
ABSTRACT
RNA interference (RNAi)-mediated gene silencing can be used to control specific insect pest populations. Unfortunately, the variable efficiency in the knockdown levels of target genes has narrowed the applicability of this technology to a few species. Here, we examine the current state of knowledge regarding the miRNA (micro RNA) and siRNA (small interfering RNA) pathways in insects and investigate the structural variability at key protein domains of the RNAi machinery. Our goal was to correlate domain variability with mechanisms affecting the gene silencing efficiency. To this end, the protein domains of 168 insect species, encompassing the orders Coleoptera, Diptera, Hemiptera, Hymenoptera, and Lepidoptera, were analysed using our pipeline, which takes advantage of meticulous structure-based sequence alignments. We used phylogenetic inference and the evolutionary rate coefficient (K) to outline the variability across domain regions and surfaces. Our results show that four domains, namely dsrm, Helicase, PAZ and Ribonuclease III, are the main contributors of protein variability in the RNAi machinery across different insect orders. We discuss the potential roles of these domains in regulating RNAi-mediated gene silencing and the role of loop regions in fine-tuning RNAi efficiency. Additionally, we identified several order-specific singularities which indicate that lepidopterans have evolved differently from other insect orders, possibly due to constant coevolution with plants and viruses. In conclusion, our results highlight several variability hotspots that deserve further investigation in order to improve the application of RNAi technology in the control of insect pests.
1. Introduction
Even in the age of genome editing, the discovery of small non-coding RNAs (sncRNAs) represents one of the most exciting frontiers in molecular biology and biotechnology. Molecular pathways related to these molecules were first described in Caenorhabditis elegans [Citation1,Citation2], plants [Citation3] and Drosophila melanogaster [Citation4], with a focus on the regulation of gene expression and viral infections [Citation5–8].
Specifically in insects, sncRNAs can be categorized into three main families based on their size and the RNA interference (RNAi) pathway that generates them: (i) micro RNAs (miRNAs), which are 22-nucleotide endogenous sncRNAs that participate in the regulation of gene expression via degradation or translational repression of mRNAs [Citation9,Citation10]; (ii) small interfering RNAs (siRNAs), which vary around 21 nucleotides in length and can be generated from either exogenous or endogenous double-stranded RNA (dsRNA) to counteract viral infections [Citation11]; and finally (iii) piwi-interacting RNAs (piRNAs), which are sncRNAs spanning 25–31 nucleotides in length that interact with PIWI-related proteins and are required for processes ranging from the maintenance of germline stem cells in flies to retro-transposon silencing in eukaryotes [Citation12,Citation13]. For biotechnological purposes that target host-parasite interactions, miRNAs- and siRNAs-based approaches are the most widely adopted.
The characterization of the miRNA and siRNA pathways in D. melanogaster coupled with the mass sequencing of genomes and transcriptomes from several insect species have led to the wide use of the RNAi technology in the development of biotechnological resources aimed at controlling the populations of insect pests and virus vectors [Citation14–17] (, see Supplementary Text ST1 – The miRNA and siRNA pathways in insects: An overview). However, the efficiency of RNAi knockdown is highly variable across insect orders, especially due to differences in the delivery, processing, and stability of sncRNAs. In the case of agriculture-driven RNAi-based technologies, delivery can be achieved either through the use of transgenic plants expressing long dsRNAs, artificial miRNAs (amiRNAs), or through topical sncRNA administration (e.g., naked or nanoparticle-borne dsRNA/amiRNA) [Citation14,Citation18–23]. The main disadvantage of transgenic plant-based approaches is that sncRNAs are processed by the plant RNAi machinery prior to their delivery. For effective dsRNA uptake by insect cells, the optimal size of dsRNA ranges from 100 to 200 nucleotides; in contrast, after pre-processing by the plant’s RNAi machinery, what remains for herbivorous insects are Argonaute-coupled single-stranded siRNAs and low levels of intact transgenic sncRNA, which jeopardizes efficient gene knockdown [Citation24–27]. This problem can be solved by the transgenic expression of sncRNA in plastids, such as chloroplasts. Chloroplasts are present in large numbers in plant cells (approximately 100 per leaf cell, depending on plant species) and display a compact genome that lacks classical elements of the RNAi machinery. Thus, sncRNAs expression in chloroplasts can provide high levels of intact transgenic sncRNA to the target insect population, thereby increasing the silencing efficiency [Citation25,Citation26]. On the other hand, the technical complications related to non-transgenic RNAi-based approaches, such as the use of sncRNA nanocapsules, can be exemplified by the difficulty in choosing the best polymer for nanoparticle preparation. Delivery of sncRNA must be efficient while keeping the dsRNA molecule intact; in parallel, the production method must be low cost and adverse effects, such as high toxicity, must not be observed in non-target species.
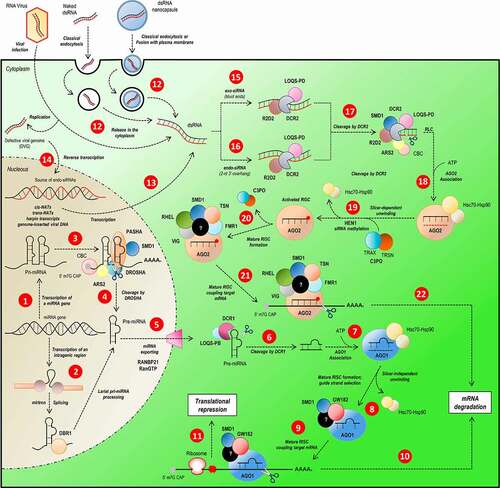
Figure 1. Overview of miRNA/siRNA gene silencing pathways in D. melanogaster. The sncRNAs can be categorized in three groups, according to their size and the processing pathway they participate. The miRNAs (22 nucleotides) and siRNAs (21 nucleotides) follow independently processing pathway for gene silencing by translational repression or mRNA degradation. The miRNA biogenesis starts with the transcription of a primary transcript (pri-miRNA) with some structural peculiarities (hairpin loop domains, 5ʹ cap and poly-A tail) (step 1). Intragenic regions can generate miRNAs; the loop present on spliceosome is recognized and processed by DBR1 (step 2), generating a pre-miRNA. The pri-miRNA loops are recognized by the DROSHA-PASHA complex associated with ARS2, CBC and SMD1, essential proteins in complex recruitment and pri-miRNA structural elements recognition (step 3). The pri-miRNA is cleaved by DROSHA (step 4) and the pre-miRNA exported to the cytoplasm by RANBP21 (step 5). In cytoplasm, the pre-miRNA is processed by DCR1 (step 6) in association with LOQS-PB and its loop is removed, generating a double-strand miRNA which is loaded on AGO1 (step 7), where one strand of miRNA duplex is selected as mature miRNA (step 8) and will constitute a mature RISC complex (step 9), which attaches to target mRNA directed by miRNA-mRNA base pairing, culminating in mRNA degradation (step 10) or translational repression (step 11). Unlike miRNA pathway, who biogenesis follows an endogenous-starting pathway, the siRNA starts, mainly, from an exogenous dsRNA source (as virus or some artificial source) or an endogenous-alternative source of dsRNA incorporated on host cell genome (steps 12, 13 and 14). According to the origin of dsRNA, it follows different, but remarkably similar, processing-pathways. The long exogenous dsRNA (exo-siRNA) is recognized by R2D2-DCR2 complex (step 15) and endo-siRNA is recognized by a complex of R2D2, DCR2 and LOQS-PD in association (step 16). Both siRNAs are cleaved by DCR2 stimulated by ARS2 and SMD1 (step 17) and associated with AGO2 (step 18). The selection of the guide-strand of mature siRNA is optimized by the association of AGO2 with the C3PO complex and its stabilization is acquired by siRNA methylation by HEN1 (step 19) until the mRNA target attachment. The mature RISC complex formation is dependent of the association of many proteins which enhance mRNA recognition and structure-changing, such as RHEL, SMD1, TSN and FMR1 (step 20). Once mRNA is attached to the mature RISC complex (step 21), the gene silencing is reached by mRNA degradation (step 22)
Since sncRNA are mainly delivered to insects through nutrient absorption, the stability of exogenous sncRNAs in the insect midgut and haemolymph is another important factor that must be considered for successful gene knockdown. Several studies involving different species and insect orders have shown the presence of more than one nuclease isoform capable of degrading exogenous dsRNA (dsRNAses) in both the midgut and haemolymph [Citation28–34]. These dsRNAses are highly stable (acting on acidic pHs) and do not present sequence specificity. In addition, transcriptional repression of these enzymes shows, in most cases, a considerable increase in the RNAi-mediated silencing efficiency of target insect populations [Citation28–34]. Recently, a study involving lepidopteran species demonstrated the presence of a specific dsRNAse, REase, whose activity was associated with the low efficacy of RNAi-based gene silencing observed in this insect order [Citation35].
A third factor to consider when evaluating gene-silencing efficiency in insects is the uptake and transport of sncRNA across insect cells, the latter of which is a crucial feature of systematic RNAi. In C. elegans, such a process is mediated by the proteins SID1 and SID2 (Systemic RNA Interference-Deficient 1 and 2), which are transmembrane proteins responsible for binding and internalizing long sncRNAs; SID2 mediates tissue-specific endocytosis of exogenous sncRNA present in the intestine of C. elegans, whereas SID1 mediates vesicle release of sncRNAs into the cytoplasm and acts as a transmembrane channel that directly imports sncRNAs from tissues other than the intestine [Citation36,Citation37]. Even though the RNAi response as a cellular mechanism is highly conserved among eukaryotes, the systemic aspect of it is not. This situation can be observed among species of different insect orders, insofar as no orthologues of C. elegans SID2 protein have been identified and possible orthologues of SID1 protein (SID1-like proteins; SIL) are generally associated with cholesterol transport rather than with sncRNA uptake [Citation38–40]. Consistent with these observations, previous studies involving D. melanogaster and Tribolium castaneum have shown that exogenous sncRNA uptake in these two insect species occurs through the clathrin-dependent endocytosis pathway. Exogenous long sncRNAs are recognized by a membrane receptor (scavenger receptor) and later internalized into endosome vesicles, which in turn fuse tardily with lysosomes [Citation24,Citation41]. To become available to the RNAi machinery in the cytosol, the dsRNA needs to escape from the early-to-late endosomes before they fuse with lysosomal compartments [Citation42]. Problems during the release of sncRNA into the cytoplasm can lead to their accumulation in vesicles, which dramatically reduces the RNAi-mediated silencing efficiency, as observed in studies with Spodoptera frugiperda Sf9 cells [Citation43].
In light of the factors aforementioned, we hypothesized that the variability present within the core proteins of the insect RNAi machinery may also influence the success of RNAi-mediated gene silencing to control insect pests. Herein, we report a thorough in silico analysis of key proteins of the miRNA and siRNA pathways identified in genomes and transcriptomes from species of five different insect orders (Coleoptera, Diptera, Hemiptera, Hymenoptera and Lepidoptera). In particular, we focused on dissecting the sequence and structure variability present at the functional domains which compose the eight core proteins of the miRNA and siRNA pathways (AGO1-2, DCR1-2, DROSHA, LOQS, PASHA and R2D2). Given that proteins never function in isolation, and to put our analyses into context, we additionally present a compact and updated overview regarding the mechanisms of miRNA and siRNA biogenesis in the Supplementary Materials (Supplementary Text ST1 – The miRNA and siRNA pathways in insects: An overview). Our results identified several variability hotspots that might be associated to the different sensitivities to gene silencing mechanisms exhibited by insects. We found that all substantial variability hotspots can be mapped to loop regions within the functional domains of the RNAi core proteins (while milder variability is present in some of the secondary structural elements). We discuss the possible implications of the different locations and biochemical composition of these loops, as well as some of the idiosyncrasies pertaining to specific insect orders. Finally, our analysis revealed that some proteins that were thought to be lacking specific domains actually harbour them; furthermore, these domains appear to retain their canonical structures with very few exceptions that amount to loop regions.
2. Methods
2.1. Database construction and phylogenetic analysis
The selection of proteins involved in insect miRNA and siRNA machinery was made according to previous studies with the model species D. melanogaster. The selection of 149 genomes and 20 transcriptomes (168 different species) belonging to the 5 insect orders analysed in this study (Coleoptera, Diptera, Hemiptera, Hymenoptera, and Lepidoptera) was made according to the following parameters: (i) agronomic importance, including insect pests, as well as virus vectors; (ii) genomes and transcriptomes with a completeness value greater or equal than 95% obtained by analysis with the BUSCO software (version 3; genome and protein modes; insect dataset odb9) [Citation44]; (iii) genomes with high N50 values. Model species with the most advanced genomes were chosen for each insect order and used as reference to search for orthologues in insects within the same order. The selected model species were: Coleoptera: T. castaneum; Diptera: D. melanogaster; Hemiptera: Bemisia tabaci; Hymenoptera: Apis melífera and Lepidoptera: Manduca sexta. Ortholog selection of the 8 selected proteins (AGO1-2, DCR1-2, DROSHA, LOQS, PASHA and R2D2) in genomes was made using the NCBI’s Basic Local Alignment Search Tool for proteins (BLASTp; in BLAST package; version 2.8) [Citation45] with default parameters and e-value threshold of 10-5 through the Best Bidirectional Hit (BBH) methodology with modifications [Citation46]. Due to the high level of duplication present in hexapod genomes [Citation47], we evaluated the best hit in BBH analysis in order to prevent the loss of orthologues [Citation48,Citation49]. Regarding the transcriptomes, the initial search for orthologues was made with tBLASTn from the NCBI BLAST package [Citation50]. Once the possible orthologues were selected, the open read frames (ORFs) were predicted for each transcript with the ORF finder tool [Citation51] and the correct ORF was selected and translated in the correct frame with the same tool. Thus, all subsequent phylogenetic and structural analyses were performed with the predicted protein sequences from all genomes and transcriptomes. All data concerning genomes and transcriptomes, and the ID of all selected sequences are summarized in Table S1. The protein sequences deduced from transcriptomes assembled in our lab (Anthonomus grandis, Diatraea saccharalis, Hypothenemus hampei and Telchin licus licus) are available in PDF format (Supplementary data). The protein sequences from other Metazoa phyla used for phylogenetic analysis (; Chordata, Cnidaria, Nematoda and Platyhelminthes) were selected with the same BBH pipeline used for selection of insect sequences (see Table S2). In addition, the initially selected orthologues were quality-filtered according to the following criteria: (i) all selected protein sequences should start with methionine and their corresponding gene must end with a stop codon; (ii) the alignment coverage between the model species (query) and the target species (subject) should be greater or equal than 80%. Subsequently, each selected protein was submitted for annotation of domains, which was performed locally using the Hidden Markov Models tool with default parameters (HMMER; version 3.2) [Citation52] against the Protein family (Pfam) database (version 32.0 with 17,929 domain families), as well as the online platform Simple Modular Architecture Research Tool (SMART; version 8.0; http://smart.embl-heidelberg.de/) in normal mode including the option Outlier homologues and homologues of known structure [Citation53]. Posteriorly, the protein domains limits were manually curated using multiple sequence alignments and protein structures from the Protein Data Bank (PDB; https://www.rcsb.org). Prior to phylogenetic analysis both complete proteins and their individual domains were aligned separately using the MAFFT software (version 7.402, via Conda repository) with – auto option, and then manually curated [Citation54]. Regarding protein domains, extremely discrepant sequences were removed from later analysis since they can represent errors in genome/transcriptome assemblies and do not have sufficient quality for this phylogenetic analysis. Spurious sequences or poorly aligned regions identified from all multiple alignments from complete proteins and domains were removed with trimAl software (version 1.2) with – gt value equal to 0.5 (columns with gaps in at least 50% of the sequences were eliminated) [Citation55]. The curated multiple alignments were submitted for phylogenetic analysis using the Maximum Likelihood method The software used for such analyses was Randomized Accelerated Maximum Likelihood (RAxML; version 8.2.12) with options –# autoMRE (the software decided how many bootstrap replicates must be run) and –m PROTGAMMAAUTO (the fittest protein substitution model was selected by the software) [Citation56]. The obtained phylogenetic trees were analysed, curated and annotated using the online tool Interactive Tree Of Life (iTOL; version 4; https://itol.embl.de/), where all phylogenetic trees presented in this study are deposited [Citation57]. The phylogenetic trees of the complete proteins (AGO1-2, DCR1-2, DROSHA, LOQS, PASHA and R2D2) are available as Supplementary material in TRE format.
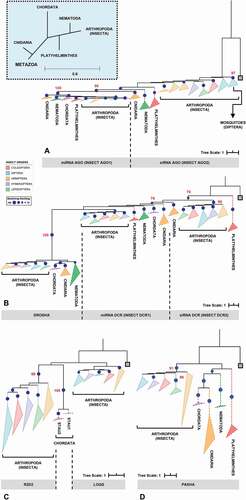
Figure 2. Phylogenetic analysis of the main RNAi machinery core elements in five different insect orders. (A-D) phylogenetic trees (Maximum Likelihood) showing the relationship among complete proteins from the basic core of miRNAs and siRNAs pathways in five insect orders (Coleoptera, Diptera, Hemiptera, Hymenoptera and Lepidoptera, represented by coloured triangles – full lines). (A) AGO proteins; (B) RNAse III proteins (DCR1-2 and DROSHA); (C) DCR partners (LOQS and R2D2; dsrm-containing proteins); and (D) PASHA. The grey square on each phylogenetic tree represents the selected outgroup: (A) Exiguobacterium sp. ACQ71053.1 (bacteria); (B) Batrachochytrium dendrobatidis XP_006676691.1 (fungi); (C) Homo sapiens NP_599150.1 (TARBP2); and (D) Rhodamnia argentea XP_030526936.1 (plant). The cut-off value for bootstrap was 70 (represented by dark blue circles). The big blue square (dashed line) on the top represents the evolutionary relationship expected for each Metazoa phylum presented on the analysis. The dashed triangles represent the outgroup phyla (purple – Chordata; orange – Cnidaria; green – Nematoda; and red – Platyhelminthes). All phylogenetic tree files (.tre) can be found in Supplementary Section, as well as the selected species and the respective protein IDs (see Tables S1 and S2)
2.2. Relative evolutionary rate inference
Site-wise relative evolutionary rates (K) are essential for computational molecular evolution and variability analysis. To investigate these evolutionary rates, the curated alignments and phylogenetic trees of all complete proteins and individual domains were used as input for the program Likelihood Estimation of Individual Site Rates (LEISR), which is implemented in the software package Hypothesis Testing Using Phylogenies (HyPhy; version 2.5.1) and used for calculating the evolution rate directly from protein data [Citation58,Citation59]. LEISR was run in protein mode with LG as the protein substitution model [Citation60] and four-category discrete gamma distribution to optimize branch lengths. The raw data were normalized with the average of all individual K values obtained for each site and box plots of the evolutionary rates were generated to assess the data distribution.
2.3. Sequence clusterization
Given that structure is much more conserved than sequence, modelling all proteins would implicate in a redundant effort. To eliminate the redundancy, proteins were repeatedly clustered using identity cut-offs; after every round of clusterization, the largest sequence of each cluster was chosen as the representative of that cluster. We created a non-redundant dataset of sequences for each type of domain (e.g. PAZ/PAZ-like), wherein the domain sequences within each dataset could have originated from different classes of proteins (e.g., DCR1, DCR2, DROSHA, AGO1 and AGO2). Each of these datasets were first clustered using 95% identity cut-off to eliminate near redundant domain sequences and then using 55% identity as cut-off in the CD-HIT suite web-server [Citation61]; 55% identity is considered a safe threshold to guarantee structure-function relationship between homologous proteins. Clusters containing only one sequence were regarded as outliers. If after these two clusterization steps the quantity of non-outlier clusters (those with two or more sequences) were bigger than 25 (square the number of insect orders evaluated), new rounds of clusterization were performed using continuously smaller identity cut-offs (in 5% steps). Once the number of non-outlier clusters reduced to at most 25, clusters were manually verified. The representative sequence of clusters comprising non-redundant, non-outlier domain sequences from each insect order were selected for homology modelling and structural assessment.
2.4. Structure-based sequence alignment and homology modelling
The structure-based sequence alignment of domains was performed in the following way: the representative cluster sequences were submitted to the SAS [Citation62], LOMETS [Citation63], FFAS [Citation64], GeneSilico [Citation65], MMseq2 [Citation66] and SEEKQUENCER (https://sysimm.org/seekquencer/) servers with the purpose of finding templates for homology modelling. The most recurrent structures appearing in the results from these servers were selected as templates. The templates were structurally aligned using the sequence-independent mode of the MaxCluster program (http://www.sbg.bio.ic.ac.uk/~maxcluster/index.html) and also by means of the POSA server [Citation67]. The superimposed structures outputted from MaxCluster and POSA were used to generate two refined structure-based MSA by employing the STACATTO program [Citation68]; sequence fragments that were not present in the structures were removed (e.g., 6BUA had large portions of its sequence unresolved in the pdb file). We compared the structure-based sequence alignments originating from the superposition of both methods and, where divergent, manually selected the one that best captured our visual inspection of the superposed structures. Thus, at the end of this step, we were equipped with a curated structure-based sequence alignment of the template structures for each domain. The representative sequences of each domain were aligned to the curated structure-based MSA via the ‘MAFFT – addfragments’ algorithm [Citation69] and an all-vs-all identity matrix was calculated using UGENE [Citation70]. The representative sequences were individually modelled using the template structure with which they shared the highest identity and at least 85% coverage (when the latter condition was not satisfied, the highest coverage was used regardless of the identity); to this end, a pairwise target-template alignment was submitted as input to the SWISSMODEL server [Citation71]. The best quality model originating from the representative sequences of each domain were chosen for posterior structure analyses (e.g., RNA-binding sites).
2.5. Multiple sequence alignments
The alignment of the remaining non-representative sequences from each domain (Figures S5-S32) were performed through two steps. First, we generated individual protein alignments for each group of insect order and domain subunit using a combination of the TCOFFEE and Probcons algorithm in the TCOFFEE server [Citation72]. For example, an individual alignment can encompass the sequences from the second RIIID subunit of DCR1 proteins from coleopterans, while another can encompass the first RIIID subunit of DCR1 proteins from coleopterans. This step is important to better align loop regions from each domain. The individual alignments were then sequentially merged with the parent alignment containing the template and representative sequences by means of the MAFFT –merge algorithm [Citation69]. Given that the sequences have been previously clustered, every group of sequences within an individual alignment has at least one representative sequence in the parent alignment. Since the merge of an alignment can influence how the next one will be merged, the order in which the alignments were merged corresponded to their representative sequence’s identity to the template structure. Thus, the alignment bearing sequences from the cluster with the highest identity to one of the template structures was added first, and then the alignment with highest average identity to the previously merged alignment was added next and so forth. This hierarchical procedure guarantees a better alignment of loop regions by gradually decreasing the identity of groups of sequences. The canonical (Q, I, Ia, Ib, Ic, II, III, IV, IVa, V, Va and VI) and non-canonical (IVb) conserved-sequence motifs were identified in the Helicase domains using the MEME suite [Citation73]; these motifs are important for ATP binding and hydrolysis, RNA binding, and for the communication between the ATP and RNA binding sites. All protein domain alignments are available as Supplementary material in FASTA format.
2.6. Statistical analysis
Statistical analyses of K values were performed using the median test for non-parametric data. To assess the normality of the data, a Kolmogorov-Smirnov test was performed before [Citation74]. All statistical tests were made by using the software IBM SPSS Statistics© version 25 (https://www.dmss.com.br/produtos/statistics/statistics1.html).
3. Results & Discussion
3.1. Phylogenetic overview of whole protein sequences
To identify potential sources of variability in the insect RNAi machinery, an in silico screening was performed through phylogenetic and structural analyses of both the complete proteins and their individual protein domains. Thus, a total of 1,211 sequences representing the core proteins of the insect siRNA and miRNA pathways were selected, namely the proteins AGO1, AGO2, DCR1, DCR2, DROSHA, LOQS, PASHA and R2D2. These proteins were chosen because they are directly associated with dsRNA processing and considerably influence the efficiency of RNAi-mediated gene silencing events, particularly those induced by environmentally introduced RNAs (environmental RNAi). Furthermore, many of the domains present in these proteins have at least one representative atomic structure deposited in the RCSB Protein Data Bank [Citation75]. This allowed us to produce structure models of homologous sequences and to map any variation onto their three-dimensional context within the protein’s structure. We identified representatives of all eight core proteins in species of the five insect orders we proposed to study: Coleoptera (e.g., beetles), Diptera (e.g., mosquitos and flies), Hemiptera (e.g., cicadas and bugs), Hymenoptera (e.g., bees and wasps) and Lepidoptera (e.g., butterflies and moths). This verified that both pathways are ubiquitous in insects [Citation76]. After the identification of orthologues by the BBH approach, the first important observation was the presence of putative paralogues of some of the core proteins in species of specific insect orders; specifically, we observed paralogues for AGO1 (in Hemiptera), AGO2 (in Diptera, Hemiptera, and Hymenoptera), LOQS (in Diptera, specifically in the Anopheles and Bactrocera genera) and PASHA (in Hemiptera) (Table S1).
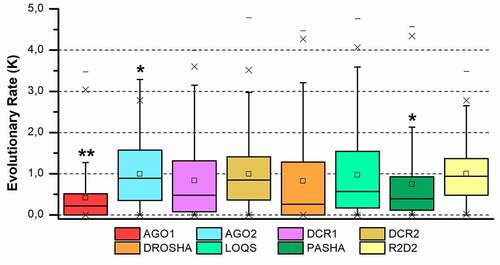
Phylogenetic analysis of the eight complete proteins revealed topologies consistent with the insect tree-of-life proposed by Misof et al. [Citation77] for the five insect orders analysed (). Moreover, such an analysis also enabled us to evaluate the phylogenetic relationships between proteins that perform similar functions, mainly because they share the same functional domains and probably the same ancestor. Four distinct maximum likelihood phylogenetic trees were produced for this purpose: (i) one including AGO1 and AGO2 proteins (); (ii) another comprising RNAse or RIIID-bearing endonucleases (DCR1-2 and DROSHA) (); (iii) the third consisting of insect-exclusive LOQS and R2D2, which are composed of double-stranded RNA-binding motif (dsrm) domains (); and (iv) the last consisting of DROSHA’s partner protein, PASHA (). Insect AGO1 proteins formed a monophyletic group (bootstrap value: 100), with shorter branches and thus less variability than AGO2 proteins. The phylogenetic reconstruction of metazoan AGO proteins shown in corroborates previous phylogenetic studies that show two conserved AGO proteins between basal metazoans (represented here by cnidarians) and invertebrates (arthropods and nematodes), while Chordata phylum maintained only one type of AGO, closer to insect AGO1 [Citation78]. Note that the Nematocera AGO2 (e.g, species of the Aedes and Anopheles genera) clustered in a clade separate from the other dipterans (; bootstrap value: 97). This observation is extremely relevant in studies aimed at controlling the population of these viral vectors because of the ‘mutualistic’ relationship between mosquitoes and viruses and the importance that the AGO2 protein has in the siRNA-mediated response to viral infection. RIIID endonucleases showed a characteristic pattern in which DCR1 and DROSHA proteins clustered in the same monophyletic clade, which was divided into two subclades, one for each protein class (bootstrap value for insect DROSHA clade: 100), whereas insect DCR2 proteins formed a separate monophyletic clade (bootstrap value: 95). These findings corroborate the hypothesis that DROSHA proteins may have evolved from the duplication of a common DCR ancestor and later specialized in the miRNA pathway [Citation79–81]. Overall, we observed that sequences of AGO1-2, DCR1-2 and DROSHA clustered according to their protein class; e.g., all AGO1 sequences formed a monophyletic clade rather than clustering with AGO2 sequences from the species to which they belong. This corroborates a canonical model of evolution in which the lineage-specific duplication of these proteins occurred, at least, before the speciation of insects [Citation82]. However, robust support exists for a model in which the duplication of these genes occurred during deep metazoan diversification, concomitant with the origin of multicellularity and long before the diversification of the Arthropoda [Citation83,Citation84]. Coupled with these analyses, the distribution of evolutionary rate (K value) for each protein family confirmed what was observed in the phylogenetic trees, wherein AGO1 orthologues showed the lowest variability among the eight core proteins (p = 0.013); in contrast, the AGO2 and DCR2 orthologues displayed the highest K values (p = 0.031 and 0.049, respectively) ().
Figure 3. Evolutionary rate evaluation of the main RNAi machinery core elements in five different insect orders. The graph shows the distribution of the evolutionary rate (K value) in each alignment position for all protein classes analysed. Box plot interpretation: The line in the middle of the box represents the median (mid-point of the data). Each part of the box divided by the median line represents 25% of the data distribution. In this way, the box represents 50% of the data. The unfiled small square inside the boxes represents the average value. The whiskers (upper and lower) represent scores outside of the 50% represented by the box. The region delimited by each whisker until the limit of the box represents respectively 25% (lower whisker) and 95% (upper whisker) of the data. The dashes (-) at the ends represent the maximum and minimum values. The ‘exes’ (x) represent outliers. The number of asterisks (*) indicates a statistically significant difference according to the non-parametric median test among insect orders (* p ≤ 0.05; ** p ≤ 0.01; *** p ≤ 0.001)
Among the protein families classified as double-stranded RNA-binding proteins (dsRBPs), LOQS and R2D2, which are found exclusively in arthropods and considered essential for RNAi-mediated gene silencing in insects, appear to have evolved distinctly from other metazoan proteins of this class [Citation85]. Our phylogenetic analysis () showed both LOQS and R2D2 in different monophyletic clades (bootstrap value for insect R2D2 clade: 98), with R2D2 being more closely related to the Staufen proteins (STAU) of the Chordata phylum. Initially characterized in D. melanogaster, STAU proteins are widely distributed in several phyla in the Metazoa kingdom and can participate in both the transport and silencing of mRNAs, as well as in the control of their translation [Citation86,Citation87].
Across most of the domains we analysed, lepidopterans presented the highest phylogenetic distance compared to the other insect orders, especially in the analyses involving proteins of the siRNA machinery (AGO2, DCR2 and R2D2 proteins; ). Specifically, regarding the high variability, and even absence, of R2D2 in the Lepidoptera (note the long branch in , R2D2 clade), some studies suggest that the function of this protein may be carried out by LOQS in species of this order [Citation88]. In summary, phylogenetic analyses of complete proteins showed highly conserved elements in the insect miRNA machinery when compared to the significantly more variable siRNA proteins. It is noteworthy that this variability is mainly observed across different insect orders but is remarkably reduced among species of the same order. This observation is important because most of the knowledge related to RNAi-mediated gene silencing in insects was initially obtained in studies involving D. melanogaster and later transferred to other insect species. Our analyses suggest that even though the primary domain functions are conserved within the miRNA and siRNA pathways, each insect order, or even species, may present idiosyncrasies that influence the RNAi-mediated gene silencing efficiency (e.g., virus vectors). This premise is an important factor to be considered when RNAi is exploited as a biotechnological tool.
Upon observing variability between insect orders in our phylogenetic analyses, two questions need to be addressed: (i) are there ‘variability hotspots’ within the sequences of each of the core RNAi proteins? and (ii) if so, is the hotspot region and its respective variability sufficient to cause structural and functional differences that could explain the RNAi efficiency/sensitivity in a given insect species? To answer these questions, it is important (and easier) to analyse the individual functional domains that make up the eight core proteins. Thus, we performed individual analyses of each domain by employing optimized structure-based sequence alignments, which are arguably more accurate than sequence-based alignments and also mitigate potential phylogenetic errors that may arise when examining the evolutionary history of said domains. Furthermore, structure-based sequence alignments allow us to use the calculated evolutionary rate of all sites in a domain’s sequence to confidently pinpoint variability hotspots and conserved regions. The evolutionary rate of a given site informs us about the significance of the different amino acid substitutions at that position and allows direct comparison between other sites or regions (since the values are normalized). Thus, the detection of variability hotspots and, conversely, of slowly evolving sites is important for mapping functionally significant regions onto the three-dimensional structure of a domain; the structure, on the other hand, allows us to associate regions that are otherwise distant from each other at the sequence level but in close proximity within the three-dimensional and, therefore, functional context.
3.2. Domain architecture of core RNAi proteins
To analyse the intrinsic variability of each protein domain, our first step was to identify all known functional domains present in each of the eight coreproteins of all 168 insect species. This step was initially achieved by annotating domains using HMM profiles from the Pfam database and then performing a data survey of protein structures deposited in the PDB that are involved in RNA interference. Bioinformatics analyses typically rely on the automatic annotation of domains using specialized databases, such as Pfam, CDD and SMART. While false-positive hits are uncommon during these annotations, the same cannot be said about false negatives – these may result from indels, domain insertion, gene truncations or sequence saturation (excess of mutations) present in the query sequence. Notably, the atomic structures of proteins involved in miRNA biogenesis indicate the presence of domains that are not readily detected by automatic annotation databases, such as the Platform-PAZ-Connector domains within DROSHA (PDB ID: 5B16) and the Rhed and CTD domains in PASHA (PDB ID: 3LE4) [Citation80,Citation89]. Even though structural data for some of these domains have been available for a while now, recent papers still fail to acknowledge them due to their reliance on automatic domain annotation servers [Citation90,Citation91]. By thoroughly analysing these protein structures, as well as reviewing their associated papers and comparing our results with the DASH database [Citation92], we were able to not only confidently expand the initial annotation using HMM profiles but also to define the precise boundaries of all annotated domains within each of our selected sequences. In total, 20 different domains were identified in the eight core RNAi proteins: ArgoL1 (PF08699.8), ArgoL2 (PF16488.3), ArgoMid (PF16487.3), ArgoN (PF16486.3), Helicase domain (DEAD/ResIII; PF00270.27/PF04851.13, Hel2i, Helicase C; PF00271.29, and Pincer), Dicer Dimer (PF03368.12), Double-Stranded RNA-binding Motif (dsrm; PF00035.24), Piwi, Argonaute and Zwille (PAZ; PF02170.20, and PAZ-Like), P Element Induced Wimpy Testis (Piwi; PF02171.15), Ribonuclease III (RIIID; PF00636.24, and RIIID-like; PF14622.4), RNA-binding haem domain (Rhed), C-terminal domain (CTD), Platform, Connector and Staufen C-terminal domain (hereafter named Staufen C; PF16482.3) ( and Figure S1-S4).
Figure 4. Protein domains from RNAi core proteins. (A) In-scale diagram of protein domains identified in silico in the classes of analysed proteins. (B-I) Distribution of the evolutionary rate (K value) of each identified domain for all protein: (B) AGO1; (C) AGO2; (D) R2D2; (E) DCR1; (F) DCR2; (G) DROSHA; (H) LOQS and (I) PASHA. Asterisks (*) show statistical analysis of the data distribution of each domain compared to the complete protein (grey boxes). The number of asterisks (*) indicates statistically significant difference according to the non-parametric median test among insect orders (* p ≤ 0.05; ** p ≤ 0.01; *** p ≤ 0.001). Box plot interpretation: The line in the middle of the box represents the median (mid-point of the data). Each part of the box divided by the median line represents 25% of the data distribution. In this way, the box represents 50% of the data. The unfiled small square inside the boxes represents the average value. The whiskers (upper and lower) represents scores outside of the 50% represented by the box. The region delimited by each whisker until the limit of the box represents respectively 25% (lower whisker) and 95% (upper whisker) of the data. The dashes (-) at the ends represent the maximum and minimum values. The ‘exes’ (x) represent outliers
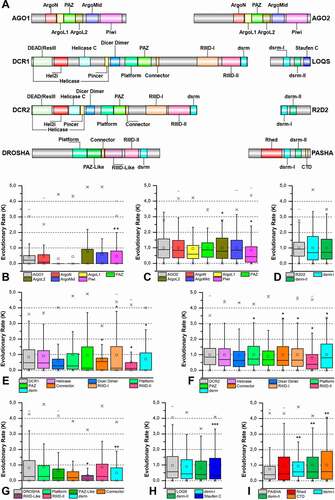
The analysis of K values for individual domains showed that those involved in the miRNA pathway presented lower K values than the ones involved in the siRNA pathway (). The AGO1 protein domains were those with the lowest K values (especially the PAZ domain; p = 0.007), while the domains of the AGO2 (e.g., ArgoL2 and PAZ domains; p = 0.038 and p = 0.041, respectively) and DCR2 proteins (e.g., Platform-Connector, RIIIDs and dsrm domains) exhibited significantly higher values (p ≤ 0.05). Considering that the K values are directly proportional to the variability levels in our analyses, we can say that the protein domains from the siRNA pathway of lepidopteran species are the most permissive to mutations (; Figures S2-S4).
Figure 5. Structural and phylogenetic analysis of dsrm domains. (A) Maximum likelihood analysis including all domains with similar structure to dsrm present in the proteins DCR1, DCR2, DROSHA, LOQS, PASHA and R2D2 from species belonging to the five insect orders (Coleoptera, Diptera, Hemiptera, Hymenoptera and Lepidoptera). Dicer Dimer and Staufen C domains were inserted on this analysis due to have high structural similarity with dsrm. Each triangle represents an insect order, according to the colour legend presented, and it is proportional to the number of branches present. The outgroup (hidden) used was the dsrm domain from human DROSHA (PDB ID: 5B16) and the bootstrap values are represented by dark blue circles (minimum 70). (B) Structural model of dsrm domain from human DROSHA (PDB ID: 5B16, B), interacting with RNA molecule, and (C) the same domain from human DROSHA superimposed with a Dicer Dimer from Arabidopsis thaliana DCL protein (PDB ID: 2KOU), highlighting the differences and similarities between these two domains. (D and E) Superposition of the models from LOQS dsrm-II and DCR2 Dicer Dimer domains, representing dsrm domains that hypothetically can interact preferentially with dsRNAs and proteins, respectively. In (D), the species that represented each insect order were: Coleoptera: T. castaneum (TC011666); Diptera: D. melanogaster (FBpp0080075); Hemiptera: B. tabaci (Bta01704); Hymenoptera: A. melífera (GB47214); and Lepidoptera: M. sexta (Msex2.00134). In (E), the species that represented each insect order were: Coleoptera: T. castaneum (TC001108); Diptera: D. melanogaster (FBpp0086061); Hemiptera: B. tabaci (Bta10685); Hymenoptera: A. melífera (GB48923); and Lepidoptera: M. sexta (Msex2.04462). In both (D) and (E) were highlighted the main variability spots
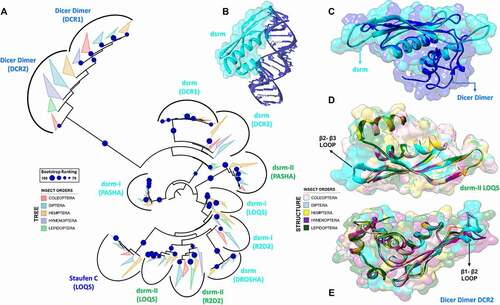
Figure 6. RNA recognition by dsrm and dsrm-like domains. (A) Canonical dsrm domains bind to one major groove and its two adjacent minor grooves by means of the β1-β2 hairpin and the N-terminal regions of helices α1 and α2. (B) The dsrm fold may present high or low affinity for dsRNA, depending on whether the conserved histidine and positively charged residues are present in the β1-β2 loop and α2 helix, respectively. Furthermore, protein-binding dsrms and dsRNA-binding dsrms display contrasting patterns of sequence conservation (see Figures S8 and S13 for complete alignment). (C) The α1-β1 loop of the Dicer Dimer domain from human Dicer (PDB ID: 5ZAK) forms two well-structured grooves which are separated by three proline residues; these proline residues are conserved in insect Dicer proteins. (D) Proposed model for the interaction of Dicer Dimer domains and ssRNA molecules. While the function of the two Dicer Dimer grooves are unknown, they present a positive electrostatic potential and are distanced such that two adjacent phosphate groups of a ssRNA backbone can be modelled to fit them (RNA template was retrieved from PDB ID: 4A36). This model was proposed to account for the Dicer Dimer’s ability to bind single-stranded nucleic acids and promote base-pairing between complementary RNA/DNA molecules in vitro [Citation104]
![Figure 6. RNA recognition by dsrm and dsrm-like domains. (A) Canonical dsrm domains bind to one major groove and its two adjacent minor grooves by means of the β1-β2 hairpin and the N-terminal regions of helices α1 and α2. (B) The dsrm fold may present high or low affinity for dsRNA, depending on whether the conserved histidine and positively charged residues are present in the β1-β2 loop and α2 helix, respectively. Furthermore, protein-binding dsrms and dsRNA-binding dsrms display contrasting patterns of sequence conservation (see Figures S8 and S13 for complete alignment). (C) The α1-β1 loop of the Dicer Dimer domain from human Dicer (PDB ID: 5ZAK) forms two well-structured grooves which are separated by three proline residues; these proline residues are conserved in insect Dicer proteins. (D) Proposed model for the interaction of Dicer Dimer domains and ssRNA molecules. While the function of the two Dicer Dimer grooves are unknown, they present a positive electrostatic potential and are distanced such that two adjacent phosphate groups of a ssRNA backbone can be modelled to fit them (RNA template was retrieved from PDB ID: 4A36). This model was proposed to account for the Dicer Dimer’s ability to bind single-stranded nucleic acids and promote base-pairing between complementary RNA/DNA molecules in vitro [Citation104]](/cms/asset/8e4a755e-3259-4fd1-9795-9b8d2cf65c70/krnb_a_1861816_f0006_oc.jpg)
Figure 7. Structural and phylogenetic analysis of PAZ and Platform domains. (A and B) Maximum likelihood analysis of the PAZ domain presents in the proteins AGO1, AGO2, DCR1, DCR2 and DROSHA (PAZ-like) (A) and Platform (B) domain presents in the proteins DCR1, DCR2 and DROSHA, both from species belonging to the five insect orders (Coleoptera, Diptera, Hemiptera, Hymenoptera and Lepidoptera). Each triangle represents an insect order, according to the colour legend presented, and it is proportional to the number of branches present. The outgroup (hidden) used to the PAZ domain tree was human DCR1 (PDB ID: 4NGD) and the Platform tree was human DROSHA (PDB ID: 5B16). The bootstrap values are represented by dark blue circles (minimum 70). (B-F) Superposition of the models from AGO and DCR PAZ domains, highlighting the main variability spots. No model was found for modelling the PAZ-like domain from DROSHA proteins. In (B), the species that represented each insect order were: Coleoptera: T. castaneum (TC005857); Diptera: D. melanogaster (FBpp0294043); Hemiptera: B. tabaci (Bta01840); Hymenoptera: A. melífera (GB48208); and Lepidoptera: M. sexta (Msex2.06997). In (C), the species that represented each insect order were: Coleoptera: T. castaneum (TC011525); Diptera: D. melanogaster (FBpp0075312); Hemiptera: B. tabaci (Bta00938); Hymenoptera: A. melífera (GB50955); and Lepidoptera: M. sexta (Msex2.05578). In (D), the species that represented each insect order were: Coleoptera: T. castaneum (TC001750); Diptera: D. melanogaster (FBpp0083717); Hemiptera: B. tabaci (Bta12886); Hymenoptera: A. melífera (GB44595); and Lepidoptera: M. sexta (Msex2.10734). In (E), the species that represented each insect order were: Coleoptera: T. castaneum (TC001108); Diptera: D. melanogaster (FBpp0086061); Hemiptera: B. tabaci (Bta10685); Hymenoptera: A. melífera (GB48923); and Lepidoptera: M. sexta (Msex2.04462). (F) Illustrative representation of Platform-PAZ-Connector domains from human DCR 5ZAK PDB model
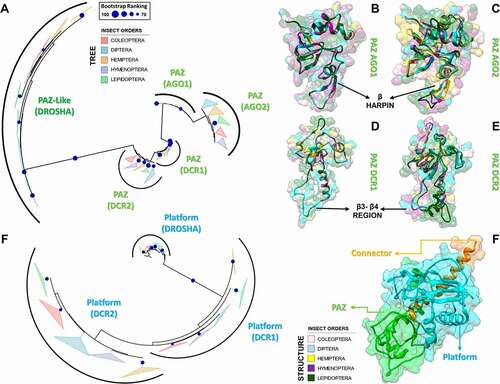
Figure 8. Variabilities within the PAZ and Platform domains. (A) Model for 5ʹ-phosphate recognition in the DCR2 PAZ domain of D. melanogaster. Three residues were mutated in the template structure (PDB ID: 4NH6) to simulate the Drosophila PAZ domain’s ability to recognize 5ʹ-phosphate in vitro in DCR2. Drosophila species lack W1013 in DCR2; we speculate that substituting H982 for either Asp or Glu will repel the phosphate towards a putative phosphate-binding pocket formed by the Arthropod-specific and Drosophila-specific mutations D981R and H994R, respectively. We labelled with asterisk (*) the mutations according to their positions in the DCR2 PAZ domain alignment, shown in Figure S20. W1013 was only identified in DCR1 proteins and can be found at position 116 of Figure S19. (B) Our analyses of K values revealed that PAZ domains typically accumulate mutations in three segments that form a solvent-exposed flat surface on the three-dimensional structure of AGO, DCR and DROSHA proteins. A distinctive groove at the opposite face of this surface was observed, adjacent to the canonical 3ʹ-overhang binding site of PAZ domains. Plants and lepidopterans display a distinctive positively-charged insertion in the N-terminal segment, suggesting their PAZ domains may bind RNA in a different orientation. (C) Comparison between the canonical phosphate-binding pocket of human DCR (blue ellipsis; PDB ID: 4NH6) and the putative phosphate-binding pocket we found in human DROSHA (green ellipsis; PDB ID: 5B16); this feature is also present in insects. Except for H982 (PAZ domain), all residues displayed in white colour refer to the Platform domain of human DCR. The insect equivalents to R778, R780 and R811 can be found at positions 21, 23 and 54 in Figure S23, while the equivalent to H982 can be found at position 85 in Figure S20. Except for R622 (Platform domain), all residues displayed in green colour refer to the DROSHA-specific insertion within the α2-α3 loop of the first Ribonuclease-III (RIIID) subunit of human DROSHA. The insect equivalents to R903, N905, F906 and R914 can be found at positions 15, 17, 18 and 26 in Figure S27, while the equivalent to R622 can be found at position 62 in Figure S24. The yellow ellipsis depicts the estimated location of Giardia lamblia’s putative phosphate-binding pocket. (D) Comparison between the canonical phosphate-binding pocket of human DCR (blue ellipsis; PDB ID: 4NH6) and the putative phosphate-binding pocket we found in G. lamblia DCR (glDCR; yellow ellipsis; PDB ID: 2QVW). The cavity forming the putative binding pocket is extremely well structured: two glutamate residues (E94 and E267 in glDCR) maintain four positively-charged residues coordinated around a central negatively-charged nucleus (R39, K270, R312 and R318). An additional histidine (H92 in glDCR) can potentially participate in the pocket insofar as E94 is repelled by an incoming phosphate. Except for R312 and R318 (RIIID-I subunit), all residues displayed in yellow colour refer to the Platform domain of glDCR. The green ellipsis depicts the estimated location of human DROSHA’s putative phosphate-binding pocket. Information regarding white-coloured residues is described in C. (E) Depiction of important features we identified in DROSHA proteins. The hydrophobic residues that comprise most of the hydrophobic groove are clustered into a single segment (residues 645–681), which is also conserved in insect DCR1 and DCR2 proteins (positions 81–112 in Figures S22 and S23); however, lepidopteran DCR1 and plant Dicer-like (DCL) proteins differ by displaying distinctive positively-charged residues in this region. Similar to what we observed for the PAZ domain, several mutation-prone segments of the Platform domain sequence are common to the DCR1, DCR2 and DROSHA proteins. Furthermore, we observed that these common mutation-prone segments cluster on the three-dimensional structure of the Platform domain to form a contiguous surface. The nature of this mutation-prone surface is unclear
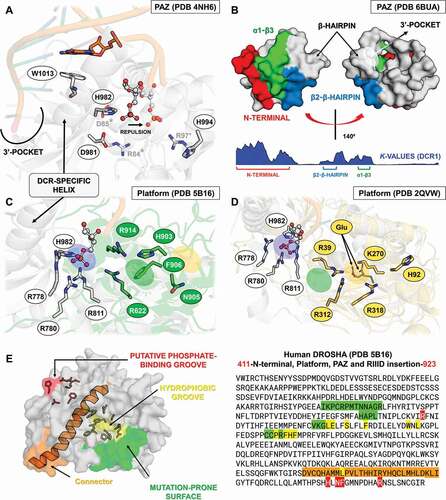
Figure 9. Structural and phylogenetic analysis of Ribonuclease III domain. (A) Maximum likelihood analysis of the two subunits (I and II) of Ribonuclease III domain (RIIID) present in the proteins DCR1, DCR2 and DROSHA from species belonging to the five insect orders (Coleoptera, Diptera, Hemiptera, Hymenoptera and Lepidoptera). The first subunit found in the DROSHA protein differs from the others, being then called RIIID-like. Each triangle represents an insect order, according to the colour legend presented, and it is proportional to the number of branches present. The outgroup (hidden) used was the RIIID domain from human DCR1 (PDB ID: 5ZAK) and the bootstrap values are represented by dark blue circles (minimum 70). (B-D) Superposition of the RIIID and RIIID-like domains from DCRs (B and C) and DROSHA (D) proteins, highlighting the main variability spots (α5-α6 loop in both RIIID-I and RIIID-II from DCR1-2, and RIIID-II from DROSHA, as well as α2-α3 loop in RIIID-like from DROSHA; see also Figures S25-S30). In (B), the species that represented each insect order were: Coleoptera: T. castaneum (TC001750); Diptera: D. melanogaster (FBpp0083717); Hemiptera: B. tabaci (Bta12886); Hymenoptera: A. melífera (GB44595); and Lepidoptera: M. sexta (Msex2.10734). In (C), the species that represented each insect order were: Coleoptera: T. castaneum (TC001108); Diptera: D. melanogaster (FBpp0086061); Hemiptera: B. tabaci (Bta10685); Hymenoptera: A. melífera (GB48923); and Lepidoptera: M. sexta (Msex2.04462). In (D), the species that represented each insect order were: Coleoptera: T. castaneum (TC016208); Diptera: D. melanogaster (FBpp0087926); Hemiptera: B. tabaci (Bta10972); Hymenoptera: A. melífera (GB49096); and Lepidoptera: M. sexta (Msex2.00504)
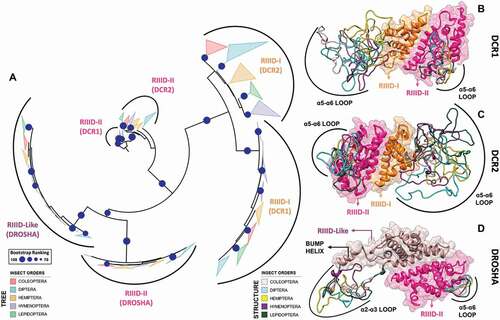
Figure 10. Variabilities within the Ribonuclease-III domain (RIIID). (A) Depiction of all the different features we found in insect RIIIDs; this was achieved by superposing the second RIIID subunit (in blue) of human DCR (PDB ID: 5ZAK) onto the first RIIID subunit (in green) of human DROSHA (PDB ID: 5B16). The Platform domain of human DROSHA was kept in the image (green transparency) to show how the Connector helix acts as surrogate for helix α1 in the first RIIID subunit of DCR and DROSHA proteins. The Bump helix is a unique feature of DROSHA proteins, which display a long insertion in the α2-α3 loop. The Loop helix is typically found in the α5-α6 loops of RIIIDs belonging to DCR proteins. The mutation-prone surface was identified in insects and is composed by the C-terminal regions of helices α3, α5 and α7. In human DROSHA, this region has been shown to bind the C-terminal tail of PASHA at two different positions, depending on which of the two RIIID subunits the binding event occurs. (B) Overview of RIIID features in the context of DCR proteins. The Loop helix from RIIID-II interacts with the Hel1 and Dicer Dimer domains. The N-terminal region flanking the Loop helix makes extensive contact with the α2-α3 loop of RIIID-II, while the flanking C-terminal region can potentially interact with the Hel2 subdomain when DCR is in the ATP-bound conformation, or with dsRNA being threaded through the Helicase domain. The α1 helix of RIIID-II is prone to accumulate mutations and located opposite to the catalytic sites; this region forms a solvent-exposed surface in-between the Hel1 domain and the rest of RIIID-II. In RIIID-I, a mutation-prone, solvent-exposed surface is formed by the C-terminal region of α2 and the unresolved region between α5 and the ‘Loop helix’. Just for illustrative purposes, a dsRNA molecule was modelled onto the structure of human DCR using the dsRNA from PDB 6BU9 as template
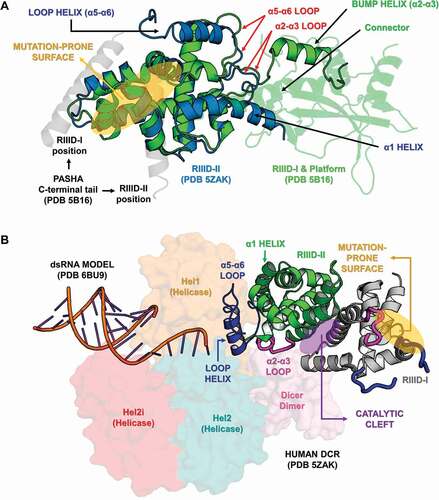
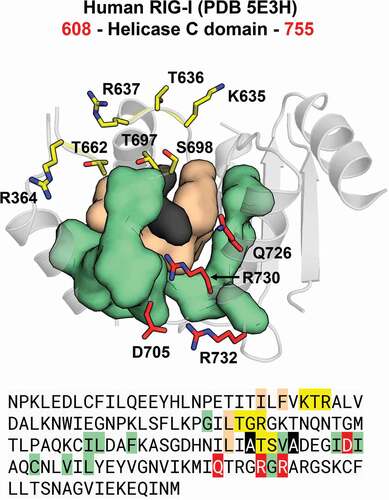
Figure 11. Structural and phylogenetic analysis of Helicase domain. (A) Maximum likelihood analysis of the complete Helicase domain present in the proteins DCR1 and DCR2 from species belonging to the five insect orders (Coleoptera, Diptera, Hemiptera, Hymenoptera and Lepidoptera). Each triangle represents an insect order, according to the colour legend presented, and it is proportional to the number of branches present. The outgroup (hidden) used was the Helicase domain from human DCR1 (PDB ID: 5ZAK) and the bootstrap values are represented by dark blue circles (minimum 70). (B and C) Superposition of the models from DCR Helicase domains, highlighting the main variability spots. Specifically in the DCR1 Helicase models (B), lepidopteran and dipteran-specific loops (β6-α7 and β13-α18 regions, respectively), as well as α14–β9 loop (identified in all insect orders) were highlighted (see also Figure S31). In (B), the species that represented each insect order were: Coleoptera: T. castaneum (TC001750); Diptera: D. melanogaster (FBpp0083717); Hemiptera: B. tabaci (Bta12886); Hymenoptera: A. melífera (GB44595); and Lepidoptera: M. sexta (Msex2.10734). In (C), the species that represented each insect order were: Coleoptera: T. castaneum (TC001108); Diptera: D. melanogaster (FBpp0086061); Hemiptera: B. tabaci (Bta10685); Hymenoptera: A. melífera (GB48923); and Lepidoptera: M. sexta (Msex2.04462). (D) Illustrative representation of Helicase domain from human RIG-I (PDB ID: 5E3H), where its four functional subdomains were highlighted: olive green – DEAD/ResIII (Hel1); red – Hel2i; dark blue – Helicase C (Hel2); and light brown – Pincer. RNA molecule is represented in cyan blue colour. The recognition sites of ATP hydrolysis and binding as well as RNA binding are represented by red and yellow circles, respectively
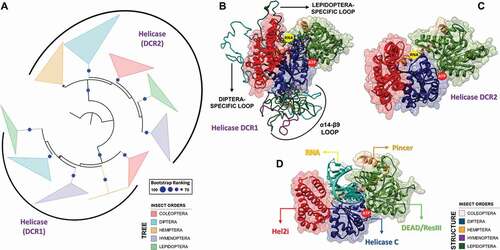
Figure 12. Communication hub for the ATP– and RNA-binding site in RIG-I-like helicases. A network of hydrophobic interactions is arranged around two main amino acid residues (in black). The first layer of hydrophobic residues to interact with the core residues is composed by four residues (in beige) that span motifs IV, IVa and V in insect DCR proteins (see Figures S31 and S32). The second layer is composed by eleven residues (in olive) that span motifs Va and VI, as well as a hitherto undescribed region which we designated as motif IVb. Together, these two layers coordinate the positioning of the ATP– and RNA-binding residues (in red and yellow, respectively). This coordination is important because for translocation and/or unwinding to occur on the dsRNA substrate, the ATP-binding event must communicate with the RNA-binding event (and vice-versa). In insect DCR proteins, the residues participating in this hub are also conserved, which suggests that a similar mechanism for the communication between the ATP – and RNA-binding sites may apply to viral and RNAi-related helicases (see blue, black, grey, red and yellow circles in Figures S31 and S32)
Next, we further analysed five protein domains whose functions are relevant to the biogenesis of sncRNAs and which presented regions with characteristic variability (high or low K values). The following domains were selected: (i) dsrm, which interacts with dsRNA molecules and is present in DCR1-2, DROSHA, LOQS, PASHA and R2D2 proteins [Citation86,Citation93]; (ii) PAZ domain, which actively participates in the selection and correct orientation of miRNA/siRNA strands in AGO proteins, which is also crucial for the discrimination and length fidelity of substrates in DCR proteins [Citation94–96]; (iii) Platform domain, which recognizes the 5ʹ phosphate moiety of dsRNA substrates and acts as a scaffold for the PAZ domain in DCR and DROSHA proteins [Citation80]; (iv) RIIID domain, identified in DCR1-2 and DROSHA proteins, which displays exquisite cleavage specificity towards A-form dsRNA molecules [Citation97–99]; and (v) Helicase domain, present in DCR proteins, which interacts with other RNAi-related proteins (e.g., LOQS) in order to modulate the specificity of DCR2 for dsRNA substrates of the endo– or exo-siRNA pathways [Citation100–103].
3.2.1. Variability within dsrm and dsrm-like domains
We identified the canonical dsrm domain in most proteins and found it to be present in either one copy (DCR1-2, DROSHA and PASHA) or two copies (LOQS and R2D2) (; ). Due to its structural similarity (α-β-β-β-α topology), we classified the Dicer Dimer and Staufen C domains as dsrm-like domains, although previous studies have shown that they can interact with ssRNA and other proteins (such as DCR2) [Citation102,Citation104]. The dsrm domain yielded, by far, the highest e-values in our HMM-Pfam analysis, which demonstrates some sequence variability among the orthologues that have been annotated and deposited in public databases. This high variability may be the reason why several studies have failed to detect the C-terminal dsrm domain present in DCR1 proteins, even though it is highly conserved across insects (). Interestingly, the dsrm domains from different proteins of the miRNA machinery (DCR1, DROSHA, and PASHA) showed a highly conserved primary structure across all of the insect orders we analysed, especially when compared to the proteins of the siRNA machinery (; ).
In general, despite exhibiting a conserved structure, we found that dsrm domains display a remarkable sequence variability in the loop between the strands β1 and β2, a region that has been shown to directly interact with the dsRNA minor groove () [Citation105]. We observed several amino acid substitutions at this site (Figures S5-S16), as well as several insertions of neutral and positively charged amino acids, mainly in species of the Anopheles genus and Lepidoptera order (Figures S10 and S13, respectively). The plasticity we observed for the β1-β2 loop () may directly influence the interaction of these domains with dsRNA and consequently impact the efficiency/sensitivity of RNAi-mediated gene silencing.
The dsrm domains exhibit two different functions: they bind dsRNA molecules and/or facilitate protein–protein interactions, primarily in association with DCR, mammalian PKR or through the formation of dimers [Citation106–108]. According to our analysis, dsrm domains that bind to dsRNA (e.g., those common to LOQS-PB and LOQS-PD) display contrasting variability hotspots compared to dsrm-like domains that are predicted to bind to proteins (e.g., Dicer Dimer and Staufen C). While we found dsRNA-binding dsrms to accumulate most of their mutations in the β1 strand and β2-β3 loop (and marginally at the end of α2 helix) ( and S13), protein-binding dsrm-like domains accumulate most mutations in the β1-β2 and β3-α2 loops (and marginally at the beginning of α1 helix) ( and S16). The dsrm fold is highly conserved across animals and plants, and our observations corroborate previous studies, which show that dsrm-dsRNA interaction occurs primarily through two interfaces: (i) a canonical histidine, present on the β1-β2 loop, which inserts the dsRNA minor groove; and (ii) a cluster of basic residues at the beginning of α2, which stabilize the dsRNA backbone at an adjacent major groove [Citation109,Citation110]. Thus, it stands to reason that dsRNA-binding dsrms should not accumulate mutations in these regions, which would directly affect their capability to bind dsRNA molecules (stabilizing selection). In accordance with this reasoning, Dias et al. [Citation111] have shown that concerted amino acid substitutions in the dsrm β1-β2 loop and α2 region have been responsible for repeated gains and losses of dsRNA affinity during the evolution of animal and plant double-stranded RNA-binding proteins (dsRBPs), and these regions are therefore considered ‘hotspots’ for ‘tinkering’ with dsrm-dsRNA interactions. Furthermore, the authors show that changes in dsrm-RNA affinity occurred often and could produce significant shifts in Kd through specific structural mechanisms: either by establishing/interfering with the critical histidine-RNA contact or by altering dsrm-dsRNA polar contacts within the β1-β2 loop and α2 region. Thus, if dsRNA-binding dsrms are to avoid these drastic shifts in affinity, as can be concluded from the low evolutionary rates we observed in these regions, it is likely that the stabilizing selection acting on the β1-β2 loop and α2 region is maintained through disruptive (purifying) selection. Conversely, protein-binding dsrms do not require the maintenance of dsRNA-binding residues (e.g., histidine) in these hotspots and, accordingly, are able to accumulate many of the ‘tinkering’ mutations reported by Dias et al. [Citation111] without apparent fitness cost. It would seem that these amino acid substitutions are responsible for the domain’s distinctive loss of dsRNA-binding affinity relative to that of canonical double-stranded RNA-binding domains (dsRBDs). This observation raises the question of whether the same reasoning could be applied to putative protein-binding regions of dsrms; i.e., will dsRNA-binding dsrms accumulate more mutations in protein-binding regions, as opposed to protein-binding dsrms are under purifying selection in the same regions? Hence, the contrasting pattern of evolutionary rates that we observed in the sequences of dsRNA– and protein-binding dsrms may provide us with a map for the identification of protein-binding interfaces in dsrms. Dias et al. [Citation111] pointed out that although dsrms have been shown to directly mediate interactions with DCRs in animals and plants [Citation108,Citation112], the extent to which dsrm-dsRNA and dsrm-protein binding may involve evolutionary trade-offs in specialization is not clear. It appears from our results that the ‘trade-offs’ are significant despite different regions being involved with each type of interaction. The three-dimensional structure of dsrms shows that these regions are on opposite sides of the domain’s long axis, which led us to propose a model wherein dsrm domains display two interaction-prone surfaces: one specialized in dsRNA recognition and another capable of binding proteins. The putative protein-binding surface () is composed by the β1 strand, β2-β3 loop (including half of each β-strand) and the C-terminus of α2 helix (e.g., DCR2’s Dicer Dimer and LOQS’ Staufen C domains; Figures S7 and S16, respectively); in some cases, the participation of β1 in protein binding appears to be relegated in preference to the α1-β1 loop (e.g., DCR1’s Dicer Dimer domain) (Figure S6). Nevertheless, we found that the β2-β3 loop contains a conserved (L/M)P(X)2–3(S/C) motif in the Dicer Dimer and Staufen C domains of DCR1-2 and LOQS-PB, respectively (see alignment positions 39, 40 and 44 in ). Considering these observations, we hypothesized that other dsrm domains might also share a similar pattern of accumulated mutations depending on whether they bind protein or dsRNA molecules. Accordingly, all other dsrm domains fell under the dsRNA-binding pattern, with the exception of the second dsrm subunit (dsrm-II) from PASHA. In this case, the prediction was slightly ambiguous, as mutations have accumulated in a large region that encompasses both the β1 strand and the β1-β2 loop (Figure S14); however, since most of the insect species retain the dsRNA-binding histidine residue in the β1-β2 loop and the positively charged residues in the N-terminus of α2 helix, we believe this dsrm domain may have a higher affinity for dsRNA while also interacting with proteins via the β2-β3 loop and the C-terminus of helix α2. An extensive literature review allowed to confirm that our predictions for the Dicer-Dimer and Staufen C domains were, in fact, accurate. The Staufen C-like domain from human TRBP [a dsRBP that partners with human DCR (hsDCR) and is equivalent to LOQS-PD in Drosophila; PDB ID: 4WYQ] was shown to bind the Helicase Hel2i domain via the β1 strand, β2-β3 loop and the C-terminus of α2 helix, all regions displaying low evolutionary rates and which we predicted to bind proteins (Figure S16) [Citation108]. The cryo-EM reconstruction of hsDCR (PDB ID: 5ZAK) also enabled us to perform a comparative assessment of the Dicer Dimer protein–binding interface: it binds the junction between the RIIIDs and the Helicase domain mainly by means of its α1-β1 and β2-β3 loops, confirming our prediction and suggesting it shares at functional similarity with its counterpart in Drosophila DCR1. However, we found the predicted binding of α2 was relegated in preference to the α3 helix (a unique feature of Dicer Dimer domains, which have an additional C-terminal extension containing two helices) [Citation113]. The Dicer Dimer has also been shown to bind single-stranded nucleic acids and to promote base-pairing between complementary RNA/DNA molecules in vitro [Citation104]. Thus, we also investigated whether the α1-β1 and β2-β3 loops from hsDCR could display other potential interaction surfaces. Strikingly, we found that the α1-β1 loop creates a flat surface on which two well-structured grooves are exposed (). These grooves are maintained and separated from each other through three conserved proline residues that are aligned in between them (see alignment positions 18, 27 and 47 in Figures S6 and S7). Both grooves are of sufficient size to accommodate phosphate anions, so we experimented modelling a single-stranded RNA (ssRNA) fragment onto the Dicer Dimer domain. The distance between the centre of both grooves fits the exact distance between two adjacent phosphate oxygens of an A-form RNA backbone (). While this finding is very promising, it is still unclear whether our model can accurately predict the nature of dsrm binding partners (i.e., either protein or nucleic acid) or even be extrapolated to dsrm domains outside the miRNA and siRNA pathways. Further investigations are needed to validate this model and effectively determine the structural interface of dsrm-dsrm, dsrm-protein and dsrm-ssRNA contacts.
Based on the study of Dias et al. [Citation111], we were also able to make predictions about the affinity of dsrm domains participating in the RNAi machinery. If a dsRNA-binding dsrm presented both the canonical histidine residue in β1-β2 and positively charged residues in α2, we categorized it as ‘high affinity’; accordingly, if a dsrm lacked both of these characteristics, we categorized it as ‘low affinity’ (). We did not make assumptions about dsrms lacking just one of the characteristics, which boiled down to the two dsrms from R2D2 (Figures S10 and S15, respectively). Thus, the putative dsrm domains that we predicted to bind to dsRNA with high affinity were the dsrm II from PASHA (Figure S14) and dsrms I and II from LOQS (Figures S8 and S13, respectively), while those predicted to bind with low affinity were the dsrm I from PASHA (Figure S9) and the C-terminal dsrms from DROSHA, DCR1 and DCR2 (Figures S5, S11 and S12, respectively). In the case of DROSHA and DCR1, the presence of mismatches, small bulges and loops in the pri-miRNA and pre-miRNA substrates might explain the lack of high-affinity residues in their dsrm domains; more importantly, it has been experimentally demonstrated that the C-terminal dsrm domain of DROSHA shows low affinity for dsRNA and that the insertion of LTLR(T/S)(M/V)(D/E) residues between α1 and β1 is important for this recognition (Figure S5) [Citation114]. As for DCR2 dsrm (Figure S12), the indication that it binds with low affinity to dsRNA is somewhat surprising; given its specialized role in antiviral RNAi, we would expect the C-terminal dsrm of DCR2 to bind dsRNA with high affinity, especially since we could not make affinity predictions on the dsrms of its partner protein, R2D2. While it might be the case that our prediction is entirely wrong, the lack of alternative highly conserved residues (Figure S12) in the three canonical RNA-binding regions (N-terminus of α1, β1-β2 loop and C-terminus of α2) further supports the low-affinity binding of DCR2 dsrm to dsRNA.
3.2.2. Variability within PAZ and PAZ-like domains
The PAZ domains within proteins of the miRNA machinery (AGO1 and DCR1) displayed low variability between the insect species we analysed (both p values lower than 0.05) (; Figures S17-S21); however, we found that the PAZ-like domain from DROSHA contains a large insertion where the canonical β-hairpin module is predicted to be located (alignment positions 46–80; in DCR1-2, the β-hairpin is found between β2 and α1, while in AGO1-2 it is found between β3 and α3). The β-hairpin region is part of the 3ʹ-pocket and interacts directly with the terminal 2-nt 3ʹ-overhang via a conserved aromatic residue that establishes a π-stacking interaction between DCR proteins and the last nitrogenous base [Citation115]; this residue is classically a phenylalanine, which shows a preference for binding to U or G [Citation116]. We found that phenylalanine can also be substituted by a tyrosine or histidine, in the PAZ-like domain of DROSHA (alignment position 56 in Figure S21). Specifically, the 3ʹ-pocket in DCR1-2 is composed of three main regions of the PAZ domain: (i) the loop between β1-β2 (β2-β3 in AGO1-2), (ii) the β-hairpin region + α1 (α3 in AGO1-2), and (iii) the β4 strand (β7 in AGO1-2) (Figures S19 and S20) [Citation115]. Remarkably, although we observed these regions might display increased evolutionary rates in both AGO and DCR proteins, they all retain the canonical residues (or similar) responsible for the recognition of the 2-nt 3ʹ-overhang (YR-29, FP-53, F60, YY-64, KY-68, and QIL-125; see Figure S19, 4NGD sequence). This finding corroborates the notion that 3ʹ dsRNA recognition is an ancestral characteristic of PAZ domains [Citation84]. The PAZ domain may also participate in 5ʹ-phosphate recognition together with the Platform domain [Citation115]. However, this characteristic is only observed in DCR proteins and is enabled due to a DCR-specific insertion between β3 and β4 (equivalent to β6 and β7 in the PAZ domain of AGO1-2; Figures S17 and S18). This insertion can form a dsRNA-interacting helix that is not critical for DCR processing, but has been associated with the release and transfer of the cleaved dsRNA molecule into AGO proteins () [Citation115]. In DCR2, we found that the PAZ residues that potentially interact with the 5ʹ-phosphate (positions H85, S87, R89, and R96 of 4NGD sequence in Figures S19 and S20) display considerable variability when compared to DCR1, as illustrated by their contrasting evolutionary rates (Figure S20, the region between β3 and β4). This observation may reflect the fact that siRNA biogenesis in insects is mediated by the Helicase domain in DCR2, which preferentially recognizes long dsRNAs (≥38 bps) without the requirement of a specific 5ʹ terminal structure (i.e., it is permissive to blunt or 5ʹ-non-monophosphorylated ends); in contrast, miRNA biogenesis is mediated by the PAZ domain in DCR1, which evolved to specifically recognize the 2 nt 3ʹ-overhang and 5ʹ-monophosphorylated ends of short dsRNAs (<38 bps) [Citation117]. Thus, while the DCR-specific insertion in the PAZ domain may mediate the release/transfer of the product in both DCR1 and DCR2 [Citation117], the conservation of key residues that we observed in DCR1 correlates with its role in the specific recognition of 5ʹ-monophosphorylated ends, as exemplified by the 5ʹ counting rule’ observed during the pre-miRNA cleavage carried out by human and Drosophila DCR1 [Citation118].
Interestingly, in vitro studies have shown that the DCR2 PAZ domain of Drosophila species has regained the ability to specifically recognize the 5ʹ-phosphate [Citation96,Citation119]. We observed that this Drosophila domain bears mutations at sites adjacent to those typically participating in the 5ʹ-phosphate recognition carried out by the DCR1 PAZ domain. We believe that these mutations might explain how 5ʹ-phosphate recognition takes place in vitro in Drosophila DCR2. The aforementioned sites bearing these mutations can be seen in the sequence alignment of the DCR2 PAZ domain at position 84, which is conserved in all arthropods and adjacent to H85 of the 4NGD sequence, and position 97, which is conserved only in Drosophila and it is adjacent to R96 (Figure S20). Mutating both of these residues to alanine in DCR2 have been shown to block the in vitro cleavage of small dsRNAs (30-bp) bearing a 5ʹ-monophophorilated end [Citation117]; in vivo, however, this activity is inhibited by R2D2 and by physiological concentrations (25 mM) of inorganic phosphate [Citation101]. Nevertheless, DCR2 from Drosophila species appear to be an exception rather than a rule with regard to 5ʹ-phosphate recognition; first, only drosopholids display an arginine at position 97 (Figure S20); second, the ability to cleave small pre-miRNAs in vitro necessarily requires a phosphate at the 5ʹ end, which differs from the activity of Drosophila DCR1 that can cleave both 5ʹ-monophosphate and 5ʹ-hydroxyl pre-miRNA substrates (containing 2 nt 3ʹ-overhangs) [Citation117]. We speculate that the mandatory requirement for 5ʹ-monophosphate is likely the result of another Drosophila-specific mutation, (E/D)85 (Figure S20), which we argue is needed to repel the negatively charged phosphate group and redirect it towards the slightly relocated phosphate pocket formed by R97 in Drosophila DCR2 (); in human and insect DCR1, the role of redirecting the 5ʹ end towards the phosphate pocket is performed by a tryptophan or arginine residue present in the DCR-specific insertion within the PAZ domain (see position 116 in Figure S19), which stacks with one of the terminal nitrogenous bases via their indole or guanidino group and causes a bifurcation of the RNA double helix () [Citation115]. We found that insect DCR2 lacks either of these residues (position 117, Figure S20). Furthermore, DCR1 requires a flexible (thermodynamically unstable) 5ʹ terminus to efficiently bifurcate the dsRNA and recognize its 5ʹ end [Citation115,Citation118]. Accordingly, the repulsion of 5ʹ-monophosphate by E or D at position 85 could simulate a thermodynamically unstable terminus and allow the substrate to be accommodated in the 5ʹ pocket (). Hence, novel structural mechanisms that nevertheless resemble the canonical 5ʹ-phosphate-binding pocket of DCR1 may allow other arthropods to regain the ability of DCR2 to recognize 5ʹ-phosphate specifically.
A general trend revealed by our analyses of evolutionary rates in PAZ domains is that its N-terminal region is highly variable independently of the protein, including DROSHA (Figures S17-S21); the N-terminal region ends at the first structural element (310-helix) in DCR proteins (equivalent to α1 in AGO1-2). Curiously, this region maps to a solvent-exposed flat surface composed by two other evolutionary-prone sequence segments (): the region between β2 and the β-hairpin (β3-β4 loop in AGO1-2) and the loop between α1 and β3 (α3-β6 loop in AGO1-2) (Figures S17-S21). Therefore, mutations appear to have accumulated within the same surface patch, indicating that this might be a variability hotspot for positive selection. Moreover, the PAZ-like domain from DROSHA harbours almost all of its variability in this surface region, although the putative α1-β3 loop is conserved (thereby creating a central conserved patch within the surface; see positions 90–98 in Figure S21). While the function of this surface is unclear, we observed it forms a distinctive groove at its opposite face, which suggests that PAZ-like domain can bind to specific moieties; this groove is also adjacent to the 3ʹ-overhang binding site of the PAZ domain (). In accordance with our hypothesis, it has been shown that Dicer-like (DCL) proteins from plants harbour a lineage-specific insertion in the N-terminal region, which was responsible for an evolutionary increase in the affinity of the PAZ domain for RNA molecules [Citation119]; in DCL1, this insertion is longer and contains several positively charged residues. Because of these observations, it has been proposed that plant DCLs may bind RNA in a different orientation than animal DCRs [Citation119]. This hypothesis is corroborated by the fact that DCL1 performs both pri-miRNA and pre-miRNA processing in plants, functions that are carried out separately in animals by DROSHA and DCR1, respectively [Citation120]. Curiously, we observed that lepidopteran species differ from all other insect orders by displaying a positively charged insertion at the N-terminal region of their DCR1 PAZ domain, similar to the one found in plants (Figure S19). This raises the question of whether lepidopteran DCR1 may also bind to dsRNA in a different orientation, which might explain the different sensitivities to gene silencing mechanisms exhibited by this order of insects [Citation121]. Alternatively, we hypothesize that the high evolutionary rates at the flat surface opposing the groove might allow the continuous selection of new potential species-specific partner proteins that reduce the free energy of the microprocessor complex ().
3.2.3. Variability within Platform domain
The Platform domain in insect DCR1 is important for the production of 22-nucleotide RNAs from double-stranded RNA precursors (miRNAs) by establishing the distance of the cleavage site from the 5ʹ end. In hsDCR, the interaction with the 5ʹ end of RNA molecules is mediated by a phosphate-binding pocket present in the region known as the Platform-PAZ-Connector cassette. Mutations in this pocket prevent correct miRNA biogenesis [Citation118]. In accordance with our previous observation that the PAZ domain from DCR2 does not retain the canonical 5ʹ-phosphate-binding residues, we also confirmed that the insect DCR2 Platform domain has a modified phosphate-binding pocket displaying sequence variability (; Figures S22-S24; compare positions R21, R23, and R54 from the 5ZAK sequence in Figure S23). This further corroborates that the initial recognition of 5ʹ end in dsRNA substrates is not performed by the Platform and PAZ domains in DCR2. Accordingly, DCR2 initially recognizes the dsRNA substrate via its Helicase domain, which threads the polynucleotide double-helix until it ‘hits’ the PAZ and Platform domains at the opposite extremity of the microprocessor, thereby allowing the catalytic RIIIDs to proceed with processive cleavages in the transiently stabilized substrate [Citation122]. It should be noted that this model also predicts the possibility that the RIIID intradimer may cleave the substrate before it reaches the PAZ domain (generating fragments <20 nt), which has indeed been demonstrated for DCR2 in D. melanogaster [Citation103]. As for DROSHA, we found its 5ʹ-pocket has been slightly relocated (~ 8.7 Å) in the template structure PDB ID: 5B16). While it bears in common with DCR1’s phosphate-binding pocket the arginine residue between strands β4 and β5 (R62; in Figure S24, 5B16 sequence), the two arginine residues from loop β1-β2 have been relegated in preference of H15 and R26 from the DROSHA-specific insertion within the α2-α3 loop of the first RIIID subunit ( and S27) [Citation80]. The latter arginine residue is located in the so-called ‘Bump helix’ and is conserved in all insect species investigated, while the histidine has been substituted by either an arginine or lysine residue (Figure S27). Additionally, a conserved asparagine and a phenylalanine are also found in the putative 5ʹ-phosphate pocket (; NF-18 in Figure S27, 5B16 sequence). Until now, the recognition of the 5ʹ-phosphate by DCR1 proteins has been regarded as a lineage-specific acquisition by metazoans (animals), largely due to the belief that DCR from Giardia lamblia (which is basal to metazoan DCRs) lacks much of the Platform and Connector domains and appears to only bind the 3ʹ end of its RNA target [Citation119,Citation123]. Contrary to this notion, we found that G. lamblia DCR (glDCR) displays most of the structural elements present in animal DCRs. Therefore, we hypothesized that the 5ʹ-phosphate pocket had not been identified previously because it could also be slightly relocated, resembling the one we found in DROSHA proteins. To investigate this issue, we have extracted the Platform domain from human DROSHA and superposed it onto the Platform domain of the full-length glDCR structure (PDB ID: 2QVW). Strikingly, we found a protuberant cavity in glDCR at approximately 7.1 Å from where we found the putative 5ʹ-phosphate pocket in DROSHA (and at ~ 15.1 Å from the canonical DCR1 pocket; ). Furthermore, we found this cavity to be extremely well structured: two glutamate residues (E94 and E267 in glDCR) maintain four positively charged residues coordinated around a central negatively charged nucleus (; R39, K270, R312 and R318). An additional histidine (H92 in glDCR) can potentially participate in the pocket insofar as E94 is repelled by an incoming phosphate. Interestingly, R39 is located between β4 and β5 strands of the Platform domain in glDCR, just like the conserved arginine residues within the 5ʹ-phosphate pocket of human and insect DCR1 and DROSHA. Thus, our analyses suggest that this region’s role in binding phosphate is likely more ancestral than previously reported [Citation119]. Noteworthy, we also found unique similarities between the putative glDCR and metazoan DROSHA 5ʹ-phosphate-binding pockets, such as the participation of residues from the α2-α3 loop of the first RIIID subunit (R312 and R318); the RIIID loop in glDCR is intermediate in length to the DROSHA-specific insertion and the short loop found in metazoan RIIIDs. This implies that either DROSHA is evolutionarily closer to the ancestral eukaryote DCR than both DCR1 and DCR2 or that DROSHA acquired this characteristic independently and represents a potential case of molecular-evolutionary convergence. It should be noted that we also looked for an alternative 5ʹ-phosphate-binding pocket in the Platform domain of DCR2 proteins by plotting conserved residues onto the structure of D. melanogaster DCR2 (PDB ID: 6BUA) and analysing its surface. However, we did not find any alternative cluster of positively charged residues and our investigation indicates that insect DCR2 has a degenerate 5ʹ-phosphate-binding pocket arranged in similar position to the one found in DCR1 proteins (Figures S22 and S23). In agreement with our observation, it has been shown that mutating DCR2 by reintroducing residues present in the 5ʹ pocket of DCR1 Platform domain (e.g., R21, R23, and R54 of 5ZAK sequence; Figure S23) can rescue high-affinity binding of DCR2 to 5ʹ-phosphate [Citation119].
A general trend we identified in the Platform domains from DCR and DROSHA is the presence of four common variability hotspots, which form an extensive surface adjacent to a pronounced hydrophobic groove (). Considering the structure of DROSHA, the regions that comprise this surface are the following: the N-terminal tail (first 12 residues of the domain), the β3-β4 loop (equivalent to β2-β3 loop in DCR1-2), the N-terminal half of α1 helix, and the loop preceding β6 (loop pre-β6) (Figure S22-S24). The loop pre-β6 is very flexible and it is located nearest to the hydrophobic groove, which is formed by residues LE-86, S89, F93, W102, L104, P117, FHF-121, and L863 (see 5B16 sequence in Figure S24; L863 is not depicted in the alignment and it is part of the Connector helix in the same PDB 5B16). The nature of this hydrophobic groove is unclear, but it is positioned symmetrically opposite to the 5ʹ-phosphate pocket in the long axis of the Connector helix, resembling a mirror image (). All of the residues forming the hydrophobic groove, except for L863, are concentrated on the segment straddling the C-terminal half of α1 to the N-terminal half of β6 ( and S24). The hydrophobic residues in this segment are also conserved in insect DCR1-2 proteins (positions 81–112 in Figures S22 and S23). Intriguingly, this region contains a unique insertion in plant DCL proteins and has been specifically pinpointed, alongside an insertion in the PAZ domain, as primarily responsible for increasing the affinity of the Platform domain for RNA molecules in DCLs. In particular, the plant-specific insertion in the Platform domain is rich in positively charged residues and has been proposed to bind to the 5ʹ-phosphate [Citation119]. Thus, the hydrophobic groove that we found in animal DROSHA and DCR proteins may turn out to be completely remodelled with positive charges in plant DCL proteins. Additionally, the remodelled groove is positioned on the same face as the plant-specific insertion in the PAZ domain, which also forms a distinctive groove. We previously mentioned that lepidopteran species also harbour a positively charged insertion in the DCR1 PAZ domain, similar to the insertion found in plant DCL1. While the same is not true regarding the presence of a Platform insertion in the α1-β5 segment (which forms the hydrophobic groove), we found that the DCR1 Platform domain from lepidopteran species also displays distinctive positively charged residues in this region, which largely contrasts with what we observed in species from all other insect orders (Figure S22). Altogether, it is tempting to speculate that DCR1 from lepidopterans is capable of binding RNA substrates in a similar fashion as plant DCL1, which may involve recognizing nucleic acids in a different orientation than that found in other animal DCR1 proteins. The implications of this idiosyncrasy, however, are unclear, especially since DCR1 from lepidopterans also retains the conserved residues that form the canonical 5ʹ-phosphate and 3ʹ-overhang pockets in the Platform and PAZ domains, respectively. Since plant DCL1 can process both pri-miRNA and pre-miRNA substrates [Citation120], it is perhaps the case that Lepidoptera DCR1s can also bind to two different substrates. This matter requires further investigation.
3.2.4. Variability within RIIID and RIIID-like domains
Two copies of the RIIID domain (RIIID-I and RIIID-II) have been identified in DCR1-2 and DROSHA proteins, wherein each one acts as a different subunit capable of cleaving one of the dsRNA strands (; Figures S25-S30). Analyses of crystallographic structures have revealed that the canonical topology is composed of seven α-helices (Figures S25-S30). In DCR1-2 and DROSHA, the second RIIID subunit displays the canonical 7-helix structure, while the first subunit lacks the α1 helix, which is instead surrogated by the C-terminal end of the Connector helix (). Apart from this peculiarity, all other secondary structural elements of RIIIDs from DCR1-2 and DROSHA superimpose well to each other and maintain a well-defined hydrophobic core (RMSD = 0.58 Å; ). Conversely, the loops between helices α2-α3 and α5-α6 show remarkable variation in size and sequence identity (Figures S25-S30); for example, DROSHA displays a distinct RIIID-I subunit (known as the RIIID-like domain), which bears a large insertion between the α2 and α3 helices (; Figure S27). Both loops are located less than six residues from the first catalytic residues of helices α3 (positions E514 and D55 in Figure S29) and α6 (positions D156 and E159 in Figure S29). Thus, it appears that these regions may play pivotal roles in the catalytic mechanism of proteins harbouring RIIID domains. In Homo sapiens DCR (hsDCR), the α5-α6 loop from RIIID-I has been identified as a minimal binding site for the interaction with human AGO proteins, i.e., the polypeptide comprising only α5-α6 loop from hsDCR was able to interact with all members of human Argonaute proteins [Citation124]. Furthermore, the α5-α6 loop sequence was shown to be highly conserved among vertebrate DCR proteins but appears to have significantly changed during the evolution of their non-vertebrate orthologues [Citation124]. In agreement with these findings, we observed that the insect loops are shorter than those from vertebrates and display low sequence identity between different orders. One explanation for the evolutionary divergence of the α5-α6 loop in insects is the existence of DCR proteins which interact with different AGO proteins, something that is not observed in vertebrates [Citation125]. It has also been suggested that the α5-α6 loop of RIIID-I helps to align or direct the dsRNA substrates into the enzyme’s active sites, reason for which it was named the ‘Positioning loop’ in Giardia DCR [Citation126]. Nonetheless, the function of α5-α6 loop remains to be assessed in insects, and further investigation is needed to confirm whether it mirrors the roles described for human or Giardia DCRs [Citation124,Citation126]. A general trend we observed concerning this loop region is that the RIIID-II subunit exhibits shorter loops (45–52 residues) than the RIIID-I subunit (70–118 residues), which accounts for the majority of the second subunit’s reduced length. The only exceptions to this are DCR2 from dipterans, suborder Brachycera (e.g., Drosophila species), wherein the RIIID-II subunits have α5-α6 loops as large as those from RIIID-I (on average 80 and 97 residues, respectively), and DCR1 from lepidopterans, in which the RIIID-I subunits have α5-α6 loops as small as those from RIIID-II (on average 54 and 47 residues, respectively). A second general trend we observed is the strictly conserved amino acid composition of α5-α6 loops in RIIID-II from all DCR1 proteins, wherein 25–28% of the residues are negatively charged (particularly Asp). Interestingly, this conservation occurs even in dipterans of suborder Nematocera (e.g., Aedes and Anopheles genera) and ticks (Ixodidae family; Arthropoda outgroup), in which the α5-α6 loops are larger (61–65 residues) than the average length of those observed for RIIID-II subunits (~50 residues). In human DCR, we found that the α5-α6 Loop helix from RIIID-II (position 100–150 in Figures S27 and S28; 5ZAK sequence) interacts with the DEAD/ResIII (Hel1) and Dicer Dimer domains (). Furthermore, we identified that the N-terminal region flanking the Loop helix makes extensive contact with the α2-α3 loop of RIIID-II and that the flanking C-terminal region can potentially interact with the Helicase C subdomain when DCR is in the ATP-bound conformation, or with dsRNA being threaded through the Helicase domain (). The details how these interactions may influence the DCR mechanism deserves more attention than we can give here, but it is important to point out that regions enriched in negatively charged residues play special biological roles: they may regulate gene expression [Citation127–129], mimic the phosphate backbone of nucleic acids [Citation130,Citation131], and bind metal ions [Citation132] or specific domains [Citation133]. While most D/E-rich repeats are predicted to be unstructured, as was observed for both α5-α6 loops in the RIIIDs of human DCR (PDB ID: 5ZAK), peptides composed solely of either Asp or Glu residues have been shown to adopt the structure of a polyproline-II helix; this suggests that a local structure can be attributed to unfolded or disordered D/E-rich regions. Polyproline-II helices, like β strands, exhibit an extended conformation that facilitates binding to partner molecules [Citation134]. Although the presence of proline residues are not necessary for the formation of polyproline-II helices, they are the most preferred residues within the composition of this secondary structure; in their absence, glycine, polar and charged residues are preferred [Citation134–136]. We observed that, in addition to displaying larger-than-average D/E-rich loops, DCR1 RIIID-II subunits from Nematocera dipterans also present the highest Gly content among all α5-α6 loops, further suggesting that this region can adopt the structure of a polyproline-II helix.
We investigated the regions displaying higher variability by mapping the sequences and evolutionary coefficients of insect RIIIDs to their homologous domains within the structure of human DCR and DROSHA proteins (PDB IDs: 5ZAK and 5B16; Figures S25-S30). As in our previous analyses of other domains, we found that regions accumulating more mutations are generally clustered on the three-dimensional structure and form contiguous solvent-exposed surfaces. For example, the C-terminal regions of α3, α5 and α7 form a contiguous solvent-exposed surface in both RIIID subunits of DCR1 and DCR2 (). In DROSHA, this surface has been shown to interact with the C-terminal tail of PASHA () [Citation80]. Furthermore, the Loop helix and subsequent unresolved region extending towards α6 (Figures S25-S30) are also adjacent to this solvent-exposed surface (). In RIIID-I of DCR2, an additional mutation-prone, solvent-exposed surface is formed by the C-terminal region of α2 and the unresolved region between α5 and the Loop helix ( and S26). Interestingly, the same two regions are also prone to mutations in RIIID-II of DCR1and DCR2, but they do not form solvent-exposed surfaces; rather, they co-participate in intradomain interactions with the Helicase and Dicer Dimer domains (Figures S28 and S29). Finally, we found that the α1 helix of RIIID-II is prone to accumulate mutations; this region forms a solvent-exposed surface in-between the RIIID-II and DEAD/ResIII (Hel1) domains, located opposite to the catalytic cleft (). While this surface has no known or apparent function, the α1 helix appears to be important for maintaining the DEAD/ResIII domain in a relatively fixed position relative to the catalytic domains ().
3.2.5. Variability within the Helicase domain
Dicers can be classified as RIG-I-like proteins due to their harbouring an RNA Helicase domain at the N-terminus; in particular, RIG-I-like proteins differ from other RNA helicases because they exhibit a large insertion between the two canonical Helicase subdomains, DEAD/ResIII and Helicase C (aka RecA-like domains) [Citation137]. According to Sinha and co-workers [Citation103], the structure of the Helicase domain from D. melanogaster DCR2 (dmDCR2) is composed by four functional subdomains: DEAD/ResIII (aka Hel1), Hel2i (the large insertion found in RIG-I-like proteins), Helicase C (aka Hel2) and Pincer (; Figures S31-S32) [Citation103]. With respect to the cryo-EM structure of dmDCR2, the Hel1 and Hel2 domains, along with Pincer, could be fitted into the electron density map as a single rigid body. On the other hand, the Hel2i domain had to be fitted as a separate rigid body. In most RIG-I-like helicases, the functional domains perform activities that are intrinsic to ATP-driven translocases [Citation137]. Whether translocation on the dsRNA substrate is also coupled with unwinding of the helix is still unclear for most RIG-I-like proteins. According to Jankowsky & Fairman-Williams [Citation137], six conserved-sequence motifs of RIG-I-like helicases are important for ATP binding and hydrolysis (Q, I, II, III, Va and VI) and five are important for RNA binding (Ia, Ic, IV, IVa and V) [Citation137]. Among the conserved-sequence motifs that we identified in the DEAD/ResIII subdomain of DCR1, those related to RNA binding are degenerate compared to those related to ATP binding and hydrolysis. For example, motif Ia, which typically harbours conserved residues that establish side-chain contacts with RNA, is almost completely disfigured, and motif Ic displays variations in the canonical RNA-binding residue that characterizes RIG-I-like helicases (Figures S31) [Citation137]. We also noticed that the Lepidoptera order does not display the canonical glutamine residue in motif Q (Figures S31); as such, the Helicase domain of species within this order is likely able to hydrolyse any of the four NTPs (in contrast, glutamine introduces specific contacts that select for the adenine base). On the other hand, all of the conserved-sequence motifs that we found in the DCR2 Helicase domain displayed the canonical ATP– and RNA-binding residues (Figure S32). For translocation and/or unwinding to occur on the dsRNA substrate, the ATP-binding event must communicate with the RNA-binding event (and vice-versa). However, the ATP – and RNA-binding sites are separated by ~30 Å and it is still unclear how this communication is established between them [Citation138]. Recent evidence has identified two positions within motif V that are critical for communication between the ATP-binding pocket and the RNA-binding cleft in the closely related family of viral DExH helicases (aka NS3/NPH-II family) [Citation139]. Interestingly, these positions, which predominantly display a threonine and serine (T407 and S411) that interact with each other, displayed the highest residue variability across motif V of all flavivirus NS3 helicases. Overall, Du Pont et al. [Citation139] showed that removing the polar groups with H-bonding potential from positions T407 and S411 (see blue circles in Figures S31 and S32) increases the helicase turnover rate, especially in the latter position, but have opposite effects by either improving (T407) or reducing (S411) the affinity for dsRNA substrates in the presence of ATP. In particular, we found that the presence of non-polar group at position T407 (such as methyl or thiol) is important for coordination of four hydrophobic residues that influence the ATP– and RNA-binding residues in NS3 helicases (). We observed that the hydrophobic nature of these residues, as well as the presence of a non-polar group at the T407-equivalent position, are also conserved in the Helicase domains of insect DCR proteins (see black circles in Figures S31 and S32). This suggests that a similar mechanism for the communication between the ATP– and RNA-binding sites may apply to viral and RNAi-related helicases. The four hydrophobic residues coordinated around the insect T407 and S411 counterparts, henceforth denominated iT407 and iS411 for the sake of simplicity, are distributed across motifs IV, IVa and V, but we found they further coordinate a second layer of eleven conserved hydrophobic residues in the structure of RIG-I-like helicases (PDB ID: 5E3H). These residues span motifs Va and VI in insect DCR proteins, as well as a non-motif region between motifs IVa and V (; see grey circles in Figures S31 and S32). In RID-I-like helicases, this non-motif region is conserved and also harbours important RNA-binding residues (PDB IDs: 5E3H and 4A36). Hence, we have designated this region as motif IVb. We found that this second layer of hydrophobic residues can directly influence the positions of the ATP– and RNA-binding residues (red and yellow circles in Figures S31 and S32, respectively); thus, the central position occupied by iT407 in this network of hydrophobic contacts appears to play an important role in regulating the translocation and/or unwinding activity of DCR helicases by indirectly coordinating residues at both binding sites (). In particular, we found that the ATP-binding residues regulated by iT407 and iS411 (motifs Va and VI) are all conserved in DCR1 and DCR2, but the RNA-binding residues (motifs IV and IVa) are somewhat degenerate in DCR1. Thus, at least where the translocation and/or unwinding mechanisms are concerned, DCR2 binds to dsRNA in a more conserved manner.
We also noticed that, while present in DCR1, the canonical ATP-binding residues of motifs I (Walker A) and II (Walker B) display some variability and might render ATP hydrolysis less efficient in this protein, especially in lepidopteran species (Figures S31). In addition, the Lepidoptera order displays a large insertion that extends motif III in the DEAD/ResIII subdomain of DCR1 ( and S31); motif III has been implicated in sensing both the ATP-hydrolysis state and nucleic acid-binding event in some SF1 and SF2 helicases [Citation140,Citation141]. We also identified a dipteran-specific insertion between the Helicase C and Pincer subdomains of DCR1 ( and S31). While the function of this insertion is elusive, it is placed in a privileged position to interact with or block any dsRNA molecule binding to the DCR1 Helicase domain (). This peculiarity of dipterans indicates that D. melanogaster might not be the best model for studying RNAi in insects. With regards to with DCR2, all five insect orders studied here display a large insertion between helix α14 and strand β9 of the DCR1 Helicase C subdomain ( and S31). Again, the function of this insertion remains elusive, but we noticed that it is located near the Dicer Dimer domain in the structure of human DCR structure and in a privileged position to interact with the stem loop of pre-miRNAs in both the open and closed states of this enzyme (PDB IDs: 5ZAL, 5ZAM, and 5ZAK) [Citation113]. Furthermore, this insertion abuts the ATP-binding site and may interfere with the helicase turnover activity (). Overall, our data indicate that the DCR1 Helicase domain of insects is capable of hydrolysing ATP efficiently but binds to dsRNA through a less conserved mechanism, which may explain the lower affinity of this domain for siRNA precursors. The large insertions we observed in the DCR1 Helicase domain could have originated by recombination of a long DNA fragment into the locus that encodes an ancestral DCR1 ortholog, thereby leading this enzyme to specialize in the processing of pre-miRNAs molecules [Citation142].
4. Conclusions & final remarks
The in silico integration of the data presented in this study sheds light on the variability of domains within the RNAi machinery of five insect orders. We confirmed the universality of the RNAi mechanism in insects, as orthologues of the eight core proteins were identified in species of all five orders. All species are expected to have the basic elements of both the miRNA and siRNA machinery, but due to the fragmentation and incompleteness of a large number of publicly available genomes, as well as limitations in the methodologies for detection of divergent orthologues, some elements were not detected in several of the selected species. Thus, it is essential that future analyses be performed using curated databases harbouring well-assembled genomes/transcriptomes and using more than one method for ortholog detection. In this regard, we have now established well-defined sequence limits and better HMM profiles for annotating functional domains of the RNAi machinery in insects, which should greatly facilitate the identification of homologous proteins in both new and old genomes/transcriptomes. The structure-based sequence alignments that were generated using our methodology provide better inputs for phylogenetic inference and structure-function analyses of RNAi-related proteins. Unfortunately, the available structural data for insect proteins, especially those belonging to the RNAi machinery, are mostly limited to model species, such as D. melanogaster. Thus, further studies with non-model insect species are needed to allow for ample functional analyses of insect proteins. In particular, considering the RNAi pathway, it is imperative that more structural models with atomic resolution be solved in order for us to answer questions about the intricacies of this mechanism in insects. Nonetheless, our results show that considerable variability exists in elements of the RNAi machinery, all of which can potentially affect the efficiency of gene silencing triggered by exogenous RNA.
Regulation mechanisms of the siRNA pathway have coevolved with viral infections, and among the insect orders studied here, lepidopterans have been shown to be the most susceptible to viral attacks (approximately 80% of the species), followed by the dipterans (9%), coleopterans (5%), hymenopterans (4%) and hemipterans (1%) [Citation143]. One can argue that this observation correlates with the efficiency of a given order in controlling viral infections through RNAi-mediated mechanisms; if true, lepidopterans would be expected to show the lowest efficiency. Intriguingly, our phylogenetic analyses have clearly shown that, in practically all domains analysed, the Lepidoptera order has the greatest evolutionary distance compared to the other orders. This corroborates previous reports that underscore the different efficiencies displayed by lepidopteran species during exogenous dsRNA-mediated gene knockdown. The variability and phylogenetic distance that we observed may be evidence that sufficient idiosyncrasies exist in the RNAi machinery of Lepidoptera to set them apart from other insects. Coleopterans are generally susceptible to RNAi and display higher silencing efficiency than lepidopterans, which are generally recalcitrant to RNAi. This has led to the recently approved commercialization of a new genetically modified crop event (MON87411) wherein the heterologous production of Bt toxins was coupled with the expression of dsRNA molecules in order to control the western corn rootworm (Diabrotica virgifera virgifera, LeConte; Coleoptera: Chrysomelidae) [Citation144]. Our analyses highlighted several variability hotspots within the core elements of the RNAi machinery, thereby enabling us to compare the data between non-efficient lepidopterans and those coleopteran species that exhibit acceptable silencing efficiencies. Four of the five domains we analysed displayed differences which could explain the contrasting gene silencing efficiency between Coleoptera and Lepidoptera species: (i) dsrm; (ii) Helicase; (iii) PAZ; and (iv) RIIID. While these differences are readily apparent, most of them were found in proteins pertaining to the miRNA pathway, which, in theory, should not cause major disturbances in RNAi-mediated gene knockdown. Nevertheless, core RNAi enzymes from the miRNA and piRNA pathways have also been shown to participate in the exogenous RNAi responses of Bombyx mori (Lepidoptera), Leptinotarsa decemlineata (Coleoptera), and D. melanogaster (Diptera) [Citation145]. Additionally, the miRNA pathway has been shown to play a role in the modulation of gene expression in response to viral infection in mammals [Citation146], as well as to produce miRNAs that target specific sites of the viral genome [Citation147]. It was even demonstrated that DROSHA, which acts upon pri-miRNAs in the nucleus, can be recruited to the cytoplasm in response to virus infections, where it has been proposed to cleave viral RNA secondary structures or host cytoplasmatic RNA hairpins [Citation148]. Therefore, we cannot exclude the possibility that differences in proteins of the miRNA pathway may somehow influence RNAi-mediated gene silencing sensitivity in lepidopterans. With that said, most of the variability displayed across insects were present in the loop regions of domains. The structure of large flexible loops is difficult to resolve; accordingly, most of them are not represented in the publicly available structural data, thereby limiting the quality of the homology models that can be generated. Nevertheless, these regions can significantly influence the activity of the proteins whereupon they are inserted; for example, they can modify substrate affinity, block catalytic sites, or even interact with other proteins. Hence, both in vitro and in silico studies aiming to characterize these regions are essential to completely elucidate the mechanism of action of the core RNAi proteins we analysed.
A marked difference was found in the dsrm-II domain of LOQS-PB, wherein lepidopterans display an insertion, V(N/A)RR, in the β1-β2 loop region (Figure S13). As previously mentioned in section 3.2.1, the dsrm β1-β2 loop binds to the minor groove of dsRNA and greatly affects the affinity for this substrate. In particular, the lepidopteran-specific insertion adds positive charges to this loop, which may increase the number of contacts made with the phosphate backbone and thereby improve the affinity of the DCR1 microprocessor for pre-miRNA. Alternatively, the insertion can extend the distance between the guanine-binding histidine in loop β1-β2 and the sequence-specific binding residue from helix α1, which will affect the size of the dsRNA regions that are specifically recognized by the dsrm domain [Citation149].
Compared to their coleopteran orthologues, the DCR1 Helicase domains of lepidopterans display a large insertion between β6 (motif III) and α7 in the DEAD/ResIII subdomain (Figure S31). Insertions in this region are common in other families of SF1 and SF2 helicases and have been implicated in the communication between the ATP– and RNA-binding sites [Citation140,Citation141]. In addition, we showed that lepidopterans lack the canonical Q residue in the eponymous Q motif (Figure S31), giving rise to the intriguing possibility that the DCR1 Helicase domain of Lepidoptera may hydrolyse NTPs other than ATP. In parallel, the insertion between α14 and β9 in the Helicase C subdomain of DCR1, which protrudes towards the ATP-binding site (), displays many order-specific sequence segments that suggest the existence of a convoluted mechanism underlying the DCR1 helicase activity (Figures S31). This insertion can be considered the major difference between the Helicase domains of DCR1 and DCR2 and likely plays an important role in how this domain engages substrates in both proteins. While coleopteran species display the shortest α14-β9 insertions among all insect DCR1 proteins that we evaluated, lepidopterans display the longest; however, the role of this region in the processing of pre-miRNAs, or even siRNA precursors, remains to be explained.
Lepidopterans display an insertion of 3–5 amino acids in the β4-β5 loop (β-hairpin module) of the PAZ domain from AGO2 proteins (Figure S18). The β-hairpin module recognizes the 3ʹ end of dsRNA molecules that are loaded onto AGO proteins. Therefore, this insertion can modify how lepidopterans interact with and load dsRNA during formation of the RISC complex [Citation150]. With respect to the DCR1 PAZ domain, lepidopterans have acquired a positively charged insertion at the N-terminal region; intriguingly, this insertion is similar to the N-terminal region of plant DCL1 proteins (Figure S19). As we have previously mentioned, this insertion could lead to lepidopteran DCR1 interacting with dsRNA in a different orientation compared to coleopteran DCR1, thereby triggering downstream variations in the gene knockdown efficiency. In parallel, the regions interacting with dsRNA in the PAZ domain of DCR2 proteins can also be considered an important source of variability between coleopterans and lepidopterans. Unlike AGO PAZ domains, the DCR counterpart harbours an insertion between β3-β4 (β6-β7 in AGO proteins) that is rich in polar and positively charged residues (Figure S20). In the X-ray structure of the Platform-PAZ cassette of human DCR (PDB ID: 4NGD), this insertion is important for stabilizing the DCR-dsRNA complex and forms a helical structure (α2 in the PAZ domain of DCR1-2; Figures S19 and S20) that is associated with the release and transfer of the cleaved dsRNA molecule onto AGO proteins [Citation115]. In coleopterans, this insertion is shorter and has a more positive residual charge than its lepidopteran orthologues, which might result in a stronger interaction of this domain with the dsRNA backbone. Consequently, the PAZ domain of coleopterans might confer higher thermodynamic stability to the DCR2 microprocessor, allowing higher delivery rates of siRNAs to AGO proteins.
The most relevant regions of variability between the endonuclease domains of DCR proteins were found in the RIIID-I domain, more precisely in the α5-α6 loop (Figures S25 and S26). As mentioned before, this loop is responsible for the interaction of human DCR with AGO proteins and may also be involved in the catalytic mechanism [Citation124]. DCR1 RIIID-I domains from lepidopterans exhibit the most divergent α5-α6 loops among all species analysed, displaying a large deletion after the Loop helix (Figure S25). This deletion may beget divergent DCR1-AGO1 interactions in lepidopterans compared to insects from other orders. Similarly, the DCR2 RIIID-I domains of lepidopterans maintain a conserved 4-residue signature in the α5-α6 loop, ExE(P/K), that differentiate them from all other analysed species. The importance of this signature in the DCR2 mechanism is unclear, but its potential involvement in Lepidoptera RNAi efficiency should be investigated nonetheless (Figure S26).
It is also known that viral infections may leave ‘scars’ in the host insect genome, the so-called endogenous viral elements (EVEs). Accordingly, EVEs related to transposons, baculoviruses and bracoviruses (viruses of parasitic wasps) can be found integrated in lepidopteran genomes [Citation151]. As previously mentioned (Supplementary Text ST1), defective viral genomes (DVGs) can be retro-transcribed into viral DNA (vDNA) and incorporated into the host genome as an EVE; these will then act as an immunological memory by providing an additional substrate to help boost the RNA interference response through the siRNA pathway, potentially promoting viral persistence in insects [Citation152]. Moreover, in addition to giving rise to endogenous viral siRNAs (vsiRNAs) via DCR2-LOQS-PD processing (; step 16), EVEs also produce viral piRNAs (vpiRNAs) that contribute to the antiviral response via the piRNA pathway [Citation153,Citation154]. In this regard, EVEs are widespread in arthropod genomes and commonly give rise to PIWI-interacting RNAs that can potentially play a role in the antiviral response [Citation155]. Interestingly, Cui and Holmes [Citation156] have also presented evidence that EVEs with high similarity to plant viruses are integrated in the genomes of mosquitoes, fruit flies, bees, ants, silkworm, pea aphid, Monarch butterfly and wasps. We have found that lepidopterans carry a plant-like, positively charged insertion at the N-terminal region of the DCR1 PAZ domain, suggesting that RNA recognition by DCR1 in this order may function similarly to that related with plant DCL1 (Figure S19). Furthermore, the Platform domain from lepidopterans, like those from plants, also display a cluster of positively charged residues that are positioned adjacent to the PAZ N-terminal insertion. Why does lepidopteran DCR1 harbour similar characteristics to those of plant DCL1? These observations are particularly interesting given that more than 70% of all agricultural pests are insects in the order Lepidoptera [Citation35]. Indeed, much of the Lepidoptera diversity can be attributed to the radiation of species in association with flowering plants: they represent the single most diverse lineage of organisms to have primarily evolved dependent upon angiosperm plants, and their numbers exceed those of the other major plant-feeding insects, such as those belonging to the orders Heteroptera, Homoptera, and Coleoptera (Chrysomeloidea and Curculionoidea) [Citation157]. One hypothesis for the similarities between plants and Lepidoptera is that lepidopteran DCR1 can recognize plant pri– or pre-miRNAs that are ingested during feeding and then further process them to regulate the expression of their own genes, particularly those associated with countering the plant’s defence mechanisms. This may provide a way for the insect to fine-tune the expression of certain genes in accordance with the plant’s miRNA-mediated response to predation. To test this hypothesis, one would need to compare the complementarity of the 5ʹ and 3ʹ-UTR regions of plant and lepidopteran mRNAs to the sequence of plant miRNAs that are overexpressed during insect feeding. This hypothesis also raises the question of whether lepidopterans have also evolved to take advantage of plant-produced vsiRNAs or vpiRNAs to defend themselves from plant viruses that can be ingested. If similar EVEs associated with plant viruses are present in the genomes of both plants and lepidopterans, then the plant-produced piRNAs or endo-siRNAs related to those EVEs, which are potentially being used to modulate a viral infection or transposable element, may also be used to trigger specific responses in the insect. What is clear is that lepidopterans engage different RNAi-related mechanisms in response to viral infections, and these mechanisms appear to differ from those involved with the responses found in other insects [Citation158]. For example, while DCR2 predominantly targets viral dsRNA during the infection of B. mori with its eponymous Cytoplasmic Polyhedrosis Virus (BmCPV), an unknown RNAse has also been linked to the origins of vsiRNA biogenesis and distribution, and an additional pathway is triggered in response to viral mRNA derived from a specific segment of the viral genome [Citation158]. Irrespective of the reason, these similarities between plant DCL1 and Lepidoptera DCR1 certainly merit further investigation.
While EVEs can encode functional proteins, for the most part, they become inactive over the course of evolution [Citation159,Citation160]. Nevertheless, these elements can retain some advantageous characteristics, which, among other functions, can act to suppress other viral infections (some viruses produce antivirals proteins to overcome competition) Antivirals proteins encoded in endogenous vDNA can therefore equip the host with tools capable of turning a fatal viral infection into a latent infection. Alternatively, endogenous vDNA may also encode viral suppressors of RNAi (VSRs), which can weaken the host antiviral defence to turn an otherwise acute infection (in which the host eliminates the virus) into a persistent infection. The main modes of action for viral suppressors of RNAi are: (i) binding to the dsRNA substrate, which prevents cleavage by DCR2; (ii) binding to siRNA, which prevents loading into RISC; (iii) degrading the siRNA molecule; and (iv) direct interaction with DCR2 or AGO2, which prevents their actions [Citation161]. Thus, both the antivirals and VSRs encoded in endogenous vDNA may influence the sensitivity of insects to RNAi-mediated gene silencing. In D. melanogaster, the expression of two insect VSRs and three out of six plant VSRs inhibited siRNA responses associated with viral RNA and injected dsRNA, suggesting that some viral suppressors can negatively impact the RNAi efficiency in some systems [Citation162]. Given the large number of viruses that infect Lepidoptera species, it is reasonable to speculate that EVEs derived from DVGs may generate molecules capable of, for example, binding to DCR2 or siRNAs and preventing their loading into RISC [Citation145]. In sum, we found clear distinctions between domains from coleopterans and lepidopterans. While these variations alone cannot irrefutably explain the differences that have been observed in RNAi-mediated gene silencing efficiency between these orders, they underscore specific regions that should be addressed to better understand the RNAi mechanism in these insects.
Our results also highlight an important factor to be considered when evaluating the efficiency of RNAi-mediated gene silencing in insects: the structural stability of the DROSHA-pri-miRNA, DCR1-pre-miRNA and DCR2-dsRNA complexes. It is important to note that structural stability (i.e., persistence of interactions, or robustness) is fundamentally different from thermodynamic stability (i.e., binding free energy, or ΔGbind). In the case of enzymes, such as DCR, structural stability speaks about the need of keeping the substrate in place for efficient catalysis, while thermodynamic stability refers to the affinity of the enzyme for its substrate. Consequently, the higher the structural stability of the aforementioned complexes (i.e., the longer the substrate remains correctly positioned in the binding site), the higher the turnover rate of miRNA and siRNA produced. In this regard, the presence of elements that increase the structural stability of these microprocessors complexes is vital for an effective response of the RNAi machinery. Studies have shown that he Staufen C protein, unique to members of the Coleoptera order, is an important factor in the development of insect resistance to RNAi [Citation163]. This protein contains multiple domains harbouring the dsRBD fold, some of which have been shown to bind to dsRNA. Due to this structural characteristic, as well as the involvement of this protein in the DCR2-mediated processing of dsRNA into siRNAs, one can hypothesize that Staufen C confers structural stability to the DCR2 microprocessor in coleopterans. Therefore, it is important to identify other dsRNA-binding proteins that may also contribute positively to increasing the efficiency of dsRNA processing in insects, which should provide a better understanding of the RNAi silencing mechanism or even be used as a biotechnological tool.
Another important factor to be considered is how insects detect the presence of viruses since viral dsRNA (as well as exogenous dsRNAs) can be considered an important pathogen-associated molecular pattern (PAMP) [Citation164]. In addition to the viral control mediated by RNAi, there are several other signalling pathways capable of controlling viral infections, mainly by triggering insect innate immune responses, among which we can highlight: (i) JAK-STAT, which regulates the downstream production of effector molecules, such as antimicrobial peptides (AMPs) [Citation164,Citation165]; and (ii) IMD and TOLL, which are NF-κB-related pathways in which the final transcriptional factors responsible for signal transduction are Relish (Rel1 and Rel2) and Dorsal/Dif, respectively [Citation164,Citation166]. Not surprisingly, these three signal transduction pathways display crosstalk between each other, wherein the signal is transduced by protein kinases and culminates in the regulation of several target genes/proteins. In this context, DCR proteins, specifically DCR2, can be considered pathogen recognition receptors (PRRs) involved in the detection of viral infections in insects [Citation164]. A study involving D. melanogaster infected with Drosophila C Virus (DCV) showed the participation of DCR2 Helicase domain in viral dsRNA recognition, which in turn stimulated the expression of antiviral genes through the upregulation of a cysteine-rich peptide, Vago, which acts in a similar way to mammalian RIG-I-like sensors [Citation142,Citation167]. This mechanism involving DCR2 was also characterized in the Culex quinquefasciatus mosquito in response to the West Nile Virus (WNV), but some differences were observed when compared to the response displayed by D. melanogaster [Citation167,Citation168]. The presence of viral dsRNA is detected by the DCR2 Helicase domain, and the Rel2 transcription factor of C. quinquefasciatus induces the expression of the vago gene via TNF receptor-associated factor (TRAF). Thereafter, similar to what occurs in Drosophila, the secreted CxVago peptide induces the JAK-STAT-mediated antiviral response [Citation167,Citation169]. In short, this mosquito’s immune response can be considered a crosstalk between the RNAi, JAK-STAT and IMD pathways [Citation170]. The central role played by the DCR2 Helicase domain in activating molecular signalling during antiviral responses, including exogenous dsRNA, highlights the importance of identifying variability within this ‘hub’ domain (Figure S32). We hypothesize that some of the variabilities we identified in DCR2 may produce yet unknown consequences in the Vago-mediated activation of the JAK-STAT pathway, or even in the biogenesis of DVGs [Citation171]. No studies have yet reported the characterization of the JAK-STAT pathway in lepidopterans. It is also possible that other uncharacterized pathways may operate during the antiviral response of lepidopterans [Citation158].
In parallel, studies have shown that the low efficiency of RNAi-mediated gene silencing in some insect species can be directly associated with the expression levels of miRNA/siRNA elements, which may provide a partial explanation for the differences in RNAi efficiency observed in Lepidoptera. For example, it is known that the expression levels of the translin gene (a component of the C3PO complex) are very low in B. mori and M. sexta cells, and in addition, some lepidopterans exhibit almost undetectable levels of the R2D2 transcript, even during viral infections [Citation145]. Studies that overexpressed elements of insect RNAi machinery (AGO2 and DCR2) in lepidopteran cells reinforce these observations since they considerably increased the RNAi-mediated antiviral response [Citation172]. However, why is there such variation in the expression of insect RNAi-related genes? How does this regulation occur? It is known that in D. melanogaster, the transcription factor Forkhead box O (dFOXO) upregulates the expression of important genes in the RNAi pathway, such as AGO2 and DCR2 [Citation173]. Following on the participation of dFOXO in responses related to metabolic changes and its relationship with multiple stress responses, a recent study has identified the participation of insulin in the antiviral response of insect vectors [Citation174]. Insulin-mediated dFOXO repression inhibits the RNAi response (by suppressing the transcription of genes encoding the AGO2 and DCR2 proteins) and, in parallel, activates the JAK-STAT pathway [Citation174]. Could the insulin-mediated response be predominant in lepidopterans, thus culminating in the repression of genes related to the RNAi pathway? Considering that the signalling pathways mediated by the Vago peptide and insulin are distinct, even though both converge to achieve an antiviral response mediated by JAK-STAT, and the fact that all these findings have also been validated in lepidopterans, we hypothesize that mutations in the receptors that sense viral infections and/or exogenous dsRNA, such as the Helicase domain, may be related to the predominance of an insulin-mediated response in some species of this insect order. Although speculative at this point, this hypothesis, associated with the data presented here, may help explain the low efficiency of RNAi-mediated gene silencing in Lepidoptera.
Considering the application of RNAi as a biotechnological tool, one question lingers: is it possible to universally apply RNAi-mediated gene silencing to control insect pest populations? The data presented here show that we are likely to fail if we generalize the application of RNAi-mediated gene silencing based on the restricted studies of a few model organisms. We have pinpointed some intriguing peculiarities within the functional domains of the RNAi machinery that must be addressed using a more species-specific approach in order to understand the nuances of differences associated with RNAi mechanisms in insects. For example, dipterans of suborders Brachycera and Nematocera show markedly different characteristics across all of the domains we analysed, implying that studies on D. melanogaster may not provide a solid framework for understanding RNAi in Aedes aegypti, and vice-versa. Besides, small modifications to the experimental design can considerably increase the efficiency of exogenous dsRNA-mediated gene silencing in specific species. Recent studies have shown that for two lepidopteran species (Helicoverpa armigera and Ostrinia furnicalis), the presence of GGU nucleotides in exogenously administered dsRNA considerably increases siRNA production due to cleavage by DCR2, downstream of this motif. On the other hand, the same study showed that in T. castaneum, a member of the Coleoptera order, dsRNA was cut downstream of more diverse sites, such as AAG, GUG, and GUU [Citation175]. In light of these reports, it is crucial to decipher how DCR2 recognizes the motifs upstream of the cleavage sites, as this would significantly improve the design of exogenous dsRNAs and considerably increase the efficiency of gene knockdown, especially in lepidopteran species.
Overall, it can be concluded that studies focusing on the genetic and structural variability of the core RNAi proteins are crucial to better understand how insects fine tune their RNAi-mediated development and antiviral response, which will ultimately drive how we design adapted biotechnological tools for the control of insect pest populations.
AUTHORS CONTRIBUTIONS
MFGS and EGJD were the lead researchers for all the work. FBMA and DM-S produced and analysed all data and wrote the draft manuscript. MF, LLPM, VJVM, CVM, and DDVN participated in the structural analysis of protein domains. DM-S and JARGB designed the structure-based sequence alignment protocol. All authors provided inputs to the manuscript, revised, and approved the final version.
Supplemental Material
Download Zip (113.4 MB)Acknowledgments
FBMA is grateful to CAPES for a PhD endowment (88882.157206/2017-01). MFGS is grateful for financial support from EMBRAPA, UCB, UnB, CNPq, PlantStress Biotech - INCT, CAPES, and FAPDF.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplemental data
Supplemental data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Fire A, Xu S, Montgomery MK, et al. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998 Feb;391(6669):806–811.
- Olsen PH, The AV. lin-4 regulatory RNA controls developmental timing in Caenorhabditis elegans by blocking LIN-14 protein synthesis after the initiation of translation. Dev Biol. 1999 Dec;216(2):671–680.
- Napoli C, Lemieux C, Jorgensen R. Introduction of a chimeric chalcone synthase gene into Petunia results in reversible co-suppression of homologous genes in trans. Plant Cell. 1990 Apr;2(4):279–289.
- Kavi HH, Fernandez H, Xie W, et al. Genetics and biochemistry of RNAi in Drosophila. Curr Top Microbiol Immunol. 2008;320:37–75.
- Li H, Li WX, Ding SW. Induction and suppression of RNA silencing by an animal virus. Science. 2002 May;296(5571):1319–1321.
- Chow J, Kagan JC. The fly way of antiviral resistance and disease tolerance. Adv Immunol. 2018 Sep;140:59–93.
- Leggewie M, Schnettler E. RNAi-mediated antiviral immunity in insects and their possible application. Curr Opin Virol. 2018 Nov;32:108–114.
- Swevers L, Liu J, Smagghe G. Defense mechanisms against viral infection in Drosophila: rNAi and non-rNAi. Viruses. 2018 May;10(5):230.
- Sempere LF, Freemantle S, Pitha-Rowe I, et al. Expression profiling of mammalian microRNAs uncovers a subset of brain-expressed microRNAs with possible roles in murine and human neuronal differentiation. Genome Biol. 2004 Feb;5:R13.
- Mallory A, Vaucheret H. Form, function, and regulation of Argonaute proteins. Plant Cell. 2010 Dec;22(12):3879–3889.
- Okamura K, Lai EC. Endogenous small interfering RNAs in animals. Nat Rev Mol Cell Biol. 2008 Sep;9(9):673–678.
- Lin H, Spradling AC. A novel group of pumilio mutations affects the asymmetric division of germline stem cells in the Drosophila ovary. Development. 1997 Jun;124:2463–2476.
- Brennecke J, Aravin AA, Stark A, et al. Discrete small RNA-generating loci as master regulators of transposon activity in Drosophila. Cell. 2007 Mar;128(6):1089–1103.
- Joga MR, Zotti MJ, Smagghe G, et al. RNAi efficiency, systemic properties, and novel delivery methods for pest insect control: what we know so far. Front Physiol. 2016 Nov;7:553.
- Airs PM, Bartholomay LC. RNA interference for mosquito and mosquito-borne disease control. Insects. 2017 Jan;8(1): 4.
- Mamta B, Rajam MV. RNAi technology: a new platform for crop pest control. Physiol Mol Biol Plants. 2017 Jul;23(3):487–501.
- Zhang J, Khan SA, Heckel DG, et al. Next-generation insect-resistant plants: rNAi-mediated crop protection. Trends Biotechnol. 2017 Jul;35(9):871–882.
- Yu N, Christiaens O, Liu J, et al. Delivery of dsRNA for RNAi in insects: an overview and future directions. Insect Sci. 2013 Feb;20(1):4–14.
- Agrawal A, Rajamani V, Reddy VS, et al. Transgenic plants over-expressing insect-specific microRNA acquire insecticidal activity against Helicoverpa armigera: an alternative to Bt-toxin technology. Transgenic Res. 2015 Oct;24(5):791–801.
- Yogindran S, Rajam MV. Artificial miRNA-mediated silencing of ecdysone receptor (EcR) affects larval development and oogenesis in Helicoverpa armigera. Insect Biochem Mol Biol. 2016 Jul;77:21–30.
- Saini RP, Raman V, Dhandapani G, et al. Silencing of HaAce1 gene by host-delivered artificial microRNA disrupts growth and development of Helicoverpa armigera. PLoS One. 2018 Mar;13(3):e0194150.
- Sharath Chandra G, Asokan R, Manamohan M, et al. Enhancing RNAi by using concatemerized double-stranded RNA. Pest Manag Sci. 2019 Feb;75:506–514.
- Whitten MM. Novel RNAi delivery systems in the control of medical and veterinary pests. Curr Opin Insect Sci. 2019 Feb;34:1–6.
- Ulvila J, Parikka M, Kleino A, et al. Double-stranded RNA is internalized by scavenger receptor-mediated endocytosis in Drosophila S2 cells. J Biol Chem. 2006 May;281(20):14370–14375.
- Jin S, Singh ND, Li L, et al. Engineered chloroplast dsRNA silences cytochrome p450 monooxygenase, V-ATPase and chitin synthase genes in the insect gut and disrupts Helicoverpa zea larval development and pupation. Plant Biotechnol J. 2015 Mar;13(3):435–446.
- Bally J, Fishilevich E, Bowling AJ, et al. Improved insect-proofing: expressing double-stranded RNA in chloroplasts. Pest Manag Sci. 2018 Aug;74(8):1751–1758.
- Wang K, Peng Y, Fu W, et al. Key factors determining variations in RNA interference efficacy mediated by different double-stranded RNA lengths in Tribolium castaneum. Insect Mol Biol. 2019;28(2):235–245.
- Liu J, Swevers L, Iatrou K, et al. Bombyx mori DNA/RNA non-specific nuclease: expression of isoforms in insect culture cells, subcellular localization and functional assays. J Insect Physiol. 2012 Aug;58(8):1166–1176.
- Wynant N, Santos D, Verdonck R, et al. Identification. Schistocerca gregaria. Insect Biochem. Mol. Biol: functional characterization and phylogenetic analysis of double stranded RNA degrading enzymes present in the gut of the desert locust; 2014 Jan.
- Almeida Garcia R, Lima Pepino Macedo L, Cabral Do Nascimento D, et al. Nucleases as a barrier to gene silencing in the cotton boll weevil. PLoS One. Anthonomus Grandis. 2017 Dec;12:e0189600.
- Spit J, Philips A, Wynant N, et al. Knockdown of nuclease activity in the gut enhances RNAi efficiency in the Colorado potato beetle, Leptinotarsa decemlineata, but not in the desert locust, Schistocerca gregaria. Insect Biochem Mol Biol. 2017 Jan;81:103–116.
- Peng Y, Wang K, Fu W, et al. Biochemical comparison of dsRNA degrading nucleases in four different insects. Front Physiol. 2018 May;9:624.
- Prentice K, Smagghe G, Gheysen G, et al. Nuclease activity decreases the RNAi response in the sweetpotato weevil Cylas puncticollis. Insect Biochem Mol Biol. 2019 Jul;110:80–89.
- Song H, Fan Y, Zhang J, et al. Contributions of dsRNases to differential RNAi efficiencies between the injection and oral delivery of dsRNA Locusta migratoria. Pest Manag Sci. 2019 Jun;75(6):1707–1717.
- Guan R-B, Li H-C, Fan Y-J, et al. A nuclease specific to lepidopteran insects suppresses RNAi. J Biol Chem. 2018 Apr;293(16):6011–6021.
- Winston WM, Molodowitch C, Hunter CP. Systemic RNAi in C. elegans requires the putative transmembrane protein SID-1. Science. 2002 Mar;295:2456–2459.
- McEwan DL, Weisman AS, Hunter CP. Uptake of extracellular double-stranded RNA by SID-2. Mol Cell. 2012 Sep;47(5):746–754.
- Tomoyasu Y, Miller SC, Tomita S, et al. Exploring systemic RNA interference in insects: a genome-wide survey for RNAi genes in Tribolium. Genome Biol. 2008 Jan;9(1):R10.
- Méndez-Acevedo KM, Valdes VJ, Asanov A, et al. A novel family of mammalian transmembrane proteins involved in cholesterol transport. Sci Rep. 2017 Aug;7(1):7450.
- Vélez AM, Fishilevich E. The mysteries of insect RNAi: a focus on dsRNA uptake and transport. Pestic Biochem Physiol. 2018 Oct;151:25–31.
- Xiao D, Gao X, Xu J, et al. Clathrin-dependent endocytosis plays a predominant role in cellular uptake of double-stranded RNA in the red flour beetle. Insect Biochem Mol Biol. 2015 May;60:68–77.
- Dominska M, Dykxhoorn DM. Breaking down the barriers: siRNA delivery and endosome escape. J Cell Sci. 2010 Apr;123(8):1183–1189.
- Yoon JS, Gurusamy D, Palli SR. Accumulation of dsRNA in endosomes contributes to inefficient RNA interference in the fall armyworm, Spodoptera frugiperda. Insect Biochem Mol Biol. 2017 Sep;90:53–60.
- Waterhouse RM, Seppey M, Simão FA, et al. BUSCO applications from quality assessments to gene prediction and phylogenomics. Mol Biol Evol. 2018 Mar;35(3):543–548.
- Altschul SF, Madden TL, Schäffer AA, et al. PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997 Sep;25(17):3389–3402.
- Zhang M, Leong HW. Bidirectional best hit r-window gene clusters. BMC Bioinf. 2010 Jan;11(Suppl 1):S63.
- Li Z, Tiley GP, Galuska SR, et al. Multiple large-scale gene and genome duplications during the evolution of hexapods. Proc Natl Acad Sci USA. 2018 May;115(18):4713–4718.
- Dalquen DA, Dessimoz C. Bidirectional best hits miss many orthologs in duplication-rich clades such as plants and animals. Genome Biol Evol. 2013;5(10):1800–1806.
- Ward N, Moreno-Hagelsieb G. Quickly finding orthologs as reciprocal best hits with BLAT, LAST, and UBLAST: how much do we miss? PLoS One. 2014 Jul;9(7):e101850.
- Gertz EM, Yu Y-K, Agarwala R, et al. Composition-based statistics and translated nucleotide searches: improving the TBLASTN module of BLAST. BMC Biol. 2006 Dec;4(1):41.
- Rombel IT, Sykes KF, Rayner S, et al. FINDER: a vector for high-throughput gene identification. Gene. 2002 Jan;282(1–2):33–41.
- Eddy SR. A new generation of homology search tools based on probabilistic inference. Genome Inform. 2009 Oct;23:205–211.
- Letunic I, Bork P. 20 years of the SMART protein domain annotation resource. Nucleic Acids Res. 2018 Jan;46(D1):D493–D496.
- Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013 Apr;30(4):772–780.
- Capella-Gutiérrez S, Silla-Martínez JM, Gabaldón T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics. 2009 Aug;25(15):1972–1973.
- Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014 May;30(9):1312–1313.
- Letunic I, Bork P. Interactive tree of life (iTOL) v4: recent updates and new developments. Nucleic Acids Res. 2019 Jul;47(W1):W256–W259.
- Sydykova DK, Jack BR, Spielman SJ, et al. Measuring evolutionary rates of proteins in a structural context. [version 2; peer review: 4 approved]. F1000Res. 2017 Oct;6:1845.
- Spielman SJ, Kosakovsky Pond SL. Relative evolutionary rate inference in HyPhy with LEISR. PeerJ. 2018 Feb;6:e4339.
- Le SQ, Gascuel O. An improved general amino acid replacement matrix. Mol Biol Evol. 2008 Jul;25(7):1307–1320.
- Huang Y, Niu B, Gao Y, et al. a web server for clustering and comparing biological sequences. Bioinformatics. 2010 Mar;26(5):680–682.
- Milburn D, Laskowski RA, Thornton JM. Sequences annotated by structure: a tool to facilitate the use of structural information in sequence analysis. Protein Eng. 1998 Oct;11(10):855–859.
- Wu S, Zhang Y. LOMETS: a local meta-threading-server for protein structure prediction. Nucleic Acids Res. 2007 May;35(10):3375–3382.
- Jaroszewski L, Li Z, Cai X, et al. FFAS server: novel features and applications. Nucleic Acids Res. 2011 Jul;39(suppl):W38–44.
- Kurowski MA, Bujnicki JM. GeneSilico protein structure prediction meta-server. Nucleic Acids Res. 2003 Jul;31(13):3305–3307.
- Mirdita M, Steinegger M, Söding J. MMseqs2 desktop and local web server app for fast, interactive sequence searches. Bioinformatics. 2019 Aug;35(16):2856–2858.
- Li Z, Natarajan P, Ye Y, et al. POSA: a user-driven, interactive multiple protein structure alignment server. Nucleic Acids Res. 2014 Jul;42(W1):W240–5.
- Shatsky M, Nussinov R, Wolfson HJ. Optimization of multiple-sequence alignment based on multiple-structure alignment. Proteins. 2006 Jan;62(1):209–217.
- Katoh K, Frith MC. Adding unaligned sequences into an existing alignment using MAFFT and LAST. Bioinformatics. 2012 Dec;28(23):3144–3146.
- Okonechnikov K, Golosova O, Fursov M. UGENE team. Unipro UGENE: a unified bioinformatics toolkit. Bioinformatics. 2012 Apr;28(8):1166–1167.
- Waterhouse A, Bertoni M, Bienert S, et al. MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 2018 Jul;46(W1):W296–W303.
- Armougom F, Moretti S, Poirot O, et al. Expresso: automatic incorporation of structural information in multiple sequence alignments using 3D-Coffee. Nucleic Acids Res. 2006 Jul;34(Web Server):W604–8.
- Bailey TL, Boden M, Buske FA, et al. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 2009 Jul;37(Web Server):W202–8.
- Wallot S, Leonardi G. Deriving inferential statistics from recurrence plots: a recurrence-based test of differences between sample distributions and its comparison to the two-sample Kolmogorov-Smirnov test. Chaos. 2018 Aug;28(8):085712.
- Burley SK, Berman HM, Bhikadiya C, et al. RCSB Protein Data Bank: biological macromolecular structures enabling research and education in fundamental biology, biomedicine, biotechnology and energy. Nucleic Acids Res. 2019 Jan;47(D1):D464–D474.
- Rubio M, Maestro JL, Piulachs M-D BX. Conserved association of Argonaute 1 and 2 proteins with miRNA and siRNA pathways throughout insect evolution, from cockroaches to flies. Biochim Biophys Acta Gene Regul Mech. 2018 Apr;1861(6):554–560.
- Misof B, Liu S, Meusemann K, et al. Phylogenomics resolves the timing and pattern of insect evolution. Science. 2014 Nov;346(6210):763–767.
- Wynant N, Santos D, Vanden Broeck J. The evolution of animal Argonautes: evidence for the absence of antiviral AGO Argonautes in vertebrates. Sci Rep. 2017 Aug;7(1):9230.
- Cerutti H, Casas-Mollano JA. On the origin and functions of RNA-mediated silencing: from protists to man. Curr Genet. 2006 Aug;50(2):81–99.
- Kwon SC, Nguyen TA, Choi Y-G, et al. Structure of human DROSHA. Cell. 2016 Jan;164(1–2):81–90.
- Moran Y, Agron M, Praher D, et al. The evolutionary origin of plant and animal microRNAs. Nat Ecol Evol. 2017 Feb;1(3):27.
- de Jong D, Eitel M, Jakob W, et al. Multiple dicer genes in the early-diverging metazoa. Mol Biol Evol. 2009 Jun;26(6):1333–1340.
- Kosik KS. MicroRNAs and cellular phenotypy. Cell. 2010 Oct;143(1):21–26.
- Mukherjee K, Campos H, Kolaczkowski B. Evolution of animal and plant dicers: early parallel duplications and recurrent adaptation of antiviral RNA binding in plants. Mol Biol Evol. 2013 Mar;30(3):627–641.
- Murphy D, Dancis B, Brown JR. The evolution of core proteins involved in microRNA biogenesis. BMC Evol Biol. 2008 Mar;8(1):92.
- St Johnston D, Brown NH, Gall JG, et al. A conserved double-stranded RNA-binding domain. Proc Natl Acad Sci USA. 1992 Nov;89(22):10979–10983.
- Wickham L, Duchaîne T, Luo M, et al. Mammalian staufen is a double-stranded-RNA- and tubulin-binding protein which localizes to the rough endoplasmic reticulum. Mol Cell Biol. 1999 Mar;19:2220–2230.
- Dowling D, Pauli T, Donath A, et al. Phylogenetic origin and diversification of RNAi pathway genes in insects. Genome Biol Evol. 2016 Dec;8:3784–3793.
- Senturia R, Faller M, Yin S, et al. Structure of the dimerization domain of DiGeorge critical region 8. Protein Sci. 2010 Jul;19(7):1354–1365.
- Davis-Vogel C, Van Allen B, Van Hemert JL, et al. Identification and comparison of key RNA interference machinery from western corn rootworm, fall armyworm, and southern green stink bug. PLoS One. 2018 Sep;13(9):e0203160.
- Sharma C, Mohanty D. Sequence- and structure-based analysis of proteins involved in miRNA biogenesis. J Biomol Struct Dyn. 2018;36(1):139–151.
- Rozewicki J, Li S, Amada KM, et al. MAFFT-DASH: integrated protein sequence and structural alignment. Nucleic Acids Res. 2019 Jul;47:W5–W10.
- Burd CG, Dreyfuss G. Conserved structures and diversity of functions of RNA-binding proteins. Science. 1994 Jul;265(5172):615–621.
- Cerutti L, Mian N, Bateman A. Domains in gene silencing and cell differentiation proteins: the novel PAZ domain and redefinition of the Piwi domain. Trends Biochem Sci. 2000 Oct;25(10):481–482.
- Hall TMT. Structure and function of argonaute proteins. Structure. 2005 Oct;13(10):1403–1408.
- Kandasamy SK, Fukunaga R. Phosphate-binding pocket in Dicer 2 PAZ domain for high-fidelity siRNA production. Proc Natl Acad Sci USA. 2016 Dec;113(49):14031–14036.
- Blaszczyk J, Tropea JE, Bubunenko M, et al. Crystallographic and modeling studies of RNase III suggest a mechanism for double-stranded RNA cleavage. Structure. 2001 Dec;9(12):1225–1236.
- Conrad C, Ribonuclease RR. III: new sense from nuisance. Int J Biochem Cell Biol. 2002 Feb;34(2):116–129.
- Blaszczyk J, Gan J, Tropea JE, et al. Non-catalytic assembly of ribonuclease III with double-stranded RNA. Structure. 2004 Mar;12(3):457–466.
- Ye X, Paroo Z, Liu Q. Functional Anatomy of the Drosophila MicroRNA-generating Enzyme. J Biol Chem. 2007 Sep;282(39):28373–28378.
- Cenik ES, Fukunaga R, Lu G, et al. Phosphate and R2D2 restrict the substrate specificity of Dicer-2, an ATP-driven ribonuclease. Mol Cell. 2011 Apr;42(2):172–184.
- Trettin KD, Sinha NK, Eckert DM, et al. Loquacious-PD facilitates Drosophila Dicer-2 cleavage through interactions with the helicase domain and dsRNA. Proc Natl Acad Sci USA. 2017 Sep;114(38):E7939–E7948.
- Sinha NK, Iwasa J, Shen PS, et al. Dicer uses distinct modules for recognizing dsRNA termini. Science. 2018 Jan;359(6373):329–334.
- Kurzynska-Kokorniak A, Pokornowska M, Koralewska N, et al. Revealing a new activity of the human Dicer DUF283 domain in vitro. Sci Rep. 2016 Apr;6(1):23989.
- Gan J, Tropea JE, Austin BP, et al. Structural insight into the mechanism of double-stranded RNA processing by ribonuclease III. Cell. 2006 Jan;124(2):355–366.
- Laraki G, Clerzius G, Daher A, et al. Interactions between the double-stranded RNA-binding proteins TRBP and PACT define the Medipal domain that mediates protein-protein interactions. RNA Biol. 2008 Jun;5(2):92–103.
- Yang SW, Chen H-Y, Yang J, et al. Structure of Arabidopsis Hyponastic Leaves 1 and its molecular implications for miRNA processing. Structure. 2010 May;18(5):594–605.
- Wilson RC, Tambe A, Kidwell MA, et al. Dicer-TRBP complex formation ensures accurate mammalian microRNA biogenesis. Mol Cell. 2015 Feb;57(3):397–407.
- Ryter JM, Schultz SC. Molecular basis of double-stranded RNA-protein interactions: structure of a dsRNA-binding domain complexed with dsRNA. Embo J. 1998 Dec;17(24):7505–7513.
- Vuković L, Koh HR, Myong S, et al. Substrate recognition and specificity of double-stranded RNA binding proteins. Biochemistry. 2014 Jun;53(21):3457–3466.
- Dias R, Manny A, Kolaczkowski O, et al. Convergence of domain architecture, structure, and ligand affinity in animal and plant RNA-binding proteins. Mol Biol Evol. 2017 Jun;34(6):1429–1444.
- Kurihara Y, Takashi Y, Watanabe Y. The interaction between DCL1 and HYL1 is important for efficient and precise processing of pri-miRNA in plant microRNA biogenesis. RNA. 2006 Feb;12(2):206–212.
- Liu Z, Wang J, Cheng H, et al. Cryo-EM structure of human dicer and its complexes with a pre-miRNA substrate. Cell. 2018 May;173(5):1191–1203.e12.
- Zhang X, Li P, Lin J, et al. The insertion in the double-stranded RNA binding domain of human Drosha is important for its function. Biochim Biophys Acta Gene Regul Mech. 2017 Dec;1860(12):1179–1188.
- Tian Y, Simanshu DK, Ma J-B, et al. A phosphate-binding pocket within the platform-PAZ-connector helix cassette of human Dicer. Mol Cell. 2014 Feb;53(4):606–616.
- Wilson KA, Holland DJ, Wetmore SD. Topology of RNA-protein nucleobase-amino acid π-π interactions and comparison to analogous DNA-protein π-π contacts. RNA. 2016 May;22:696–708.
- Fukunaga R, Colpan C, Han BW, et al. Inorganic phosphate blocks binding of pre-miRNA to Dicer 2 via its PAZ domain. Embo J. 2014 Feb;33(4):371–384.
- Park J-E, Heo I, Tian Y, et al. Dicer recognizes the 5ʹ end of RNA for efficient and accurate processing. Nature. 2011 Jul;475(7355):201–205.
- Jia H, Kolaczkowski O, Rolland J, et al. Increased affinity for RNA targets evolved early in animal and plant Dicer lineages through different structural mechanisms. Mol Biol Evol. 2017 Dec;34(12):3047–3063.
- Zhu J-K. Reconstituting plant miRNA biogenesis. Proc Natl Acad Sci USA. 2008 Jul;105(29):9851–9852.
- Terenius O, Papanicolaou A, Garbutt JS, et al. RNA interference in Lepidoptera: an overview of successful and unsuccessful studies and implications for experimental design. J Insect Physiol. 2011 Feb;57(2):231–245.
- Lau P-W, Guiley KZ, De N, et al. The molecular architecture of human Dicer. Nat Struct Mol Biol. 2012 Mar;19(4):436–440.
- MacRae IJ, Doudna JA. An unusual case of pseudo-merohedral twinning in orthorhombic crystals of Dicer. Acta Crystallogr Sect D, Biol Crystallogr. 2007 Sep;63:993–999.
- Sasaki T, Shimizu N. Evolutionary conservation of a unique amino acid sequence in human DICER protein essential for binding to Argonaute family proteins. Gene. 2007 Jul;396(2):312–320.
- Maillard PV, van der Veen AG, Poirier EZ, et al. Slicing and dicing viruses: antiviral RNA interference in mammals. Embo J. 2019 Apr;38(8):e100941.
- MacRae IJ, Zhou K, Doudna JA. Structural determinants of RNA recognition and cleavage by Dicer. Nat Struct Mol Biol. 2007 Oct;14(10):934–940.
- Oliver D, Sheehan B, South H, et al. The chromosomal association/dissociation of the chromatin insulator protein Cp190 of Drosophila melanogaster is mediated by the BTB/POZ domain and two acidic regions. BMC Cell Biol. 2010 Dec;11(1):101.
- Kumar A, Lualdi M, Loncarek J, et al. Loss of function of mouse Pax-Interacting Protein 1-associated glutamate rich protein 1a (Pagr1a) leads to reduced Bmp2 expression and defects in chorion and amnion development. Dev Dyn. 2014 Jul;243(7):937–947.
- Hsu TI, Lin SC, Lu PS, et al. MMP7-mediated cleavage of nucleolin at Asp255 induces MMP9 expression to promote tumor malignancy. Oncogene. 2015 Feb;34(7):826–837.
- Putnam CD, Tainer JA. Protein mimicry of DNA and pathway regulation. DNA Repair (Amst). 2005 Dec;4(12):1410–1420.
- Wang H-C, Ho C-H, Hsu K-C, et al. DNA mimic proteins: functions, structures, and bioinformatic analysis. Biochemistry. 2014 May;53(18):2865–2874.
- Kinoshita S, Katsumi E, Yamamoto H, et al. Molecular and functional analyses of aspolin, a fish-specific protein extremely rich in aspartic acid. Mar Biotechnol. 2011 Jun;13(3):517–526.
- Scartezzini P, Egeo A, Colella S, et al. Cloning a new human gene from chromosome 21q22.3 encoding a glutamic acid-rich protein expressed in heart and skeletal muscle. Hum Genet. 1997 Mar;99:387–392.
- Kumar P, Bansal M. Structural and functional analyses of PolyProline-II helices in globular proteins. J Struct Biol. 2016 Sep;196(3):414–425.
- Adzhubei AA, Sternberg MJE, Makarov AA. Polyproline-II helix in proteins: structure and function. J Mol Biol. 2013 Jun;425(12):2100–2132.
- Morgan AA, Rubenstein E. Proline: the distribution, frequency, positioning, and common functional roles of proline and polyproline sequences in the human proteome. PLoS One. 2013 Jan;8(1):e53785.
- Jankowsky E, Fairman-Williams ME. Chapter 1. an introduction to RNA helicases: superfamilies, families, and major themes. In: Jankowsky E, editor. RNA Helicases. Cambridge: Royal Society of Chemistry; 2010. p. 1–31.
- Mastrangelo E, Bolognesi M, Milani M. Flaviviral helicase: insights into the mechanism of action of a motor protein. Biochem Biophys Res Commun. 2012 Jan;417(1):84–87.
- Du Pont KE, Davidson RB, McCullagh M, et al. Motif V regulates energy transduction between the flavivirus NS3 ATPase and RNA-binding cleft. J Biol Chem. 2020 Feb;295(6):1551–1564.
- Caruthers JM, McKay DB. Helicase structure and mechanism. Curr Opin Struct Biol. 2002 Feb;12(1):123–133.
- Papanikou E, Karamanou S, Baud C, et al. Helicase Motif III in SecA is essential for coupling preprotein binding to translocation ATPase. EMBO Rep. 2004 Aug;5(8):807–811.
- Deddouche S, Matt N, Budd A, et al. The DExD/H-box helicase Dicer-2 mediates the induction of antiviral activity in drosophila. Nat Immunol. 2008 Dec;9:1425–1432.
- Swevers L, Vanden Broeck J, Smagghe G. The possible impact of persistent virus infection on the function of the RNAi machinery in insects: a hypothesis. Front Physiol. 2013 Nov;4:319.
- Dias NP, Cagliari D, Dos Santos EA, et al. Insecticidal gene silencing by RNAi in the neotropical region. Neotrop Entomol. 2020 Feb;49(1):1–11.
- Cooper AM, Silver K, Zhang J, et al. Molecular mechanisms influencing efficiency of RNA interference in insects. Pest Manag Sci. 2019 Jan;75(1):18–28.
- Claycomb JM. Ancient endo-siRNA pathways reveal new tricks. Curr Biol. 2014 Aug;24(15):R703–15.
- Trobaugh DW, Klimstra WB. MicroRNA regulation of RNA virus replication and pathogenesis. Trends Mol Med. 2017;23(1):80–93.
- Shapiro JS. Processing of virus-derived cytoplasmic primary-microRNAs. Wiley Interdiscip Rev RNA. 2013 Aug;4(4):463–471.
- Masliah G, Barraud P, Allain FH-T. RNA recognition by double-stranded RNA binding domains: a matter of shape and sequence. Cell Mol Life Sci. 2013 Jun;70:1875–1895.
- Song -J-J, Liu J, Tolia NH, et al. The crystal structure of the Argonaute 2 PAZ domain reveals an RNA binding motif in RNAi effector complexes. Nat Struct Biol. 2003 Dec;10:1026–1032.
- Drezen J-M, Josse T, Bézier A, et al. Impact of lateral transfers on the genomes of Lepidoptera. Genes (Basel). 2017 Nov;8(11):315.
- Vignuzzi M, López CB. Defective viral genomes are key drivers of the virus-host interaction. Nat Microbiol. 2019 Jun;4:1075–1087.
- Guo Z, Li Y, Ding S-W, et al. A-based antimicrobial immunity. Nat Rev Immunol. 2019;19(1):31–44.
- Kolliopoulou A, Santos D, Taning CNT, et al. PIWI pathway against viruses in insects. Wiley Interdiscip Rev RNA. 2019 Jun;10(6):e1555.
- Ter Horst AM, Nigg JC, Dekker FM, et al. Endogenous viral elements are widespread in arthropod genomes and commonly give rise to PIWI-interacting RNAs. J Virol. 2019 Mar;93(6):e02124-18.
- Cui J, Holmes EC. Endogenous RNA viruses of plants in insect genomes. Virology. 2012 Jun;427(2):77–79.
- Powell JA. Lepidoptera: moths, butterflies. In: Resh V, Cardé R, editors. Encyclopedia of Insects. 2nd ed ed. USA: Elsevier; 2009. p. 559–587.
- Zografidis A, Van Nieuwerburgh F, Kolliopoulou A, et al. Viral small-RNA analysis of Bombyx mori larval midgut during persistent and pathogenic cytoplasmic polyhedrosis virus infection. J Virol. 2015 Nov;89(22):11473–11486.
- Lavialle C, Cornelis G, Dupressoir A, et al. Paleovirology of ‘ syncytins’, retroviral env genes exapted for a role in placentation. Philos Trans R Soc Lond B, Biol Sci. 2013 Sep;368(1626):20120507.
- Ryabov EV. Invertebrate RNA virus diversity from a taxonomic point of view. J Invertebr Pathol. 2017;147:37–50.
- Mongelli V, Saleh M-C. Bugs are not to be silenced: small RNA pathways and antiviral responses in insects. Annu Rev Virol. 2016 Sep;3(1):573–589.
- Berry B, Deddouche S, Kirschner D, et al. Viral suppressors of RNA silencing hinder exogenous and endogenous small RNA pathways in Drosophila. PLoS One. 2009 Jun;4(6):e5866.
- Yoon J-S, Mogilicherla K, Gurusamy D, et al. Double-stranded RNA binding protein, Staufen, is required for the initiation of RNAi in coleopteran insects. Proc Natl Acad Sci USA. 2018 Aug;115(33):8334–8339.
- Kingsolver MB, Huang Z, Hardy RW. Insect antiviral innate immunity: pathways, effectors, and connections. J Mol Biol. 2013 Dec;425(24):4921–4936.
- Brown S, Hu N, Hombría JC. Identification of the first invertebrate interleukin JAK/STAT receptor, the Drosophila gene domeless. Curr Biol. 2001 Oct;11(21):1700–1705.
- Gottar M, Gobert V, Michel T, et al. The Drosophila immune response against Gram-negative bacteria is mediated by a peptidoglycan recognition protein. Nature. 2002 Apr;416(6881):640–644.
- Paradkar PN, Duchemin J-B, Voysey R, et al. Dicer-2-dependent activation of Culex Vago occurs via the TRAF-Rel2 signaling pathway. PLoS Negl Trop Dis. 2014 Apr;8(4):e2823.
- Cheng G, Liu Y, Wang P, et al. Mosquito defense strategies against viral infection. Trends Parasitol. 2016 Mar;32(3):177–186.
- Paradkar PN, Trinidad L, Voysey R, et al. Secreted Vago restricts West Nile virus infection in Culex mosquito cells by activating the Jak-STAT pathway. Proc Natl Acad Sci USA. 2012 Nov;109(46):18915–18920.
- Sim S, Jupatanakul N, Dimopoulos G. Mosquito immunity against arboviruses. Viruses. 2014 Nov;6(11):4479–4504.
- Poirier EZ, Goic B, Tomé-Poderti L, et al. Dicer-2-dependent generation of viral DNA from defective genomes of RNA viruses modulates antiviral immunity in insects. Cell Host Microbe. 2018 Mar;23(3):353–365.e8.
- Santos D, Wynant N, Van den Brande S, et al. Insights into RNAi-based antiviral immunity in Lepidoptera: acute and persistent infections in Bombyx mori and Trichoplusia ni cell lines. Sci Rep. 2018 Feb;8(1):2423.
- Spellberg MJ, Marr MT. FOXO regulates RNA interference in Drosophila and protects from RNA virus infection. Proc Natl Acad Sci USA. 2015 Nov;112(47):14587–14592.
- Ahlers LRH, Trammell CE, Carrell GF, et al. Insulin potentiates JAK/STAT signaling to broadly inhibit flavivirus replication in insect vectors. Cell Rep. 2019 Nov;29(7):1946–1960.e5.
- Guan R, Hu S, Li H, et al. in vivo dsRNA cleavage has sequence preference in insects. Front Physiol. 2018 Dec;9:1768.