
ABSTRACT
One important task of eukaryotic cells is to translate only mRNAs that were correctly processed to prevent the production of truncated proteins, found in neurodegenerative diseases and cancer. Nuclear quality control of splicing requires the SR-like proteins Gbp2 and Hrb1 in S. cerevisiae, where they promote the degradation of faulty pre-mRNAs. Here we show that Gbp2 and Hrb1 also function in nonsense mediated decay (NMD) of spliced premature termination codon (PTC)-containing mRNAs. Our data support a model in which they are in a complex with the Upf-proteins and help to transmit the Upf1-mediated PTC recognition to the transcripts ends. Most importantly they appear to promote translation repression of spliced transcripts that contain a PTC and to finally facilitate degradation of the RNA, presumably by supporting the recruitment of the degradation factors. Therefore, they seem to control mRNA quality beyond the nuclear border and may thus be global surveillance factors. Identification of SR-proteins as general cellular surveillance factors in yeast will help to understand the complex human system in which many diseases with defects in SR-proteins or NMD are known, but the proteins were not yet recognized as general RNA surveillance factors.
Introduction
All cellular processes depend on the correct and effective translation of mRNAs into proteins. Eukaryotic cells control the correctness of the mRNAs both in the nucleus and in the cytoplasm [Citation1–5]. While the nuclear quality control recognizes rather structural defects of the transcripts, the cytoplasmic quality control detects incorrect open reading frames through the decoding capability of the ribosome. Structural defects include transcripts that have retained their introns or mRNAs that are uncapped or non-polyadenylated [Citation4,Citation5]. Such defects can create problems in translation, as both transcript ends are usually connected in the cytoplasm to allow a repeated cycling of the ribosomes on transcripts to increase translation efficiency [Citation6].
Several proteins contribute to generating correctly matured mRNAs [Citation4,Citation7]. However, one group of proteins is particularly important for the nuclear mRNA surveillance in yeast, because their absence results in the leakage of faulty mRNAs into the cytoplasm [Citation8]. These guard proteins include Npl3, Gbp2, Hrb1 and Nab2, the first three of which are highly homologous with human serine arginine (SR)-proteins [Citation5]. Among them, Gbp2 and Hrb1 preferentially bind to transcripts that undergo splicing, as they interact with late splicing factors [Citation9]. In case splicing does not occur correctly, Gbp2 and Hrb1 recruit the TRAMP-complex, which fetches the nuclear exosome to degrade the faulty transcript. On correctly spliced mRNAs Gbp2 and Hrb1 interact with Mex67-Mtr2 (TAP-p15 in human) instead, promoting nuclear export [Citation9]. Other guard proteins control different maturation steps, but they operate similarly in principle. Proper packaging of the RNA into a ribonucleoparticle (RNP) supports transit through the nuclear pore complex (NPC) [Citation4,Citation5]. At the NPC, the nuclear basket protein Mlp1 controls proper Mex67-coverage of the guard proteins on the mRNA [Citation4,Citation5]. Thus, also in the absence of Mlp1, faulty transcripts are not retained in the nucleus and leak into the cytoplasm [Citation9,Citation10].
Remarkably, while Mex67 is removed upon transport, the guard proteins remain bound on the transcript until translation [Citation11]. This suggests that they may have additional functions in the cytoplasm and might continue their roles as quality control factors. In particular, because after dissociation of Mex67 they are free for new interactions. However, such a cytoplasmic quality control function for the guard proteins has not been explored to date. In contrast to the nuclear quality control system, the cytoplasmic quality control checks the encoded sequence. During translation, intact or faulty open reading frames can be distinguished. In this way, broken mRNAs that lack a stop codon or mRNAs with strong secondary structures that stall the ribosome are eliminated by the no-stop- (NSD) or no-go-decay (NGD), respectively [Citation7]. Another severe defect is premature termination, often resulting from improper splicing and transcripts that escaped nuclear quality control [Citation1,Citation12]. PTC-containing transcripts are recognized and eliminated by the nonsense-mediated decay (NMD). In its centre are the Upf-proteins. Upon interacting with eRF1 and eRF3 at the terminating ribosome, they signal the cell to inhibit translation and degrade affected mRNAs [Citation1,Citation3,Citation13]. Upf1 is the central ATP-dependent RNA helicase and required for the recognition of PTC-containing mRNAs. Upf2 and Upf3 support NMD by formation of a Upf1-Upf2-Upf3 complex signalling degradation [Citation1,Citation3,Citation13].
How NMD is initiated is still debated. In metazoans, Upf2 and Upf3 bind to the exon-junction complex (EJC) downstream of the PTC, and are thought to communicate somehow with the ribosome to trigger RNA degradation [Citation1,Citation3,Citation13]. Interestingly, some human SR proteins, which are known to be important mediators of splicing, are also associated with the EJC [Citation14]. However, their function in NMD is rather nebulous, not the least because of their upstream role in splicing, which is difficult to study independently from their potential subsequent function in NMD. Although EJCs are characteristic for multicellular organisms and have not been discovered in S. cerevisiae, a similar system was described in yeast, involving Hrp1. Hrp1 is a yeast RNA-binding protein that binds to a downstream sequence element (DSE), originally identified in the PGK1 mRNA. Hrp1 was shown to interact with the Upf-proteins when the PGK1 contained a PTC [Citation15]. Another proposed trigger of NMD is a long 3ʹ-untranslated region (UTR). A long distance between the terminating ribosome and the poly(A) tail impedes the interaction between the poly(A) binding protein Pab1 (PABP in human) and the terminating ribosome, which under regular conditions promotes efficient termination. In this model, multiple copies of Upf1 are distributed on the RNA in a loosely bound fashion and removed by the passing ribosome. However, when termination occurs prematurely, Upf1-binding is stabilized. As Pab1 is far away, formation of a stable Upf1-2-3 complex is promoted and NMD is elicited [Citation1,Citation16].
While the exact mechanism of NMD activation is relatively vague, our understanding of the downstream events is even less clear. It seems evident that PTC-containing mRNAs are translationally repressed and mainly degraded from the 5ʹ ends in yeast [Citation17]. Yet, how this is mediated is currently unclear. It is known that the ATPase activity of Upf1 is required to disassemble the ribosome and allow complete degradation of the transcript [Citation18,Citation19]. It is also known that Upf1 can be found in a complex with decay enzymes [Citation20]. Dcp1 and Dcp2 are required for decapping of NMD targets and Xrn1 for the subsequent 5ʹ to 3ʹ exonucleolytic RNA decay. However, how the degradation factors are recruited from the PTC to the distant ends of the transcript is unclear. Although the 5ʹ-end mediated degradation pathway is mainly used, decay can also occur from the 3ʹ-end via the exosome and its cytoplasmic co-factor complex, containing Ski2 [Citation1,Citation21]. In metazoans degradation of the recognized NMD targets is supported by additional factors, such as SMG6, which cleaves NMD-transcripts endonucleolytically, and SMG5-SMG7, which bridge interactions between Upf1 and degrading enzymes at the PTC [Citation1,Citation22]. In yeast, similar auxiliary factors are poorly understood, and likely more factors participate in NMD than currently known.
Our study presented here suggests that the nuclear and the cytoplasmic mRNA quality control systems may be coupled. We found the nuclear guard proteins Gbp2 and Hrb1 to be players in NMD. They continue their guarding function, originally discovered in the nucleus, further in the cytoplasm. For a subset of targets, those that contained intron sequences, these SR-like proteins appear to help repress translation upon detection of a PTC and to recruit the cytoplasmic degradation machineries to the faulty transcript. Importantly, by bridging the Upf1-bound PTC to the 5ʹ end of the mRNA Gbp2 and Hrb1 may help to transmit the signal of the mRNA defect directly to the starting point of translational repression and degradation.
Materials and methods
Saccharomyces cerevisiae strains used in this study are listed in Table S1, plasmids in Table S2 and oligonucleotides in Table S3. Yeast strains and plasmids were generated by conventional methods. Yeast strains were cultivated in standard media at 25°C and harvested in log phase at 1–3 × 107 cells/ml or OD600 0.5–1.3.
Method details
Induction of NMD reporters with galactose responsive promoters
For induction of NMD reporters under the control of the GAL1 promoter, yeast cells were grown in media containing sucrose instead of glucose. The promoter was induced by addition of 2% galactose for 2 hours before harvesting, with the following exceptions: In reporters with the endogenous CBP80 and DBP2 promoters were used. In the NMD reporter was induced for 20 min after which transcription was stopped with 2% glucose. Cells were harvested after another 30 min of growth. In Fig. A, C, G DBP2PTC was not induced, cells were grown in sucrose to maintain a low transcription rate. In , E, G, I CBP80PTC was induced for 4 h.
Figure 1. The nuclear guard proteins Gbp2 and Hrb1 show features of NMD factors. (A) Protein aggregation is increased in cells lacking GBP2 and HRB1. Localization of RFP-tagged Hsp104 is shown in the indicated strains that were grown to the logarithmic growth phase at 25°C and shifted to 37°C for 1 h. (B) Cells that contain Hsp104-RFP foci were counted and the percentage of cells with aggregates is shown. 300 cells were counted per experiment and error bars represent the standard deviation between different experiments. n = 3 (wild type and upf1∆ n = 6). (C) Scheme of the used reporter constructs. See also Fig S1A. (D) Gbp2 and Hrb1 do not function in the Upf1-mediated decay of the intron-less PGK1 transcript. PTC-containing transcripts were expressed by 2 h galactose induction and monitored by qPCR. Newly generated DBP2PTC and CBP80PTC reporters were expressed in wild type and upf1∆ to compare them to the established PGK1PTC reporter. n = 3 (PGK1PTC),n= 5 (DBP2PTC, CBP80PTC). (E) Gbp2 and Hrb1 are required for the effective degradation of the PTC-containing, spliced DBP2PTC and CBP80PTC transcripts. Transcripts were expressed using their endogenous promoters. qPCRs from RNA of the indicated strains were carried out in the presence or the absence of the PTC and are shown in relation. The average wild type level of PTC-containing NMD reporter per PTC-less reporter was set to 1 and other data are shown in relation. n = 4 and n = 4 (gbp2∆ hrb1∆ n = 7), respectively. See also Fig S1B. (F) The binding of Upf1 to the PTC-containing reporter RNA is independent of Gbp2 and Hrb1. RNA-co-immunoprecipitation (RIP) experiments of Upf1-GFP were carried out in the indicated strains and the amount of bound PTC-reporter transcript was normalized to RPS6A mRNA. n = 8 (gbp2∆ hrb1∆), n = 4 (upf2∆). See also Fig S1C. (G) The binding of Gbp2 and Hrb1 to CBP80PTC and endogenous NMD substrates is reduced in the absence of Upf1. RIP experiments with Gbp2 and Hrb1 were done in wild type and upf1∆ cells and qPCR results are shown. RNA levels were normalized to 21S rRNA. CBP80PTC: Gbp2 n = 7, Hrb1 n = 6; GCR1: n = 6; HNT1: Gbp2 n = 7, Hrb1 n = 5. See also Fig S1D
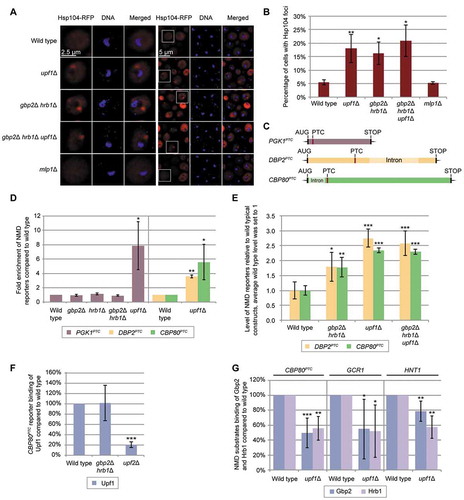
Figure 2. Gbp2 and Hrb1 are involved in translation repression of NMD targets. (A) Translation of the DBP2PTC reporter results in a truncated protein, shown on a western blot. Zwf1 served as a loading control. (B) Translation of the 5ʹ-proximal PTC-containing CBP80PTC reporter results in the expression of the full-length protein, shown on a western blot. (C-F) Proper translational repression of the DBP2PTC requires both Gbp2 and Hrb1 and proper translational repression of the CBP80PTC requires Gbp2. Expression of DBP2PTC (C) and CBP80PTC (E) in the indicated strains was monitored by western blot analysis. (D, F) Protein expression of independent experiments shown in (C) and (E) were quantified. MYC-Dbp2PTC (D) and MYC-Cbp80PTC (F) signals were normalized to the loading control and the relative reporter RNA level (Fig S2B, S2C). The standard deviation of upf1Δ cells is 4.1 and 6.5, respectively. n = 5. (G) The translational repression activity of Gbp2 and Hrb1 requires Upf1. Expression of the PTC-containing reporter transcripts is shown in upf1∆ and upf1∆ gbp2∆ hrb1∆ cells. The asterisk indicates a band of Gbp2. (H) Protein expression shown in (G) was quantified as in (D) and (F). n = 4. (I) Known RGG motif translational repressors do not suppress translation of PTC-containing transcripts. Expression of the CBP80PTC was compared in the indicated strains on western blots. See also Fig S2E. (J) Protein level of three independent experiments, one of which is shown in (I), was quantified. MYC-Cbp80PTC signals were normalized to the loading control Zwf1. Results for gbp2Δ and upf1Δ are replotted from previous experiments for comparison (Fig S2C)
Co-Immunoprecipitation (IP)
GFP fusion proteins were purified using GFP-Trap_A beads (Chromotek, gta-400) or GFP-selector beads (Nanotag Biotechnologies, N0310), following the manufacturer’s instructions. Cell pellets were lysed in 1x volume cold PBSKMT buffer (137 mM NaCl, 5.7 mM KCl, 10 mM KH2PO4, 2 mM Na2HPO4 2.5 mM MgCl2, 0.5% Triton X-100) with protease inhibitor (5 μl per 100 μl cell pellet, Merck, 11,697,498,001). One pellet-volume of glass beads (0.4–0.6 mm) was added and cells were lysed in a FastPrep-24 (MP Biomedicals) at 4 m/s for 30 s twice. Glass beads and cell debris were removed by centrifugation at 16000x g for 1 min at 4°C and the supernatant was further cleared by centrifugation at 16000x g for 10 min at 4°C. Approx. 2% of the cleared lysate was kept as lysate sample for western blot analysis. The remaining lysate was incubated with equilibrated GFP-Trap_A beads (Chromotek) () or GFP-selector beads (Nanotag Biotechnologies) (–D) for 2 h at 4°C. Where indicated, 200 μg/ml RNase A was added. The conditions for RNA removal were verified by qPCR. The beads were washed 4–8 times with PBSKMT and resuspended in SDS sample buffer (125 mM Tris – pH 6.8, 4% (w/v) SDS, 20% (v/v) glycerol, 0.05% (w/v) Bromophenol blue and 5% (v/v) 2-mercaptoethanol)). The complete eluate and the lysate samples were used for SDS-PAGE and western blot analysis with the indicated antibodies (GFP (GF28R, Thermo Fischer Scientific, MA5-15256) 1/50000, Zwf1 (Merck, A9521) 1/50000, Tdh1 (GA1R, Thermo Fischer, MA5-15738) 1/50000, Hem15 (U. Mühlenhoff) 1/5000, Gbp2 (self-made) 1/50000, Hrb1 (self-made) 1/20000, c-MYC (9E10, Santa Cruz, Sc-40) 1/750, HA (F-7, Santa Cruz, sc-7392) 1/750, Grx4 (U. Mühlenhoff) 1/1000.
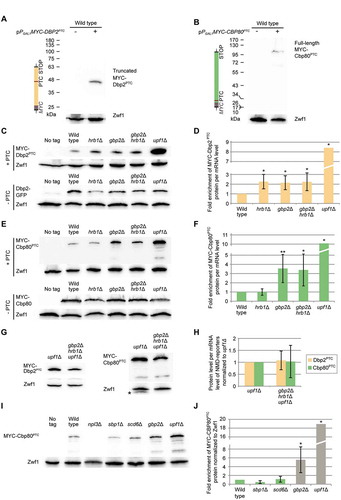
Figure 3. Gbp2 and Hrb1 seem to take part in NMD in the cytoplasm. (A) Gbp2 and Hrb1 co-precipitate with all three Upf proteins. Western blot analysis of co-IPs of Gbp2 and Hrb1 with GFP-tagged Upf1, Upf2 and Upf3 are shown. GFP-tagged Upf-proteins were not detectable in the lysates. Hem15 served as a negative control. (B) The interaction of Gbp2 and Upf1 increases when the ATPase activity of the helicase is defective. A western blot of a Upf1-GFP and upf1-DE572AA-GFP IP and Gbp2 and Hrb1 co-precipitation is shown. (C) The binding of Gbp2 and Hrb1 with upf1-DE572AA shown in (B) was quantified from independent experiments. n = 7. The signal intensities of the Gbp2 and Hrb1 bands were related to the corresponding Upf1- or upf1-DE572AA-GFP pull-down signals. (D) Upf1 and Gbp2 physically interact. Split-GFP experiments with the indicated plasmids are shown. Cells expressing either N-GFPsplit or C-GFPsplit alone were used as negative controls. The experiments were performed in xrn1∆ cells to reduce the degradation of PTC-containing transcripts after NMD initiation. If indicated, pPGAL1:CBP80PTC was induced for 2 h to increase the presence of PTC-mRNAs. The signal of 100 cells was quantified per experiment. n = 3. (E) Both Gbp2 and Hrb1 mislocalize to the cytoplasm when PTC-containing transcripts cannot be degraded efficiently. GFP-tagged Gbp2 and Hrb1 were localized by fluorescence microscopy in wild type, xrn1∆ and upf1∆ xrn1∆ cells in the presence or absence of the indicated PTC-reporter plasmids. Cell cultures were split in two and expression of the reporter constructs was induced for 2 h in one sample. (F) Quantification of the experiments shown in (E). Error bars represent the standard deviation between independent experiments with 100–200 analysed cells per experiment. n = 3. See also Fig S3D
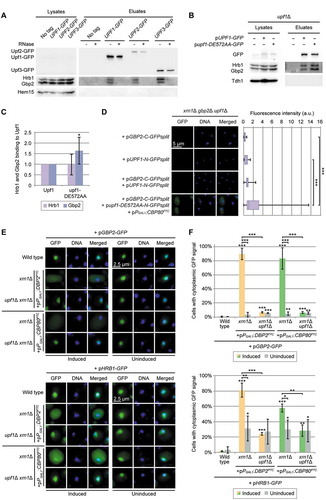
Figure 4. Hrb1 is involved in the recruitment of the 5ʹ-end degradation machinery. (A) Gbp2 and Hrb1 co-precipitate with Dcp1. Western blots of co-IPs of Gbp2 and Hrb1 with Dcp1-GFP are shown. The asterisks indicate bands of Hrb1 from previous detection with the Hrb1 antibody. Tdh1 served as a negative control. For the RNase treated IP the cells were treated with 1% formaldehyde for 10 min at 25°C. After the precipitation, proteins were de-crosslinked for 20 min at 95°C. Dcp1-GFP was not detectable in the lysates. See also Fig S4B and S4C. (B) The interaction of Upf1 and Dcp1 is promoted by Gbp2 and Hrb1. Co-IPs of Upf1-HA with GFP-tagged Dcp1 in GBP2 HRB1 and gbp2∆ hrb1∆ cells are shown on western blots. All cells are deleted for UPF1, and express pUPF1-HA. The asterisk indicates an unspecific cross-reaction with the GFP antibody. Dcp1-GFP was not detectable in the lysates. Tdh1 served as a negative control. (C) Quantification of seven independent co-IPs shown in (B). Signal intensities of the Upf1-HA bands were related to the corresponding Dcp1-GFP pull-down signals. (D) The binding of Dcp1 to a PTC-containing transcript is disturbed in the HRB1 knock out. Dcp1 RIP experiments and subsequent qPCRs were carried out in the indicated strains. All strains express genomic DCP1-GFP. n = 5 (hrb1∆ n = 6). Co-purified RNA levels were normalized to the endogenous wild-typical CBP80 mRNA and the total levels from whole-cell lysates. Dashed lines indicate the level of wild type and average level of upf1∆. See also Fig S4D. (E) Xrn1 interacts with Gbp2 and Hrb1. Gbp2 and Hrb1 co-IPs with Xrn1-GFP are shown on western blots. Hem15 served as a negative control. (F) Xrn1 recruitment to PTC-containing substrates is Upf1- but not Gbp2- or Hrb1-dependent. Xrn1 RIP experiments and subsequent qPCRs with the PTC-containing reporter are shown in the indicated strains. All strains express genomic XRN1-GFP. n = 3. See also Fig S4E. (G) Decapping of CBP80PTC RNA is defective without Gbp2 or Hrb1. RNA was isolated in the indicated strains containing the CBP80PTC reporter. A sample of this RNA was used for in vitro Xrn1 digestion, which can only degrade decapped RNAs. CBP80PTC and endogenous CBP80 were detected after Xrn1 digestion via qPCR and normalized to control samples without Xrn1 digestion. n = 6 (upf1∆ n = 4)
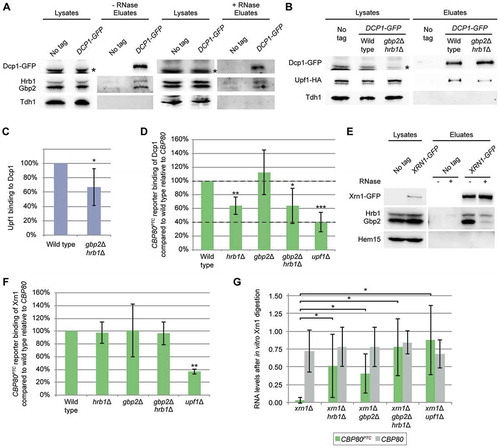
Figure 5. Gbp2 and Hrb1 are involved in the recruitment of the 3ʹ-end degradation machinery. (A) Ski2 co-precipitates Gbp2 and Hrb1. Western blot of Gbp2 and Hrb1 co-IPs with Ski2-GFP is shown. Ski2-GFP was not detectable in the lysate. See also Fig S5B. (B) Proper interaction of Upf1 and Ski2 requires Gbp2 and Hrb1. Upf1-HA co-IPs with Ski2-GFP are shown on a western blot in the indicated strains. All cells are deleted for UPF1, and express pUPF1-HA. (C) The Ski2 and Upf1 interaction shown in (B) was quantified. Signal intensities of Upf1-HA bands were related to the corresponding Ski2-GFP pull-down signals from three independent co-IPs. (D) Gbp2 and Hrb1 promote the Ski2 interaction with the CBP80PTC transcript. Ski2 RIP experiments and subsequent qPCRs with the PTC-containing reporter are shown in the indicated strains. All strains express genomic SKI2-GFP. n = 6 (gbp2∆ n = 8, gbp2∆ hrb1∆ n = 7). Dashed lines indicate the level of wild type and average level of upf1∆. See also Fig S5C
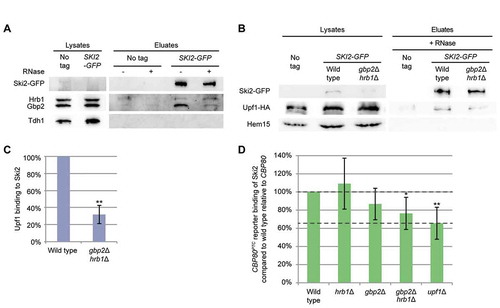
Figure 6. Gbp2 and Hrb1 might help to transmit the Upf1-mediated PTC alert to the 5ʹ-end of the mRNA. (A) Gbp2 and (B) Hrb1 interact with each other and themselves. Co-IPs of differently tagged and untagged Gbp2 and Hrb1 versions upon RNase treatment are shown. Hem15 served as a negative control. Gbp2-GFP was not always detectable in the lysates. The asterisks indicate Gbp2 bands. (C) Gbp2 and Hrb1 interact with eIF4E and eIF4G. Co-IP of Gbp2 and Hrb1 with GFP tagged versions of the 5ʹ mRNA-binding proteins is shown. The asterisks indicate Hrb1 (top) and Gbp2 (bottom) bands. (D) The Upf1 interaction with eIF4G is significantly reduced in gbp2∆ hrb1∆ upon RNase treatment. Co-IP of Upf1 with eIF4G is shown in the indicated strains. pPGAL1:CBP80PTC was induced for 2 h. All cells express pUPF1-HA. (E) Quantification of IP experiments shown in (D). Signal intensities of the Upf1-HA bands were related to the corresponding eIF4G-GFP pull-down signals. Upf1-HA signals without RNase treatment were quantified using less-exposed figures than shown in Fig. 6D. No RNase n = 5, + RNase n = 3. See also Fig S6
For formaldehyde crosslinking (), yeast cells were treated with 1% formaldehyde for 10 min at 25°C prior to harvesting. The formaldehyde was quenched by adding 0.5 M glycine. Immunoprecipitation was performed as described above with 20 min decrosslinking at 95°C in SDS sample buffer before gel loading.
Yeast cell lysis for western blot analysis
For experiments shown in , log phase yeast cells were lysed in SDS sample buffer (125 mM Tris – pH 6.8, 4% (w/v) SDS, 20% (v/v) glycerol, 0.05% (w/v) Bromophenol blue and 5% (v/v) 2-mercaptoethanol) with one pellet volume (or 200 µl for smaller cell pellets) of glass beads (0.4–0.6 mm) and heated at 95°C for 5 min. Glass beads and cell debris were removed by centrifugation at 16000x g for 1 min. The supernatant was used for western blot experiments. For experiments shown in , yeast cell cultures were split before harvesting. One half was used for western blot analysis, the other half for RNA isolation (see below).
Quantification of western blot signals
Western blot signals were quantified with the Bio-1D software (Vilber Lourmat) by measuring the optical densities of western blot bands. With the Bio-1D software, images were selected in which the analysed signal intensities were not saturated. Background subtraction was performed using the rolling ball method and setting a detection threshold.
Fluorescence microscopy
Logarithmic yeast cells were fixated with 2.6% formaldehyde and immediately harvested by centrifugation at 3500x g for 5 min at 4°C. The cells were washed once with 0.1 M potassium phosphate buffer pH 6.5, once with P solution (0.1 M potassium phosphate buffer pH 6.5, 1.2 M Sorbitol) and resuspended in P solution. The cells were incubated 15 min on polylysine coated microscope slides and excess cells were removed. The cells were permeabilized with 0.5% Triton X-100 in P-solution for approx. 1 min and washed once with P solution and once with Aby wash 2 (0.1 M Tris – pH 9.5, 0.1 M NaCl). DNA was stained with DAPI (1 µg/ml in Aby wash 2) for 5 min and washed three times for 5 min with Aby wash 2. Microscope slides were dried and the cells mounted in 40% (v/v) glycerol, 20% (v/v) PBS (137 mM NaCl, 2.7 mM KCl, 10 mM KH2PO4, 2 mM Na2HPO4 and 1% (w/v) n-propyl gallate). Microscopy images were taken with a Leica AF6000 microscope and a LEICA DFC360FX camera with the LEICA AF 2.7.3.9 software. In , Z-stacks (10 images, 0.2 µm) were deconvoluted (blind, 3 iterations, with the LEICA AF 2.7.3.9 software).
Split GFP analysis
Proteins of interest were fused with either the N-terminal (amino acids 1–155) or the C-terminal (amino acids 156–239) part of eGFP. Fluorescence microscopy experiments were performed as described above, except cells were treated with 1.5% formaldehyde for fixation. For quantification of the fluorescence signal, 100 cells from each strain from each experiment were randomly chosen and the mean signal intensity of each cell was measured using Image J. Significant differences between strains were calculated by comparing all 300 signal intensity values.
RNA co-immunoprecipitation (RIP)
Immunoprecipitation (IP) was performed essentially as described above followed by RNA isolation. A no-tag control was always used to verify RNA-binding to the precipitated proteins. RIP experiments shown in , G. were performed without UV crosslinking. In the other RIP experiments () protein-RNA complexes were crosslinked by UV irradiation. For this the cells were treated two times for 3.5 min (0.6 J/cm in a 50 ml suspension in a 15 cm petri dish) with 254 nM UV light on a cold metal block, with light shaking in between. For RIP experiments the cells were lysed in two pellet volumes RIP buffer (150 mM NaCl, 2 mM MgCl2, 0.2 mM PMSF, 0.5 mM DTT, 0.2% (v/v) Triton X-100, 25 mM Tris/HCl – pH 7.5) with protease inhibitor (5 μl per 100 μl cell pellet, Merck, 11697498001) and RNase inhibitor (0.12 μl/100 μl pellet-volume RiboLock, Thermo Scientific, EO0381). Approx. 2% of the cleared lysate was kept as lysate sample for western blot analysis and 5–10% lysate was used as lysate sample for RNA isolation. DNaseI was added to the lysate sample (14 Kunitz units per 100 μl, Qiagen, 79256) and to the remaining RIP sample (6.5 Kunitz units per 100 μl). GFP tagged proteins were precipitated with GFP-Trap_A beads (Chromotek) ( (Gbp2)) or GFP-Selector beads (Nanotag Biotechnologies) () and the beads were washed 5–7 times with RIP buffer. Approx. 20% of the beads were resuspended in SDS sample buffer and used for western blot analysis. The remaining beads were used for RNA isolation with TRIzol (Thermo Fisher Scientific, 15596018), following the manufacturer’s instructions. In UV-crosslinked RIP experiments, the beads were washed two more times with proteinase K buffer (50 mM NaCl, 0.5 mM DTT, 0.2% Triton X-100, 50 mM Tris/HCl pH 7.5) and resuspended in 100 µl proteinase K buffer. Afterwards, 0.5% SDS, 5 mM EDTA and 80 μg (lysate sample) or 40 μg (eluate sample) Proteinase K was added and incubated 90 min at 55°C with shaking prior to RNA isolation. DNA was further removed with the TURBO DNA-free DNase kit (Thermo Fischer Scientific, AM1907).
Total RNA isolation
Total RNA was isolated from log phase yeast cultures using the NucleoSpin® RNA isolation kit (Macherey-Nagel, 740,955). DNA was further removed with the TURBO DNA-free DNase kit (Thermo Fischer Scientific, AM1907).
Reverse transcription and quantitative PCR
One microgram total RNA or 50–100 ng eluted RNA was reverse transcribed with the Maxima First Strand cDNA Synthesis Kit (Thermo Fisher Scientific, EP0741) or the FastGene Scriptase II kit (NIPPON Genetics, LS63). Reverse transcription was performed with either Oligo (dT)18 ( (Gbp2), ) or random primers (). qPCR was performed in triplicates, using the qPCRBIO SyGreen Mix Lo-ROX (NIPPON Genetics, PB20.11–50) in a CFX Connect 96FX2 qPCR cycler (BIO RAD).
Quantification and statistical analysis
All experiments were performed in independent biological replicates as indicated in the figure legends. All bar graphs show arithmetic mean values and error bars illustrate the standard deviation of biological replicates. P-values were calculated by unpaired, two tailed, homo- or heteroscedastic Student’s t-test and are indicated by * (p < 0.05), ** (p < 0.01) and *** (p < 0.001). The number of biological replicates is indicated as ‘n’ in the figure legends.
Results
The nuclear guard proteins Gbp2 and Hrb1 show features of NMD factors
Gbp2 and Hrb1 are established nuclear quality control proteins that shuttle with the mRNA into the cytoplasm [Citation9,Citation23]. Thus, it seems conceivable that they might also participate in the cytoplasmic mRNA quality control. It was shown earlier that defects in the NMD, NGD and NSD pathways result in increased protein aggregation due to progressive abnormal association of misfolded proteins in insoluble protein structures, central to the pathology of neurodegenerative diseases such as Alzheimer’s and Parkinson’s [Citation24]. In their studies, Jamar et al. analysed protein aggregation of RFP tagged Hsp104 by visualizing and quantifying fluorescent Hsp104-RFP foci, which increased when mRNA surveillance is defective, such as in upf1∆ cells. We used their established Hsp104-RFP microscopy assay and compared foci formation in wild type and upf1∆ cells to the situation in the gbp2∆ hrb1∆ double knock out strain. While in wild type these protein aggregates were only visible in ~5% of the cells, both upf1∆ and gbp2∆ hrb1∆ strains showed dot-like protein aggregates in more than 15% of the cells, comparable with the values obtained in the original publication (). This accumulation didn’t significantly increase in the triple knock out strain gbp2∆ hrb1∆ upf1∆, suggesting that these proteins might act in one pathway. As Gbp2 and Hrb1 are involved in the nuclear surveillance, we investigated whether defects in nuclear quality control factors per se lead to increased protein aggregation due to an increased leakage of defective mRNAs, which might overwhelm the cytoplasmic surveillance systems. For this we analysed cells that were deleted for the NPC gatekeeper MLP1. As shown in , mlp1∆ showed no increased protein aggregations of Hsp104, indicating that the nuclear escape of faulty mRNAs is not sufficient to create protein aggregates. This suggests that Gbp2 and Hrb1 might have a yet undiscovered function in the cytoplasmic quality control.
Because Gbp2 and Hrb1 are nuclear quality control factors for splicing, a processing step that is a source for PTCs when not carried out correctly, we investigated whether Gbp2 and Hrb1 might function in NMD. We used the well-established reporter assay in which PTC-containing PGK1 transcript is highly expressed using a galactose-inducible promoter [Citation25] (, S1A). However, while an ~8-fold increase of the PGK1PTC mRNA was detectable in upf1∆ by qPCRs from the isolated total RNA, no increased level was measured for gbp2∆ or hrb1∆ or the double mutant (). This suggests that Gbp2 and Hrb1 are not involved in the NMD-induced degradation of this intron-less reporter transcript. However, these guard proteins are preferentially loaded onto spliced pre-mRNAs and have roles in nuclear mRNA quality control specific for these transcripts [Citation9], which reminds of the EJC in metazoans. Thus, a function of these proteins in cytoplasmic quality control may also be specific to the subset of mRNAs that are spliced. Therefore, we constructed two intron-containing reporter mRNAs (, S1A). We chose the DBP2 gene, which possesses a long open reading frame before the intron sequence and placed the PTC upstream of the intron, which is rather atypical for yeast, but represents the frequently found EJC-model in human cells. Additionally, we selected the CBP80 gene in which the intron sequence is located very close to the AUG start codon, which is common in yeast, and placed the PTC shortly downstream of the intron. We detected the steady-state RNA levels of these intron-containing reporters, expressed upon galactose induction, and observed a ~ 3.6-fold (DBP2PTC) and ~5.6-fold (CBP80PTC) increase in upf1∆ compared to wild type (), showing that they are targeted for the Upf1-dependent NMD pathway under wild-typical conditions.
We next asked if deletion of GBP2 and HRB1 would have an effect on the intron-containing DBP2PTC and CBP80PTC reporters. It should be noted that Gbp2 and Hrb1 are nuclear retention factors and are dispensable for mRNA export and splicing [Citation9]. Further, to exclude effects from their nuclear quality control function, we used wild-typical reporters as controls that are identical but lack the PTCs (Fig S1A). As PTCs can only be recognized during translation, the difference between PTC-containing and PTC-less reporters have to be a consequence of mRNA stability through the NMD pathway. We expressed these reporters using the transcripts’ endogenous promoters in order to reflect natural conditions as much as possible. Results of qPCR analyses show that the NMD reporters are enriched ~2.8-fold (DBP2PTC) and ~2.3-fold (CBP80PTC) in upf1∆ (). Interestingly, with a ~ 1.8-fold enrichment for both reporters, gbp2∆ hrb1∆ cells showed approximately half of the upf1∆ effect. In the absence of Upf1, the additional loss of Gbp2 and Hrb1 had no further effect on either reporter (, S1B). Together, these observations suggest that Gbp2 and Hrb1 may act in the Upf1-mediated pathway on transcripts derived from intron-containing genes.
Upf1 is stabilized on NMD targets [Citation26]. Studies on human UPF1 showed that NMD factors that are relevant for the initial detection of NMD targets, such as UPF2, are important for the interaction of UPF1 with PTC-mRNAs [Citation27]. To investigate whether Gbp2 and Hrb1 might affect the initial detection of NMD or rather act after Upf1 has triggered the pathway, we carried out similar experiments and tested via RNA-co-immunoprecipitation (RIP) whether the interaction of Upf1 with the CBP80PTC reporter is affected in the absence of Gbp2 and Hrb1. As an internal control to rule out NMD unrelated effects, we normalized reporter RNA levels to an endogenous wild-typical mRNA. All precipitated RNA levels were normalized to their relative levels from whole-cell lysates. We found that, unlike upf2∆, the double knock out of GBP2 and HRB1 did not affect the binding of Upf1 to the CBP80PTC NMD reporter (, S1C). In contrast, about 50% less PTC-containing CBP80 was bound to either Gbp2 or Hrb1 when Upf1 was missing (, S1D). This suggests that Gbp2 and Hrb1 are likely not involved in NMD substrate recognition and Upf1 recruitment, but might rather help in downstream events of NMD. When Upf1 is absent and PTCs are not identified as false, Gbp2 and Hrb1 may dissociate earlier from the NMD reporter during normal rounds of translation, resulting in the observed decreased association.
To analyse the relevance of Gbp2 and Hrb1 to NMD in physiological conditions, we studied the binding of these proteins to natural NMD substrates. We chose the intron-containing GCR1 and HNT1 transcripts that were identified as putative NMD targets in a genome wide analysis [Citation20]. We found that interactions of Gbp2 and Hrb1 with these natural NMD substrates were reduced when UPF1 was deleted, similar to the findings with the reporter construct (). Together, our first results uncover the involvement of Gbp2 and Hrb1 in NMD and suggest that they likely act downstream of Upf1 in the effective elimination of a subset of NMD targets (presumably those that are spliced).
Gbp2 and Hrb1 repress translation of NMD reporter transcripts
NMD prevents the expression of prematurely terminated and thus potentially harmful proteins after PTC detection by two distinct, yet intertwined, mechanisms: a) the degradation of the transcripts, and b) the repression of further translation initiation on such faulty mRNAs. For NMD it is known that this repression requires Upf1, as NMD substrates show an increased translation in upf1∆ cells [Citation17]. To investigate whether our intron-containing PTC-reporter constructs also undergo translational repression, we analysed their expression in wild type and upf1∆ cells. For this purpose, we created variants of the reporter mRNAs that encode N-terminally MYC-tagged proteins (Fig S1A), which allowed detection, even if they were only translated up to the PTC. With western blot analyses, we show that translation of the MYC-DBP2PTC reporter was terminated at the PTC, resulting in a 45 kDa truncated protein (), which was substantially enriched when Upf1 was missing (). Likewise, a Upf1-dependent translational repression of MYC-CBP80PTC was also observed (). Interestingly, the translated product of MYC-CBP80PTC was not terminated at the PTC, but rather at the original stop codon, producing a full-length protein (). However, translation of CBP80PTC in wild type was 10-fold lower than in upf1∆ (), comparable to other described NMD reporters [Citation17,Citation20,Citation28], entailing that the PTC is indeed recognized by the NMD machinery efficiently. Moreover, the read-through product in upf1∆ is still several magnitudes lower than the normal PTC-less CBP80 translation (Fig S2A), suggesting that read through of the PTC is extremely rare in wild type cells and presumably occurs only when NMD fails and not vice versa. Consistently, PTCs are described to be susceptible to readthrough, especially when NMD is impaired [Citation29]. In fact, the widely used PGK1PTC reporter showed an identical behaviour when fused to an N-terminal MYC-tag (Fig S2D) and although the PTC is apparently read through in upf1∆ cells, the mRNA remains susceptible to NMD [Citation30]. In line with this, it was recently demonstrated that each round of translation has an equal probability to initiate NMD [Citation31]. Thus, read through of a PTC by one ribosome does not render the mRNA immune to NMD in subsequent rounds of translation.
To study whether the guard proteins Gbp2 and Hrb1 also function in translational repression of the NMD targets, we analysed the expression of PTC-containing reporter constructs in the single and double knock out strains. To obtain an estimate of the translation rate of the reporters, we measured the relative RNA level in each strain by qPCR (Fig S2B, S2C) and related the protein signals to the respective RNA levels. We found that in the case of the DBP2PTC reporter in which the PTC is in the middle of the transcript, both Gbp2 and Hrb1 were necessary for functional translational repression, as their absence increased translation more than two-fold (). Interestingly, in case of the rather yeast-typical CBP80PTC reporter in which the PTC is shortly after the start codon, only Gbp2 seemed to be relevant with its absence leading to a ~ 5.5-fold increase in protein level (Fig S2C) and a ~ 3.5-fold increase of protein per mRNA (). The effects are PTC-dependent, as protein levels of PTC-less reporters remain similar in all strains (). Consistently, such Gbp2- and/or Hrb1-mediated translational repression was not observed with the PGK1PTC reporter that was derived from an unspliced gene (Fig S2D). To manifest that this translational repression is Upf1-dependent, we compared the NMD reporter translation obtained in the upf1∆ strain with that detected in the gbp2∆ hrb1∆ upf1∆ triple knock out strain. Similar to the effect on PTC-containing mRNA degradation, the loss of Gbp2 and Hrb1 had no further effect if Upf1 was absent (). The measured protein levels in our analyses may also be affected by differences in protein stability, since Upf1 also causes destabilization of the nascent polypeptide [Citation28,Citation32]. Nevertheless, this is also a Upf1-mediated effect as part of NMD, hence the observed effects of Gbp2 and Hrb1 on protein level per PTC-mRNA are most probably effects within the NMD pathway. Further, the full-length MYC-Cbp80 protein appears to be a consequence of failed NMD at the PTC followed by normal translation termination at the regular stop codon. It seems unlikely that Upf1 causes destabilization of such normal translation products, indicating that the difference in MYC-Cbp80 levels resulted essentially from differences in translation. Therefore, while the effects of Gbp2 and Hrb1 on DBP2PTC may partially be a consequence of protein stability, we have to assume that Gbp2 is indeed involved in the translation repression of the CBP80PTC reporter.
Both guard proteins contain a serine/arginine (SR)-rich domain, which is also comprised of several arginine/glycine/glycine (RGG)-motifs. The RGG domain was described to be important for a group of proteins, Scd6, Sbp1 and Npl3, involved in inhibition of translation initiation by directly binding eIF4G via their RGG-motifs [Citation33–35]. This makes Gbp2 and Hrb1 potential candidates for NMD-dependent translation repressors. However, to investigate if the known RGG-motif translation repressors can also inhibit the translation of NMD targets, we investigated the expression of our reporters in the respective knock out strains. As shown in , the absence of none of the three proteins increased the translation of the NMD reporter, suggesting that translational repression of NMD substrates could be a specific function of Gbp2 and Hrb1. Interestingly, protein expression in npl3∆ was completely abolished (undetectable even with long exposure times) while the RNA level was ~10% of that in wild type cells (Fig S2E), suggesting a more general function for Npl3 in translation, which would fit to its proposed role in ribosomal subunit joining [Citation33]. The fact that RGG domain-containing Gbp2 and Hrb1 specifically affected translational repression of DBP2PTC and CBP80PTC but not of the PGK1PTC reporter raises the possibility that these proteins may directly repress translation on specific NMD substrates downstream of Upf1.
Although in previous analyses we observed relatively mild effects on the RNA levels of our intron-containing reporters (), more significant effects were seen on the protein level. Functional Upf1 reduced the amount of translated protein from the reporters on average ~19- and ~25-fold (Fig S2B, S2C), comparable to other established NMD reporters [Citation17,Citation20,Citation28]. As a quality control pathway, one of the main functions of NMD is the repression of aberrant protein production and in this regard, NMD seems to function normally on the CBP80PTC and DBP2PTC reporters.
Gbp2 and Hrb1 presumably take part in NMD in the cytoplasm
While we could see that Gbp2 and Hrb1 are relevant for NMD on our reporter constructs on both the RNA and protein-level, it is unclear if the two proteins are physically involved in NMD in the cytoplasm. To investigate whether Gbp2 and Hrb1 physically interact with the Upf-proteins, we carried out co-immunoprecipitation (co-IP) analyses. GFP-tagged Upf1, Upf2 and Upf3 were pulled down from yeast cell lysates and the co-precipitation of Gbp2 and Hrb1 was investigated using specific antibodies (Fig S3A, S3B). Both guard proteins co-purified with all three Upf-proteins (), although the interactions were sensitive to RNase. This could mean that the proteins are present on the same RNA but not in the same complex, or that the interactions occur only when Gbp2 and Hrb1 are bound to RNA. To further understand the interactions between these proteins, we performed co-IP experiments with strains expressing the wild-type or an ATP-hydrolysis defective mutant of Upf1, upf1-DE572AA (). In the upf1-DE572AA mutant, RNA-binding of upf1 is not affected [Citation36], but the ribosome cannot disassemble after NMD has been initiated and the Xrn1-mediated 5ʹ decay stops at the stalled ribosome, resulting in accumulation of a 3‘ decay fragment [Citation18,Citation19]. Moreover, several NMD factors showed increased co-purification with mutant upf1 on the decay fragments in human cells [Citation18]. To test the functionality of the UPF1- and upf1-DE572AA-GFP plasmids, we transformed upf1∆ cells and analysed cell growth on cycloheximide-containing plates (Fig S3C). UPF1 deletion was shown to result in increased sensitivity of the cell to the translation inhibitor [Citation37,Citation38], an effect that was attributed to the fact that NMD is translation-dependent. This growth defect could be rescued by the wild-typical UPF1- but not the upf1-DE572AA-GFP plasmid (Fig S3C). Subsequently, we found that co-precipitation of Gbp2 with upf1-DE572AA selectively increased more than 1.5-fold compared to wild-type Upf1 (). Since Hrb1 did not show an increased association, it cannot be an unspecific enrichment of general RNA-binding proteins. This suggests that Gbp2 is likely still bound to the RNA decay fragments, while Hrb1 might dissociate at an earlier point in time.
To gain further insight into the interaction between Upf1 and Gbp2, we used the split-GFP system, which allows detection of transient protein-protein interactions [Citation39]. The proteins of interest were expressed with N-terminal or C-terminal parts of GFP. In case of close proximity, the GFP fragments assemble and emit fluorescent light in living cells [Citation39]. Although no significant amount of GFP-signal was detectable under wild-typical conditions, clear GFP-signals were measured in the presence of elevated levels of NMD substrates when the C-terminal GFP (C-GFPsplit) was tagged to Gbp2 and N-terminal GFP (N-GFPsplit) fused with the upf1-DE572AA mutant (). This shows that Gbp2 comes into close proximity with Upf1 in the cell, presumably in the same complex at the site of the PTC, as the upf1-DE572AA protein is stalled there. However, this analysis suggests also that such complexes are low abundant and rather labile in wild-typical situations, possibly due to the immediate degradation of the PTC-containing mRNA and the simultaneous disassembly of the associated protein complexes.
To get further evidence for a cytoplasmic involvement of the guard proteins in NMD, we impaired NMD at an earlier point in time, by deletion of XRN1, to prevent the initial 5ʹ-degradation and analysed, whether this delay would visibly affect re-import of Gbp2 and Hrb1 into the nucleus. Clearly, in the presence of increased levels of NMD-substrates, we detected both guard proteins in the cytoplasm of xrn1∆ (). To ascertain that this is indeed caused by NMD we additionally deleted UPF1. In fact, the cytoplasmic localization of both guard proteins disappeared in xrn1∆ when Upf1 was absent (), despite the reporter levels being even higher in these cells (Fig S3D), suggesting an NMD-specific effect. In agreement, overexpression of PTC-less reporters did not result in the cytoplasmic localization of either Gbp2 or Hrb1 (Fig S3E). This shows that ongoing NMD delays the nuclear reimport of Gbp2 and Hrb1, possibly because the proteins remain associated with the RNAs that hold out for NMD degradation. Together, these results imply that both Gbp2 and Hrb1 are present in NMD-complexes. The stronger mislocalization of Gbp2 and its persistent binding to stalled Upf1-complexes furthermore supports the idea that Hrb1 might leave the NMD-identified mRNA earlier than Gbp2.
Hrb1 promotes the recruitment of the 5ʹ-degradation machinery to NMD targets
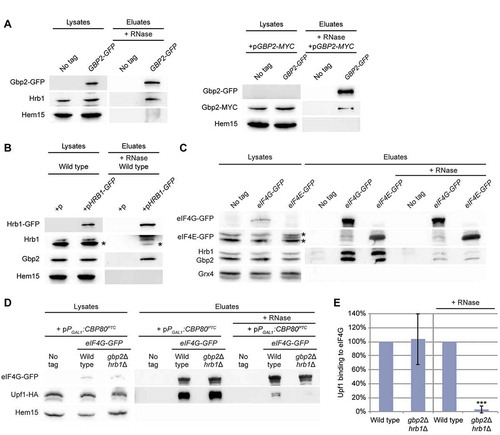
A nuclear function of the guard proteins is to load the degradation machinery to faulty transcripts and in this way initiate their elimination [Citation5,Citation9]. It is conceivable that Gbp2 and Hrb1 might have a similar function in the cytoplasm. Degradation of NMD-targets mainly depends on the Dcp1/Dcp2-mediated de-capping and the subsequent Xrn1-mediated exonucleolytic RNA decay [Citation18,Citation19,Citation25,Citation40]. Dcp1, as well as Dcp2, co-purified with Upf1-bound complexes that also contain other decay factors [Citation20]. Therefore, we first investigated whether Gbp2 and Hrb1 interact with Dcp1. Co-IPs with GFP-tagged Dcp1 showed an interaction of Dcp1 with both Gbp2 and Hrb1 (, S4A). While we initially observed that the co-precipitation of Gbp2 with Dcp1 was lost upon RNase treatment (Fig S4B), this co-precipitation was visible under crosslinking conditions with formaldehyde () in which effective RNA removal was verified via qPCR (Fig S4C).
To ensure that the interaction of the guard proteins with Dcp1 is relevant for NMD, we investigated whether the Dcp1-Upf1 interaction was affected by the absence of the two guard proteins. Indeed, their interaction was reduced to ~67% in the gbp2∆ hrb1∆ strain (). This could indicate that Dcp1 is not properly targeted to NMD-substrates when Gbp2 and Hrb1 are missing. While our studies on the CBP80PTC and DBP2PTC reporters suggest that the Upf1-mediated degradation is diminished approximately by half in gbp2∆ hrb1∆ (, S1B), we can only see an average reduction of one third in the overall Upf1-Dcp1 interaction. However, this analysis was performed without expressing an NMD reporter and relies on the interaction of Upf1 with Dcp1 on endogenous NMD targets. As Gbp2 and Hrb1 appear to be relevant for a subset rather than all NMD targets, a milder effect would be expected in this analysis. Consequently, we would expect stronger effects by directly analysing the Dcp1 binding to an NMD target that is affected by Gbp2 and Hrb1. Indeed, RIP-experiments revealed a significantly reduced binding of Dcp1 to CBP80PTC when the guard proteins were missing (, S4D). Loss of Gbp2 and Hrb1 reduces Dcp1 binding approximately half as much as Upf1, agreeing with our analysis shown in . Interestingly, while the single knock out of HRB1 showed the same decrease in Dcp1 recruitment to the NMD-target as the double knock out, we detected no effect for gbp2∆. This supports a model in which mostly Hrb1 is involved in proper Dcp1 recruitment to a subset of NMD targets.
RNAs with removed caps are substrates for Xrn1, which also physically interacts with both Gbp2 and Hrb1 (, S4A) and Upf1 [Citation20]. The interaction between Hrb1 and Xrn1 remained intact upon addition of RNase A, while the interaction of Gbp2 strongly decreased, suggesting that Gbp2 may require RNA binding for interaction with the 5ʹ-degradation machinery. Nevertheless, the Hrb1-Xrn1 interaction was RNase insensitive, which indicates a physical complex of Hrb1 and the 5ʹ degradation machinery. Interestingly, subsequent RIP-experiments of Xrn1 to the NMD-targets revealed that the interaction of Xrn1 was unaffected in gbp2∆ hrb1∆ (, S4E), suggesting that Xrn1 is not recruited by the guard proteins, but might rather wait in the NMD-complexes for uncapped substrates. As it cannot degrade capped RNAs, there is no necessity for a regulated recruitment of Xrn1. To further test if decapping, and thereby Xrn1 degradation, is defective without Gbp2 and Hrb1, we performed an in vitro Xrn1 digestion experiment. We observed that Xrn1 readily degrades CBP80PTC RNA purified from cells deleted for XRN1 (), indicating that the purified reporter RNAs are mostly decapped. The additional deletion of GBP2, HRB1 or UPF1 strongly impairs the in vitro degradation, suggesting that decapping is defective in these strains. This effect is PTC dependent, as wild-typical CBP80 showed no differences between the mutants (). The remaining fractions of the CBP80PTC RNA vary strongly in the mutant strains in this analysis. This doesn’t allow quantitative comparison between the different mutants; however, all mutants do appear to have an obvious decapping defect compared to the xrn1∆ single mutant in which the PTC-reporter RNA was consistently removed almost completely throughout all repetitions. The in vitro Xrn1 digestion of CBP80PTC indicates that decapping of this reporter is also defective in gbp2∆ cells (), although Dcp1 recruitment was unaffected (). Presumably, this is the consequence of Gbp2’s involvement in translation inhibition (), as active translation initiation counteracts decapping [Citation41].
Together, our findings suggest that Hrb1 functions in the NMD-induced 5ʹ degradation of PTC-containing mRNAs by promoting recruitment of Dcp1. Once de-capping is initiated, Hrb1 probably leaves the PTC-containing transcript, while Gbp2 is still part of the Upf1-complex.
Gbp2 and Hrb1 help to recruit the 3ʹ-end degradation machinery
In addition to the major 5ʹ degradation pathway, the Ski-complex and the cytoplasmic exosome degrade NMD targets from the 3ʹ-end [Citation21,Citation25,Citation40]. Co-IPs with Ski2-GFP revealed physical interactions with both guard proteins, which persisted when RNase A was added (, S5A). However, the interaction with Gbp2 was again decreased, suggesting that RNA binding enables protein interaction (, S5B). That these interactions could be relevant for NMD is shown in the co-IP experiment between Ski2 and Upf1, where a ~ 70% decreased interaction between these proteins was observed when Gbp2 and Hrb1 were missing (). To analyse whether Ski2 recruitment is promoted by the two guard proteins, we compared its binding to the CBP80PTC transcript in RIP-experiments. The absence of Upf1 resulted only in a ~ 30% decrease in the interaction of Ski2 with the NMD-target (, S5C), which likely reflects the subordinate role of the 3ʹ-mediated degradation of NMD targets [Citation1,Citation21]. Interestingly, in the absence of Gbp2 and Hrb1, the interaction of Ski2 with the NMD-target was more than 20% decreased, more than half of the effect in upf1∆, suggesting that the two guard proteins likely promote Ski complex recruitment. As Gbp2 is in close contact with Upf1 and shows an increased binding in stalled Upf-complexes, it might play a more important role in NMD-induced 3ʹ-mediated mRNA degradation.
Gbp2 and Hrb1 may help connect the 5ʹ-end with the PTC
The discovered functions of Gbp2 and Hrb1 in translational repression and NMD-mediated degradation of the target RNAs occur at the ends of the transcripts, while detection of the PTC happens within the open reading frame. To communicate premature termination to the transcript ends, the pathway must be able to bridge this distance. In human cells Upf1 was already suggested to contact the 5ʹ end somehow [Citation42]. However, so far it was not possible to get a clear picture. In order to investigate whether Gbp2 and Hrb1 could contribute to forming a higher ordered structure of the mRNA, we first checked if the two proteins can interact with each other. By using differently tagged guard proteins in co-IPs we were able to show that Gbp2 interacts with Hrb1 independently of RNA and both proteins interact with themselves (). Secondly, we analysed their ability to contact the 5ʹ cap through interaction with the cap-binding proteins eIF4E and eIF4G. Co-IPs showed physical interactions of both cap-binders with Gbp2 and Hrb1 (, S6A). As the guard proteins associate with both Upf1 and eIF4G, we tested if they would promote an interaction between these two proteins. With co-IP experiments, we could detect a physical interaction between eIF4G and Upf1, but it seemed not to be affected in the absence of Gbp2 and Hrb1 (Fig. S6B). We then overexpressed the CBP80PTC reporter to enhance NMD in the cells, and observed that the eIF4G-Upf1 interaction was evidently reduced in gbp2Δ hrb1Δ when RNase was added (). This indicates that eIF4G and Upf1 probably bind to the same transcript independently of the two guard proteins, as shown by the unchanged co-purification without RNase treatment. However, their direct physical interaction is likely promoted by Gbp2 and Hrb1, as in the absence of RNA these proteins were less co-purified in gbp2Δ hrb1Δ than in wild type.
The signals with RNase treatment were close to the detection limit. Therefore, the actual reduction might be smaller than suggested by the quantified values (, rightmost bar). Nonetheless, a significant decrease of the interaction was evident, hinting at a possible role of Gbp2 and Hrb1 in transferring the information that a PTC was detected to the ends of the mRNA. Through interactions with each other and themselves, multiple copies of Gbp2 and Hrb1 at different positions on the RNA may promote formation of mRNP structures that bring proteins along the mRNA into spatial proximity.
Taken together, our findings indicate that Gbp2 and Hrb1 are involved in NMD. Similar to their guarding function in the nucleus, where they recruit the export receptor Mex67 upon successful splicing or, instead, the degradation machinery when splicing fails, they monitor gene expression also in the cytoplasm: From correct mRNAs, they dissociate during early translation [Citation11], but in case of Upf1-mediated detection of a PTC, the guard proteins remain mRNA bound, promote repression of new rounds of translation and presumably the recruitment of degradation machineries (). We propose a model in which the guard proteins bridge the PTC-bound Upf-complex to the 5ʹ-end of the transcript, thereby facilitating the information flow of the need for rapid translational repression and exonucleolytic degradation to the place of action. Thus, their guarding function continues in the cytoplasm after nuclear quality control.
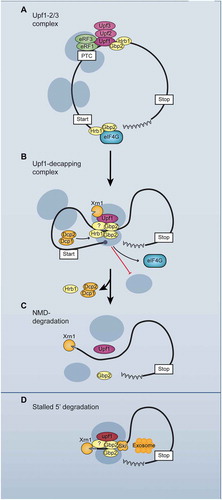
Figure 7. Model for the functions of the guard proteins Gbp2 and Hrb1 in NMD. (A) Gbp2 and Hrb1 are bound to the translated mRNA with a preference towards the 5ʹ UTR, where the introns are located in yeast. Upf1 binds to a PTC and is joined by Upf2 and Upf3 forming the Upf1-2/3 complex. (B) Through interactions with themselves and possibly additional factors, the guard proteins help to restructure the RNP and transmit the PTC recognition from Upf1 to the 5ʹ end of the mRNA, where they inhibit translation initiation. Hrb1 promotes Dcp1 recruitment to the Upf1-decapping complex. Decapping can occur after translation inhibition and dissociation of translation initiation factors. Xrn1 binds to the Upf1-decapping complex independently of Gbp2 and Hrb1. (C) After decapping, Xrn1 can degrade the mRNA. Hrb1 dissociates upon decapping or the onset of Xrn1-mediated degradation. Gbp2 dissociates when the helicase activity of Upf1 detaches the ribosome from the PTC. (D) In the minor 3ʹ-5ʹ degradation pathway, Gbp2 recruits the Ski complex to Upf1. The Ski complex facilitates degradation by the exosome
Discussion
The splicing guard proteins are also cytoplasmic mRNA surveillance factors
Gbp2 and Hrb1 were identified as nuclear quality control factors [Citation9]. Both guard proteins accompany the mRNAs into the cytoplasm and remain bound during translation [Citation11,Citation23,Citation43], which might be relevant for the cytoplasmic surveillance system, similar to the EJC in humans, where the nuclear information from splicing is preserved in the cytoplasm. Indeed, after constructing intron-containing reporter genes, we could identify a role of Gbp2 and Hrb1 as auxiliary factors in NMD (), which also appears to be relevant for endogenous NMD targets under natural physiological conditions (, , and ). Since Gbp2 and Hrb1 are involved in the regulation of nuclear mRNA export, we could consider the possibility that the nuclear export of reporter RNAs is impaired. However, it was shown that Gbp2 and Hrb1 can retain RNAs in the nucleus but are no mRNA export factors, as their loss shows no mRNA export defects [Citation8,Citation23,Citation43]. Moreover, we can see increased protein levels translated from the reporter constructs in gbp2∆ hrb1∆ cells (), thus the reporters appear to be efficiently exported from the nucleus. Because Gbp2 and Hrb1 also affect degradation of mRNAs in the nucleus [Citation9], we would not be able to identify cytoplasmic-specific effects from RNA half-life measurements. Therefore, we had to rely on steady-state RNA levels and relation to PTC-less control reporters initially to demonstrate effects that are specific to the cytoplasm and to NMD. Nevertheless, the in vitro Xrn1 digestion experiment showed clearly that indeed degradation of the reporter construct is defective in cells depleted of GBP2 and HRB1 ().
The observation that Gbp2 and Hrb1 only affected the intron-containing reporters but not the (intron-less) PGK1 reporter might be explained by the fact that Gbp2 and Hrb1 only stably bind to spliced transcripts. In yeast only 5% of all genes contain introns, but since many of them are highly expressed, such as genes encoding ribosomal proteins, 25% of all mRNAs are spliced [Citation44]. Thus, intron-containing transcripts could contribute to a considerable portion of NMD targets. If Gbp2 and Hrb1 are indeed involved in NMD, specifically for spliced targets, this would also include correctly spliced transcripts when premature termination is caused by other means. It would, however, also be a failsafe mechanism to remove incorrectly spliced transcripts that escaped nuclear quality control. Previously shown severe sickness or lethality of gbp2∆ hrb1∆ cells when splicing is affected [Citation9] may be a consequence of the two proteins removing aberrant transcripts in the nucleus and the cytoplasm. That said, it is possible that Gbp2 and Hrb1 affect a subset of transcripts that is defined by other RNA features than splicing. Similarly, it was shown that Ebs1 and Nmd4, potential yeast homologs of human SMG5-7, have partial effects on NMD compared to Upf1 [Citation20,Citation45] and are presumably involved on a subset of targets. Further, cases of NMD have been reported that are independent of Upf2 and Upf3 [Citation46,Citation47], supporting the idea that as auxiliary factors, Gbp2 and Hrb1 may affect only a subgroup of NMD substrates. This also suggests that more players act in NMD and likely multiple factors together contribute to efficient NMD.
Gbp2 and Hrb1 could be precursors of the EJC
We have shown earlier that the stable transcript association of Gbp2 and Hrb1 is a consequence of splicing [Citation9]. Further, their rather 5ʹ proximal binding pattern of mRNAs correlates with the typical intron position in yeast [Citation48,Citation49]. Thus, these SR-like proteins might represent precursors of the human EJC. In fact, several human shuttling SR-proteins were reported to be part of the EJC [Citation14]. Moreover, reports have demonstrated effects of this group of proteins on NMD, but the mechanisms are not understood. For example, overexpression of either SRSF1 or SRSF2 induces NMD [Citation50]. Furthermore, SRSF1 was suggested to induce NMD indirectly by promoting translation [Citation51,Citation52], but also directly by contacting Upf1 [Citation53]. Together, these findings from metazoans suggest that SR-proteins are involved in NMD, but their exact cytoplasmic functions remain rather nebulous. Also, up to date only the SR-proteins from yeast were described as nuclear guard proteins that prevent the leakage of faulty transcripts into the cytoplasm. But metazoan shuttling SR-proteins were also noticed as nuclear export factors, because they promote splicing and the subsequent recruitment of the Mex67 homolog TAP for nuclear export, similar to Gbp2 and Hrb1 [Citation54,Citation55]. Thus, yeast and human shuttling SR-proteins show many similarities in their behaviour and future studies are required to define roles of the human SR-proteins as potential nuclear guard proteins and specify their role as cytosolic NMD-factors, either as part of the EJC or as additional and independent regulators of NMD.
NMD-target degradation
We discovered a function of the guard proteins in degradation of NMD-targets, which is dependent on Upf1 (). In fact, both SR-proteins co-precipitated with all three Upf-proteins (). Endogenous NMD events, which are normally rare, measurably increased the association of Gbp2 with mutant upf1 (), and this association could further be enhanced by the overexpression of an NMD substrate (). As the interaction of Gbp2, but not Hrb1 increases in the presence of the stalled upf1-DE572AA complex and is detectable with the split GFP system, we suggest a direct physical contact between Upf1 and Gbp2 that is very transient under normal conditions. In this late NMD-complex, Hrb1 might already have fulfilled its function at the 5ʹ-end and left the NMD-substrate. Similarly, some but not all human NMD factors are enriched in the mutant upf1-complex [Citation18].
Such Gbp2- and Hrb1-containing NMD complexes, whose formation depends on Upf1, are further supported by the cytoplasmic localization of the usually nuclear guard proteins at steady state. In cells where NMD-substrates accumulate, such as in xrn1∆ cells, both guard proteins were enriched in the cytoplasm (). Importantly, for this accumulation Upf1 is required, which clearly indicates that cytoplasmic function of these guard proteins is linked to NMD. Further, we found physical interactions of these proteins with cytoplasmic degradation factors (, and , S4B, S5B). Notably, the interactions of Hrb1 appear rather RNase resistant, while Gbp2 shows reduced interactions upon RNase treatment. This could suggest that Gbp2 can only fold properly to interact with the 5ʹ degradation machinery when bound to RNA. In line with that, a crosslinking reagent was required for a visible interaction between Dcp1 and Gbp2 with RNase treatment (, S4B, S4C). Since co-precipitation of Gbp2 and Hrb1 was visible with the tested degradation factors upon RNase treatment, this suggests that Gbp2 and Hrb1 associate with the degrading complexes and are not simply present on the same RNA.
One reason why the NMD-targets are stabilized in the absence of the two guard proteins might be that the proteins help to recruit RNA degrading factors in the cytoplasm ( and ), similar to their nuclear quality control function [Citation9]. There, they recruit the nuclear Mtr4 protein, a part of the TRAMP-complex, which is a co-factor for the nuclear exosome. In the cytoplasm, Gbp2 and Hrb1 are required for the effective recruitment of Ski2, which is the cytoplasmic counterpart of Mtr4, a highly homologous RNA-helicase that is necessary for the exosomal RNA degradation [Citation56]. Given that Gbp2 accumulated on PTC-containing transcripts on which 5ʹ degradation stalled due to the upf1-DE572AA mutant, Ski2 might only act after the ribosome is dissociated. Ribosome dissociation upon utilizing the ATPase activity of Upf1 might lead to rearrangements of the NMD complex and allow Gbp2 to promote Ski2-mediated degradation. Hrb1 also seems to be relevant, although it doesn’t seem to accumulate on the 3ʹ degradation fragments in upf1-DE572AA ().
For the main degradation pathway from the 5ʹ-end [Citation17,Citation25,Citation40], our results show that Hrb1 is required for efficient Dcp1 recruitment (). Nevertheless, the in vitro Xrn1 digestion of CBP80PTC indicates that decapping of this reporter is also defective in gbp2∆ (), although Dcp1 recruitment was unaffected (). In addition to a potential, combined action of Gbp2 and Hrb1 in structuring the RNP, Gbp2 appears to promote access of the decapping enzyme to the cap concomitantly through its function in translation initiation inhibition. Such roles have also been suggested for the RGG proteins Sbp1 and Scd6 [Citation35,Citation57]. This is interesting, because it shows for the first time that the highly homologous guard proteins Gbp2 and Hrb1 affect the same pathway but do so via different mechanisms.
Translational repression of NMD-substrates
For NMD it is not only important to degrade a faulty transcript, but also to repress new rounds of translation in order to prevent the expression of potentially toxic truncated proteins. Upf1 was shown to repress translation of NMD-targets [Citation20,Citation25,Citation32] and we found that the guard proteins are also involved in the translational repression of NMD substrates that are intron-containing (). The proteins had no influence on translation when Upf1 was missing or no PTC was present, suggesting that this effect is NMD-specific (). The fact that NMD seems to have a much greater effect on the protein level than on the RNA level of the new NMD reporters might reflect the fact that the main function of this quality control pathway is to prevent the production of potentially harmful polypeptides. This makes the removal of the PTC-containing mRNA rather subordinate as long as the cell effectively prevents the protein production. That said, NMD was also described to function in regulation of RNA levels for certain targets apart from quality control [Citation1,Citation3]. There, regulation of the RNA stability per se is presumably the main function.
Both guard proteins were previously detected to be associated with polysomes [Citation11]. Also, Gbp2 was found to accumulate in P-bodies, in which RNAs accumulate and are translationally repressed after starvation [Citation58]. However, they have not been analysed for their potential to repress translation. Interestingly, both Gbp2 and Hrb1 contain arginine, glycine, glycine (RGG)-repeat motifs that have the potential to inhibit translation initiation. Other RGG-containing proteins, Scd6, Sbp1 and Npl3, were shown to interact with eIF4G via the RGG-motif and inhibit translation in vivo and in vitro [Citation11,Citation34,Citation35]. Gbp2 and Hrb1 also interact with the cap-binder eIF4E and its interacting scaffolding protein eIF4G (), but in contrast to Npl3, Sbp1 and Scd6, they appear to specifically be involved in the translation of NMD-substrates (), suggesting that Gbp2 and Hrb1 are potentially specific translational repressors of their bound NMD-targets.
Gbp2 and Hrb1 transmit the PTC-recognition alert to the transcript ends
How the Upf-proteins, bound to the PTC, communicate to the ends of the transcripts that translation on this mRNA should be suppressed and degradation initiated was unclear. At least Gbp2 gets into close proximity with PTC-bound Upf1 () and both Gbp2 and Hrb1 associate with the 5ʹ- and 3ʹ-degradation machineries ( and ) as well as the cap-binding eIF4E and eIF4G (). Furthermore, the proteins interact with each other and themselves (). These characteristics make them excellent candidates for establishing contact between the PTC-bound Upf-proteins and the 5ʹ end of the transcript. RNA commonly folds into variable secondary structures and restructuring of mRNA promoted by protein-protein interactions has also been demonstrated previously [Citation59]. By such RNP complex rearrangements the alert for PTC-recognition could be transmitted to the 5ʹ-end, where the consequential repression of translation initiation and mRNA degradation are executed. We found indeed a significant reduction of the RNA-independent interaction between Upf1 and eIF4G in the absence of the guard proteins, which supports our model that Gbp2 and Hrb1 mediate the connection of the PTC with the 5ʹ-end of the transcript, thereby bringing the PTC-alert to the site where further action is required ().
Taken together, we have identified the nuclear splicing guard proteins Gbp2 and Hrb1 as auxiliary NMD-factors for intron-containing transcripts. Upon detection of a PTC by Upf1, they seem to be involved in directing this information to the ends of the transcript, translational repression and degradation of the faulty RNA (). Their splicing-mediated binding to transcripts appears analogous to the loading of EJCs in higher eukaryotes and it is tempting to speculate that they might be the yeast counterpart or precursor of the EJC. Most importantly, to date human SR-proteins have not been in the focus of nuclear and cytoplasmic mRNA quality control. However, due to the fact that these proteins are mutated in many neurodegenerative diseases and cancer (http://www.cbioportal.org/), further understanding of their functions in human would provide valuable knowledge for the future. In particular, human SR-proteins are bona fide splicing factors, which can indirectly affect NMD, and the expression of some SR-proteins is auto-regulated via the NMD pathway [Citation60–64], making it complicated to sort out the function of these proteins in mRNA quality control. The identification of the yeast SR-proteins Gbp2 and Hrb1 not only as nuclear but also cytoplasmic quality control factors, required for the degradation and translational repression of PTC-containing transcripts and connecting both surveillance mechanisms in the cell, offers new perspectives for the understanding of human SR-proteins and related diseases.
Supplemental Material
Download Zip (5.6 MB)Acknowledgments
We thank K.E. Baker, R. Lill, L.A. Megeney, U. Mühlenhoff, R. Parker and P.A. Silver for providing plasmids or antibodies.
Disclosure statement
The Authors declare no competing interests.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- He F, Jacobson A. Nonsense-Mediated mRNA Decay: degradation of Defective Transcripts Is Only Part of the Story. Annu Rev Genet. 2015;49(1):339–366.
- Inada T. The Ribosome as a Platform for mRNA and Nascent Polypeptide Quality Control. Trends Biochem Sci. 2017;42(1):5–15.
- Kurosaki T, Popp MW, Maquat LE. Quality and quantity control of gene expression by nonsense-mediated mRNA decay. Nat Rev. 2019;20:406–420.
- Soheilypour M, Mofrad MRK. Quality control of mRNAs at the entry of the nuclear pore: cooperation in a complex molecular system. Nucleus. 2018;9(1):202–211.
- Zander G, Krebber H. Quick or quality? How mRNA escapes nuclear quality control during stress. RNA Biol. 2017;14(12):1642–1648.
- Amrani N, Ghosh S, Mangus DA, et al. Translation factors promote the formation of two states of the closed-loop mRNP. Nature. 2008;453(7199):1276–1280.
- Doma MK, Parker R. RNA quality control in eukaryotes. Cell. 2007;131(4):660–668.
- Zander G, Hackmann A, Bender L, et al. mRNA quality control is bypassed for immediate export of stress-responsive transcripts. Nature. 2016;540(7634):593–596.
- Hackmann A, Wu H, Schneider UM, et al. Quality control of spliced mRNAs requires the shuttling SR proteins Gbp2 and Hrb1. Nat Commun. 2014;5(1):3123.
- Galy V, Gadal O, Fromont-Racine M, et al. Nuclear retention of unspliced mRNAs in yeast is mediated by perinuclear Mlp1. Cell. 2004;116(1):63–73.
- Windgassen M, Sturm D, Cajigas IJ, et al. Yeast shuttling SR proteins Npl3p, Gbp2p, and Hrb1p are part of the translating mRNPs, and Npl3p can function as a translational repressor. Mol Cell Biol. 2004;24(23):10479–10491.
- Shoemaker CJ, Green R. Translation drives mRNA quality control. Nat Struct Mol Biol. 2012;19(6):594–601.
- Karousis ED, Muhlemann O. Nonsense-Mediated mRNA Decay Begins Where Translation Ends. Cold Spring Harb Perspect Biol. 2019;11(2):a032862.
- Singh G, Kucukural A, Cenik C, et al. The cellular EJC interactome reveals higher-order mRNP structure and an EJC-SR protein nexus. Cell. 2012;151(4):750–764.
- Gonzalez CI, Ruiz-Echevarria MJ, Vasudevan S, et al. The yeast hnRNP-like protein Hrp1/Nab4 marks a transcript for nonsense-mediated mRNA decay. Mol Cell. 2000;5(3):489–499.
- Amrani N, Ganesan R, Kervestin S, et al. A faux 3ʹ-UTR promotes aberrant termination and triggers nonsense-mediated mRNA decay. Nature. 2004;432(7013):112–118.
- Muhlrad D, Parker R. Recognition of yeast mRNAs as “nonsense containing” leads to both inhibition of mRNA translation and mRNA degradation: implications for the control of mRNA decapping. Mol Biol Cell. 1999;10:3971–3978.
- Franks TM, Singh G, Lykke-Andersen J. Upf1 ATPase-dependent mRNP disassembly is required for completion of nonsense- mediated mRNA decay. Cell. 2010;143(6):938–950.
- Serdar LD, Whiteside DL, Baker KE. ATP hydrolysis by UPF1 is required for efficient translation termination at premature stop codons. Nat Commun. 2016;7(1):14021.
- Dehecq M, Decourty L, Namane A, et al. Nonsense-mediated mRNA decay involves two distinct Upf1-bound complexes. EMBO J. 2018;37(21):e99278.
- Mitchell P, Tollervey D. An NMD pathway in yeast involving accelerated deadenylation and exosome-mediated 3ʹ–>5ʹ degradation. Mol Cell. 2003;11(5):1405–1413.
- Unterholzner L, Izaurralde E. SMG7 acts as a molecular link between mRNA surveillance and mRNA decay. Mol Cell. 2004;16(4):587–596.
- Windgassen M, Krebber H. Identification of Gbp2 as a novel poly(A)+ RNA-binding protein involved in the cytoplasmic delivery of messenger RNAs in yeast. EMBO Rep. 2003;4(3):278–283.
- Jamar NH, Kritsiligkou P, Grant CM. Loss of mRNA surveillance pathways results in widespread protein aggregation. Sci Rep. 2018;8(1):3894.
- Muhlrad D, Parker R. Premature translational termination triggers mRNA decapping. Nature. 1994;370(6490):578–581.
- Johansson MJ, He F, Spatrick P, et al. Association of yeast Upf1p with direct substrates of the NMD pathway. Proc Natl Acad Sci U S A. 2007;104(52):20872–20877.
- Kurosaki T, Li W, Hoque M, et al. A post-translational regulatory switch on UPF1 controls targeted mRNA degradation. Genes Dev. 2014;28(17):1900–1916.
- Kuroha K, Tatematsu T, Inada T. Upf1 stimulates degradation of the product derived from aberrant messenger RNA containing a specific nonsense mutation by the proteasome. EMBO Rep. 2009;10(11):1265–1271.
- Keeling KM, Bedwell DM. Suppression of nonsense mutations as a therapeutic approach to treat genetic diseases. Wiley Interdisciplinary Reviews: RNA. 2011;2(6):837–852.
- Maderazo AB, Belk JP, He F, et al. Nonsense-containing mRNAs that accumulate in the absence of a functional nonsense-mediated mRNA decay pathway are destabilized rapidly upon its restitution. Mol Cell Biol. 2003;23(3):842–851.
- Hoek TA, Khuperkar D, Lindeboom RGH, et al. Single-Molecule Imaging Uncovers Rules Governing Nonsense-Mediated mRNA Decay. Mol Cell. 2019;75(2):324–39 e11.
- Kim WK, Yun S, Kwon Y, et al. mRNAs containing NMD-competent premature termination codons are stabilized and translated under UPF1 depletion. Sci Rep. 2017;7(1):15833.
- Baierlein C, Hackmann A, Gross T, et al. Monosome formation during translation initiation requires the serine/arginine-rich protein Npl3. Mol Cell Biol. 2013;33(24):4811–4823.
- Rajyaguru P, She M, Parker R. Scd6 targets eIF4G to repress translation: RGG motif proteins as a class of eIF4G-binding proteins. Mol Cell. 2012;45(2):244–254.
- Segal SP, Dunckley T, Parker R. Sbp1p affects translational repression and decapping in Saccharomyces cerevisiae. Mol Cell Biol. 2006;26(13):5120–5130.
- Weng Y, Czaplinski K, Peltz SW. Genetic and biochemical characterization of mutations in the ATPase and helicase regions of the Upf1 protein. Mol Cell Biol. 1996;16(10):5477–5490.
- Estrella LA, Wilkinson MF, Gonzalez CI. The Shuttling Protein Npl3 Promotes Translation Termination Accuracy in Saccharomyces cerevisiae. J Mol Biol. 2009;394(3):410–422.
- Leeds P, Wood JM, Lee BS, et al. Gene products that promote mRNA turnover in Saccharomyces cerevisiae. Mol Cell Biol. 1992;12(5):2165–2177.
- Magliery TJ, Wilson CG, Pan W, et al. Detecting protein-protein interactions with a green fluorescent protein fragment reassembly trap: scope and mechanism. J Am Chem Soc. 2005;127(1):146–157.
- He F, Jacobson A. Upf1p, Nmd2p, and Upf3p regulate the decapping and exonucleolytic degradation of both nonsense-containing mRNAs and wild-type mRNAs. Mol Cell Biol. 2001;21(5):1515–1530.
- Coller J, Parker R. Eukaryotic mRNA decapping. Annu Rev Biochem. 2004;73(1):861–890.
- Maquat LE, Hwang J, Sato H, et al. CBP80-promoted mRNP rearrangements during the pioneer round of translation, nonsense-mediated mRNA decay, and thereafter. Cold Spring Harb Symp Quant Biol. 2010;75:127–134.
- Hacker S, Krebber H. Differential Export Requirements for Shuttling Serine/Arginine-type mRNA-binding Proteins. J Biol Chem. 2004;279(7):5049–5052.
- Ares M Jr., Grate L, Pauling MH. A handful of intron-containing genes produces the lion’s share of yeast mRNA. RNA. 1999;5(9):1138–1139. (New York, NY).
- Luke B, Azzalin CM, Hug N, et al. Saccharomyces cerevisiae Ebs1p is a putative ortholog of human Smg7 and promotes nonsense-mediated mRNA decay. Nucleic Acids Res. 2007;35(22):7688–7697.
- Chan WK, Huang L, Gudikote JP, et al. An alternative branch of the nonsense-mediated decay pathway. EMBO J. 2007;26(7):1820–1830.
- Gehring NH, Kunz JB, Neu-Yilik G, et al. Exon-junction complex components specify distinct routes of nonsense-mediated mRNA decay with differential cofactor requirements. Mol Cell. 2005;20(1):65–75.
- Baejen C, Torkler P, Gressel S, et al. Transcriptome Maps of mRNP Biogenesis Factors Define Pre-mRNA Recognition. Mol Cell. 2014;55(5):745–757.
- Tuck AC, Tollervey D. A Transcriptome-wide Atlas of RNP Composition Reveals Diverse Classes of mRNAs and lncRNAs. Cell. 2013;154(5):996–1009.
- Zhang Z, Krainer AR. Involvement of SR proteins in mRNA surveillance. Mol Cell. 2004;16(4):597–607.
- Sanford JR, Gray NK, Beckmann K, et al. A novel role for shuttling SR proteins in mRNA translation. Genes Dev. 2004;18(7):755–768.
- Sato H, Hosoda N, Maquat LE. Efficiency of the pioneer round of translation affects the cellular site of nonsense-mediated mRNA decay. Mol Cell. 2008;29(2):255–262.
- Aznarez I, Nomakuchi TT, Tetenbaum-Novatt J, et al. Mechanism of Nonsense-Mediated mRNA Decay Stimulation by Splicing Factor SRSF1. Cell Rep. 2018;23(7):2186–2198.
- Huang Y, Steitz JA. SRprises along a messenger’s journey. Mol Cell. 2005;17(5):613–615.
- Müller-McNicoll M, Botti V, de Jesus Domingues AM, et al. SR proteins are NXF1 adaptors that link alternative RNA processing to mRNA export. Genes Dev. 2016;30(5):553–566.
- Johnson SJ, Jackson RN. Ski2-like RNA helicase structures: common themes and complex assemblies. RNA Biol. 2013;10(1):33–43.
- Nissan T, Rajyaguru P, She M, et al. Decapping activators in Saccharomyces cerevisiae act by multiple mechanisms. Mol Cell. 2010;39(5):773–783.
- Buchan JR, Muhlrad D, Parker R. P bodies promote stress granule assembly in Saccharomyces cerevisiae. J Cell Biol. 2008;183(3):441–455.
- Aibara S, Gordon JM, Riesterer AS, et al. Structural basis for the dimerization of Nab2 generated by RNA binding provides insight into its contribution to both poly(A) tail length determination and transcript compaction in Saccharomyces cerevisiae. Nucleic Acids Res. 2017;45(3):1529–1538.
- Lejeune F, Maquat LE. Mechanistic links between nonsense-mediated mRNA decay and pre-mRNA splicing in mammalian cells. Curr Opin Cell Biol. 2005;17(3):309–315.
- Sikorski RS, Hieter P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics. 1989;122:19–27.
- Becker D, Hirsch AG, Bender L, et al. Nuclear Pre-snRNA Export Is an Essential Quality Assurance Mechanism for Functional Spliceosomes. Cell Rep. 2019;27(11):3199–214 e3.
- Lee RE, Brunette S, Puente LG, et al. Metacaspase Yca1 is required for clearance of insoluble protein aggregates. Proc Natl Acad Sci U S A. 2010;107(30):13348–13353.
- Fabrizio P, Dannenberg J, Dube P, et al. The evolutionarily conserved core design of the catalytic activation step of the yeast spliceosome. Mol Cell. 2009;36(4):593–608.