- Wadler CS, Vanderpool CK. A dual function for a bacterial small RNA: SgrS performs base pairing dependent regulation and encodes a functional polypeptide. Proc Natl Acad Sci USA. 2007;104:20454–20459.
- Bronesky D, Wu Z, Marzi S, et al. Staphylococcus aureus RNAIII and its regulon link quorum sensing, stress responses, metabolic adaptation, and regulation of virulence gene expression. Annu Rev Microbiol. 2016;8:299–316.
- Kaito C, Saito Y, Ikuo M, et al. Mobile genetic element SCCmec-encoded psm-mec RNA suppresses translation of agrA and attenuates MRSA virulence. PLoS Pathog. 2013;9:e1003269.
- Mangold M, Siller M, Roppenser B, et al. Synthesis of group A streptococcal virulence factors is controlled by a regulatory RNA molecule. Mol Microbiol. 2004;53:1515–1527.
- Licht A, Preis S, Brantl S. Implication of CcpN in the regulation of a novel untranslated RNA (SR1) in B. subtilis. Mol Microbiol. 2005;58:189–206.
- Heidrich N, Chinali A, Gerth U, et al. The small untranslated RNA SR1 from the B. subtilis genome is involved in the regulation of arginine catabolism. Mol Microbiol. 2006;62:520–536.
- Gimpel M, Heidrich N, Mäder U, et al. A dual function sRNA from B. subtilis: SR1 acts as a peptide encoding mRNA on the gapA operon. Mol Microbiol. 2010;76:990–1009.
- Gimpel M, Preis H, Barth E, et al. SR1 – a small RNA with two remarkably conserved functions. Nucleic Acids Res. 2012;40:11659–11672.
- Gimpel M, Brantl S. Dual-function sRNA-encoded peptide SR1P modulates moonlighting activity of B. subtilis GapA. RNA Biol. 2016;13:916–926.
- Irnov I, Sharma CM, Vogel J, et al. Identification of regulatory RNAs in Bacillus subtilis. Nucleic Acids Res. 2010;38:6637–6651.
- Brantl S, Nuez B, Behnke D. In vitro and in vivo analysis of transcription within the replication region of plasmid pIP501. Mol Gen Genet. 1992;234:105–112.
- Commichau FM, Rothe FM, Herzberg C, et al. Novel activities of glycolytic enzymes in Bacillus subtilis: interactions with essential proteins involved in mRNA processing. Mol Cell Proteomics. 2009;8:1350–1360.
- Mars RA, Mendonça K, Denham EL, et al. The reduction in small ribosomal subunit abundance in ethanol-stressed cells of Bacillus subtilis is mediated by a SigB-dependent antisense RNA. Biochim Biophys Acta. 2015;1853:2553–2559.
- Heidrich N, Moll I, Brantl S. In vitro analysis of the interaction between the small RNA SR1 and its primary target ahrC mRNA. Nucleic Acids Res. 2007;35:331–346.
- Preis H, Eckart RA, Gudipati RK, et al. CodY activates transcription of a small RNA in Bacillus subtilis. J Bacteriol. 2009;191:5446–5457.
- Newman JA, Hewitt L, Rodrigues C, et al. Dissection of the network of interactions that links RNA processing with glycolysis in the Bacillus subtilis degradosome. J Mol Biol. 2012;416:121–136.
- Lehnik-Habrink M, Schaffer M, Mäder U, et al. RNA processing in Bacillus subtilis: identification of targets of the essential RNase Y. Mol Microbiol. 2011;81:1459–1473.
- Shahbabian K, Jamalli A, Zig L, et al. RNase Y, a novel endoribonuclease, initiates riboswitch turnover in Bacillus subtilis. EMBO J. 2009;28:3523–3533.
- Yao S, Bechhofer D. Initiation of decay of Bacillus subtilis rpsO mRNA by endoribonuclease RNase Y. J Bacteriol. 2010;192:3279–3286.
- DeLoughery A, Lalanne JB, Losick R, et al. Maturation of polycistronic mRNAs by the endoribonuclease RNase Y and its associated Y-complex in Bacillus subtilis. Proc Natl Acad Sci USA. 2018;115:E5585–E5594.
- Jahn N, Preis H, Wiedemann C, et al. BsrG/SR4 from Bacillus subtilis – the first temperature-dependent type I toxin-antitoxin system. Mol Microbiol. 2012;83:579–598.
- Müller P, Jahn N, Ring C, et al. A multistress responsive type I toxin-antitoxin system: bsrE/SR5 from Bacillus subtilis. RNA Biol. 2016;13:511–523.
- Kubatova N, Pyper DJ, Jonker HRA, et al. Rapid biophysical characterization and NMR spectroscopy structural analysis of small proteins from Bacteria and Archaea. Chembiochem. 2019. DOI:10.1002/cbic.201900677
- Lehnik-Habrink M, Lewis RJ, Mäder U, et al. RNA degradation in Bacillus subtilis: an interplay of essential endo- and exoribonucleases. Mol Microbiol. 2012;84:1005–1017.
- Nurmohamed S, Vincent HA, Titman CM, et al. Polynucleotide phosphorylase activity may be modulated by metabolites in Escherichia coli. J Biol Chem. 2011;286:14315–14323.
- Carpousis AJ. The RNA degradosome of Escherichia coli: an mRNA-degrading machine assembled on RNase E. Annu Rev Microbiol. 2007;61:71–87.
- Kawamura F, Doi RH. Construction of a Bacillus subtilis double mutant deficient in extracellular alkaline and neutral proteases. J Bacteriol. 1984;160:442–444.
- Brantl S, Behnke D. Characterization of the minimal origin required for replication of the streptococcal plasmid pIP501 in Bacillus subtilis. Mol Microbiol. 1992;6:3501–3510.
- Hanahan D. Studies on transformation of Escherichia coli with plasmids. J Mol Biol. 1983;166:557–580.
- van Ooij C, Losick R. Subcellular localization of a small sporulation protein in Bacillus subtilis. J Bacteriol. 2003;185:1391–1398.