
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
Bacterial small-RNA (sRNA) sequences are functional RNAs, which play an important role in regulating the expression of a diverse class of genes. It is thus critical to identify such sRNA sequences and their probable mRNA targets. Here, we discuss new procedures to identify and characterize sRNA and their targets via the introduction of an integrated online platform ‘PresRAT’. PresRAT uses the primary and secondary structural attributes of sRNA sequences to predict sRNA from a given sequence or bacterial genome. PresRAT also finds probable target mRNAs of sRNA sequences from a given bacterial chromosome and further concentrates on the identification of the probable sRNA-mRNA binding regions. Using PresRAT, we have identified a total of 66,209 potential sRNA sequences from 292 bacterial genomes and 2247 potential targets from 13 bacterial genomes. We have also implemented a protocol to build and refine 3D models of sRNA and sRNA-mRNA duplex regions and generated 3D models of 50 known sRNAs and 81 sRNA-mRNA duplexes using this platform. Along with the server part, PresRAT also contains a database section, which enlists the predicted sRNA sequences, sRNA targets, and their corresponding 3D models with structural dynamics information.
Introduction
Small-RNA (sRNA) based regulation of mRNA sequences play a central role in various bacterial gene expressions in response to environmental changes [Citation1,Citation2]. These sRNA sequences are extensively studied in context with different bacterial regulatory processes such as quorum sensing [Citation3], the formation of biofilms [Citation4], expression of diverse virulence factors [Citation5], iron uptake [Citation6], etc. In the prokaryotic system, these functional RNA regulators exist as small transcripts about 30–600 nucleotides long and modulate the translation and stability of mRNAs by binding with them [Citation7]. Binding regions of these sRNA sequences with their cognate mRNAs are not fixed. Some sRNA sequences primarily bind at the ribosome binding site (RBS) of their target mRNA sequences [Citation1] while other sRNA sequences modulate translation of target genes by attaching at distant locations [Citation1,Citation7]. The antisense mechanisms exhibited by the sRNA sequences to modulate their targets are the key to understand the function of these RNA regulators [Citation1,Citation2,Citation7]. The significance of these sRNA sequences and their targeted genes in bacteria has driven the research community to identify and more of these sRNA sequences and their targets concerning various biological functions. Different experimental methods such as RNA-seq, tiling arrays, sRNA pulse expression, immunoprecipitation are available to detect sRNA sequences and their targets. Still, they are tedious, expensive, and the outputs of these experiments also need exhaustive filtering [Citation8–10]. Hence, computational approaches can augment the experimental methods [Citation11] in the detection of sRNA sequences and their targets.
Comparative genomics are a standard computational practice to find sRNA sequences within a bacterial genome [Citation12]. QRNA [Citation13] and Intergenic Sequence Inspector [Citation14] programs implemented a comparative genomics approach to find sRNAs. Programs like RNAz [Citation15] and sRNApredict [Citation16] use the conserved RNA secondary structure information to calculate the thermodynamic energy for sRNA prediction. GLASSgo [Citation17] combined an iterative BLAST strategy with pairwise identity filtering and structure-based clustering to find sRNA homologs from scratch. Some of the programs also utilize the promoter and terminator sequence signal annotations for sRNA prediction. sRNAscanner [Citation18] applies a position weighted matrix of sRNA specific transcriptional signals, and sRNAfinder [Citation19] utilizes a generalized probabilistic model that integrates primary sequence data, transcript expression data, microarray experiments, and conserved RNA structure information to predict sRNA genes. Diverse sets of methods also utilize different machine learning approaches [Citation20,Citation21], antisense RNA transcript expression profiling [Citation22], and motif-based discovery [Citation23] algorithms to predict sRNA sequences.
Identification of sRNA target genes is also important and represents a challenging task. Experimental procedures, including genetic screens [Citation24], knockouts studies [Citation25], microarray analysis [Citation26], and other systematic proteomic investigations [Citation27], are implemented to identify sRNA target genes. However, these methods are expensive, time-consuming, and extremely tedious. Therefore, the development of sensitive and accurate computational approaches would aid and complement the experimental analysis. The first step in sRNA target prediction is to search for a complementary region between the sRNA and its target mRNA sequence. Comparing to microRNA and its target mRNA interaction, which requires a generally conserved 6–8 nucleotide long seed region for initiation of binding, the sRNA-mRNA binding region is relatively more extended and more evolutionary flexible [Citation28]. Non-Watson-Crick base-pairing plays a key role in sRNA-mRNA target interaction and programs such as TargetRNA [Citation26], GUUGle [Citation29] utilizes both Watson-Crick and non-Watson-Crick base pairing models to find sRNA targets and their interacting regions. RNAcofold [Citation30] computes the hybridization energy and base-pairing arrangement between a pair of RNA molecules, while RNAplex [Citation31] calculates the same hybridization energy using modified thermodynamic energy models. RNAup [Citation32] utilizes the thermodynamics of RNA-RNA interactions and the hybridization energy, while IntaRNA [Citation33] incorporates binding site accessibility information for searching the best interaction with minimum extended hybridization energy. SPOT [Citation34] incorporated existing computational tools to search for sRNA binding sites and utilizes experimental data for filtering.
Despite the availability of various sRNA and its target prediction algorithms, the sensitivity remains quite low (20–49%) [Citation11]. Hence, it is fair to assume that many more sRNA sequences and their target genes remain undiscovered, particularly in less studied prokaryotic organisms. Here, in this article, we introduce an integrated platform with an alternative approach to identify bacterial small-RNA and their target genes from a given nucleotide sequence and a whole bacterial chromosome. This online platform, PresRAT (Prediction of sRNA and their Targets) integrates two separate but related procedures of sRNA and their target identification into a simple and effective single dais.
Apart from identifying the sRNA sequences and their targets, a key feature of this platform is to provide sRNA and sRNA-mRNA duplex structures generated via three dimensional (3D) modelling and further refined through molecular dynamics simulation. Predicting the accurate 3D structure of RNA from the primary sequence is still a fundamental and challenging problem in computational biology [Citation35]. There are few notable programs such as iFoldRNA [Citation36], FARNA/FARFAR [Citation37,Citation38], MC-Sym [Citation39], MANIP [Citation40], 3DRNA [Citation41], RNA2D3D [Citation42], NAST [Citation43], and ASSEMBLE [Citation44] try to tackle the existing bottlenecks of RNA structure prediction and improve the algorithms for RNA 3D structure generation. As per the latest RCSB protein databank [Citation45] statistics, there are 1528 experimentally solved 3D structures of RNA representing only ~0.01% of the whole deposited structures. However, there are no experimentally solved 3D structures available for sRNA only apart from a few conjugated with chaperone proteins [Citation46–50]. PresRAT uses the RNA2D3D [Citation42] program to generate an initial RNA model followed by extensive refinement of the structure using GROMACS v4.6.3 molecular dynamics simulation package [Citation51].
The validation and benchmarking results of the PresRAT for sRNA, sRNA-target, and sRNA-mRNA binding regions suggest improvements compared to those achieved by other available programs. In this current study, we have studied 292 bacterial genomes to identify a total of 21,286 and 44,923 sRNA sequences from genic and non-genic regions of the respective genomes. We also have identified 2447 potential targets for 50 unique sRNA sequences from 13 bacterial genomes. Further, we have shown the structural details of 50 sRNA and 81 sRNA-mRNA duplex 3D models through molecular dynamics simulation studies. All these procedures and information are automated and integrated into the PresRAT platform freely available at http://www.hpppi.iicb.res.in/presrat.
Results
PresRAT: the webserver interfaces and available programs
PresRAT webserver and the database sections are built using CGI-Perl scripts. Integrated CGI-Perl scripts allow the users to utilize this webserver efficiently. The webserver is freely available at http://www.hpppi.iicb.res.in/presrat. The PresRAT webserver section is divided into four parts to accomplish different tasks. The first two parts are called PredsRNA and PredGsRNA. PredsRNA interface can be used to identify sRNA sequences one at a time while the PredGsRNA interface directs the user to identify potential sRNA sequences from different bacterial sequences. The next two parts of the PresRAT webserver compromises the sRNA target identification protocols. The PredTar Interface helps to identify interacting nucleotides between a pair of sRNA and mRNA sequence, and PredWG performs the function of sRNA target identification from a given bacterial genome. The following subsections briefly describe the methodology and the results of each of these parts of the PresRAT server.
PredsRNA: prediction of sRNA from its sequence and secondary structural attributes
PredsRNA uses sequence and secondary structural information of existing sRNA and non-sRNA sequences to calculate a combined score to predict novel sRNA sequences. In sequence analysis, a directional (5ʹ->3ʹ) dinucleotide Sequencescore is first calculated for the input nucleotide sequence using Log Odds (LOD) ratio matrices (Please refer the detailed sRNA prediction methodology in Supplementary File 1 and Figure S1). We also calculate a U-richness score that reflects the presence of uracil (U) nucleotides at the 3ʹ end of the sRNA sequence. PresRAT effectively uses the secondary structure information by first calculating a set of local minima conformations of the provided RNA sequence, followed by estimating the two different scores, namely ABEscore and ALEscore. ABEscore and ALEscore indicate the Average Base Energy and Average Loop Energy of local minima secondary structure conformations. The final combined score i.e. sRNAscore, is calculated as follows:
The combined sRNAscore (details of the scoring schemes can be found in Supplementary File 1) is used as the final scoring scheme for filtering the true positive sRNA instances from the true negative ones. Equally weighted bacterial small-RNA (sRNA) feature set of Sequencescore, Urichscore, ABEscore, and ALEscore described in the Supplementary File 1 offers a simple but powerful discriminating scoring scheme to filter out sRNA sequences from non-sRNA sequences. All the individual feature sets can differentiate an sRNA from a non-sRNA sequence, as demonstrated in Figure S2.
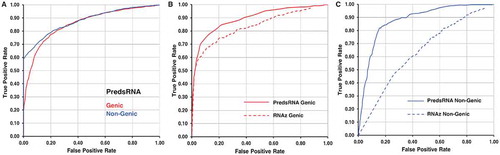
For benchmarking and validation of sRNA prediction by PresRAT, a set of non-redundant 1174 sRNA (Additional file1), 5870 genic, and 5870 non-genic non-sRNA sequences were randomly collected from 54 different bacterial species (Additional file 2). Receiver operating characteristic (ROC) statistics were calculated by comparing the sRNA to non-sRNA sRNAscore in a 1:5 ratio. The ROC statistics generated using the sRNAscore distribution demonstrate sensitivity values of 27.7%, 49.6%, 65.0% and 77.5% at 1.0% 5.0%, 10.0%, and 20.0% error rates, against non-sRNA sequences collected from genic regions whereas sensitivities of 60.3%, 64.6%, 70.1%, and 78.7% were obtained at 1.0% 5.0%, 10.0%, and 20.0% error rates against non-sRNA sequences collected from non-genic parts of the bacterial chromosomes ( and Additional file 3), respectively. A comparative study against RNAz [Citation15] program with PredsRNA also showed better performance in predicting sRNA sequences as compared to genic non-sRNA sequences (). However, it is also important to mention that RNAz requires evolutionary information to predict functional RNA sequences, and thus it is restricted only to homologous RNA sequences in bacterial chromosomes. Therefore, the final comparison was performed on a limited set of commonly predicted sRNA (334 sequences), genic (1670), and non-genic (1670) non-sRNA sequences by the two programs.
Figure 1. Receiver operating characteristic curve for sRNA prediction. Panel A shows the ROC curve for sRNA prediction performance by the PredsRNA program in genic and non-genic category. A total of 1174 sRNA, 5870 genic, and 5870 non-genic non-sRNA sequences were considered to measure the performance. Panel B and C shows the performance of sRNA prediction by PredsRNA and RNAz programs in genic and non-genic categories, respectively. A total of common 334 sRNA, 1670 genic, and 1670 non-genic non-sRNA sequences were considered to measure the performance
PredsRNA page accepts query nucleotide sequence and based on its sRNAscore and predicts whether the given sequence is likely to be sRNA or not above a user-defined confidence level. PredsRNA result page provides a minimum free energy secondary structure of the probable sRNA sequence. PredsRNA also provides a useful option of building the three-dimensional (3D) structural model of the predicted sRNA sequence via the PredsRNA3D module, which generates 3D sRNA models using RNA2D3D [Citation42] and RNAfold [Citation52Citation53Citation54] programs, respectively. Following the generation of a 3D sRNA structure, a molecular dynamics (MD) simulation is performed on the RNA structure using the GROMACS [Citation51] MD simulation package (details of the structural analysis protocol can be found at Supplementary File 1). The initial and final structures are provided from the MD simulation trajectory file along with other necessary information.
PredGsRNA: prediction of sRNA sequences within the bacterial chromosome
PredGsRNA aims to predict potential sRNA sequences within a bacterial genome. PredGsRNA applies a heuristic approach to find potential sRNA sequences from the bacterial genome. Briefly, PredGsRNA first extracts the 300 nucleotides starting 5ʹ and ending 3ʹ regions of both the genic and non-genic sequences (Figure S3 in Supplementary File 1), followed by a filtering step to exclude sequences containing an exclusive set of 8mer nucleotide sequences (Figure S4 in Supplementary File 1). The retained sequences are then subjected to sRNAscorecalculation. The result page of PredGsRNA contains the sRNA sequences predicted from the genic sequences and non-genic sequences of the bacterial genome, respectively. The corresponding result page of this program consists of the top 100 predicted sRNA sequences along with their start and end loci, sRNAscore, and p-values. Similarly, a pictorial representation of the predicted sRNA sequences on a genome browser view is also provided for better visualization (please refer to the Supplementary File 1 for details of this approach).
PredGsRNA is used to predict new probable sRNA sequences from the 292 bacterial genomes (Additional file 4) to identify a total of 21,286 and 44,923 hypothetical sRNA sequences from genic and non-genic parts of the bacterial genomes (Additional file 5). sRNAscanner [Citation18], a separate program for bacterial chromosomal sRNA detection, is used for comparative analysis. sRNAscanner uses a transcriptional signal based computational method for sRNA detection and detected only 114 unique sRNA sequences. In comparison, our protocol identified a total of 219 sRNA sequences out of 1174 known sRNA sequences (Additional file 6 and Figure S5). Hence, PredGsRNA demonstrates ~2 fold higher sensitivity over existing program in identification of known sRNA sequences from the whole bacterial genome. However, PredGsRNA is a greedy and heuristic approach to search sRNA sequences from bacterial genomes. The method is restricted to search the sRNA sequences within the 300bps of genic and non-genic feature. The users are encouraged to use other methods like sRNAscanner to complement sRNA sequence searching from a bacterial genome.
PredTAR: prediction of sRNA-mRNA target binding regions
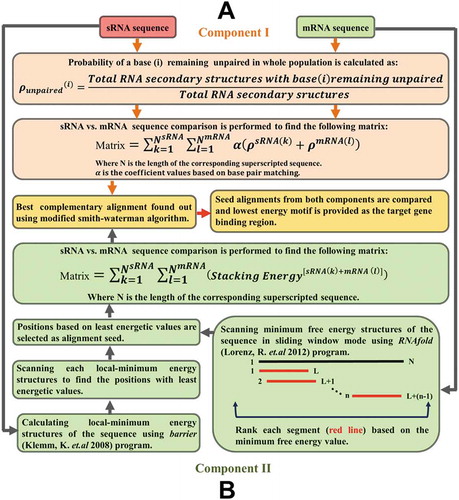
PredTAR aims to predict potential binding regions between bacterial sRNA and their target mRNA binding regions. In short, PredTAR combines two different approaches to find the sRNA-mRNA target binding region. The first approach (Component I) uses the base un-pairing probability of both the sRNA and its target mRNA sequence (). The second approach (Component II) () utilizes the local minima energy structures of sRNA and the minimum free energy structure of the target mRNA to find the potential binding region (Please refer to Supplementary File 1 and Figure S6 for details). The webserver output page provides the alignment of the given sRNA and mRNA sequences along with scores and duplex energy values.
Figure 2. Prediction of sRNA targets. Panel A demonstrates the sRNA-mRNA target binding region identification protocol (Component I) of PredTAR. The main aim of this protocol is to identify the un-pairing probability of each base from both sRNA and mRNA sequences and utilize this probability information to identify the binding region. Panel B describes the Component II protocol used by PresRAT to identify the sRNA-mRNA target binding region. In this protocol, the energetic value of both sRNA and mRNA secondary structures are taken into account to identify binding regions
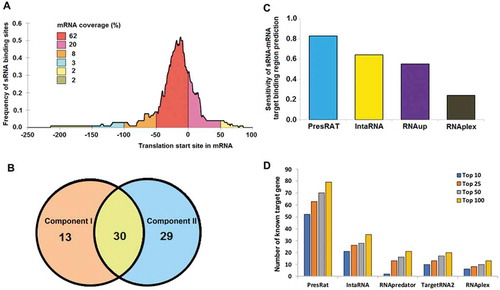
Experimentally known binding regions in 88 out of 91 sRNA and their respective target mRNA sequences (Additional file 7) were used to validate the Component I and II algorithms. The selection of these 88 binding regions was made based on the frequency distribution of the sRNA binding site on mRNA sequences relative to its translation initiation site (). The frequency distribution of the 91 sRNA binding sites on mRNA sequences reveals that in 97% of the cases, the sRNA binding site is located within the −250 to +100 window region of a target mRNA (‘0’ being the translation start site). The Component I and Component II could correctly identify the binding region for 49% and 67% of the true sRNA-mRNA pair within the window mentioned above. The Venn diagram in shows the overlap of correctly predicted sRNA-mRNA binding regions by the two algorithms. A 74% (65 out of 88) sensitivity was achieved when a region with the lowest free energy of sRNA-mRNA binding by either of the algorithms was considered, while 82% (72 out of 88) sensitivity () was achieved when both the common and uniquely predicted binding regions were considered. A comparative analysis () with other published methods (RNAplex [Citation31], RNAup [Citation32], and IntaRNA [Citation33], respectively) shows a much better performance of PredTAR in predicting sRNA-mRNA targets (a complete list of predicted sRNA-mRNA binding regions is provided in Additional file 8).
Figure 3. sRNA target finding performance. Panel A shows the frequency distribution of sRNA binding sites in the 91 mRNA sequences. In 98% of the cases, it is found that the sRNA binding site is present within the window of −250 to +100 nucleotides (0 being the translation initiation site) of the target gene. Panel B shows the overlap of correctly predicted target by the PresRAT Component I and II protocols, respectively. Panel C shows the PresRAT sensitivity in predicting the correct sRNA-mRNA target binding regions in 88 samples compared to other programs. Panel D shows the number of true sRNA target gene identification through whole-genome search by PresRAT compared to other existing programs
PredTAR also provides a useful option of building the three-dimensional structural (3D) model of the sRNA-mRNA bound duplex region via the Preddup3D module, which generates the duplex model using RNA2D3D [Citation42] program for the secondary structure information followed by molecular dynamics (MD) simulation. Details of the structural analysis can be found in Supplementary File 1. The initial and final structures are provided from the MD simulation trajectory file along with other necessary information.
PredWG: prediction of sRNA target genes and binding regions within a bacterial genome
PredWG aims to predict potential bacterial sRNA targets within a bacterial genome. The details about this approach are documented in Supplementary File 1. The result page of PredWG contains the list of the predicted target genes along with individual alignment and the corresponding duplex energy value of the binding region. Similarly, a pictorial representation of the predicted sRNA sequences on a genome browser view is also provided for better visualization.
A performance evaluation using 91 known sRNA-mRNA target pairs, PredWG achieved 86% sensitivity in predicting the correct target genes (within the top 100 predictions/genome) within bacterial genomes. The other competing programs, like IntaRNA [Citation33], RNAPredator [Citation55], TargetRNA2 [Citation56], and RNAplex [Citation31], achieved sensitivity values of 38%, 23%, 22%, and 14%, respectively () (complete list of targets found by the various programs are provided in Additional file 9). For a fair comparison, a similar search window (−250 to +100 of mRNA translation start site) length was maintained for all the programs. Within the top 100 predictions for individual sRNA sequences, PredWG predicted 2447 novel mRNA targets with a similar binding energy distribution profile (p values < 0.05) (Figure S7) as that of the known 91 sRNA-mRNA interactions. The complete list of the identified target mRNAs for 50 sRNA sequences along with their corresponding functions, is provided in the Additional file 10. We also performed a Gene Ontology (GO) [Citation57] based molecular functional network of E. coli known sRNA sequences (18 sequences) with their known (27 unique targets) and predicted mRNA target sequences (192) (Figure S8A). The molecular functional network suggests the involvement of functions such as binding, catalysis, and transporter activities. Additionally, a smaller subset of the predicted target genes also offers unique molecular functions like electron transport, guanyl-nucleotide exchange, and other regulator activities (Figure S8B).
PresRAT: the database interface
PresRAT database section enlists the predicted sRNA sequences, sRNA targets, and the corresponding 3D models of selected sRNAs and sRNA-mRNA duplex structures. Predicted sRNA sequences from 292 bacterial genomes are accessible to users from a drop-down selection menu provided on the database page. Similarly, the list of predicted target genes for a given sRNA within a bacterial genome is readily accessible in a similar manner. The database also harbours mRNA targets of 50 known sRNA sequences from 13 bacterial genomes along with their details.
Exhaustive structural analysis was performed to predict the 3D structures of the known sRNA sequences and their corresponding binding regions with the target genes. Three-dimensional modelling of sRNA sequences and the target binding regions using their current secondary structure information provides the opportunity to gain structural insight, which in turn may become quite valuable in RNA based therapeutics development. There are 50 sRNA models in the PresRAT database. All the 50 sRNA and 81 unique sRNA-mRNA bound duplex models are explicitly refined using the GROMACS package through one-nanosecond of molecular simulation runs. The final models with other structural details are available to users in the database section. Further methodical details and validation results can be found in Supplementary File 1.
Discussion
Here, we introduce a web-based platform PresRAT to identify and analyse bacterial small-RNAs using both individual and genomic sequence level searches. The platform integrates a combination of scoring schemes developed based on primary and secondary structural attributes of small-RNAs. Using PresRAT, we have scanned a total of 292 bacterial genomes to identify 21,286 and 44,923 potential sRNA sequences. One of the key features of this platform is the generation of sRNA three-dimensional models and their dynamic analysis. For in-depth structural analysis, the availability of energy minimized and refined three-dimensional models of sRNAs are essential. PresRAT provides an option for automated model building and molecular dynamics simulation of sRNA in the presence of water. Using this automated procedure, we have successfully built 50 different small-RNA structures and analysed some of their basic properties calculated from their dynamics data (Figure S9 in Supplementary File 1). Comparison of our server derived model of DsrA and the NMR structure of the SL1 of the DsrA (pdb code: 5WQ1) [Citation46] shows high similarities (RMSD: 2.75 Å) between the structures, indicating reliability of our 3D model (Figure S10 in Supplementary File 1). However, the NMR structure covers only a short fraction of the DsrA sequence.
A set of 81 experimentally known sRNA-mRNA duplex binding regions was modelled using the Preddup3D module and the resultant models were further stabilized via MD simulations (Figure S11 in Supplementary File 1). We also investigated the influence of Hfq on four different sRNA sequences using this platform (please refer to the Supplementary File 1 and Figure S12 for more details). In corroboration with previous experimental results, our molecular dynamics simulations demonstrated a major influence of Hfq binding in the regulation of the flexibility and conformations of the bound and unbound sRNA sequences. PresRAT enables a user to search for probable target mRNAs of sRNA sequences from a given bacterial chromosome and further concentrate on the identification of the probable mRNA target binding regions. For identification of target binding regions, PresRAT uses base un-pairing probability information of sRNA-mRNA sequences and local minima of sRNA secondary structures. Both the approaches combined with other additional steps provide a simple but effective way of identification of target mRNA sequences from the whole bacterial chromosomes. In this way, we have scanned 13 bacterial genomes and identified 2447 novel mRNA target sequences for existing 50 sRNA sequences. The identified target mRNAs for a given sRNA are provided in the database section of PresRAT along with their interacting nucleotide region and their related functional information. Existing model generation and molecular dynamics simulation procedure are extended to build the energy minimized and refined three-dimensional models of the sRNA-mRNA duplex regions, along with their structural characterization. Our study revealed that in general, these duplex regions are thermodynamically unfavourable, which indicates the involvement of other critical factors for the generation of stable sRNA-mRNA complexes.
Author contributions
A.C. collected and organized the data, developed the program, webserver and drafted the manuscript. K.K collected data, worked on the web server and drafted the manuscript. S. C. drafted the manuscript and coordinated the project. All authors read and approved the final manuscript.
Supplemental Material
Download Zip (5.1 MB)Acknowledgments
The Council of Scientific and Industrial Research (CSIR), Government of India supported the study through funding grant BSC0121. K.K. acknowledges Department of Biotechnology (DBT), Goverment of India for fellowship.
Disclosure statement
The author(s) declare no competing financial interests.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
References
- Storz G, Vogel J, Wassarman KM. Regulation by small RNAs in bacteria: expanding frontiers. Mol Cell. 2011;43(880–891). DOI:10.1016/j.molcel.2011.08.022
- Modi SR, Camacho DM, Kohanski MA, et al. Functional characterization of bacterial sRNAs using a network biology approach. Proc Natl Acad Sci U S A. 2011;108:15522–15527.
- Liu X, Zhou P, Wang R. Small RNA-mediated switch-like regulation in bacterial quorum sensing. IET Syst Biol. 2013;7(182–187). DOI:10.1049/iet-syb.2012.0059
- Mika F, Hengge R. Small regulatory RNAs in the control of motility and biofilm formation in E. coli and Salmonella. Int J Mol Sci. 2013;14:4560–4579.
- Geissmann T, Possedko M, Huntzinge E, et al. Regulatory RNAs as mediators of virulence gene expression in bacteria. Handb Exp Pharmacol. 2006;(173):9–43.
- Payne SM, Wickoff EE, Murphy ER et al. Iron and pathogenesis of Shigella: iron acquisition in the intracellular environment. Biometals. 2006;19(173–180). DOI:10.1007/s10534-005-4577-x
- Waters LS, Storz G. Regulatory RNAs in bacteria. Cell. 2009;136:615–628.
- Croucher NJ, Thomson NR. Studying bacterial transcriptomes using RNA-seq. Curr Opin Microbiol. 2010;13:619–624.
- Li W, Ying X, Lu Q, et al. Predicting sRNAs and their targets in bacteria. Genomics Proteomics Bioinformatics. 2012;10:276–284.
- Sharma CM, Vogel J. Experimental approaches for the discovery and characterization of regulatory small RNA. Curr Opin Microbiol. 2009;12:536–546.
- Lu X, Goodrich-Blair H, Tjaden B. Assessing computational tools for the discovery of small RNA genes in bacteria. RNA. 2011;17:1635–1647.
- Sridhar J, Gunasekaran P. Computational small RNA prediction in bacteria. Bioinform Biol Insights. 2013;7:83–95.
- Rivas E, Klein RJ, Jones TA, et al. Computational identification of noncoding RNAs in E. coli by comparative genomics. Curr Biol. 2001;11:1369–1373.
- Pichon C, Felden B. Intergenic sequence inspector: searching and identifying bacterial RNAs. Bioinformatics. 2003;19:1707–1709.
- Washietl S, Hofacker IL, Stadler PF. Fast and reliable prediction of noncoding RNAs. Proc Natl Acad Sci U S A. 2005;102:2454–2459.
- Livny J, Fogel MA, Davis BM, et al. sRNAPredict: an integrative computational approach to identify sRNAs in bacterial genomes. Nucleic Acids Res. 2005;33:4096–4105.
- Lott SC, Schäfer RA, Mann M, et al. GLASSgo - Automated and reliable detection of sRNA homologs from a single input sequences. Front Gen. 2018;9:124.
- Sridhar J, Narmada SR, Sabarinathan, et al. sRNAscanner: a computational tool for intergenic small RNA detection in bacterial genomes. PLoS One. 2010;5:e11970.
- Tjaden B. Prediction of small, noncoding RNAs in bacteria using heterogeneous data. J Math Biol. 2008;56:183–200.
- Carter RJ, Dubchak I, Holbrook SR. A computational approach to identify genes for functional RNAs in genomic sequences. Nucleic Acids Res. 2001;29:3928–3938.
- Saetrom P, Sneve R, Kristiansen KI, et al. Predicting non-coding RNA genes in Escherichia coli with boosted genetic programming. Nucleic Acids Res. 2005;33:3263–3270.
- Herbig A, Nieselt K. nocoRNAc: characterization of non-coding RNAs in prokaryotes. BMC Bioinformatics. 2011;12:40.
- Salari R, Aksay C, Karakoc E, et al. smyRNA: a novel Ab initio ncRNA gene finder. PLoS One. 2009;4:e5433.
- Mandin P. Genetic screens to identify bacterial sRNA regulators. Methods Mol Biol. 2012;905:41–60.
- Harris JF, Micheva-Viteva S, Li N, et al. Small RNA-mediated regulation of host-pathogen interactions. Virulence. 2013;4:785–795.
- Tjaden B, Goodwin SS, Opdyke JA, et al. Target prediction for small, noncoding RNAs in bacteria. Nucleic Acids Res. 2006;34:2791–2802.
- Ramachandran R, Stevens AM. Proteomic analysis of the quorum-sensing regulon in Pantoea stewartii and identification of direct targets of EsaR. Appl Environ Microbiol. 2013;79:6244–6252.
- Backofen R, Hess WR. Computational prediction of sRNAs and their targets in bacteria. RNA Biol. 2010;7:33–42.
- Gerlach W, Giegerich R. GUUGle: a utility for fast exact matching under RNA complementary rules including G-U base pairing. Bioinformatics. 2006;22:762–764.
- Bernhart SH, Tafer H, Mückstein U, et al. Partition function and base pairing probabilities of RNA heterodimers. Algorithms Mol Biol. 2006;1:3.
- Tafer H, Hofacker IL. RNAplex: a fast tool for RNA-RNA interaction search. Bioinformatics. 2008;24(2657–2663). DOI:10.1093/bioinformatics/btn193
- Muckstein U, Tafer H, Hackermuller J, et al. Thermodynamics of RNA-RNA binding. Bioinformatics. 2006;22:1177–1182.
- Busch A, Richter AS, Backofen R. IntaRNA: efficient prediction of bacterial sRNA targets incorporating target site accessibility and seed regions. Bioinformatics. 2008;24:2849–2856.
- King AM, Vanderpool CK, Degnan PH. sRNA target prediction organizing tool (SPOT) integrates computational and experimental data to facilitate functional characterization of bacterial small RNAs. mSphere. 2019;4:e00561–18.
- Sripakdeevong P, Beauchamp K, Das R. Why can’t we predict RNA structure at atomic resolution? Nuc Acids Mol Bio. 2011;27:43–65.
- Sharma S, Ding F, Dokholyan NV. iFoldRNA: three-dimensional RNA structure prediction and folding. Bioinformatics. 2008;24:1951–1952.
- Das R, Baker D. Automated de novo prediction of native-like RNA tertiary structures. Proc Natl Acad Sci U S A. 2007;104:14664–14669.
- Das R, Karanicolas J, Baker D. Atomic accuracy in predicting and designing noncanonical RNA structure. Nat Methods. 2010;7:291–294.
- Parisien M, Major F. The MC-Fold and MC-Sym pipeline infers RNA structure from sequence data. Nature. 2008;452:51–55.
- Massire C, Westhof E. MANIP: an interactive tool for modelling RNA. J Mol Graph Model. 1998;16(197–205):197–255.
- Zhao Y, Huang Y, Gong Z, et al. Automated and fast building of three-dimensional RNA structures. Sci Rep. 2012;2:734.
- Martinez HM, Maizel JV Jr., Shapiro BA. RNA2D3D: a program for generating, viewing, and comparing 3-dimensional models of RNA. J Biomol Struct Dyn. 2008;25:669–683.
- Jonikas MA, Radmer RJ, Laederach A, et al. Coarse-grained modeling of large RNA molecules with knowledge-based potentials and structural filters. RNA. 2009;15:189–199.
- Jossinet F, Ludwig TE, Westhof E. Assemble: an interactive graphical tool to analyze and build RNA architectures at the 2D and 3D levels. Bioinformatics. 2010;26(2057–2059). DOI:10.1093/bioinformatics/btq321
- Rose PW, Bi C, Bluhm WF, et al. The RCSB protein data bank: new resources for research and education. Nucleic Acids Res. 2013;41:D475–482.
- Morris ER, Hall G, Li C, et al. Structural rearrangement in an RsmA/CsrA ortholog of Pseudomonas aeruginosa creates a dimeric RNA-binding protein, RsmN. Structure. 2013;21:1659–1671.
- Sauer E. Structure and RNA-binding properties of the bacterial LSm protein Hfq. RNA Biol. 2013;10:610–618.
- Wang W, Wang L, Wu J, et al. Hfq-bridged ternary complex is important for translation activation of rpoS by DsrA. Nucleic Acids Res. 2013;41:5938–5948.
- Sauer E, Weichenrieder O. Structural basis for RNA 3ʹ-end recognition by Hfq. Proc Natl Acad Sci U S A. 2011;108:13065–13070.
- Link TM, Valentin-Hansen P, Brennan RG. Structure of Escherichia coli Hfq bound to polyriboadenylate RNA. Proc Natl Acad Sci U S A. 2009;106:19292–19297.
- Van Der Spoel D, Lindahl E, Hess B, et al. GROMACS: fast, flexible, and free. J Comput Chem. 2005;26:1701–1718.
- http://rna.tbi.univie.ac.at/cgi-bin/RNAWebSuite/RNAfold.cgi (Accessed on 25 March 2019).
- Lorenz R, Bernhart SH, Höner Zu Siederdissen C, et al. ViennaRNA Package 2.0. Algorithms Mol Biol. 2011;6:26.
- Turner DH, Mathews DH. NNDB: the nearest neighbor parameter database for predicting stability of nucleic acid secondary structure. Nucleic Acids Res. 2010;38:D280–282.
- Eggenhofer F, Tafer H, Stadler PF, et al. RNApredator: fast accessibility-based prediction of sRNA targets. Nucleic Acids Res. 2011;39:W149–154.
- Kery MB, Feldman M, Livny J, et al. TargetRNA2: identifying targets of small regulatory RNAs in bacteria. Nucleic Acids Res. 2014;42:W124–129.
- Harris MA, Clark J, Ireland A, et al. The gene ontology (GO) database and informatics resource. Nucleic Acids Res. 2004;32:D258–261.