
ABSTRACT
The mechanisms that regulate cell-cycle arrest of cardiomyocytes during heart development are largely unknown. We have previously identified Tudor staphylococcal nuclease (Tudor-SN) as a cell-cycle regulator and have shown that its expression level was closely related to cell-proliferation capacity. Herein, we found that Tudor-SN was highly expressed in neonatal mouse myocardia, but it was lowly expressed in that of adults. Using Data Base of Transcription Start Sites (DBTSS), we revealed that Tudor-SN was a terminal oligo-pyrimidine (TOP) mRNA. We further confirmed that the translational efficiency of Tudor-SN mRNA was controlled by the mammalian target of rapamycin complex 1 (mTORC1) pathway, as revealed via inhibition of activated mTORC1 in primary neonatal mouse cardiomyocytes and activation of silenced mTORC1 in adult mouse myocardia; additionally, this result was recapitulated in H9c2 cells. We also demonstrated that the downregulation of Tudor-SN in adult myocardia was due to inactivation of the mTORC1 pathway to ensure that heart growth was in proportion to that of the rest of the body. Moreover, we revealed that Tudor-SN participated in the mTORC1-mediated regulation of cardiomyocytic proliferation, which further elucidated the correlation between Tudor-SN and the mTORC1 pathway. Taken together, our findings suggest that the translational efficiency of Tudor-SN is regulated by the mTORC1 pathway in myocardia and that Tudor-SN is involved in mTORC1-mediated regulation of cardiomyocytic proliferation and cardiac development.
Introduction
Tudor staphylococcal nuclease (Tudor-SN), also known as staphylococcal nuclease domain containing 1 (SND1) or p100, is a multifunctional protein that is involved in a variety of cellular processes, such as gene transcription [Citation1], pre-mRNA splicing [Citation2], DNA-damage responses [Citation3], stress-granule assembly [Citation4], and cell-cycle regulation [Citation5]. We have previously identified that the expression level of Tudor-SN is closely related to cell-proliferation capacity. It is highly expressed in proliferating cells, such as those in the kidney, liver, and spleen, as well as within virtually all cancer cells, but is lowly expressed or is undetectable in terminally differentiated cells, such as within cardiac and skeletal muscles [Citation5]. Recently, we notice that Tudor-SN is highly expressed in neonatal myocardia which possess proliferative capacity, and is lowly expressed in adult myocardia that have exited the cell cycle. Few previous studies focused on the regulatory mechanisms of Tudor-SN expression. Sandra et al reported that the transcription factors NF-κB, Sp1, and NF-Y controlled the gene transcription activity of Tudor-SN in HepG2 cells [Citation6]. However, the mechanisms mediating downregulation of Tudor-SN in terminally differentiated cells remain unclear. In the present study, we investigated the potential molecular mechanisms underlying downregulation of Tudor-SN in terminally differentiated adult cardiomyocytes.
Although mammalian neonatal cardiomyocytes proliferate rapidly and are regenerated following injury [Citation7], the majority of cardiomyocytes permanently exit the cell cycle and lost the regenerative capacity following the first two weeks of postnatal development [Citation8]. Limited cardiomyocytic turnover may occur [Citation9–11], but it is not sufficient to repair damage and to restore contractile function following injury. Accumulating evidence has demonstrated that the withdrawal of cardiomyocytes from the cell cycle during heart development is accompanied by progressively downregulation of positive regulators, such as cyclins and their associated cyclin-dependent kinases (CDKs) [Citation12]. Meanwhile, many transcriptional factors (e.g., YAP and Meis1) participate in cardiomyocytic cell-cycle arrest via regulation of cell-cycle-related genes [Citation13,Citation14]. Correspondingly, one major strategy for reactivating the adult cardiomyocytic cell cycle is to modulate the expression of cell-cycle-related genes and their regulators [Citation15–17]. Tudor-SN promotes the G1/S phase transition through enhancing transcriptional activation of cell-cycle-related genes [Citation5]. It suggests that the downregulation of Tudor-SN during cardiac development may also be involved in the cardiomyocytic cell-cycle arrest. Therefore, investigating the mechanisms that downregulate Tudor-SN in terminally differentiated adult cardiomyocytes may help to elucidate the mechanisms of postnatal cardiomyocytic cell-cycle arrest.
In the present study, our results demonstrate that Tudor-SN is a novel terminal oligo-pyrimidine (TOP) mRNA and that its translational efficiency in cardiomyocytes is regulated by the mammalian target of rapamycin complex 1 (mTORC1) pathway. Furthermore, we reveal that Tudor-SN is involved in mTORC1-mediated regulation of cardiomyocytic proliferation. Taken together, our findings provide novel insight into the regulatory mechanisms of Tudor-SN expression and may be useful for better understanding the underlying mechanisms of how cardiomyocytes developmentally transition from rapid neonatal proliferation to postnatal cell-cycle arrest.
Materials and methods
Animals
The mice used in the present study were from a C57BL/6 J genetic background. All of the experiments with mice were approved by the Institutional Animal Care and Use Committee of Tianjin Medical University (Tianjin, China).
Cell cultures and treatments
H9c2 cells and HEK 293 T cells were purchased from American Type Culture Collection (Manassas, VA, USA). The C2C12 cell line was a generous gift from Dr. Wenyan Niu (Tianjin Medical University, China). All of the cells were cultured in Dulbecco’s modified eagle’s medium (DMEM) (06–1055-57-1ACS, Biological Industries) with 10% foetal bovine serum (FBS) (04–001-1ACS, Biological Industries) in an incubator with 5% CO2 at 37°C. The mTORC1 inhibitor (Torin 1; 14379, Cell Signalling Technology), PI3K inhibitor (Ly294002; 9901, Cell Signalling Technology), proteasome inhibitor (MG132; 474787, Sigma), and lysosome inhibitor (chloroquine; 14774, Cell Signalling Technology) were used to treat H9c2 cells according to the manufacturers’ protocols. H9c2 cells were transfected with Lipofectamine 2000 transfection reagent (11668083, Thermo Fisher Scientific) according to the manufacturer’s protocol.
Construction of plasmids and stable cell lines
The 5ʹUTR sequences of mouse and rat Tudor-SN mRNAs were separately cloned into pGL3-Control plasmids to construct luciferase-reporter plasmids. These plasmids were constructed by GENEWIZ Company (Jiangsu, China).
The cDNA sequences of Tudor-SN containing CDS or 5ʹUTR + CDS were cloned into the pLVX-IRES-Puro Vector (632183, Clontech Laboratories) with a flag tag at the 3ʹ end to construct pLVX-IRES-Puro-Tudor-SN-Flag plasmids.
Lentiviral products were produced in the supernatant of HEK293T cells after being co-transfected with the above lentiviral expression plasmids and two envelope -expression plasmids. H9c2 cells were transfected with lentiviruses for 12 h and were then treated with 2 μg/mL of puromycin for three days. Thus, different positive stable cell lines were obtained.
The H9c2 Tudor-SN-/- (KO) stable cell line was generated by a modified CRISPR/Cas9 double-nicking gene-editing system [Citation18]. A pair of sgRNAs (α: 5ʹ-CACCGTCTGGAGGCGGACCGCCCCG-3ʹ, 3ʹ-CAGACCTCCGCCTGGCGGGGCCAAA-5ʹ; β: 5ʹ-CACCGAGCTGGAAATCTCGCCCGT-3ʹ, 3ʹ-CTCGACCTTTAGAGCGGGCACAAA-5ʹ), which specifically identify the upstream and downstream sequences, respectively, within exon 2 of the Tudor-SN gene, were cloned into a pX462 vector to construct two recombinant eukaryotic expression plasmids (pX462-Tudor-SN-sgRNA-α, pX462-Tudor-SN-sgRNA-β). These two recombinant plasmids were co-transfected into H9c2 cells, and puromycin was used to screen the positive monoclonal H9c2 Tudor-SN-/- (KO) stable cell line.
Sucrose gradient analysis of polysomes
The myocardia from one-day-old (1d) and eight-week-old (8 w) mice were ground with PBS buffer and then subjected to a protease inhibitor cocktail (5892970001, Roche). Subsequently, these samples were lysed with polysome lysis buffer (1% Triton X-100, 15 mmol/L MgCl2, 0.3 mol/L NaCl, 15 mmol/L Tris-HCl PH 7.4) that was supplemented with 100 μg/mL of cycloheximide (239763, Millipore), 1 Unit/μL of RNase inhibitor (2311A, Takara), and a protease inhibitor cocktail. As our previous study described [Citation19], the total lysates were added onto the surface of a linear sucrose gradient from 5% to 50%, which was produced by Gradient Master (BioComp Instruments, Fredericton, New Brunswick). Then, the samples were ultracentrifuged in a Beckman SW41 rotor for 3 h at 39,000 rpm. The fractions were collected via a piston Gradient Fractionator (BioComp Instruments) while monitoring the optical density at 254 nm. The polysome profiles were captured with a Model 251 Gradient Profiler (BioComp Instruments) and were analysed via Excel 2013. Subsequently, RNAs were extracted from each fraction, and RT-PCR was used to analyse the mRNA distributions in different fractions.
H9c2 WT cells were treated with Torin 1 for 24 h, then the cells were incubated with 100 μg/mL of cycloheximide for 15 min. The sucrose gradient and polysome profile analysis were performed as indicated above. The primer sequences that were used for RT-PCR were as follow: Tudor-SN (mouse): forward primer 5ʹ-TGTGCCACTGTCACCATTGGAG-3ʹ, reverse primer 5ʹ-CAGCTCATCGTAGTGTGAAGACC-3ʹ; Tudor-SN (rat): forward primer 5ʹ-GCTACGCAACCAGATGGGAAA-3ʹ, reverse primer 5ʹ-CTCGTCCTTGGGGAGTCTTGT-3ʹ; eEF2 (rat): forward primer 5ʹ-CCGACTCCCTTGTGTGCAA-3ʹ, reverse primer 5ʹ-AGTTCAGGTCGTTCTCAGAGAG-3ʹ; Gapdh (mouse): forward primer 5ʹ-CATCACTGCCACCCAGAAGACTG-3ʹ, reverse primer 5ʹ-ATGCCAGTGAGCTTCCCGTTCAG-3ʹ; and β-Actin (rat): forward primer 5ʹ-CATTGCTGACAGGATGCAGAAGG-3ʹ, reverse primer 5ʹ-TGCTGGAAGGTGGACAGTGAGG-3ʹ. These primers were synthesized by the GENEWIZ Company (Jiangsu, China).
Rapid amplification of 5ʹ cDNA ends (RACE) assays
We used the First-Choice RLM-RACE Kit (Am1700m, Thermo Fisher Scientific) to detect the transcription start sites of Tudor-SN mRNA. According to the manufacture’s protocol, the RNA was treated with calf-intestine alkaline phosphatase (CIP) at 37°C for 1 h to remove free 5ʹ-phosphates, and then the RNA was treated with tobacco acid pyrophosphatase (TAP) at 37°C for 1 h to remove the cap structure from the full-length mRNA. The RNA with 5ʹ-monophosphates was ligated with RNA adapter oligonucleotides at 37°C for 1 h via T4 RNA ligases, and nested PCR was used to amplify the 5ʹ end of Tudor-SN mRNA with two provided nested primers and two specific primers for Tudor-SN mRNA. Finally, the product of each inner PCR was sequenced by GENEWIZ Company (Jiangsu, China). The specific primers for Tudor-SN mRNA in rat and mouse were as follows: the outer primer was 5ʹ-TGCGAAGGAACTCTCTAGCA-3ʹ and the inner primer was 5ʹ-AGGCCATGTGTGGAGCTGGAAA-3ʹ for rat; and the outer primer was 5ʹ-TGGCCAAGCCTTCTGCAACTA-3ʹ and the inner primer was 5ʹ-AGGCCATGTGTGGAGCTGGAAA-3ʹ for mouse.
Isolation and culturing of neonatal cardiomyocytes
Primary cardiomyocytes from 1d neonatal hearts were isolated by enzymatic disassociation. Briefly, mice were anesthetized by cooling on an ice bed for 1–2 min. Hearts were extracted and soaked in 4°C ADS buffer (0.68% NaCl, 0.476% HEPES, 0.012% NaH2PO4, 0.1% glucose, 0.04% KCl, 0.01% MgSO4, pH 7.35). The auricular appendages were removed from the heart and the rest of the tissues were cut into sections that were approximately 1 mm3. Then, the tissue was digested in ADS buffer containing 1.25 mg/mL collagenase II (DH071, Thermo Fisher Scientific) and 1.25 mg/mL pancreatin (T8003, Sigma) at 37°C while shaking for approximately 15 min. The same volume of stop buffer (90% DMEM + 10% FBS) was added to stop the digestion, and was then filtered through a 40-μm-aperture filter. Cells were centrifuged at 2,000 rpm for 5 min. Thereafter, cells were plated on cell-culture dishes and were incubated for 90 min at 37°C with 5% CO2 to remove cardiac fibroblasts. Cardiomyocytes were plated on laminin-coated cell-culture dishes and were incubated overnight at 37°C with 5% CO2 for cellular attachment. The following day, the culture medium was replaced with maintenance medium (80% DMEM + 20% FBS) supplemented with 1% penicillin/streptomycin. Finally, cardiomyocytes were used for further experiments.
Western blotting
Western blotting was performed as previously described [Citation5]. The following antibodies were used: anti-AKT (4691, Cell Signalling Technology), anti-p-AKT (4060, Cell Signalling Technology), anti-P70S6 (9202, Cell Signalling Technology), anti-p-P70S6 (9204, Cell Signalling Technology), anti-4EBP (9452, Cell Signalling Technology), anti-p-4EBP (2855, Cell Signalling Technology), anti-Igf1 (PA5-27207, Thermo Fisher Scientific), anti-Itga5 (PA5-82027, Thermo Fisher Scientific), anti-Larp4 (PA5-109902, Thermo Fisher Scientific), anti-Gng2 (PA5-49529, Thermo Fisher Scientific), Col1α1 (PA5-29569, Thermo Fisher Scientific), Col3α1 (PA5-34787, Thermo Fisher Scientific), anti-GAPDH (5174S, Cell Signalling Technology), and anti-β-Actin (A5316, Sigma). The anti-Tudor-SN antibody was generated against the SN4 domain (amino acids 507–674) of Tudor-SN by the Institute of Medical Technology at the University of Tampere (Tampere, Finland). ImageJ (2X) software (National Institutes of Health, Bethesda, MD, USA) was used to analyse protein-band densities.
RNA extraction and quantitative real-time PCR (qRT-PCR)
Total RNA was extracted from cardiac tissues and cells by using Trizol reagent (15596018, Thermo Fisher Scientific) according to the manufacture’s protocol. Synthesis of cDNA was achieved by using the Revert Aid First-Strand cDNA Synthesis Kit (K1622, Thermo Fisher Scientific) according to the manufacturer’s protocol. Quantitative real-time PCR (qRT-PCR) was performed by using the Fast Start Universal SYBR Green Master Mix (4913850001, Roche) on a Step One Real-Time PCR System (ABI-StepOne Plus, Thermo Fisher Scientific). The primer sequences using for qRT-PCR were as follows (those for Tudor-SN [mouse and rat] and Gapdh [mouse and rat] were the same as described above): Igf1 (mouse) forward primer 5ʹ- GTGGATGCTCTTCAGTTCGTGTG-3ʹ, reverse primer 5ʹ-TCCAGTCTCCTCAGATCACAGC-3ʹ; Igf2 (mouse) forward primer 5ʹ- CTTCAGTTTGTCTGTTCGGACCG-3ʹ, reverse primer 5ʹ-GTGGCACAGTATGTCTCCAGGA-3ʹ; Fgf12 (mouse) forward primer 5ʹ-GGAGAAGGCTATCTCTACAGCTC-3ʹ, reverse primer 5ʹ-CTGATTCCTGCTGGCGATACAG-3ʹ; Pdgfa (mouse) forward primer 5ʹ-CTGGCTCGAAGTCAGATCCACA-3ʹ, reverse primer 5ʹ-GACTTGTCTCCAAGGCATCCTC-3ʹ; Itga5 (mouse) forward primer 5ʹ-ACCTGGACCAAGACGGCTACAA-3ʹ, reverse primer 5ʹ-CTGGGAAGGTTTAGTGCTCAGTC-3ʹ; Itga8 (mouse) forward primer 5ʹ-CCGATTTGCTGTTCCTCGCCTT-3ʹ, reverse primer 5ʹ-GACCTGAGCAATGGCAGTGATG-3ʹ; Larp4 (mouse) forward primer 5ʹ-GCCAAGTAGTCTTGAATCCTCTC-3ʹ, reverse primer 5ʹ-TGGTGTTGGAGACTGCGTCTGA-3ʹ; Gng2 (mouse) forward primer 5ʹ-GCCAGGAAGCTGGTAGAACA-3ʹ, reverse primer 5ʹ-AGGGTCTTCCTTGGCATGTG-3ʹ; Lamb1 (mouse) forward primer 5ʹ- CTTCTCCCTAAACGGGTTTTCTG-3ʹ, reverse primer 5ʹ-GCCAACAGTGAAGATGTCCAGC-3ʹ; Lama4 (mouse) forward primer 5ʹ-CAGTTTGTCCTCTACCTCGGAAG-3ʹ, reverse primer 5ʹ-CTCACAGGCTTGGAATCCAGGA-3ʹ; Thbs1 (mouse) forward primer 5ʹ-GGTAGCTGGAAATGTGGTGCGT-3ʹ, reverse primer 5ʹ-GCACCGATGTTCTCCGTTGTGA-3ʹ; Col1a1 (mouse) forward primer 5ʹ-CCTCAGGGTATTGCTGGACAAC-3ʹ, reverse primer 5ʹ-CAGAAGGACCTTGTTTGCCAGG-3ʹ; Col3a1 (mouse) forward primer 5ʹ-GACCAAAAGGTGATGCTGGACAG-3ʹ, reverse primer 5ʹ-CAAGACCTCGTGCTCCAGTTAG-3ʹ. These primers were synthesized by the GENEWIZ Company (Jiangsu, China).
Immunohistochemistry
The myocardia from neonatal (1d) to adult (8 w) mice were fixed in 10% formalin for 24 h and embedded in paraffin. Tissue sections were blocked and incubated with anti-Tudor-SN antibody. Then, sections were incubated with goat anti-mouse/rabbit-HRP secondary antibody followed by DAB staining. The images were collected under the inverted microscope (Leica DMI6000 B; Wetlar, Germany).
Immunofluorescence
Immunofluorescent assays were performed as previously described [Citation19]. Briefly, primary cardiomyocytes were fixed, permeabilized, and incubated with specific antibodies at 4°C overnight. The antibodies included anti-cardiac Troponin T (cTNT) (ab8295, Abcam), anti-BrdU (MA1-81890, Thermo Fisher Scientific), anti-p-H3 (PA5-17869, Thermo Fisher Scientific), and anti-Ki67 (PA5-19462, Thermo Fisher Scientific). After washing with PBST, samples were incubated with fluorescent secondary antibodies at 4°C for 7 ~ 8 h in the dark and were then counterstained with DAPI. The secondary antibodies included Alexa Fluor 488-coupled donkey anti-rabbit IgG (H + L) (A21206, Thermo Fisher Scientific) and Alexa Fluor 546-coupled donkey anti-mouse IgG (H + L) (A10036, Thermo Fisher Scientific) diluted in 1% BSA. Images were collected under an inverted microscope (Leica DMI6000 B; Wetlar, Germany).
Echocardiography
Echocardiographic assays were performed blindly. Mice were anesthetized with 3% isoflurane, followed by maintenance at 2% isoflurane. Mice were placed in a supine position on a warm platform to maintain body temperature at approximately 37°C. The limbs were taped onto metal electrocardiographic leads, and chest hair was removed by using hair-removal cream. Echocardiography was performed via a Visual Sonics Vevo 2000 system equipped with a 40-MHz transducer for cardiac imaging. The transducer was placed along the long axis of the left ventricle (LV) and was directed to the right side of the neck to obtain the two-dimensional LV long axis. Then, the transducer was rotated by 90 degrees, and the LV short-axis view was visualized. The two-dimensional-guided LV M-mode was recorded from the short-axis view. Echocardiographic images were analysed offline via image-processing software. At least three images were measured and averaged per mouse. The end-diastolic and end-systolic left-ventricular internal diameters (LVIDd, LVIDs) were measured from the LV short-axis view via 2D-orientated M-mode imaging. LV systolic function was estimated by fractional shortening (FS, %) according to the following formula: FS (%) = [(LVIDd – LVIDs)/LVIDd] × 100%. Ejection fraction (EF, %) was calculated by using the end-systolic and end-diastolic volumes, as follows: EF (%) = [(EDV – ESV)/EDV] × 100%. More than four mice per group were used for echocardiography.
Cell-cycle analysis
For cell-cycle analysis, H9c2 cells were seeded into 6-cm plates. Approximately 1 × 106 cells were collected and fixed in 70% ice-cold ethanol overnight. Then, cells were centrifuged at 2,000 rpm for 5 min to remove the ethanol, and the cell pellets were then washed with PBS. Then, cells were stained with 50 μg/mL propidium iodide (P4864, Sigma) together with 50 μg/mL RNase treatments. Cell-cycle distributions were detected by flow cytometry (BD FACS Verse system, San Jose, CA) and were analysed with Modfit software.
Cellular proliferation assays
H9c2 cells were seeded in quintuplicates in 96-well plates at 1,000 cells per well. Cellular proliferation was detected via Cell Counting Kit-8 (CCK-8, CK04, Dojindo) assays following five days in culture. According to the manufacturer’s protocol, after cells were seeded in plates and pre-incubated at 37°C with 5% CO2, the supernatant was removed and 100 μL of new medium containing 10 μL CCK-8 solution was added. Thereafter, the plate was incubated for 3 h in an incubator. Finally, we measured the absorbance of each sample at 450 nm via an Optimal Microplate Reader (Varioskan Flash, Thermo Fisher Scientific).
Statistical analysis
Data are presented as the mean ± standard error of the mean (SEM). Differences between groups were tested for statistical significance via unpaired two-tailed t-tests or one-way analyses of variance (ANOVAs) with Bonferroni post-hoc tests. Differences at p = 0.05 or lower were considered to be statistically significant.
Results
Silencing expression of Tudor-SN in adult mouse myocardia is regulated at the translational level
We have previously reported that the protein expression level of Tudor-SN is closely correlated to the cellular proliferative capacity. It is abundant in proliferating cells but is lowly expressed in terminally differentiated cells [Citation5]. Neonatal cardiomyocytes proliferate rapidly and gradually lose the proliferative ability around 7-days-old (7d) and exit the cell cycle at 14-days-old (14d), after which they permanently transform to terminal differentiation. We therefore investigated the expression pattern of Tudor-SN during cardiac development in mice. We isolated myocardia from 1d, 7d, 14d, 21d, and 8 w mice and detected the corresponding protein expression levels of Tudor-SN. The results () showed that Tudor-SN protein was highly expressed in 1d myocardia that proliferated rapidly, gradually downregulated as cardiomyocytes lost their proliferative ability, and was lowly expressed in 8 w mouse myocardia that were terminally differentiated. Immunohistochemical staining () also demonstrated the same temporal expression patterns of Tudor-SN protein. However, there was no significant difference at the mRNA level of Tudor-SN from 1d to 8 w mouse myocardia (). These data suggest that the decrease of Tudor-SN protein during cardiac development is likely regulated at the post-transcriptional level.
Figure 1. Silenced expression of Tudor-SN in adult mouse myocardia is regulated at the translational level. (A–B) The protein levels of Tudor-SN in 1d, 7d, 14d, 21d and 8 w mouse myocardia were detected by Western blotting and analysed by ImageJ (2X) software. Data are presented as the mean ± SEM (1d, six mice/group; 7d, three mice/group; 14d, three mice/group; 21d, two mice/group; 8 w, one mouse/group. n = 3 independent experiments), and a one-way ANOVA with Bonferroni post-hoc tests were used (*p < 0.05, **p < 0.01). (C) The heart sections from 1d, 7d, 14d, 21d and 8 w mice were detected by immunohistochemical staining with anti-Tudor-SN antibody (scale bar, 50 μm). (D) The mRNA levels of Tudor-SN in 1d, 7d, 14d, 21d and 8 w mouse myocardia were analysed by qRT-PCR. Data are presented as the mean ± SEM (n = 3 independent experiments), and a one-way ANOVA with Bonferroni post-hoc tests were used (ns, no significance). (E) Ribosomal absorption profiles of 1d and 8 w mouse myocardia lysates were analysed by sucrose-gradient centrifuge assays. Note that 40s and 60s denote the corresponding ribosomal subunits, and 80s denotes monoribosomes. (F) The Tudor-SN and Gapdh mRNA levels in different fractions from (E) were detected by RT-PCR and analysed by ImageJ (2X) software in (G). Data are presented as the mean ± SEM (1d, 10 mice/group; 8 w, two mice/group. n = 3 independent experiments), and unpaired two-tail t-tests were used (**p < 0.01, ns, no significance)
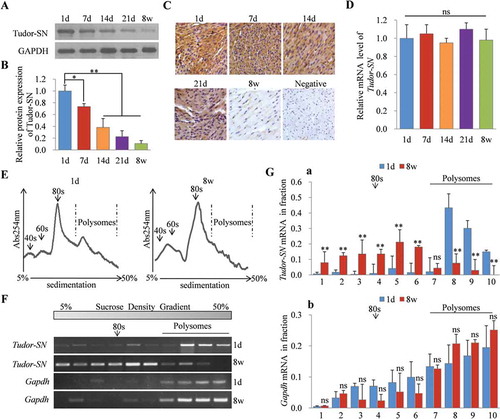
To determine translational efficiencies, we selected 1d and 8 w mouse myocardia to detect the distributions of Tudor-SN mRNA in polysome profile fractions. In polysome profile assays, the heavy polysomes distribute in the high-density sucrose, while the light monosomes and ribosomal subunits distribute in the low-density sucrose. The polysomes-associated mRNAs are efficiently translated, while the monosomes or ribosomal subunits-associated mRNAs are poorly translated. As shown in , compared with those in 1d mice, polysomes were collapsed and ribosomes were shifted out from polysomes in the myocardia of 8 w mice. This result suggests that the total translational efficiency is remarkably reduced in adult myocardia. RT-PCR () indicated that the Tudor-SN mRNA of 1d myocardia (, panel 1) was distributed in the high-density gradient and co-localized with the polysomes, which indicated that its translational efficiency was high. The Tudor-SN mRNA of 8 w myocardia (, panel 2) was distributed in the lower density gradient compared with 1d myocardia, which indicated the translational efficiency of Tudor-SN decreased. As the control, there were no significant changes in the distribution of Gapdh mRNA (, panel 3, 4) between 1d and 8 w groups. Statistical analysis () further indicated that Tudor-SN mRNA in 8 w mouse myocardia was significantly depleted from polysomes compared with that in 1d mouse myocardia (, a), and there were also no significant changes in the distribution of Gapdh mRNA between these two groups (, b). These data further verify that the inhibited expression of Tudor-SN in adult mouse myocardia is regulated at the translational level.
Tudor-SN is a novel terminal oligo-pyrimidine (TOP) mRNA
Translational regulation plays an important role in modulating gene expression. The 5ʹ TOP motif is a well-characterized cis-regulatory motif for translational control. It is initiated with cytosine immediately after the 5ʹ-cap and is followed by 4–14 oligo-pyrimidine-long tracts [Citation20]. In the present study, we identified Tudor-SN as a novel TOP mRNA. Using the Data Base of Transcription Start Sites (DBTSS) [Citation21], we found that there was a potential TOP motif in the Tudor-SN mRNA with cytosine in the transcription start site (TSS), followed by six pyrimidines in both human (NM_014390.3) and mouse (NM_019776.2) (). Furthermore, we sequenced the 5ʹ untranslated regions (UTR) of Tudor-SN mRNA via rapid amplification of 5ʹ cDNA ends (RACE) assays. Consistent with the DBTSS analysis, the sequencing data () showed the same TOP motif of Tudor-SN mRNA in both human and mouse. Additionally, the sequencing data revealed that the Tudor-SN mRNA of rat also had the TOP motif.
Figure 2. Tudor-SN is a novel TOP mRNA. (A) The TSS of Tudor-SN mRNA (red triangle) in human and mouse was determined by the DBTSS; 5ʹ TOP motifs are indicated in red, and the length of 5ʹ TOP and 5ʹ UTR were labelled. (B) The 5ʹ RACE assay was performed to further clarify the TSS and 5ʹ TOP motif of Tudor-SN mRNA in human, mouse, and rat. (C) The pGL3-Mus 5ʹUTR-luc and pGL3-Rat 5ʹUTR-luc plasmids were constructed and transfected into H9c2 cells. Then, H9c2 cells were treated with 250 nmol/L of Torin 1 for different durations and relative luciferase activities were detected. Data are presented as the mean ± SEM (n = 3 independent experiments), and a one-way ANOVA with Bonferroni post-hoc tests were used (**p < 0.01, ns, no significance). (D–E) Tudor-SN-5ʹUTR+CDS-Flag and Tudor-SN-CDS-Flag plasmids were constructed and transfected into Tudor-SN-/- H9c2 cells. Then, the cells were incubated in the absence or presence of 250 nmol/L of Torin 1 for 24 h. Exogenous Tudor-SN protein expression was detected by Western blotting and analysed by ImageJ (2X) software. Data are presented as the mean ± SEM (n = 3 independent experiments), and unpaired two-tail t-tests were used (*p < 0.05, ns, no significance)
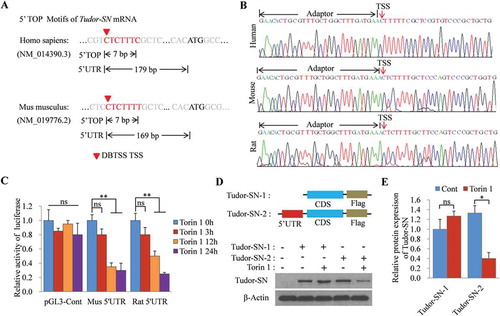
The translational efficiency of TOP mRNA is regulated by the mTORC1 pathway [Citation22,Citation23]. To investigate whether Tudor-SN is a TOP mRNA, mammalian expression plasmids containing a luciferase reporter gene with a 5ʹ-UTR TOP sequence of Tudor-SN mRNA of mouse (pGL3-Mus 5ʹUTR-luc) and rat (pGL3-Rat 5ʹUTR-luc) were constructed, and were then transfected into H9c2 cells. Then, the cells were treated with the mTORC1 inhibitor, Torin 1, at different time points as indicated in . Thereafter, luciferase reporter assays were performed to investigate whether the TOP sequence of Tudor-SN influenced the expression of luciferase. As shown in , there was no significant difference among the control group as a function of Torin 1 treatment, while the luciferase activities of pGL3-Mus 5ʹUTR-luc and pGL3-Rat 5ʹUTR-luc were significantly inhibited following Torin 1 treatment for 12 h or 24 h.
To further determine whether the TOP sequence influenced the translation of Tudor-SN mRNA, we next constructed mammalian expression plasmids containing the full-length coding sequence (CDS) of Tudor-SN without the TOP sequence (Tudor-SN-1: Tudor-SN-CDS-Flag) or with the TOP sequence (Tudor-SN-2: Tudor-SN-5ʹUTR+CDS-Flag). The H9c2 cell line was originally derived from embryonic rat ventricular tissues with expression of endogenous Tudor-SN protein. To rule out interference from endogenous Tudor- SN, we generated a Tudor-SN-/- H9c2 stable cell line (H9c2 KO) via the modified CRISPR/Cas9 double-nicking system. Then, H9c2 KO cells were transfected with Tudor-SN-CDS-Flag or Tudor-SN-5ʹUTR+CDS-Flag plasmids. After treating with Torin 1 for 24 h, ectopically expressed Tudor-SN proteins were detected by Western blotting. As shown in , Tudor-SN was efficiently expressed in cells with the Tudor-SN-CDS-Flag, but was inhibited in cells with the Tudor-SN-5ʹUTR+CDS-Flag containing the TOP sequence following Torin 1 treatment (, panel 1). Statistical analysis () further indicated that the TOP sequence significantly influenced Tudor-SN protein expression following Torin 1 treatment. These data demonstrate that Tudor-SN is a novel TOP mRNA.
The translational efficiency of Tudor-SN mRNA in H9c2 cells is regulated by the mTORC1 pathway
To further clarify that Tudor-SN is a real mTORC1 target mRNA, we detected the expression of endogenous Tudor-SN in H9c2 cells following treatment with Torin 1. As shown in , the phosphorylation levels of 4EBP (, panel 3) and P70S6 (, panel 5), which are downstream targets of mTORC1, were reduced dramatically and were barely detectable after Torin 1 treatment. This result indicated that the mTORC1 activity was efficiently inhibited by Torin 1. Meanwhile, the protein expressions of Tudor-SN were significantly reduced in a time-dependent manner, at both 12 and 24 h (, panel 1 and ), whereas no obvious change was observed at the mRNA level (). The same results were obtained when the cells were treated with the PI3K inhibitor, Ly294002 (Fig. S1A–C), or starved by serum and amino-acid deprivation (Fig. S1D–F), which also inhibited the activity of mTORC1 pathway. In addition, H9c2 cells were treated with isoproterenol (ISO), which activates mTORC1. As shown in Fig. S1G, ISO treatment enhanced the phosphorylation of 4EBP (Fig. S1G, panel 3) and P70S6 (Fig. S1G, panel 5). Accordingly, the protein expressions of Tudor-SN were increased as ISO stimulation was prolonged (Fig. S1G, panel 1 and Fig. S1H), but no obvious effect was found at the mRNA level (Fig. S1I). These results confirm that the expression of Tudor-SN is regulated by the mTORC1 pathway.
Figure 3. Protein expression of Tudor-SN in H9c2 cells is regulated by the mTORC1 pathway. (A–C) H9c2 cells were treated with 250 nmol/L of Torin 1 for 0, 3, 12, or 24 h. The protein levels of Tudor-SN and other indicated proteins of the mTORC1 pathway were detected by Western blotting; the band intensity of Tudor-SN was analysed by ImageJ (2X) software, and the mRNA level of Tudor-SN was analysed by qRT-PCR. Data are presented as the mean ± SEM (n = 3 independent experiments), and a one-way ANOVA with Bonferroni post-hoc tests were used (**p < 0.01, ns, no significance). (D) The ribosomal absorption profiles of H9c2 cells in the absence or presence of 250 nmol/L of Torin 1 for 24 h were analysed by sucrose-gradient centrifuge assays. Note that 40s and 60s denote the corresponding ribosomal subunits, while 80s denotes monoribosomes. (E–F) The distributions of Tudor-SN, eEF2, and β-Actin mRNA levels in different fractions from (D) were detected by RT-PCR and analysed by ImageJ (2X) software. Data are presented as the mean ± SEM (n = 3 independent experiments), and unpaired two-tail t-tests were used (**p < 0.01, ns, no significance). (G–H) H9c2 cells were treated with 250 nmol/L of Torin 1 for 24 h, together with the proteasome inhibitor, MG132 (10 μmol/L), or the lysosome inhibitor, chloroquine (100 μmol/L), in the last 8 h of the total 24 h incubation. The protein expression of Tudor-SN was detected by Western blotting and analysed by ImageJ (2X) software. Data are presented as the mean ± SEM (n = 3 independent experiments), and a one-way ANOVA with Bonferroni post-hoc tests were used (*p < 0.05, **p < 0.01)
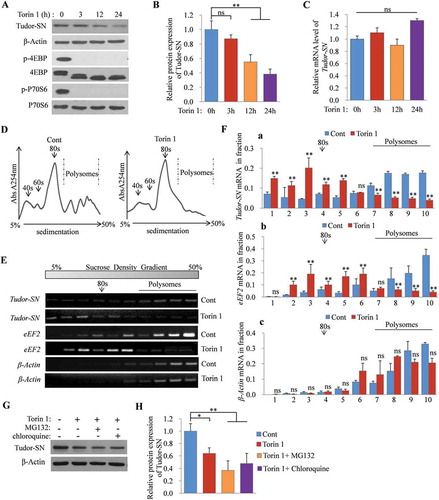
Moreover, we detected the distribution of Tudor-SN mRNA in polysome profile fractions in H9c2 cells in the absence or presence of Torin 1 treatment for 24 h. The polysome profile assay revealed that Torin 1 impaired the total translational efficiency, as the polysomes collapsed and the ribosomes shifted out from the polysomes (). Meanwhile, RT-PCR showed that the Tudor-SN mRNA of parental H9c2 cells (Cont) (, panel 1) distributed in the high-density gradient and co-localized with the polysomes, which indicated that its translational efficiency was high, while Torin 1 strongly reduced polysome loading of Tudor-SN mRNA in H9c2 cells (, panel 2) compared with that in the parental cells. We also detected the distribution of eEF2 mRNA, which is a well-known TOP mRNA. The result showed that eEF2 mRNA was also significantly shifted out from polysomes after Torin 1 treatment (, panels 3 and 4). As the control, there were no significant changes in β-Actin mRNA (, panels 5 and 6) distributions with or without Torin 1 treatment. Statistical analysis () further indicated that Tudor-SN and eEF2 mRNA were significantly depleted from polysomes after Torin 1 treatment (, a–b), and no significant changes in β-Actin mRNA were found (, c). These results indicate that inhibition of mTORC1 reduces the translational efficiency of Tudor-SN mRNA. To clarify that the decrease of Tudor-SN protein caused by mTORC1 inhibition was not due to protein degradation, H9c2 cells were treated with Torin 1 for 24 h and were co-treated with the proteasome inhibitor, MG132, or the lysosome inhibitor, chloroquine, in the last 8 h of the 24 h Torin 1 treatment. The results showed that the expression of Tudor-SN was reduced after Torin 1 treatment, even in the presence of MG132 or chloroquine treatment, which indicated that the reduced expression of Tudor-SN (, panel 1) was not due to ubiquitin-mediated proteasomal degradation or lysosomal degradation (, H). These data reveal that the translational efficiency of Tudor-SN mRNA is regulated by the mTORC1 pathway.
Decreased expression of Tudor-SN in adult mouse myocardia is caused by the low activity of the mTORC1 pathway
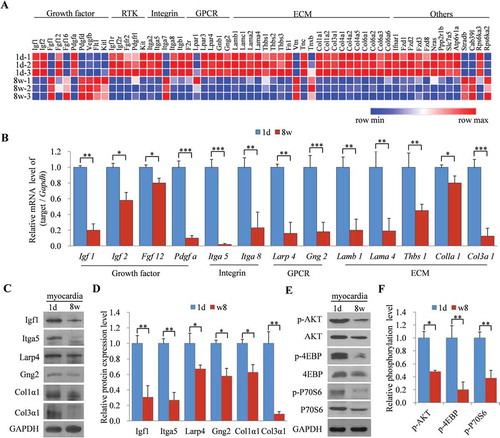
To determine the potential mechanisms for the dynamically reduced expression of Tudor-SN controlled by the mTORC1 pathway in myocardia during cardiac development, we analysed the transcriptomic profiles of upstream regulators and downstream targets of the mTORC1 pathway based on our previous microarray-chip data (Affymetrix Mouse Gene 1.0 ST) of myocardia from 1d and 8 w mice [Citation24]. Pathway enrichment analysis illustrated the differentially expressed genes that were involved in the mTORC1 pathway (Fig. S2). The corresponding heat map revealed that most of these genes were downregulated in adult mouse myocardia compared with those in 1d mouse myocardia (), including many growth factors (GFs), receptor-tyrosine kinases (RTKs), integrins (ITG), G-protein-coupled receptors (GPCRs), and extracellular matrixes (ECM). Consistently, qRT-PCR further verified that the mRNA levels of Igf1, Igf2, Fgf12, Pdgfa, Itga5, Itga8, Larp4, Gng2, Lamb1, Lama4, Thbs1, Colla1, and Col3a1 (), which are upstream regulators of the mTORC1 pathway, were all decreased in adult mouse myocardia comparing with those in 1d mouse myocardia. We then selected Igf1, Itga5, Larp4, Gng2, Col1α1, and Col3α1 to detect their protein expression levels in 1d and 8 w mouse myocardia. The results showed that all of these proteins were downregulated in 8 w mouse myocardia compared to those in 1d mouse myocardia (, D). Moreover, the phosphorylation levels of AKT (, panel 1), 4EBP (, panel 3), and P70S6 (, panel 5) were reduced in adult mouse myocardia (, F). These results indicated that most upstream regulators of mTORC1 were downregulated in adult mouse myocardia, as well as the activity of the mTORC1 pathway. These data also demonstrate that the expression of Tudor-SN is highly correlated with the activation of the mTORC1 pathway, and suggest that the reduced expression of Tudor-SN is due to inactivation of the mTORC1 pathway in adult mouse myocardia.
Figure 4. Decreased expression of Tudor-SN in adult mice myocardia is caused by the low activity of the mTORC1 pathway. (A) Heat map of gene expression in 1d and 8 w mouse myocardia. Hierarchical cluster analysis of significantly differentially expressed genes is presented (red, high expression; blue, low expression). (B) The relative mRNA levels of 13 differentially expressed genes were analysed by qRT-PCR. Data are presented as the mean ± SEM (n = 3 independent experiments), and unpaired two-tail t-tests were used (*p < 0.05, **p < 0.01, ***p < 0.001). (C–D) The protein levels of six representative genes in (B) were detected by Western blotting and were analysed by ImageJ (2X) software. Data are presented as the mean ± SEM (n = 3 independent experiments), and unpaired two-tail t-tests were used (*p < 0.05, **p < 0.01). (E–F) The relative phosphorylation levels of AKT, 4EBP, and P70S6 in 1d and 8 w mouse myocardia were detected by Western blotting and analysed by ImageJ (2X) software. Data are presented as the mean ± SEM (n = 3 independent experiments), and unpaired two-tail t-tests were used (*p < 0.05, **p < 0.01)
The expression of Tudor-SN in mouse myocardia is regulated by the mTORC1 pathway
To further determine if the expression of Tudor-SN in myocardia is directly regulated by the mTORC1 pathway, we inhibited the activated mTORC1 pathway in primary neonatal mouse cardiomyocytes or activated the silenced mTORC1 pathway in adult mouse myocardia to determine changes in protein expression of Tudor-SN. As shown in , B, endogenous Tudor-SN was highly expressed in neonatal cardiomyocytes but was obviously reduced following Torin 1 treatment for 24 h (, panel 1). Meanwhile, the phosphorylation levels of 4EBP (, panel 3) and P70S6 (, panel 5) were significantly inhibited following Torin 1 treatment, which confirmed the inactivation of the mTORC1 pathway in cardiomyocytes. However, there were no obvious changes in the mRNA level of Tudor-SN (). The same results were also obtained when the cells were treated with the PI3K inhibitor, Ly294002 (Fig. S3A–C), or when they were starved (Fig. S3F–H). Besides, we examined the proliferative ability of cardiomyocytes by staining the cells with proliferation markers – namely, BrdU, p-H3, and Ki67 – after treating the cells with Torin 1 (, F, H) for 24 h. The results showed that the proliferative ability was high in neonatal cardiomyocytes but was dramatically decreased following Torin 1 treatment, as quantified by decreases in the numbers of BrdU+/cTNT+, p-H3+/cTNT+, or Ki67+/cTNT+ cells (, G, I). We also stained the cells with BrdU after treating them with Ly294002 or starving them by serum and amino-acid deprivation. The same results were obtained (Fig. S3D, E, I, J). To further detect the proliferative ability of cardiomyocytes, we counted the cardiomyocytes after treating the cells with or without Torin 1 or Ly294002, or after being starved for 24 h. As shown in and Fig. S3K, L, the number of cardiomyocytes was reduced when the mTORC1 pathway was inhibited. These results not only verified mTORC1 regulation of Tudor-SN expression in neonatal mouse cardiomyocytes, but also revealed a correlation of Tudor-SN with cardiomyocytic proliferation.
Figure 5. The expression of Tudor-SN in mouse myocardia is regulated by the mTORC1 signalling pathway. (A–C) 1d mouse primary cardiomyocytes were isolated and treated with 250 nmol/L of Torin 1 for 24 h. The protein levels of Tudor-SN and other indicated proteins of the mTORC1 pathway were detected by Western blotting and analysed by ImageJ (2X) software. The mRNA level of Tudor-SN was analysed by qRT-PCR. Data are presented as the mean ± SEM (n = 3 independent experiments), and unpaired two-tail t-tests were used (*p < 0.05, ns, no significance). (D, F, H) 1d mouse primary cardiomyocytes were treated with 250 nmol/L of Torin 1 for 24 h and the proliferative ability of cardiomyocytes was detected by immunofluorescent staining with anti-cardiac troponin T (cTNT, red), an antibody to mark cardiomyocytes, as well as with anti-BrdU, anti-p-H3, and anti-Ki67 (green) (antibodies for marking proliferation). DAPI (blue) was used as a nuclear counterstain. The scale bar denotes 50 μm. (E, G, I) Quantitative analysis of BrdU+/cTNT+ cells, p-H3+/cTNT+ cells, and Ki67+/cTNT+ cells per field in (D, F, H), respectively. Data are presented as the mean ± SEM (five mice/group, n = 3 independent experiments), and unpaired two-tail t-tests were used (*p < 0.05). (J) The 1d mouse primary cardiomyocytes (1 × 104 cells) were seeded in 24-well plates and treated with 250 nmol/L of Torin 1 for 24 h, then the number of cells was counted and analysed. Data are presented as the mean ± SEM (five mice/group, n = 3 independent experiments), and unpaired two-tail t-tests were used (*p < 0.05). (K–M) The 8 w mice were treated with 30 mg/kg of ISO via intraperitoneal injections for 24 h, and normal saline was used as the control. The protein expression of Tudor-SN and other indicated proteins of the mTORC1 pathway in myocardia were detected by Western blotting and analysed by ImageJ (2X) software; the mRNA level of Tudor-SN was analysed by qRT-PCR. Data are presented as the mean ± SEM (one mice/group, n = 3 independent experiments), and unpaired two-tail t-tests were used (*p < 0.05, ns, no significance). (N) Cardiac function was measured by echocardiography after mice were treated with ISO or normal saline. Data are presented as the mean ± SEM (five mice/group, n = 3 independent experiments), and unpaired two-tail t-tests were used (*p < 0.05). EF, ejection fraction; FS, fractional shortening
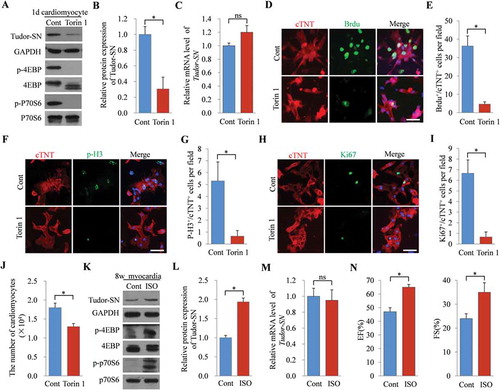
Comparing with that of the control group, the protein level of Tudor-SN was increased (, panel 1 and ) in adult mouse myocardia following ISO treatment for 24 h to artificially activate the mTORC1 pathway. Consistently, the phosphorylation levels of 4EBP (, panel 3) and P70S6 (, panel 5) were significantly enhanced. However, no significant changes in the mRNA levels of Tudor-SN were observed (). The improvement of the cardiac ejection fractions (EF%) and fraction shortening (FS%) indicated that ISO worked effectively (). All of these data demonstrate that the expression of Tudor-SN is regulated by the mTORC1 pathway in mouse myocardia.
Tudor-SN participates in mTORC1-mediated regulation of cardiomyocytic proliferation
In our previous study, we revealed that Tudor-SN is a novel regulator of the G1/S transition in carcinoma cells [Citation5]. To verify whether Tudor-SN is involved in the cell-cycle regulation of cardiomyocytes, we first analysed the cell-growth rates and cell-cycle distributions of H9c2 WT cells and H9c2 Tudor-SN KO cells. The knockout efficiency of Tudor-SN protein was detected by Western blotting (, panel 1). As shown in , the H9c2 Tudor-SN KO cells had a slower growth rate compared with that of H9c2 WT cells, as evaluated by CCK8 assays. Meanwhile, compared with those of H9c2 WT cells (G1: 62.62%, S: 31.14%), more H9c2 Tudor-SN KO cells were distributed at the G1 phase (75.01%) and fewer cells were found in the S phase (14.31%), as analysed by flow cytometry (). Statistical analysis further indicated that the difference was significant (). To further verify the effects of Tudor-SN on the cell cycle, we ectopically over-expressed Tudor-SN in H9c2 cells (Tudor-SNOE) with the pLVX-IRES-Puro-Tudor-SN-Flag plasmid (, panel 1) and detected cell-growth rates and cell-cycle distributions. As shown in , H9c2 Tudor-SNOE cells grew faster than H9c2 cells transfected with a pLVX-IRES-Puro Vector (H9c2 Vector cells), and comparing with H9c2 Vector cells (G1: 60.01%, S: 29.79%), there were more H9c2 Tudor-SNOE cells distributed in the S phase (38.28%) and fewer cells were found in the G1 phase (52.18%) (, H). These results indicate that Tudor-SN promotes cell-cycle progression and cellular proliferation of cardiomyocytes.
Figure 6. Tudor-SN participates in the mTORC1-mediated regulation of cardiomyocytic proliferation. (A) The Tudor-SN gene was knocked out by a modified CRISPR/Cas9 double-nicking gene-editing system. The protein levels of Tudor-SN in H9c2 WT cells and H9c2 Tudor-SN KO cells were detected by Western blotting. (B) The proliferative abilities of H9c2 WT and H9c2 Tudor-SN KO cells were detected by CCK8 assays. Data are presented as the mean ± SEM (n = 3 independent experiments), and unpaired two-tail t-tests were used (***p < 0.001). (C–D) The cell-cycle distributions of H9c2 WT and Tudor-SN KO cells were detected by flow cytometry and analysed by Modfit software. Data are presented as the mean ± SEM (n = 3 independent experiments), and unpaired two-tail t-tests were used (*p < 0.05, ns, no significance). (E) Tudor-SN was overexpressed in H9c2 cells (Tudor-SNOE), Vector was used as a control, and the protein expression of Tudor-SN was detected by Western blotting. (F) The proliferative abilities of H9c2 Vector and Tudor-SNOE cells were detected by CCK8 assays. Data are presented as the mean ± SEM (n = 3 independent experiments), and unpaired two-tail t-tests were used (*p < 0.05). (G–H) The cell-cycle distributions of H9c2 Vector and Tudor-SNOE cells were detected by flow cytometry and analysed by Modfit software, Data are presented as the mean ± SEM (n = 3 independent experiments), and unpaired two-tail t-tests were used (*p < 0.05, ns, no significance). (I–K) H9c2 Vector and Tudor-SNOE cells were treated with 250 nmol/L of Torin 1 for 24 h. The protein levels of Tudor-SN and other indicated proteins of the mTORC1 pathway were detected by Western blotting. Cell-cycle distributions were detected by flow cytometry and analysed by Modfit software. Data are presented as the mean ± SEM (n = 3 independent experiments), and unpaired two-tail t-tests were used (*p < 0.05, ns, no significance). (L) H9c2 Vector and Tudor-SNOE cells (1 × 104 cells) were seeded in 12-well plates and treated with 250 nmol/L Torin 1 for 24 h, the number of cells was counted and anzlyzed. Data are presented as the mean ± SEM (n = 3 independent experiments), and unpaired two-tail t-tests were used (*p < 0.05, ns, no significance)
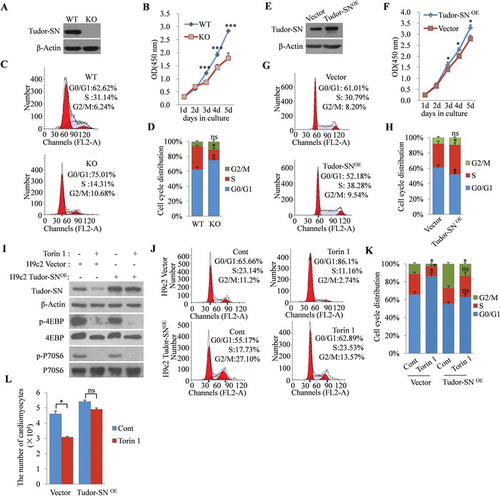
The mTORC1 pathway also regulates cardiomyocytic proliferation [Citation25]. The above results showed that Tudor-SN expression correlated with myocardia proliferative ability and mTORC1 activity. To verify whether Tudor-SN is involved in mTORC1-mediated regulation of cardiomyocytic proliferation, we inhibited mTORC1 in H9c2 Tudor-SNOE cells and H9c2 Vector cells by treating the cells with Torin 1 for 24 h and then detected cell-cycle distributions. Western blotting showed that the phosphorylation levels of 4EBP (, panel 3) and P70S6 (, panel 5) dramatically reduced, which indicated inactivation of mTORC1 (). Because only the CDS sequence of Tudor-SN was cloned into the pLVX-IRES-Puro Vector, the pLVX-IRES-Puro-Tudor-SN-Flag plasmid did not have the 5ʹ TOP sequence, and correspondingly the overexpression of Tudor-SN in H9c2 Tudor-SNOE cells was not affected by mTORC1 inhibition (, panel 1). Flow cytometry showed that mTORC1 inhibition retained more H9c2 Vector cells in the G1 phase (Cont: 65.66% to Torin 1: 86.1%), while Tudor-SN overexpression reduced G1-phase arrest, which was induced by mTORC1 inhibition (Cont: 55.17% to Torin 1: 62.89%) (, K). We also counted the H9c2 Tudor-SNOE cells and H9c2 Vector cells after treating these cells with Torin 1. As shown in , mTORC1 inhibition reduced the number of H9c2 Vector cells but had no significant influence on H9c2 Tudor-SNOE cells. These results demonstrate that Tudor-SN participates in mTORC1-mediated regulation of cardiomyocytic proliferation.
Discussion
The transition of cardiomyocytes from rapid neonatal proliferation to postnatal cell-cycle arrest is accompanied by downregulation of many cell-cycle regulators [Citation11,Citation12]. Identification of cardiomyocytic cell-cycle regulators may be helpful for revealing the mechanisms and for providing novel targets to promoting adult cardiomyocytic regeneration. In the present study, we observed a correlation of Tudor-SN expression with proliferative capacity of cardiomyocytes. Furthermore, we revealed the regulatory mechanisms of Tudor-SN in cardiomyocytes. The TOP motif is a well-known cis-regulatory motif for translational control and is located immediately downstream of transcriptional start sites [Citation20]. The translational efficiency of the TOP mRNA is exquisitely dependent on cellular proliferative ability [Citation26] and is controlled by the mTORC1 pathway [Citation22]. In the present study, we discovered that Tudor-SN is a novel TOP mRNA, as demonstrated by the following evidence: (1) Tudor-SN mRNA was found to have a cis-regulatory motif composed of a six-oligo-pyrimidine-long tract initiated with cytosine at the 5ʹ end of mRNA; (2) Tudor-SN was highly expressed in rapidly proliferating myocardia and in primary cardiomyocytes of neonatal mice, while it was dramatically decreased in adult myocardia and in Torin-1-treated primary neonatal mouse cardiomyocytes that had exited the cell cycle; and (3) the expression of Tudor-SN was correlated with the activation pattern of mTORC1. Taken together, these findings demonstrate that downregulation of Tudor-SN in adult mouse myocardia is regulated by mTORC1 at the translational level.
The mTOR pathway plays a central role in regulating many fundamental cellular processes [Citation27]. It exerts different cellular functions through forming two different multi-protein complexes, mTORC1 and mTORC2 [Citation27]. The mTORC1 pathway – which regulates protein translation, cellular proliferation, cellular metabolism, and autophagy – is essential for cardiac development in embryos and during neonatal periods [Citation28]. It not only promotes cardiomyocytic proliferation, but also controls cardiomyocytic growth [Citation25]. To ensure that heart size is in proportion to that of the rest of the body, both the proliferation and growth of cardiomyocytes gradually cease during cardiac development [Citation29,Citation30]. Here, we revealed that most upstream regulators of the mTORC1 pathway were downregulated in adult myocardia, and that the phosphorylation levels of 4EBP and P70S6 – which are downstream targets of mTORC1 – were dramatically reduced in adult myocardia. These results demonstrate that the activity of the mTORC1 pathway is gradually decreased during cardiac development to control cardiac size. Furthermore, increased mTORC1 activity is necessary for cardiac adaptation to pressure overload and development of compensatory hypertrophy [Citation25]. In the present study, we observed that Tudor-SN was highly expressed in neonatal mouse myocardia but was gradually decreased during cardiac development, which was strongly correlated with the activation pattern of mTORC1. We also found that the Tudor-SN expression was significantly reduced following inhibition of the mTORC1 pathway, and was enhanced when the mTORC1 pathway was activated in H9c2 cell lines. Moreover, we observed that upregulation of Tudor-SN was induced following mTORC1 activation in a heart compensatory-hypertrophy model. These findings confirm that the expression of Tudor-SN in myocardia is controlled by the mTORC1 pathway, suggesting that Tudor-SN may participate in mTORC1-mediated regulation of cardiac development.
Previous studies have revealed that Tudor-SN accelerates cell-cycle progression through promoting the G1/S-phase transition [Citation5]. The coincidence of Tudor-SN expression with myocardial proliferation and mTORC1 activity during cardiac development prompted us to investigate whether Tudor-SN participated in the regulation of cardiomyocytic proliferation. We revealed that Tudor-SN promoted the proliferation of H9c2 cells. Moreover, Tudor-SN influenced mTORC1-mediated regulation of H9c2 proliferation, since overexpression of Tudor-SN weakened the H9c2 cell-cycle arrest which induced by mTORC1 inhibition. Cardiomyocytic proliferative levels are high during the first postnatal week and then gradually decrease and are undetected after postnatal day 14 (P14) [Citation31]. The inactivation of the mTORC1 pathway and the downregulation of Tudor-SN during cardiac development may be involved in the cardiomyocytic cell-cycle arrest. Whether Tudor-SN participates in the regulation of neonatal cardiomyocytic proliferation and/or drives adult cardiomyocytic regeneration after myocardial injury requires further investigation.
In summary, our present findings provide a novel insight into the regulatory mechanisms of Tudor-SN expression. We found that Tudor-SN, as a novel TOP mRNA, was regulated by the mTORC1 pathway at the translational level in cardiomyocytes. Furthermore, we identified Tudor-SN as a novel regulator of cardiomyocytic proliferation, which may be helpful for better understanding the underlying mechanisms of cardiomyocytic cell-cycle arrest and providing novel potential target for reactivating the adult cardiomyocytic cell cycle.
Disclosure of potential conflicts of interest
The authors report no conflicts of interest.
Supplemental Material
Download MS Word (823 KB)Acknowledgments
We gratefully thank Dr. L. Shi (Tianjin Medical University, China) for providing the lentivirus plasmids.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
References
- Yang J, Aittomaki S, Pesu M, et al. Identification of p100 as a coactivator for STAT6 that bridges STAT6 with RNA polymerase II. Embo J. 2002;21:4950–4958.
- Yang J, Valineva T, Hong J, et al. Transcriptional co-activator protein p100 interacts with snRNP proteins and facilitates the assembly of the spliceosome. Nucleic Acids Res. 2007;35:4485–4494.
- Fu X, Zhang C, Meng H, et al. Oncoprotein Tudor-SN is a key determinant providing survival advantage under DNA damaging stress. Cell Death Differ. 2018;25:1625–1637.
- Su C, Gao X, Yang W, et al. Phosphorylation of Tudor-SN, a novel substrate of JNK, is involved in the efficient recruitment of Tudor-SN into stress granules. Biochim Biophys Acta Mol Cell Res. 2017;1864:562–571.
- Su C, Zhang C, Tecle A, et al. Tudor staphylococcal nuclease (Tudor-SN), a novel regulator facilitating G1/S phase transition, acting as a co-activator of E2F-1 in cell cycle regulation. J Biol Chem. 2015;290:7208–7220.
- Armengol S, Arretxe E, Rodriguez L, et al. NF-kappaB, Sp1 and NF-Y as transcriptional regulators of human SND1 gene. Biochimie. 2013;95:735–742.
- Porrello ER, Mahmoud AI, Simpson E, et al. Transient regenerative potential of the neonatal mouse heart. Science. 2011;331:1078–1080.
- Walsh S, Ponten A, Fleischmann BK, et al. Cardiomyocyte cell cycle control and growth estimation in vivo–an analysis based on cardiomyocyte nuclei. Cardiovasc Res. 2010;86:365–373.
- Bergmann O, Bhardwaj RD, Bernard S, et al. Evidence for cardiomyocyte renewal in humans. Science. 2009;324:98–102.
- Ali SR, Hippenmeyer S, Saadat LV, et al. Existing cardiomyocytes generate cardiomyocytes at a low rate after birth in mice. Proc Natl Acad Sci U S A. 2014;111:8850–8855.
- Senyo SE, Steinhauser ML, Pizzimenti CL, et al. Mammalian heart renewal by pre-existing cardiomyocytes. Nature. 2013;493:433–436.
- Ahuja P, Sdek P, MacLellan WR. Cardiac myocyte cell cycle control in development, disease, and regeneration. Physiol Rev. 2007;87:521–544.
- Zhou Q, Li L, Zhao B, et al. The hippo pathway in heart development, regeneration, and diseases. Circ Res. 2015;116:1431–1447.
- Mahmoud AI, Kocabas F, Muralidhar SA, et al. Meis1 regulates postnatal cardiomyocyte cell cycle arrest. Nature. 2013;497:249–253.
- Mohamed TMA, Ang YS, Radzinsky E, et al. Regulation of cell cycle to stimulate adult cardiomyocyte proliferation and cardiac regeneration. Cell. 2018;173:104–116 e112.
- Lin Z, von Gise A, Zhou P, et al. Cardiac-specific YAP activation improves cardiac function and survival in an experimental murine MI model. Circ Res. 2014;115:354–363.
- Vagnozzi RJ, Molkentin JD, Houser SR. New myocyte formation in the adult heart: endogenous sources and therapeutic implications. Circ Res. 2018;123:159–176.
- Ran FA, Hsu PD, Lin CY, et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell. 2013;154:1380–1389.
- Gao X, Fu X, Song J, et al. Poly(A)(+) mRNA-binding protein Tudor-SN regulates stress granules aggregation dynamics. Febs J. 2015;282:874–890.
- Yamashita R, Suzuki Y, Takeuchi N, et al. Comprehensive detection of human terminal oligo-pyrimidine (TOP) genes and analysis of their characteristics. Nucleic Acids Res. 2008;36:3707–3715.
- Suzuki A, Wakaguri H, Yamashita R, et al. DBTSS as an integrative platform for transcriptome, epigenome and genome sequence variation data. Nucleic Acids Res. 2015;43:D87–91.
- Thoreen CC, Chantranupong L, Keys HR, et al. A unifying model for mTORC1-mediated regulation of mRNA translation. Nature. 2012;485:109–113.
- Nandagopal N, Roux PP. Regulation of global and specific mRNA translation by the mTOR signaling pathway. Translation. 2015;3:e983402.
- Wang R, Su C, Wang X, et al. Global gene expression analysis combined with a genomics approach for the identification of signal transduction networks involved in postnatal mouse myocardial proliferation and development. Int J Mol Med. 2018;41:311–321.
- Zhang D, Contu R, Latronico MV, et al. MTORC1 regulates cardiac function and myocyte survival through 4E-BP1 inhibition in mice. J Clin Invest. 2010;120:2805–2816.
- Hamilton TL, Stoneley M, Spriggs KA, et al. TOPs and their regulation. Biochem Soc Trans. 2006;34:12–16.
- Saxton RA, Sabatini DM. mTOR signaling in growth, metabolism, and disease. Cell. 2017;169:361–371.
- Sciarretta S, Volpe M, Sadoshima J. Mammalian target of rapamycin signaling in cardiac physiology and disease. Circ Res. 2014;114:549–564.
- Heallen TR, Kadow ZA, Wang J, et al. Determinants of cardiac growth and size. Cold Spring Harb Perspect Biol. 2020;12:a037150.
- Sedmera D, Thompson RP. Myocyte proliferation in the developing heart. Dev Dyn. 2011;240:1322–1334.
- Takeuchi T. Regulation of cardiomyocyte proliferation during development and regeneration. Dev Growth Differ. 2014;56:402–409.