
ABSTRACT
Colicin D is a plasmid-encoded bacteriocin that specifically cleaves tRNAArg of sensitive Escherichia coli cells. E. coli has four isoaccepting tRNAArgs; the cleavage occurs at the 3′ end of anticodon-loop, leading to translation impairment in the sensitive cells. tRNAs form a common L-shaped structure and have many conserved nucleotides that limit tRNA identity elements. How colicin D selects tRNAArgs from the tRNA pool of sensitive E. coli cells is therefore intriguing. Here, we reveal the recognition mechanism of colicin D via biochemical analyses as well as structural modelling. Colicin D recognizes tRNAArgICG, the most abundant species of E. coli tRNAArgs, at its anticodon-loop and D-arm, and selects it as the most preferred substrate by distinguishing its anticodon-loop sequence from that of others. It has been assumed that translation impairment is caused by a decrease in intact tRNA molecules due to cleavage. However, we found that intracellular levels of intact tRNAArgICG do not determine the viability of sensitive cells after such cleavage; rather, an accumulation of cleaved ones does. Cleaved tRNAArgICG dominant-negatively impairs translation in vitro. Moreover, we revealed that EF-Tu, which is required for the delivery of tRNAs, does not compete with colicin D for binding tRNAArgICG, which is consistent with our structural model. Finally, elevation of cleaved tRNAArgICG level decreases the viability of sensitive cells. These results suggest that cleaved tRNAArgICG transiently occupies ribosomal A-site in an EF-Tu-dependent manner, leading to translation impairment. The strategy should also be applicable to other tRNA-targeting RNases, as they, too, recognize anticodon-loops.
Abbreviations: mnm5U: 5-methylaminomethyluridine; mcm5s2U: 5-methoxycarbonylmethyl-2-thiouridine
KEYWORDS:
Introduction
Transfer RNA (tRNA) is an essential adapter molecule which links a codon on an mRNA to the cognate amino acid on the ribosome and forms a tightly packed structure, in order to evade attack by cellular nucleases. An exception is anticodon-loops, a veritable ‘Achilles’ heel’ for tRNA molecules. During the past quarter of a century, several ribonucleases targeting anticodon-loops have been reported in microorganisms [Citation1]. Foremost examples are colicins E5 and D, which are plasmid-encoded bacteriocins that specifically cleave Escherichia coli tRNAs [Citation2–5]. Following their release into the medium post expression, colicins E5 and D bind to specific receptors on the surface of sensitive cells, and the C-terminal RNase domains are excised during the translocation step, yielding E5-CRD and D-CRD, respectively [Citation6,Citation7]. They then enter the cytosol of the sensitive E. coli cells and cleave the target tRNAs, leading to translation impairment. Colicin E5 cleaves tRNATyr, tRNAHis, tRNAAsp, and tRNAAsn sbetween positions 34 and 35, which are the first and second letters of the anticodon, respectively, yielding a 2′,3′-cyclic phosphate and a 5′-OH termini [Citation4]. These tRNAs possess queuine (Q), a modified base introduced by the base exchange of guanine (G) in the precursor tRNAs at position 34, and colicin E5 does not distinguish between Q and G, as it cleaves both mature and precursor tRNAs [Citation4,Citation8]. On the other hand, colicin D cleaves all tRNAArg isoacceptors, tRNAArgICG, tRNAArgCCG, tRNAArgmnm5UCU, and tRNAArgCCU; inosine (I) being a modified base of adenine (A) in the first letter of precursor tRNA. The cleavage occurs between positions 38 and 39, at the 3′ end of anticodon-loops () [Citation5]. Other examples are eukaryotic killer toxins, zymocin and PaT, encoded by cytoplasmic linear plasmids of Kluyveromyces lactis and Pichia acaciae yeasts, respectively. Zymocin cleaves tRNAGlumcm5s2UUC, tRNALysmcm5s2UUU, and tRNAGlnmcm5s2UUG of Saccharomyces cerevisiae between positions 34 and 35, leading to cell cycle arrest at the G1 phase [Citation9]. PaT arrests yeast cells at the S phase by cleaving tRNAGln between positions 34 and 35, followed by a second cleavage between positions 32 and 33 excising di-nucleotides [Citation10,Citation11].
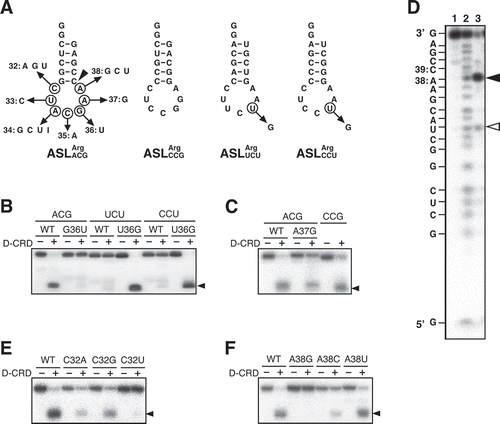
Figure 1. Comparison of the susceptibilities of E. coli tRNAArg isoacceptors to colicin D. (A) In vitro transcripts for E. coli tRNAArg isoacceptors used in this study are presented as a clover-leaf structure. All these transcripts possess G1-C72 and G2-C71 pairs to enhance the transcription efficiency. Nucleotide numbering is according to the proposal of Sprinzl [Citation12]. Arrowheads indicate the cleavage sites of colicin D. Note that the modified base inosine is introduced in the first letter of anticodon of tRNAArgACG. (B) Each of the four 3′-labelled E. coli tRNAArg transcripts was incubated with various amounts of D-CRD as indicated, and the cleavage efficiency was compared. The extent of cleavage was calculated according to the following equation. The extent of cleavage (%) = 100 × band intensity of the cleaved fragment/(band intensity of the intact tRNA + band intensity of the cleaved fragment). Arrowhead indicates the cleaved fragments
![Figure 1. Comparison of the susceptibilities of E. coli tRNAArg isoacceptors to colicin D. (A) In vitro transcripts for E. coli tRNAArg isoacceptors used in this study are presented as a clover-leaf structure. All these transcripts possess G1-C72 and G2-C71 pairs to enhance the transcription efficiency. Nucleotide numbering is according to the proposal of Sprinzl [Citation12]. Arrowheads indicate the cleavage sites of colicin D. Note that the modified base inosine is introduced in the first letter of anticodon of tRNAArgACG. (B) Each of the four 3′-labelled E. coli tRNAArg transcripts was incubated with various amounts of D-CRD as indicated, and the cleavage efficiency was compared. The extent of cleavage was calculated according to the following equation. The extent of cleavage (%) = 100 × band intensity of the cleaved fragment/(band intensity of the intact tRNA + band intensity of the cleaved fragment). Arrowhead indicates the cleaved fragments](/cms/asset/9ef11729-0d91-40ed-bd5d-0ac6f049c7cd/krnb_a_1838782_f0001_c.jpg)
tRNA cleavage has been widely observed, and not exclusively with respect to microbial toxins. PrrC, which is expressed in some E. coli strains possessing the optional prr locus, cleaves tRNALys of host cells between positions 33 and 34 upon T4 phage infection [Citation13]. This shuts down the host translational system, which prevents further propagation of the infecting phage. A toxin–antitoxin system (TA system), where the ‘toxin’ is an effector while the ‘antitoxin’ neutralizes the former’s activity, is a stress response element widely distributed in prokaryotes, leading to growth inhibition and dormancy under unfavourable conditions [Citation14]. Salmonella enterica serovar Typhimurium LT2, along with a virulence plasmid of Shigella flexneri 2a, encodes a novel toxin, VapC, that can cleave the initiator tRNAfMet between the 38 and 39 positions [Citation15]. Recently, VapC produced by Mycobacterium tuberculosis, designated as VapC-mt4, was revealed to cleave M. tuberculosis tRNAs for Ala and Ser between positions 36 and 37 or positions 35 and 36, respectively [Citation16]. Furthermore, some bacteria apply direct cell-to-cell contact for interbacterial competition, referred to as contact-dependent growth inhibition (CDI) [Citation17]. To impair the growth of other cells, the CDI+ cell forms an extracellular filamentous β-helical structure (CdiA), and the C-terminal toxin domain of CdiA (CdiA-CT) is suggested to be excised and translocated into the target CDI− cell [Citation18]. CdiA-CT, encoded by the Burkholderia pseudomallei strain E478 (CdiA-CTE478) and uropathogenic E. coli strain 536 (CdiA-CTUPEC536), cleaves specific tRNAs [Citation19,Citation20]. CdiA-CTE478 is highly homologous to E5-CRD and possesses the same substrate specificity as that of the latter molecule [Citation20]. Interestingly, a missense mutation in CdiA-CTUPEC536 abolishes tRNA cleavage activity concomitantly with the biofilm formation activity of host cells [Citation21]. Consequently, tRNA cleavage is widely distributed to regulate cell growth of its own or other cells in microorganisms.
tRNA forms a common L-shaped structure and possesses many conserved nucleotides across various species. It is therefore quite interesting how these tRNA-targeting RNases recognize their substrate tRNAs. For example, E. coli aminoacyl-tRNA synthetase for arginine (ArgRS), which shares its substrate tRNAs with colicin D, selects tRNAArgs mainly by recognizing the D-loop (nucleotide position at 20 (A)), anticodon-loop (positions at 35 (C) and 36 (G or U)), and acceptor stem (position at 73 (A or G) [Citation22–26]. Nearly the entire structure of tRNAArgs is thus required for the recognition of ArgRS [Citation27]. On the contrary, such a mode of recognition is difficult for colicin D due to the smaller size of D-CRD compared to tRNA. Previously, we showed that colicin E5 recognizes the YQUN sequence at positions 33 to 36 of anticodon-loops (or YGUN in precursor tRNAs; where Y is a pyrimidine and N is any nucleotide), and an anticodon stem-loop (ASL) moiety is sufficient for the cleavage [Citation8,Citation28]. Moreover, PrrC and zymocin γ-subunit also specifically cleave the ASL moiety of substrate tRNAs [Citation29,Citation30]. These observations suggest that they have a specialized method for recognizing substrate tRNAs using ASL, which is common in tRNA-targeting RNases. Here, we reveal via biochemical analyses together with structural modelling that colicin D recognizes the anticodon-loop and D-arm of tRNAArgs for cleavage. Thus, colicin D cleaves tRNAArgICG as a primary target. It has been assumed that this cleavage of tRNAArg by colicin D results in the exhaustion of the cytoplasmic pool of these tRNAArg species, causing impairment in protein synthesis [Citation5]. In the present study, however, we show that it is not the exhaustion of the intact tRNAArgs, but rather the emergence of cleaved tRNA molecules, specifically those of tRNAArgICG, that is more likely to be responsible for translation impairment.
Results
Anticodon-loop sequence determines the susceptibility to D-CRD
As described above, the C-terminal region of colicin D includes the RNase domain (D-CRD). In an early stage of our study, we found that the C-terminal region of colicin D starting from Asp532 has cytotoxic activity that is almost equivalent to that of full-length colicin D, based on the characterization of a variety of chimeric colicins between E5 and D (Fig. S1A). Using the purified C-terminal polypeptide, we confirmed that it retains the intrinsic cleavage activity and specificity of the full-length colicin D (Fig. S1B). This region, designated as D-CRD532, was therefore used in our study. Our previous study suggested that the susceptibility to D-CRD is not equal among the four E. coli tRNAArgs [Citation5,Citation31]. Subsequently, the substrate preference of D-CRD was examined using in vitro transcripts of tRNAArgs. tRNAArg transcripts, which do not include modified nucleotides, were efficiently cleaved by D-CRD. tRNAArgICG is the most abundant among E. coli tRNAArgs, and its transcript, tRNAArgACG, was found to be the most susceptible, followed by tRNAArgCCG, then tRNAArgUCU and tRNAArgCCU, which were only slightly susceptible (). The cleavage preference is consistent across the results observed in cells challenged by colicin D [Citation5].
The molecular mass of D-CRD532 (18,000) is smaller than that of substrate tRNA (more than 25,000), suggesting that D-CRD recognizes a limited area, including ASL as well as other tRNA-targeting RNases. Further, the susceptibility of transcripts corresponding to the ASL moieties of four tRNAArgs (ASLArgs) was examined. As shown in and C ASLArgACG and ASLArgCCG were cleaved; the cleavage site was confirmed to be between positions 38 and 39 (). On the contrary, ASLArgUCU and ASLArgCCU were not cleaved at all, even though their tRNA forms were weakly susceptible (). These results indicate that D-CRD recognizes ASL in the same manner as for the corresponding tRNA, and that the susceptibilities of the four tRNAArg isoacceptors are almost faithfully depicted by those of the ASL moiety. The lower susceptibilities of ASL compared to the full-size molecules suggest that D-CRD also recognizes some part(s) of tRNA other than ASL (compare and Fig. S2B).
Figure 2. Determination of nucleotides important for the susceptibility to D-CRD within the anticodon-loop. (A) Secondary structures of the anticodon stem-loop (ASL) transcripts for E. coli tRNAArgs used are as shown. All these ASL transcripts possess GG extension at their 5′ termini to enhance the transcription efficiency. Arrows indicate the nucleotide replacement, and nucleotide numbering is according to those of full-length tRNAs as seen in . Arrowheads correspond to the cleavage sites of ASLArgACG, as seen in Fig. 2D. (B, C, E, and F) Each of the nucleotides was replaced as indicated, and the susceptibility to D-CRD was evaluated. Arrowheads indicate the cleaved 3′ fragments. (D) Cleavage site on ASLArgACG was determined. The 5′-labelled ASLArgACG was incubated without (lane 1) or with D-CRD (lane 3). The alkaline hydrolysis ladder was loaded on lane 2. The cleavage site is depicted by a closed arrowhead, and the minor cleavage site, which probably occurs indirectly, is denoted by an open arrowhead
G36 sets tRNAArgICG as a primary target of colicin D
The results obtained with ASLArgs suggest that positive determinants as a good substrate of D-CRD are included in the anticodon-loops of ASLArgACG and ASLArgCCG. It is notable that G36 is shared between the susceptible ASLArgACG and ASLArgCCG, instead of U36 in the resistant ASLArgUCU and ASLArgCCU (). Replacement of G36 with U on ASLArgACG rendered complete resistance to D-CRD, while the introduction of G36 into ASLArgUCU and ASLArgCCU made it susceptible to D-CRD (). These results clearly indicate that G36 is a strong determinant for the susceptibility to D-CRD. Despite sharing a G36 base, ASLArgACG was more susceptible than ASLArgCCG (, C). The two ASLs have the same anticodon-loop sequence except for positions 34 and 37; ASLArgACG has A34 and A37, while ASLArgCCG has C34 and G37 (). Replacement of A37 with G decreased the susceptibility of ASLArgACG almost to the same extent seen in ASLArgCCG, indicating that D-CRD prefers adenosine to guanosine at position 37 (). However, replacement of A34 in ASLArgACG with any other nucleotides, including the modified base of inosine (I) observed in the native one, did not affect its susceptibility (Figs. S2A and S2B).
The contributions of C32, U33, C35, and A38, which are conserved among the four ASLArgs, were also examined. Replacement of U33 – which is invariable among all E. coli tRNAs – with C did not affect the susceptibility of ASLArgACG, while ASLArgACG with C35A was somewhat less susceptible (Fig. S2C). Strikingly, replacement of C32 with U (C32U), or A38 with G (A38G), conferred almost complete resistance to D-CRD on ASLArgACG, while the replacement of C32 or A38 with other nucleotides only partially reduced the susceptibility (, F). On the other hand, replacement of U32 with C on E. coli tRNAProCGG, which shows quite a low susceptibility despite possessing G36, exhibited a noticeably increased cleavage efficiency (Fig. S2D). These results indicate that C32 is a cryptic determinant of susceptibility to colicin D.
Structural insight of substrate recognition of colicin D
Based on the results obtained by our biochemical analysis, we tried to build the docking model of D-CRD∙tRNAArgICG [Citation32]. Here, the structure of D-CRD starting at the 595th residue of colicin D (D-CRD595), whose structure complexed with ImmD had been previously determined by our group [Citation31], was applied. We confirmed that tRNA cleavage activity and substrate specificity are fully retained by D-CRD595 as well as D-CRD532 [Citation31]. In most ribonucleases, histidine residues play critical roles as catalysts, with the exception of colicin E5, which has no histidine in its RNase domain [Citation33]. In colicin D, His611, out of four histidine residues in its C-terminal domain, was found to be critical in the reaction and supposed as a catalyst [Citation5].
The initial complex structure in modelling of D-CRD595 with ASL for tRNAArgICG was constructed so that His611 takes a pivotal position to interact with the 2′-OH of A38 as a general base. Then, the 2′-O nucleophilically attacks the central phosphorus of the phosphodiester bond to form a transition state, leading to the interaction of a general acid with 5′-O to facilitate its release from the phosphate [Citation5,Citation31,Citation34]. First, we considered Lys608 as a general acid, and the initial structure was constructed such that Lys608 was close to the oxygen atom of the phosphate group between A38 and C39. However, His611 was dissociated from the cleavage site after the molecular dynamics (MD) simulations. Subsequently, Lys610, another candidate for the general acid (Fig. S3A), was used instead of Lys608. In the structure, His611 was still located near the 2′-OH of A38, indicating the validity of the structure (, B). Lys608 and Lys610 made a salt bridge with the phosphate group between C39 and C40 and between A38 and C39, respectively. This situation is similar to E5-CRD of colicin E5, where two lysine residues play important roles of interacting with two consecutive phosphodiester bonds; Lys25 of E5-CRD with the scissile phosphodiester bond and Lys60 with the next one [Citation33]. Ser677 interacts with the 2′-O of C39 to form a hydrogen bond, while Tyr649 and Arg651 do so with the base of C39. Moreover, G36, which is the strongest determinant of substrate selection within the anticodon-loop, forms a hydrogen bond with Asn687. The adenosine ring at position 38 is flipped out and hydrophobically interacts with Trp679, which is consistent with the previous assumption [Citation31,Citation34].
Figure 3. Structural insight of substrate recognition and catalytic mechanism of colicin D. (A) Docking model of D-CRD595 with ASL for tRNAArgICG was shown. The structure of D-CRD595 was obtained from the D-CRD595∙ImmD complex determined by our group (PDB code: 1TFO), and ASL was generated based on the structure of yeast tRNAPhe (PDB code: 1EHZ). (B) The active site of D-CRD boxed in (A) was magnified and amino acid residues of D-CRD involved in substrate recognition/catalysis, as well as bases of G36, A38, and C39 of ASL for tRNAArgICG, were labelled. The 2′-O and 3′-O of A38 were also indicated. (C) Docking model of tRNA∙D-CRD was constructed by superposing the structure of yeast tRNAPhe onto that of ASL in ASL∙D-CRD shown in (A). Structures of ASL, tRNA, and D-CRD are coloured orange, light orange, and blue, respectively. D-arm of tRNA is labelled in red in (C)
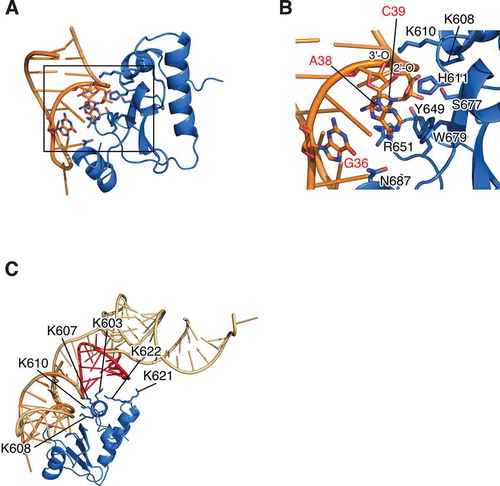
Next, the D-CRD∙tRNAArgICG model was constructed with the structure of yeast tRNAPhe. The resultant structure suggested that the positively charged area, composed of Lys603, Lys607, Lys608, and Lys610, was faced with the surface of tRNA, which is consistent with the previous discussion [Citation31,Citation34], and Lys621 and Lys622 were also located near the D-arm (). Replacement of Lys607, Lys608, Lys610, Tyr649, Arg651, Ser677, and Trp679 with Ala actually reduced the cytotoxic activity of colicin D, while K603A mutation did not have any influence on the activity; these results are consistent with the previous study (Fig. S3A) [Citation34]. N687A mutation on colicin D decreased the cytotoxicity of colicin D by about one-tenth, suggesting the contribution of Asn687 for the recognition of G36 (Fig. S3A). The importance of Lys621 and Lys622 was also examined and showed that colicin D (K622A), but not colicin D (K621A), decreased the cytotoxic activity about one-twentieth, suggesting that Lys622 is used to interact with the D-arm of substrate tRNAs (Fig. S3B).
Proportion of cleaved tRNAArg determines viability against colicin D
tRNAArgICG is the major species among the four tRNAArg isoacceptors and, as shown, tRNAArgACG transcript is the most susceptible among tRNAArgs in vitro (), implying that tRNAArgICG is a primary target of colicin D. Assuming that colicin D impairs translation of sensitive cells by depleting susceptible tRNAs [Citation5], we expected that overexpression of tRNAArgICG makes the host cells more resistant to colicin D. Besides, as shown above, replacement of C32 with U greatly decreased the susceptibility of tRNA to D-CRD (). We tried to delete the gene cluster (argV-argY-argZ-argQ), encoding all tRNAArgICG genes, in the strain expressing tRNAArgICG or tRNAArgICG (C32U) from the plasmid (pBR328-ICG or pBR328-ICG (C32U); see Supplementary Tables 1 and 2, and Supplementary Materials and Methods). Therein, we found that the resulting two strains were viable, indicating that tRNAArgICG (C32U) replaces the function of wild-type tRNAArgICG during translation (Figs. S4A and S4B). This is consistent with the result that a mutation at position 32 is acceptable to yeast tRNAArgICG [Citation35]. Each of the tRNAArgs and their C32U mutants was therefore overexpressed in E. coli cells in the hope of increasing the tolerance of wild-type host cells to colicin D. These cells were further challenged with colicin D, and the status of tRNAs as well as cell viabilities were determined. Even after challenging with colicin D, the levels of each intact tRNAArg were higher than that of the cells with an empty plasmid, due to overexpression (). However, contrary to our expectations, the overexpression of wild-type tRNAArgICG did not restore cell viability at all, while a noticeable recovery of viability was observed in cells overexpressing tRNAArgICG (C32U) (). Since tRNAArgICG (C32U) is active as well as the wild-type form in translation, these results indicate that tRNAArgICG is the primary target of colicin D in vivo. It is also suggested that an abundance of intact tRNAArgICG does not determine the cell viability after being challenged with colicin D.
Figure 4. Correlation of intracellular level of tRNAArgICG and viability of cells challenged with colicin D. (A and B) Each of the four isoaccepting tRNAArgs and their C32U mutants was overexpressed in the wild-type E. coli cells, and these cells were challenged with colicin D. Subsequently, the status of tRNA prepared from these cells was analysed with northern blot hybridization, as depicted in (A). Simultaneously, viable cell count (CFU) was calculated and represented in (B). U* indicates a modified base of mnm5U appearing in the anticodon of tRNAArgmnm5UCU. The values shown in the histogram were a mean of three independent experiments, and the error bar indicates standard error. Different letters above each data point indicate significant differences (P < 0.01; Tukey–Kramer). (C) The wild-type strain harbouring tRNAArgICG-expressing plasmid used in (A) was challenged with serially diluted colicin D, and the viability as well as status of tRNAArgICG were analysed. Cells with the empty plasmid were used as a control (leftmost lane). Error bars shown in the bottom scatter plot indicate standard error of five independent experiments. The asterisk indicates that the signal intensity of intact tRNAArgICG in the lane is almost comparable to that of the control cells with empty plasmid (leftmost lane). Arrowheads observed in (A) and (C) indicate the cleaved 3′-tRNA fragments. 5S labelled in (A) indicates a band of E. coli 5S rRNA detected as a loading control
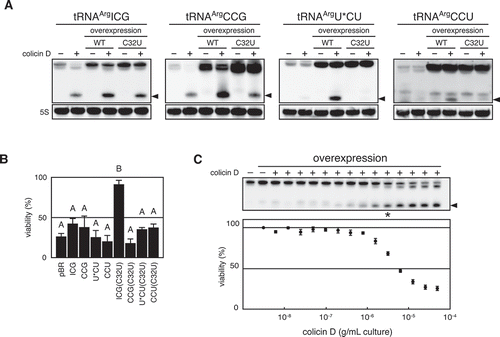
To determine the relationship between the tRNAArgICG level and cell viability, cells overexpressing tRNAArgICG, as demonstrated in , were challenged with serially diluted colicin D, and the cell viabilities along with the extent of tRNAArgICG cleavage were compared. Apparently, cell viability decreased in proportion to the decrease in intact tRNAArgICG level. However, cells maintaining almost the same tRNAArgICG level as that of control cells (a strain carrying the empty plasmid) showed significantly decreased viability after the colicin D challenge (; compare the lane labelled with an asterisk with the leftmost lane). Moreover, the viability is decreased proportionately with the accumulation of cleaved tRNAArgICG. This result indicates that the viability is not well determined by the abundance of intact tRNAArgICG, but rather by the proportion of cleaved tRNAArgICG to the intact one.
Nicked tRNAArgICG impairs translation in vitro
Cleaved tRNAArg in the sensitive cells is easily detectable with northern blot hybridization () [Citation5]. Additionally, cleaved fragments of tRNAArg transcript were not dissociated from each other in the presence of Mg2+, as was demonstrated with tRNAArgICG prepared from colicin D-challenged cells (Fig. S5). These results indicate that tRNAArgICG cleaved within an anticodon-loop still stably retains its structure in sensitive cells. This raises the possibility that the nicked tRNAArgICG itself impairs translation. To assess this hypothesis, total aminoacyl-tRNA was prepared and incubated with D-CRD to cleave tRNAArgICG. After removal of the D-CRD by phenol/chloroform extraction (Fig. S6), the total tRNA containing nicked tRNAArg was added to an E. coli in vitro translation system containing a complete set of tRNAs, and the translation activity was consequently measured. Subsequently, we found that a small amount of nicked tRNAArgICG (the ratio of nicked tRNAArgICG to intact tRNAArgICG was estimated at 1:30) dominant-negatively impairs translation, suggesting that nicked tRNA is delivered to the A-site of ribosome to effectuate the impairment of translation ().
Figure 5. Involvement of the nicked-tRNAArgICG in translation impairment. (A) Aminoacyl-total tRNA was incubated with or without D-CRD. After the removal of D-CRD by phenol/chloroform treatment, each of these total tRNAs (0.4 μg) was added to in vitro translation system containing a complete set of total tRNA (12 μg). The translation activity was evaluated by the activity of β-galactosidase synthesized in the translation system. Open and solid circles indicate the addition of aminoacyl tRNA treated with or without D-CRD, respectively, and the solid square denotes the addition of tRNA stock buffer instead. Error bars indicate standard error of three independent experiments. (B) Total aminoacyl-tRNA was incubated with D-CRD for the cleavage of tRNAArgICG, which was followed by further incubation with EF-Tu∙GTP or EF-Tu alone. The mixture was electrophoresed in native and denaturing gels, followed by northern blot hybridization with a probe specific for the 3′-fragment of tRNAArgICG to analyse TC formation (upper panel) and tRNA cleavage (lower panel). (C) Total aminoacyl-tRNA was incubated with EF-Tu in the presence or absence of GTP. Subsequently, various amounts of D-CRD were added, followed by further incubation, which was indicated by labelling it as ‘incubation.’ TC formation (upper panel) and the cleavage (lower panel) of tRNAArgICG were analysed as shown in (B). (D) Colicin D-resistant strain harbouring each of the pSTV28-derived plasmids encoding wild-type tRNAArgICG, tRNAArgICG (C32U), or tRNAArgICG (A20U) was challenged with colicin D, and their viabilities (upper histogram) as well as the status of tRNAArgICG (lower panel) were analysed. Empty pSTV28 plasmid was used as a control, which is indicated as ‘none.’ Error bar indicates standard error of seven independent experiments. Statistical significance compared to the control strain (labelled ‘none’) was determined using Student’s t-test. P-values less than 0.01 are represented as **. Closed and open arrows in (B) and (C) indicate tRNAArgICG with or without EF-Tu∙GTP, respectively, while closed arrowheads in (B), (C), and (D) indicate the 3′-fragment of cleaved tRNAArgICG.
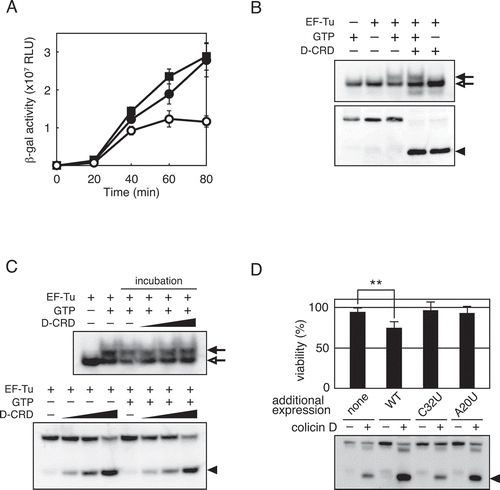
To enter the A-site, tRNA should interact with elongation factor-Tu (EF-Tu) and GTP to form a ternary complex (TC). Our structural modelling demonstrates that the 3′ acceptor stem of tRNAArg is actually free to interact with EF-Tu (). The association of EF-Tu∙GTP with nicked-tRNA was therefore further examined. The total aminoacyl-tRNA (Fig. S7) containing fully cleaved tRNAArgICG was incubated with EF-Tu, in the presence or absence of GTP. After carrying out electrophoresis in native and denaturing gels, northern hybridization was performed. Subsequently, EF-Tu∙GTP was found to form TC with nicked Arg-tRNAArgICG as well as the intact one (). The interaction of nicked Arg-tRNAArgICG with EF-Tu∙GTP was specific due to the requirement of GTP to form TC (, upper panel). These results indicate that EF-Tu does not distinguish between nicked Arg-tRNAArgICG and its intact form. Next, we assessed whether or not tRNAArgICG within TC was susceptible to D-CRD, and whether or not the TC complex was maintained after possible tRNA cleavage. The total aminoacyl-tRNA was incubated with EF-Tu∙GTP to form TC, and the TC was incubated with D-CRD. The susceptibility of tRNAArgICG was then analysed with northern blot hybridization. The tRNAArgICG integrated into TC was almost as susceptible to D-CRD as the free tRNAArgICG (, bottom panel). Moreover, TC formation was not affected by the cleavage (, upper panel). These results indicate that actions of D-CRD and EF-Tu do not compete against each other, suggesting interference of the translation process by the nicked tRNAArg species integrated in TC. It is possible that the nicked-tRNAArgICG interacts with Arginyl-tRNA synthetase (ArgRS), leading to dead-end inhibition of ArgRS and translation impairment. However, tRNA cleavage prevents tRNAArgICG from interacting with ArgRS, indicating that this possibility does not occur (Fig. S8). Another possibility is that the capture of EF-Tu by nicked-Arg-tRNAArgICG leads to exhaustion of active EF-Tu itself. If this is the case, strains lacking tufA or tufB, that is, genes coding for EF-Tu, are more sensitive to colicin D than the wild-type strain, as EF-Tu in ΔtufA or ΔtufB is less abundant compared to that of the wild-type strain. However, such a difference in the sensitivity was not observed, indicating that the exhaustion of EF-Tu, if any, is not a cause of translation impairment following the tRNA cleavage (Fig. S9).
Nicked tRNAArgICG decreases cell viability of susceptible cells in vivo
If the nicked tRNAArgICG itself impairs translation, then the cells with increased nicked tRNAArgICG will show decreased viability. As shown in Fig. S4, we demonstrated that the strain ΔargVYZQ [C32U], lacking all genomic tRNAArgICG genes while expressing tRNAArgICG (C32U) from pBR328 plasmid (pBR328-ICG (C32U); see Supplementary Table 1), was almost completely resistant to colicin D. Based on this strain, it is expected that additional expression of wild-type tRNAArgICG increases the nicked form when challenged with colicin D, leading to a decrease of viability. pSTV28 was chosen for the additional tRNA expression owing to compatibility with pBR328; however, both pSTV28 and pBR328 possess a chloramphenicol-resistance gene. We therefore replaced pBR328, the backbone plasmid for the expression of tRNAArgICG (C32U), with pBR322 (pBR322-ICG (C32U); see Supplementary Table 1) in the strain ΔargVYZQ [C32U]. Each of the pSTV28 plasmids encoding wild-type tRNAArgICG or tRNAArgICG (C32U), together with empty plasmid as a control, was then introduced to generate ΔargVYZQ [C32U/WT], ΔargVYZQ [C32U/C32U], and ΔargVYZQ [C32U/–] strains, respectively (Supplementary Tables S1 and S2), and sensitivities of the resultant strains to colicin D were examined. ΔargVYZQ [C32U/C32U], additionally expressing less susceptible tRNAArgICG (C32U), showed almost the same resistance as that of control ΔargVYZQ [C32U/–], while ΔargVYZQ [C32U/WT], additionally expressing the wild-type one, showed decreased viability (). It is notable that nicked-tRNAArgICG showed more accumulation in the ΔargVYZQ [C32U/WT] strain compared to ΔargVYZQ [C32U/–] and ΔargVYZQ [C32U/C32U] (, bottom panel). Adenine at position 20 (A20) of E. coli tRNAArg is responsible for arginine acceptance both in vitro and in vivo [Citation25,Citation36], and substitution or deletion of this moiety impairs TC formation. Strain ΔargVYZQ [C32U/A20U], additionally expressing tRNAArgICG (A20U), does not decrease viability, despite the susceptibility of tRNAICG (A20U) to cleavage (). These results indicate that TC formation is required for translation impairment and a subsequent decrease of cell viability mediated by the nicked-tRNAArgICG. Besides bands indicating the cleaved products, a band seeming to indicate the shortened tRNAArgICG is also observed just below the band corresponding to the full-length one after the colicin D challenge as well as those in , S4C, and S5B. We do not believe that the possible shortened product is involved in the translation impairment, as the accumulation does not correlate with the decrease of viability of host cells (Fig. S4C, compare lanes 2 and 4 of the left panel).
Discussion
Our results show that susceptibility of ASLs for arginine to colicin D faithfully reflects that of corresponding full-length tRNA transcripts (). Moreover, as described above, the susceptibility of tRNA transcripts is consistent with that of native tRNA [Citation5]. These validate our studies using in vitro-synthesized transcripts, and base modifications are likely to have a small impact, if any, on susceptibility to colicin D. U33C mutation on ASLArgACG does not affect susceptibility (Fig. S2C). It is known that U33 is required to form a ‘U-turn,’ a conserved structural motif found in anticodon-loops, suggesting that such a structure is not involved in susceptibility. Our structural model suggests that G36, a strong determinant for susceptibility, interacts with Asn687 (); however, colicin D mutant N687A still retains a weak cytotoxic activity (Fig. S3A). This may account for the fact that G at position 36 is not absolutely essential for the cleavage (). tRNAArgCCG, sharing G36 with tRNAArgICG, is less susceptible to colicin D compared to tRNAArgICG (). This may be because G36 and G37 of tRNAArgCCG competitively interact with N687. A38, located just at the 5′ of cleavage site, may be flipped out and hydrophobically interact with Trp679, making the 2′-OH of A38 accessible to His611. Introduction of U∙A or C∙G pairing between positions 32 and 38 of tRNA strongly decreases susceptibility (, F), which may be due to the impairment of A38 flipping. Although the D-arm is dispensable for the cleavage, ASLs alone greatly reduced susceptibility compared to corresponding tRNA transcripts. This is because the ASL moiety does not interact with Lys622 of colicin D.
Our results indicate that the nicked-tRNAArgICG together with EF-Tu is involved in translation impairment (). It is reported that 80% of tRNAs are charged in exponentially growing cells, and most of tRNAArgICG cleaved by colicin D is acylated [Citation37,Citation38]. Therefore, the most likely scenario is that nicked-tRNAArgICG is delivered to the ribosomal A-site in an EF-Tu-dependent manner, leading to translation impairment. Negative correlation of cell viability with the proportion of the nicked-tRNAArgICG suggests that frequent occurrence of A-site occupation by inactive tRNAArgICG increases the seriousness of translation impairment (). This may be why colicin D selects tRNAArgICG, the most abundant among all the tRNAArg isoacceptors in E. coli, as a primary target. Previously, Garza-Sanchez et al. supposed that ribosomal A-site is transiently occupied by nicked-tRNAArgICG in sensitive cells after the challenge of colicin D [Citation38]. They revealed that colicin D induces mRNA cleavage in the sensitive cells at two sites, at the 3′ boundary of the ribosome and at the arginine codon in the ribosomal A-site [Citation38]. The 3′ cleavage is typically observed in mRNAs where ribosomes are arrested by the occupation of tRNAs at the A-site, such as SecM-mediated ribosomal stall, while it is not observed in arginine-starved cells [Citation39,Citation40]. These results will support our insight that nicked-tRNAArgICG is delivered to the ribosomal A-site, leading to ribosome pausing. On the other hand, the latter A-site cleavage was observed in cells lacking transfer-messenger RNA (tmRNA), but not in wild-type cells. The tmRNA, in conjunction with small protein B (SmpB), serves as a ribosome rescue system referred to as trans-translation. When ribosomes are stalled on mRNA, tmRNA is recruited to the A-site with the aid of SmpB, which facilitates degradation of the nascent peptide and recycling of stalled ribosomes [Citation41]. They showed that colicin D induces the tmRNA-mediated degradation of the nascent peptide [Citation38]. Moreover, we showed that tRNA cleavage induces transient bacteriostasis of cells challenged with colicin D, which requires trans-translation [Citation42]. These results suggest that nicked-tRNAArgICG enters the ribosomal A-site competitively with the tmRNA that mediates transient dormancy.
Docking models of E5-CRD or VapC-mt4 with tRNA structure are available, and both complexes seem to interact with EF-Tu∙GTP [Citation16,Citation28]. Consequently, our model should be applicable to other tRNA-targeting RNases, wherein they recognize the anticodon-loop to avoid competing with EF-Tu. tRNAs are destined to expose their anticodon-loops in order to make base pairs with the cognate codons buried in the A- and P-sites of ribosomes. Therefore, it is reasonable that these tRNA-targeting RNases target the anticodon-loop. Our findings should add new insight into a mode of tRNA recognition that is necessary for translation impairment.
Materials & methods
Strains and plasmids
Plasmids and E. coli strains used in this study are summarized in Supplementary Tables S1 and S2, respectively.
Statistical Analysis
Statistical analyses in were performed with JMP 14.1 software (SAS Institute Japan).
Protein purification
Genes coding D-CRD532 and C-terminal His-tagged ImmD were cloned into the NcoI-BclI site of the plasmid as previously described [Citation4], and the resultant plasmid was designated as pDD532His. E. coli K-12 RR1 possessing pDD532His was grown in L-broth containing 100 μg/ml ampicillin at 37°C. When the optical density of the bacterial culture reached 0.8 at 660 nm, 0.4 μg/ml mitomycin C was added to induce the expression of D-CRD532∙ImmD complex and further cultivated for 3 h. Cells were harvested and sonicated in 20 mM potassium phosphate buffer (pH 7.0). Polyethyleneimine was added to the lysate to precipitate nucleic acids. Then, the supernatant containing 500 mM of potassium chloride was applied to an Ni-NTA agarose column equilibrated with a buffer containing 20 mM potassium phosphate (pH 7.0) and 500 mM potassium chloride. After the column was washed out, D-CRD532∙ImmD complex was eluted with buffer containing 20 mM potassium phosphate (pH 7.0), 500 mM potassium chloride and 300 mM imidazole. D-CRD532∙ImmD complex was dialysed against 9 M urea, 20 mM potassium phosphate (pH 7.0), and applied to an Ni-NTA agarose column equilibrated with the same buffer in order to capture dissociated His-tagged ImmD to the column. After the column was washed, D-CRD532 was eluted with buffer containing 9 M urea, 20 mM potassium phosphate (pH 7.0), 500 mM potassium chloride, and 20 mM imidazole. The separated D-CRD532 was purified using a Mono S column equilibrated with 20 mM Tris-HCl (pH 7.7), followed by gel filtration with Superose 12 HR.
Preparation of RNA transcripts
In vitro transcription was performed as previously described [Citation43]. To prepare tRNA transcripts, template DNA coding tRNA gene under T7 promoter was PCR-amplified. Transcription of ASL was performed as previously described [Citation8]. It should be noted that the tRNA and ASL transcripts used in this study possess G1-C72 and G2-C71 pairs and an additional GG dinucleotide at the 5′-termini, respectively, to enhance transcription efficiency. The transcribed RNAs were electrophoresed on a denaturing polyacrylamide gel, the bands were extracted, and RNAs were eluted from the gel by shaking them in water overnight at 37°C. After centrifugation, ethanol precipitation was performed to recover transcripts. All transcripts were incubated at 70°C for 10 min, followed by slow cooling to refold the structure into the original form. The 5′- or 3′-end labelling was performed as described previously [Citation4,Citation5,Citation8].
Cleavage assay
The cleavage assay using transcripts of tRNA and ASL was performed as follows: 10 pmol of each tRNA transcript was incubated with 1, 10, or 100 fmol of D-CRD, as indicated, in 20 μl of 20 mM Tris-HCl (pH 7.5), 1 mM MgCl2, 100 μg/ml BSA, and 1 mM DTT at 37ºC for 20 min. For the assay with ASL, 10 pmol of substrate was incubated with 200 fmol of D-CRD in 20 μl of 20 mM Tris-HCl (pH 7.5), 100 μg/ml BSA, and 1 mM DTT at 37ºC for 20 min. The reaction was terminated by adding an equal volume of a buffer containing 9 M urea, 0.2% XC, and 0.2% BPB, and electrophoresis was performed with a TBE gel containing 7 M urea. Extent of cleavage was calculated as follows: Extent of cleavage (%) = 100 × intensity of cleaved fragment/(intensity of intact RNA + intensity of cleaved fragment). In , trace amounts of 5′- or 3′ 32P-labelled tRNA or ASL were added to the substrate RNAs to facilitate detection.
Northern blot hybridization
Northern blot hybridization was performed according to the method described previously [Citation5]. Sequences of DNA probes used are shown in Supplementary Table S3.
Construction of strains overexpressing tRNAs
pBR322-based plasmids overexpressing each of four tRNAArgs (designated as pBR322-ICG, pBR322-CCG, pBR322-UCU, and pBR322-CCU) and those of C32U mutants used in were constructed as follows. Genes for wild-type or mutant tRNAArgs were cloned into EcoRI-SacI sites of plasmid pBSup3+(CCAOH) [Citation44], so that each of the tRNA genes was inserted between the lipoprotein lpp promoter and rrnC terminator in the same direction. The resultant plasmids confer only tetracycline resistance on the host strain, as the Ap resistance gene was disrupted in the process of plasmid construction. The resultant plasmids and pBR322 used as a control were introduced into E. coli K-12 DH5α. On the other hand, pSTV28-derived plasmids, with a pACYC184 replicon compatible with pBR322, overexpressing tRNAArgICG, tRNAArgICG (C32U), or tRNAArgICG (A20U) (designated as pSTV28-ICG, pSTV28-ICG (C32U), and pSTV28-ICG (A20U), respectively), as represented in , were constructed as follows. PstI-BamHI fragments of pBR322-ICG or pBR322-ICG (C32U) described above were excised and tRNA genes were cloned between PstI and BamHI sites of pSTV28 (Cm resistant). A20 of the tRNAArgICG gene in pSTV-ICG was replaced by T, in order to generate pSTV28-ICG (A20U). All these plasmids described above are summarized in Supplementary Table 1. Note that the expression of these tRNAs depends on the lpp promoter. First, we constructed a strain, designated as ΔargVYZQ [C32U], lacking argV-argY-argZ-argQ, a cluster encoding all tRNAArgICG genes, and expressing tRNAArgICG (C32U) from the pBR328-based plasmid instead (Supplementary Fig. S4). The detail of gene disruption was described in Supplementary Materials and Methods. The resultant strain ΔargVYZQ [C32U] (Ap, Cm, and Km resistant) was transformed with pBR322-ICG (C32U) (Tc resistant) and cultivated in L-broth containing 5 μg/ml of Tc to eliminate pBR328-ICG (C32U) (Ap and Cm resistant) by plasmid incompatibility between pBR322 and pBR328. After confirmation that pBR328-ICG (C32U) was replaced with pBR322-ICG (C32U), pSTV28-ICG, pSTV28-ICG (C32U), pSTV28-ICG (A20U), or pSTV28 as a control (Cm resistant) was introduced. The resultant strains were designated as ΔargVYZQ [C32U/WT], ΔargVYZQ [C32U/C32U], ΔargVYZQ [C32U/A20U], and ΔargVYZQ [C32U/–], respectively. These strains were cultured in the presence of Tc and Cm to maintain both pBR322- and pSTV-derived plasmids.
Measurement of viability
The transformants were grown at 37°C in L-broth containing 5 μg/ml of Tc (), or 5 μg/ml of Tc and 20 μg/ml of Cm (). When OD660 reached 0.4, they were placed on ice. Subsequently, cultivation was initiated again at 37°C for 5 min, and 2 μg/ml (), 10 μg/ml (), or the indicated amount () of colicin D∙ImmD complex was added to the medium, followed by further cultivation at 37°C for 20 min. Following this, the colony-forming unit (CFU) was counted and the viabilities were calculated as follows: Viability (%) = 100 × (CFU of colicin D-treated cells/CFU of colicin D-untreated cells).
Gel-shift assay for EF-Tu•GTP∙Arg-tRNAArg ternary complex
Aminoacylated total tRNA was prepared from E. coli cells overexpressing tRNAArgICG (represented in ) according to the previously described method [Citation45]. Purified aminoacylated total tRNA was stored in 10 mM sodium acetate (pH 4.5) and 1 mM MgCl2. Aminoacylation level of tRNAArgICG was validated via an acid-urea PAGE [Citation45]. A gene for EF-Tu was cloned between NcoI and XhoI sites of pET24d (+) so that the resultant plasmid (pET24-EF-Tu) expresses EF-Tu carrying His-tag at the C-terminal. Purification of E. coli EF-Tu and method for the gel-shift assay was performed according to the previous report, with some minor modifications [Citation46]. Four hundred pmol of purified EF-Tu was pre-incubated in 10 μl of buffer containing 40 mM Tris-HCl (pH 7.5), 52 mM NH4OAc, 8 mM Mg(OAc)2, 5 mM DTT, 1 mM GTP, 3 mM phosphoenol pyruvate, and 50 μg/ml pyruvate kinase at 37ºC for 30 min. Two hundred pmol of total aminoacyl-tRNA was incubated with 0.4 pmol of D-CRD in 6 μl of buffer containing 50 mM Tris-HCl (pH 7.5), 1 mM MgCl2, and 1 mM DTT at 37ºC for 15 min. Then, EF-Tu solution and 4 μl of aaBind buffer (150 mM Tris-HCl (pH 7.5), 195 mM NH4OAc, and 30 mM Mg(OAc)2) were added, and further incubated at 37ºC for 10 min. After the incubation, 4 μl of GSA buffer (0.05% bromophenol blue and 50% glycerol) was added to the reaction mixture, and 10 μl of solution was applied to 8% native gel buffered with 50 mM Tris-acetate (pH 6.8), 65 mM NH4OAc, 10 mM Mg(OAc)2, and 10 μM GTP. The remaining solution was mixed with an equal volume of loading solution (9 M urea, 0.2% XC, and 0.2% BPB), and then 20 μl of the solution was subjected to electrophoresis on a 7 M urea-containing 10% polyacrylamide gel. To assess the susceptibility of tRNA forming TC, EF-Tu and GTP were pre-incubated as described above. To maintain tRNA as a free-form, EF-Tu was pre-incubated without GTP. Then, 200 pmol of aminoacyl-tRNA was dissolved in 6 μl of buffer containing 50 mM Tris-HCl (pH 7.5), 1 mM MgCl2, and 1 mM DTT and 4 μl of aaBind buffer was added to the pre-incubated EF-Tu solution. Additionally, BSA was added to give a final concentration of 100 μg/ml, and the total volume of the resultant solution was 20 μl. D-CRD was diluted to 1.1, 3.3 and 10 nM with a buffer containing 50 mM Tris-HCl (pH 7.5), 1 mM MgCl2, 1 mM DTT, and 100 μg/ml BSA. Then 1 μl of each D-CRD solution was added to the EF-Tu solution and incubated at 37ºC for 15 min. After the incubation, 4 μl of GSA buffer was added, followed by electrophoresis and northern blot hybridization carried out as described above.
In vitro translation assay
PURExpress Δ (aa, tRNA) kit (New England Biolabs) was used for an in vitro translation assay. In the kit, tRNA is separately provided, whereby the ratio of nicked tRNA to intact tRNA can be determined as desired. E. coli β-galactosidase gene was cloned into NcoI-SalI site of pET24d (+) to generate pET24-LacZ; this plasmid was used as a template DNA for in vitro translation. Total aminoacyl-RNA was prepared from E coli W3110 as described above. Prepared total tRNA was incubated with D-CRD in a buffer containing 50 mM Tris-HCl (pH 7.5), 1 mM MgCl2, 1 mM DTT, and 100 μg/ml BSA at 37°C for 15 min. Phenol/chloroform extraction was performed twice to remove D-CRD and BSA. After ethanol precipitation, total tRNA was dissolved in 10 mM sodium acetate (pH 5.2) and stored at −20°C. Acylated tRNA or that incubated with D-CRD was diluted with 10 mM sodium acetate (pH 5.2), and 0.4 μg of each was mixed with 12 μg of total tRNA present in the kit, and pre-incubated in 4 μl of buffer containing 50 mM Tris-HCl (pH 7.5) and 2 mM MgCl2 at 37°C for 10 min. Subsequently, 1.2 μl of Solution A, 1.8 μl of Solution B, 0.6 μl of Amino Acid Mixture (3 mM each), and 60 ng of template DNA were added to the tRNA solution and incubated at 37°C to initiate translation. At each interval, 0.5 μl was withdrawn and diluted with 50 mM Tris-HCl (pH 7.5) and 100 μg/ml BSA. Then, 0.5 μl of diluted solution was mixed with 30 μl of Beta-Glo Assay System (Promega) and the activity of β-galactosidase was measured with Luminescencer-PSN AB2200 (ATTO).
Molecular modelling of D-CRD∙tRNA
A model structure of the E. coli tRNAArgICG ASL was generated based on the structure of S. cerevisiae tRNAPhe (PDB code: 1EHZ, positions 27–43). The bases of tRNAPhe were replaced with those of tRNAArgICG at the corresponding positions. The coordinates of D-CRD were taken from those of the D-CRD∙ImmD complex structure (PDB code: 1TFO) determined by our group [Citation31]. The model of the tRNAArgICG ASL was docked with D-CRD using the FTDock program [Citation32]. The generated complex structures were ranked according to max(d1, d2), where d1 and d2 are the distances between His611 Nε2 and A38 O2′, and between Lys610 Nζ and C39 OP1 (an oxygen atom of the phosphate group between A38 and C39), respectively, and max(x, y) is a function that returns larger x and y values. We visually inspected the higher-ranked structures, noting that the residual part of tRNA would not overlap with D-CRD, and selected several models.
We next performed molecular dynamics (MD) simulations to evaluate the stability of the selected complex models. Each model was immersed in a cubic box of water with an edge length of about 70 Å. Potassium ions were included to neutralize the system. Amber ff14SB [Citation47], ff99OL3 [Citation48,Citation49], and modrna08 [Citation50] force-field parameters were used for the protein, tRNA, and modified bases of the tRNA. The TIP3P model [Citation51] was used for water. After energy minimization and equilibration, restrained MD runs were performed for 100 ns. In these runs, distances d1 and d2 were restrained with a harmonic potential. In addition, because Lys608 Nζ was close to C40 OP1 as well as to C39 OP1, another restraint was imposed on either the distance between Lys608 Nζ and C39 OP1 or the distance between Lys608 Nζ and C40 OP1. The force constant of the harmonic potential was gradually decreased from 4000 kJ mol−1 nm−2 to 100 kJ mol−1 nm−2. Subsequently, unrestrained MD runs were performed for 1 μs. During the MD simulations, the temperature was maintained at 300 K using the velocity-rescaling method [Citation52], and the pressure was kept at 1.0 × 105 Pa using the Berendsen weak coupling method [Citation53]. Bond lengths involving hydrogen atoms were constrained using the LINCS algorithm [Citation54,Citation55] to allow the use of a large time step (2 fs). Electrostatic interactions were calculated with the particle mesh Ewald method [Citation56,Citation57]. All MD simulations were performed with Gromacs 2016.4 [Citation58], and the coordinates were recorded every 10 ps. A structural model of D-CRD complexed with tRNA was generated by superposing the phosphate backbone of the anticodon stem (positions 27–30 and 40–43) in ASL∙D-CRD model on that of the yeast tRNAPhe structure.
Author contributions
T. O and H. M. designed the study; T. O., K. T., and W. I. conducted research; T. O., K. T., W. I., T. A., M. H., and H. M. analyzed the data; T. A. performed statistical analysis; K. T. and T. T. generated structural models; T. O., K. T., T. T, and H. M. wrote the paper; H. M. supervised the study.
Supplemental Material
Download MS Word (36.3 MB)Acknowledgments
We thank Dr. Kotaro Nakanishi (The Ohio State University) for the valuable discussions. We also thank Dr. Kozo Tomita (The University of Tokyo) for providing plasmid pBSup3+(CCAOH). We would like to extend our gratitude to Dr. Takuya Ueda (Waseda University) for providing ArgRS and EF-Tu, which were used for the initial screening of experimental conditions. We would also like to acknowledge the National BioResource Project (NIG, Japan): E. coli for providing strains of Keio collection and ASKA clone.
Disclosure statement
The authors report no conflict of interest.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
References
- Ogawa T. tRNA-targeting ribonucleases: molecular mechanisms and insights into their physiological roles. Biosci Biotechnol Biochem. 2016;80:1037–1045.
- Mock M, Pugsley AP. The BtuB group col plasmids and homology between the colicins they encode. J Bacteriol. 1982;150:1069–1076.
- Timmis K, Hedges AJ. The killing of sensitive cells by colicin D. Biochim Biophys Acta. 1972;262:200–207.
- Ogawa T, Tomita K, Ueda T, et al. A cytotoxic ribonuclease targeting specific transfer RNA anticodons. Science. 1999;283:2097–2100.
- Tomita K, Ogawa T, Uozumi T, et al. A cytotoxic ribonuclease which specifically cleaves four isoaccepting arginine tRNAs at their anticodon loops. Proc Natl Acad Sci U S A. 2000;97:8278–8283.
- Cascales E, Buchanan SK, Duché D, et al. Colicin biology. Microbiol Mol Biol Rev. 2007;71:158–229.
- Chauleau M, Mora L, Serba J, et al. FtsH-dependent processing of RNase colicins D and E3 means that only the cytotoxic domains are imported into the cytoplasm. J Biol Chem. 2011;286:29397–29407.
- Ogawa T, Inoue S, Yajima S, et al. Sequence-specific recognition of colicin E5, a tRNA-targeting ribonuclease. Nucleic Acids Res. 2006;34:6065–6073.
- Lu J, Huang B, Esberg A, et al. The Kluyveromyces lactis gamma-toxin targets tRNA anticodons. RNA. 2005;11:1648–1654.
- Klassen R, Paluszynski JP, Wemhoff S, et al. The primary target of the killer toxin from Pichia acaciae is tRNA(Gln). Mol Microbiol. 2008;69:681–697.
- Meineke B, Kast A, Schwer B, et al. A fungal anticodon nuclease ribotoxin exploits a secondary cleavage site to evade tRNA repair. RNA. 2012;18:1716–1724.
- Sprinzl M, Vassilenko K. Compilation of tRNA sequences and sequences of tRNA genes. Nucleic Acids Res. 2005;33:D139–140.
- Kaufmann G. Anticodon nucleases. Trends Biochem Sci. 2000;25:70–74.
- Page R, Peti W. Toxin-antitoxin systems in bacterial growth arrest and persistence. Nat Chem Biol. 2016;12:208–214.
- Winther KS, Gerdes K. Enteric virulence associated protein VapC inhibits translation by cleavage of initiator tRNA. Proc Natl Acad Sci U S A. 2011;108:7403–7407.
- Cruz JW, Sharp JD, Hoffer ED, et al. Growth-regulating Mycobacterium tuberculosis VapC-mt4 toxin is an isoacceptor-specific tRNase. Nat Commun. 2015;6:7480.
- Aoki SK, Pamma R, Hernday AD, et al. Contact-dependent inhibition of growth in Escherichia coli. Science. 2005;309:1245–1248.
- Ruhe ZC, Low DA, Hayes CS. Bacterial contact-dependent growth inhibition. Trends Microbiol. 2013;21:230–237.
- Aoki SK, Diner EJ, de Roodenbeke CT, et al. A widespread family of polymorphic contact-dependent toxin delivery systems in bacteria. Nature. 2010;468:439–442.
- Nikolakakis K, Amber S, Wilbur JS, et al. The toxin/immunity network of Burkholderia pseudomallei contact-dependent growth inhibition (CDI) systems. Mol Microbiol. 2012;84:516–529.
- Diner EJ, Beck CM, Webb JS, et al. Identification of a target cell permissive factor required for contact-dependent growth inhibition (CDI). Genes Dev. 2012;26:515–525.
- McClain WH, Foss K. Changing the acceptor identity of a transfer RNA by altering nucleotides in a “variable pocket”. Science. 1988;241:1804–1807.
- McClain WH, Foss K, Jenkins RA, et al. Nucleotides that determine Escherichia coli tRNA(Arg) and tRNA(Lys) acceptor identities revealed by analyses of mutant opal and amber suppressor tRNAs. Proc Natl Acad Sci U S A. 1990;87:9260–9264.
- Schulman LH, Pelka H. The anticodon contains a major element of the identity of arginine transfer RNAs. Science. 1989;246:1595–1597.
- Tamura K, Himeno H, Asahara H, et al. In vitro study of E.coli tRNA(Arg) and tRNA(Lys) identity elements. Nucleic Acids Res. 1992;20:2335–2339.
- Liu W, Huang Y, Eriani G, et al. A single base substitution in the variable pocket of yeast tRNA(Arg) eliminates species-specific aminoacylation. Biochim Biophys Acta. 1999;1473:356–362.
- Stephen P, Ye S, Zhou M, et al. Structure of Escherichia coli Arginyl-tRNA synthetase in complex with tRNA. J Mol Biol. 2018;430:1590–1606.
- Yajima S, Inoue S, Ogawa T, et al. Structural basis for sequence-dependent recognition of colicin E5 tRNase by mimicking the mRNA-tRNA interaction. Nucleic Acids Res. 2006;34:6074–6082.
- Jiang Y, Blanga S, Amitsur M, et al. Structural features of tRNA(Lys) favored by anticodon nuclease as inferred from reactivities of anticodon stem and loop substrate analogs. J Biol Chem. 2002;277:3836–3841.
- Lu J, Esberg A, Huang B, et al. Kluyveromyces lactis gamma-toxin, a ribonuclease that recognizes the anticodon stem loop of tRNA. Nucleic Acids Res. 2008;36:1072–1080.
- Yajima S, Nakanishi K, Takahashi K, et al. Relation between tRNase activity and the structure of colicin D according to X-ray crystallography. Biochem Biophys Res Commun. 2004;322:966–973.
- Katchalski-Katzir E, Shariv I, Eisenstein M, et al. Molecular surface recognition: determination of geometric fit between proteins and their ligands by correlation techniques. Proc Natl Acad Sci U S A. 1992;89:2195–2199.
- Inoue-Ito S, Yajima S, Fushinobu S, et al. Identification of the catalytic residues of sequence-specific and histidine-free ribonuclease colicin E5. J Biochem. 2012;152:365–372.
- Graille M, Mora L, Buckingham RH, et al. Structural inhibition of the colicin D tRNase by the tRNA-mimicking immunity protein. EMBO J. 2004;23:1474–1482.
- Geslain R, Martin F, Camasses A, et al. A yeast knockout strain to discriminate between active and inactive tRNA molecules. Nucleic Acids Res. 2003;31:4729–4737.
- Bi K, Zheng Y, Gao F, et al. Crystal structure of E. coli arginyl-tRNA synthetase and ligand binding studies revealed key residues in arginine recognition. Protein Cell. 2014;5:151–159.
- Dittmar KA, Sørensen MA, Elf J, et al. Selective charging of tRNA isoacceptors induced by amino-acid starvation. EMBO Rep. 2005;6:151–157.
- Garza-Sanchez F, Gin JG, Hayes CS. Amino acid starvation and colicin D treatment induce A-site mRNA cleavage in Escherichia coli. J Mol Biol. 2008;378:505–519.
- Garza-Sánchez F, Janssen BD, Hayes CS. Prolyl-tRNA(Pro) in the A-site of SecM-arrested ribosomes inhibits the recruitment of transfer-messenger RNA. J Biol Chem. 2006;281:34258–34268.
- Muto H, Nakatogawa H, Ito K. Genetically encoded but nonpolypeptide prolyl-tRNA functions in the A site for SecM-mediated ribosomal stall. Mol Cell. 2006;22:545–552.
- Keiler KC, Waller PR, Sauer RT. Role of a peptide tagging system in degradation of proteins synthesized from damaged messenger RNA. Science. 1996;271:990–993.
- Sakai F, Sugita R, Chang JW, et al. Transfer-messenger RNA and SmpB mediate bacteriostasis in Escherichia coli cells against tRNA cleavage. Microbiology. 2015;161:2019–2028.
- Milligan J, Uhlenbeck O. Synthesis of small RNAs using T7 RNA polymerase. Methods Enzymol. 1989;180:51–62.
- Tomita K, Weiner A. Collaboration between CC- and A-adding enzymes to build and repair the 3ʹ-terminal CCA of tRNA in Aquifex aeolicus. Science. 2001;294:1334–1336.
- Varshney U, Lee CP, RajBhandary UL. Direct analysis of aminoacylation levels of tRNAs in vivo. Application to studying recognition of Escherichia coli initiator tRNA mutants by glutaminyl-tRNA synthetase. J Biol Chem. 1991;266:24712–24718.
- Ohtsuki T, Yamamoto H, Doi Y, et al. Use of EF-Tu mutants for determining and improving aminoacylation efficiency and for purifying aminoacyl tRNAs with non-natural amino acids. J Biochem. 2010;148:239–246.
- Maier JA, Martinez C, Kasavajhala K, et al. ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. J Chem Theory Comput. 2015;11:3696–3713.
- Pérez A, Marchán I, Svozil D, et al. Refinement of the AMBER force field for nucleic acids: improving the description of alpha/gamma conformers. Biophys J. 2007;92:3817–3829.
- Zgarbová M, Otyepka M, Sponer J, et al. Refinement of the Cornell et al. nucleic acids force field based on reference quantum chemical calculations of glycosidic torsion profiles. J Chem Theory Comput. 2011;7:2886–2902.
- Aduri R, Psciuk BT, Saro P, et al. AMBER force field parameters for the naturally occurring modified nucleosides in RNA. J Chem Theory Comput. 2007;3:1464–1475.
- Jorgensen WL, Chandrasekhar J, Madura JD, et al. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79:926–935.
- Bussi G, Donadio D, Parrinello M. Canonical sampling through velocity rescaling. J Chem Phys. 2007;126:014101.
- Berendsen HJC, Postma JPM, Vangunsteren WF, et al. Molecular-dynamics with coupling to an external bath. J Chem Phys. 1984;81:3684–3690.
- Hess B. P-LINCS: a parallel linear constraint solver for molecular simulation. J Chem Theory Comput. 2008;4:116–122.
- Hess B, Bekker H, Berendsen HJC, et al. LINCS: A linear constraint solver for molecular simulations. J Comput Chem. 1997;18:1463–1472.
- Darden T, York D, Pedersen L. Particle Mesh Ewald - AN N.LOG(N) method for Ewald sums in large systems. J Chem Phys. 1993;98:10089–10092.
- Essmann U, Perera L, Berkowitz ML, et al. A smooth particle Mesh Ewald method. J Chem Phys. 1995;103:8577–8593.
- Hess B, Kutzner C, van der Spoel D, et al. GROMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation. J Chem Theory Comput. 2008;4:435–447.