
ABSTRACT
RNA and protein are interconnected biomolecules that can influence each other’s life cycles and functions through physical interactions. Abnormal RNA–protein interactions lead to cell dysfunctions and human diseases. Therefore, mapping networks of RNA–protein interactions is crucial for understanding cellular processes and pathogenesis of related diseases. Different practical protein-centric methods for studying RNA–protein interactions have been reported, but few robust RNA-centric methods exist. Here, we developed CRISPR-based RNA proximity proteomics (CBRPP), a new RNA-centric method to identify proteins associated with an endogenous RNA of interest in native cellular context without pre-editing of the target RNA, cross-linking or RNA–protein complexes manipulation in vitro. CBRPP is based on a fusion of dCas13 and proximity-based labelling (PBL) enzyme. dCas13 can deliver PBL enzyme to the target RNA with high specificity, while PBL enzyme labels the surrounding proteins of the target RNA, which are then identified by mass spectrometry.
Introduction
RNA is bound to protein from birth to death. RNA-binding proteins (RBPs) play a pivotal role in a wide range of biological processes, including RNA transcription, processing, modification, transport, translation and stabilization [Citation1–4]. RNAs, in turn, influence protein expression, localization and interactions with other proteins [Citation5–7]. Aberrant RNA–protein interactions are related to cellular dysfunctions and human diseases [Citation3,Citation8,Citation9]. For example, myotonic dystrophy type 1 (DM1) is caused by expansion of a CTG trinucleotide repeat in the 3ʹUTR of the DMPK gene [Citation10,Citation11]. Expanded DMPK transcripts aggregate into nuclear foci, which can sequester and disrupt the normal activities of RBPs belonging to the MBNL and CELF families, thereby altering splicing of numerous pre-mRNAs [Citation10,Citation11]. Therefore, mapping networks of RNA–protein interactions is of great importance for understanding many cellular biological processes and pathogenesis of diseases.
Based on the type of molecule they start with, methods for studying RNA–protein interactions are classified into protein-centric methods and RNA-centric methods [Citation12]. Protein-centric methods start with a protein of interest and study RNAs that interact with that protein. Generally, these methods either directly purify the protein of interest to identify associated RNAs, or selectively chemically modify RNAs in a manner that relies on their association with the protein of interest. The vast majority of studies that identify RNAs bound to a given protein were accomplished by purifying protein of interest. RNA immunoprecipitation (RIP)-seq and cross-linking immunoprecipitation (CLIP)-seq are the two most commonly used methods [Citation13,Citation14]. In RIP-seq, the protein of interest is immunopurified from cell lysates, then associated RNAs are identified by sequencing [Citation14]. CLIP-seq is very similar to RIP-seq, except for an additional crosslinking step [Citation13]. In addition, targets of RNA-binding proteins identified by editing (TRIBE) and RNA Tagging can identify the RNA targets of an RBP without purifying the RBP [Citation15,Citation16]. In TRIBE, the RBP of interest, which is responsible for binding to RNAs, is fused with the catalytic domain of the RNA-editing enzyme ADAR, which irreversibly deaminates the proximal adenosine to inosine to serve as a marker during sequence analysis [Citation15]. In RNA Tagging, the RBP of interest is fused with a poly(U) polymerase, which adds poly(U) tails to bound RNAs. Tagged RNAs are then easily identified using high-throughput sequencing [Citation16]. Conversely, RNA-centric methods start with an RNA of interest and focus on proteins that bind it. In vitro methods commonly use in vitro-transcribed (IVT) RNA that contains a tag, which can be immobilized on a solid support. After the IVT RNA is immobilized to a solid support, cellular extract is added, and subsequently washing steps are carried out to remove non-specific RBPs. Finally, RBPs bound to the IVT RNA are eluted and identified. Approaches to tag IVT RNA include biotinylation [Citation17], S1 aptamer [Citation18] and Cys4 hairpin loop [Citation19]. Although these in vitro methods are timesaving and convenient, they have obvious drawbacks. On the one hand, IVT RNA do not have the same modification or structure that a given RNA has in cells. On the other hand, IVT RNA is potentially biased towards the interaction with abundant proteins in cell extracts. For in vivo methods, researchers firstly ‘freeze’ physiological RNA–protein interactions in cells by cross-linking. Then, cells are lysed and RNA-protein complexes are captured from solution. After washing to remove non-specific proteins, the bound proteins are eluted and finally identified by proteomic analysis. These methods use either UV or formaldehyde cross-linking, but differ in RNA capture approaches [Citation20–25]. For example, RNA affinity purification (RAP) [Citation20], chromatin isolation by RNA purification (ChIRP) [Citation21] and capture hybridization analysis of RNA targets (CHART) [Citation22] utilize biotinylated antisense oligonucleotide probes to pull down RNA-protein complexes. Peptide-nucleic-acid-assisted identification of RBPs (PAIR) uses peptide nucleic acid probes with cell-penetrating ability to enter cells and hybridize to RNA [Citation23]. Tandem RNA isolation procedure (TRIP) primarily purifies polyadenylated RNAs with oligo(dT) beads, followed by the capture of specific RNAs with biotinylated antisense oligonucleotide probes [Citation24]. MS2 in vivo biotin-tagged RAP (MS2-BioTRAP) utilizes the tight association between MS2 hairpin loop and MS2 coat protein (MCP) to capture RNA [Citation25]. These in vivo methods all require cross-linking and RNA-protein complexes manipulation in vitro. However, both UV and formaldehyde cross-linking have biases [Citation26–29], and the efficiency of UV cross-linking is low [Citation30]. Besides, RNA-protein complexes manipulation in vitro leads to the possible disruption of RNA–protein interactions and RNA degradation easily. Recently, two in vivo methods without cross-linking have applied PBL to study RNA–protein interactions using the MS2-MCP strategy [Citation31] or a similar strategy [Citation32]. Both methods do not require cross-linking or RNA-protein complexes manipulation in vitro, but require insertion of MS2 or BoxB stem-loop into the target RNA in advance, which may influence the structure or function of the target RNA. Therefore, developing an in vivo RNA-centric method without cross-linking, RNA-protein complexes manipulation in vitro and pre-editing of the target RNA is needed. In addition, modified proximity ligation assay (PLA) methods have been developed to study RNA–protein interactions in cells by replacing one of the primary antibody/PLA probe pairs in standard PLA with a DNA oligonucleotide probe [Citation33] or replacing one of the primary antibodies with flag-tagged multiply-labelled tetravalent RNA imaging probes (FMTRIP) [Citation34,Citation35]. These methods can detect interactions between an unmodified endogenous RNA and a binding protein in situ in fixed cells, but cannot be used to discover RBPs of an RNA or RNAs bound by a protein.
In recent years, PBL has emerged as a powerful complementary approach to classic affinity purification (AP) of multiprotein complexes in mapping of protein–protein interactions [Citation36,Citation37]. By fusing protein of interest to enzyme that generate reactive molecules, most commonly biotin, adjacent proteins are covalently labelled so that they can be isolated by streptavidin beads and subsequently identified by mass spectrometry (MS) () [Citation36,Citation37]. To date, multiple versions of the PBL enzyme have been developed, including biotin ligases (BirA*, BioID2, TurboID, miniTurbo, BASU) [Citation32,Citation38–40], and engineered ascorbate peroxidases (APEX, APEX2) [Citation41,Citation42]. The first biotin-based proximity labelling technique, BioID, was developed by the Burke group in 201238. BioID is based on promiscuous biotinylation generated by a mutant biotin ligase, BirA*. This enzyme catalyzes the conversion of biotin to biotinoyl-5ʹ-adenosine monophosphate. This highly reactive form of biotin attaches covalently to accessible lysine residues in neighbouring proteins. To capture protein complexes, researchers can simply express a bait protein fused to BirA* in cells and then incubate these cells with exogenous biotin for 18–24 hours, leading to biotinylation of endogenous proteins that are proximate to the bait. Finally, biotinylated proteins are captured by streptavidin beads for identification by mass spectrometry. In this way, the BioID method generates a ‘history’ of candidate protein–protein interactions under relatively physiological conditions in living cells. The simplicity and practicality of BioID have resulted in hundreds of applications since its introduction, in mammalian cells [Citation38,Citation43], plants [Citation44], mouse [Citation45], yeast [Citation46], and parasites [Citation47]. In 2016, the Roux group optimized BioID to create BioID2, a smaller and improved promiscuous biotin ligase [Citation39]. BioID2 requires less biotin than other enzymes, which is potentially useful in model systems where sufficient biotin supplementation is difficult. TurboID and miniTurbo were developed by directed evolution of biotin ligases in Ting lab in 2018 to increase the speed of proximity labelling, which both enable biotin labelling in 10 minutes without any issues in toxicity [Citation40]. The short labelling time required by TurboID and miniTurbo makes them suitable for studying protein–protein interactions occur over short period of time. In 2018, the Khavari group engineered a new mutant BirA* from Bacillus subtilis, termed BASU [Citation32]. They claimed that the enzyme activity and labelling time of BASU are between BioID2 and TurboID. APEX, used as a genetic tag for electron microscopy, is a 27 kDa ascorbate peroxidase that catalyzes the oxidation of biotin-phenol to short-lived biotin-phenoxyl radical in the presence of H2O241, [Citation48]. This radical reacts with accessible tyrosine residues on neighbouring proteins, leading to their biotinylation. To improve the sensitivity of APEX, the Ting group employed directed evolution to develop the more catalytically active APEX242. Both APEX and APEX2 excel at cellular compartmental proteomics, such as studying proteomic composition of mitochondria [Citation41,Citation42,Citation49]. The advantages of PBL in comparison to traditional AP methods are manifold. First, one major benefit is its ability to analyse protein–protein interactions in a native context, because covalent biotinylation occurs before cell lysis and solubilization. Second, PBL can capture weak or transient interactions that can be lost in AP approaches. Third, due to the high affinity of biotin for streptavidin, harsh conditions can be used to transform insoluble proteins into solution, so PBL can be applied to both soluble and insoluble proteins.
Figure 1. Workflow diagrams of PBL and CBRPP
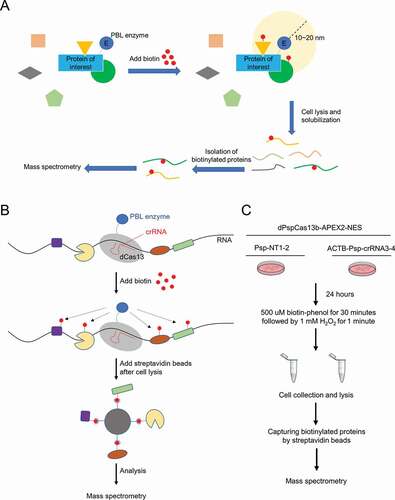
The Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)-CRISPR-associated proteins (Cas) system has revolutionized modern molecular biology [Citation50]. Many CRISPR-Cas systems have been used to develop potent research tools or powerful therapeutic tools, with Cas9 being the most widespread [Citation50–52]. While most of the utilized systems are DNA-targeting, recently more and more attention is paid to those that target RNA. Cas13 is an outlier in the CRISPR world because it targets RNA, not DNA [Citation53,Citation54]. Its ability to specifically recognize a given RNA sequence in an easily programmable way make it an ideal toolbox to manipulate RNA in live cells. Catalytically active Cas13, under the guidance of a specific CRISPR RNA (crRNA), can recognize and cleave the target transcript, leading to transcript knockdown [Citation55]. Catalytically dead Cas13 (dCas13) retains programmable RNA-binding capability, which can be utilized for RNA imaging and base editing [Citation56,Citation57]. Currently, there are several orthologs and subtypes of Cas13 that are catalytically active inside mammalian cells, including LwaCas13a [Citation53], PspCas13b [Citation57] and RfxCas13d [Citation58].
Inspired by a strategy that leveraged catalytically dead Cas9 (dCas9) to guide biotin ligase to specific genomic loci [Citation59,Citation60], we proposed that by fusing dCas13 and PBL enzyme together, dCas13, under the guidance of a specific crRNA, can act as an RNA tracker to bring PBL enzyme to the target RNA, so that there is no need to edit the target RNA in advance. Then, PBL enzyme can biotinylate the surrounding proteins of the target RNA with biotin, which avoids cross-linking and RNA-protein complexes manipulation in vitro. Finally, these biotinylated proteins can be easily enriched by streptavidin beads and identified by mass spectrometry (). We referred to this combination of CRISPR-Cas13 and PBL as CRISPR-based RNA proximity proteomics (CBRPP), a new in vivo RNA-centric method studying RNA–protein interactions without pre-editing of the target RNA, cross-linking or RNA–protein complexes manipulation in vitro. In this paper, through several improvements, we proved that CBRPP is feasible and successfully identified the RBPs of ACTB mRNA and NORAD using CBRPP.
Materials and methods
Cell culture
HEK293T (Human Embryonic Kidney 293 T) cells was obtained from ATCC. Cells were cultured in DMEM medium supplemented with 10% FBS (Gibco) and 100 U/ml Penicillin-Streptomycin in a humidified incubator at 37°C with 5% CO2.
Reagents and antibodies
PEI (764,582, Sigma-Aldrich) was used for transfection. Antibodies used in this study include the following: IgG (mouse, sc2025, Santa Cruz); anti-HA (rabbit, H6908, Sigma-Aldrich); anti-FLAG (mouse, M185-3 L, MBL); anti-beta-tubulin (mouse, HC101-01, Transgen); anti-G3BP1 (mouse, sc-365,338, Santa Cruz); HRP-conjugated Streptavidin (SA00001-0, Proteintech). The antibodies were diluted 1,000 times for immunoblots, 200 times in confocal microscopy.
Plasmid constructs
Expression constructs generated for this study were constructed by Gibson cloning (CL116, Biomed) and coding sequences entirely verified. All the mutants were constructed by Gibson cloning. Each mutant was confirmed by sequencing. All primers were ordered from Synbio Technologies. All plasmid constructs and their sequence are listed in . All crRNAs used in this paper are listed in .
Table 1. crRNAs used in this paper
Table 2. qPCR primers used in this paper
Western blot
Cells were washed with PBS and lysed by incubation on ice for 10 min with RIPA lysis buffer (50 mM Tris, 150 mM NaCl, 0.1% SDS, 0.5% sodium deoxycholate, 1% Triton X-100, protease inhibitor cocktail [C0001, Targetmol], and 1 mM PMSF). The proteins were resolved by SDS/PAGE and transferred to 0.22 µM nitrocellulose membrane (PALL), which then was incubated overnight with primary antibodies. The membrane was further incubated with the corresponding HRP-conjugated secondary antibodies and detected by enhanced chemiluminescence.
Immunofluorescence microscopy
HEK293T cells were plated and grew on coverslips with indicated treatments, washed with pre-warmed PBS, and fixed with 4% paraformaldehyde for 10 minutes. The cells were permeated with 0.5% Triton-100 for 3 min, blocked with 3% BSA for 30 minutes, washed, and incubated with primary antibodies for 1 h at 37°C. After washing, cells were stained with Alexa Fluor 488-conjugated secondary antibodies (A11029, Invitrogen) or Alexa Fluor 555-conjugated secondary antibodies (A-21,428, Invitrogen) for 1 h at 37°C, and then with DAPI (4′,6-Diamidino-2-phenylindole, Roche) for 15 min. The coverslips were washed extensively and mounted onto slides. Imaging of the cells were carried out using N-STORM5.0 microscope.
RNA extraction and quantitative reverse transcription PCR (RT-qPCR)
Total RNA from cells were isolated using total RNA extraction kit (DP419, TIANGEN). 1 µg RNA was reverse transcribed using a FastKing RT Kit (KR116, TIANGEN). Levels of the indicated genes were analysed by quantitative real-time PCR amplified using SYBR Green (Q311, Vazyme). All primers were listed in Table 3.
Generation of stable expression mammalian cell lines
For preparation of lentiviruses, HEK293T cells in 6-well plates were transfected with the lentiviral vector of interest (1,800 ng), the lentiviral packaging plasmids psPAX2 (600 ng) and pMD2.G (600 ng) and 12 µl of PEI (1 mg/ml). About 48 h after transfection the cell medium-containing lentiviruses was centrifugalized at 12,000 g for 3 min and the supernatant was harvested. HEK293 cells were then infected at ~50% confluency by lentiviruses for 48 h, followed by selection with 1 μg/ml puromycin in growth medium for 7 days. The stable transgene monoclonal cells were harvested by limiting dilution in cell pools.
Generation of tetracycline (Tet) inducible expression HEK293T cell lines
The two consecutive manipulation steps are necessary to generate HEK293T cell lines inducibly expressing gene of interest. The first step is generation of HEK293T cell line stably expressing Tet-On 3 G transactivator protein. HEK293T cells were infected at ~50% confluency by lentiviruses containing pLVX-TetO3G(rtTA)-hygr vector for 48 hours, followed by selection with 50 ug/ml hygromycin in growth medium for 7 days, and hygromycin resistant clones were selected. Several clones were picked and tested for Tet-On 3 G expression by immunoblotting. After testing for all molecular and cell biological parameters of interest, the ‘best’ Tet-On 3 G-positive clone was expanded and stored. The next step is to generate HEK293T cell lines inducibly expressing gene of interest on the basis of the ‘best’ Tet-On 3 G-positive clone. The ‘best’ Tet-On 3 G-positive clone was infected by lentiviruses containing target plasmids (Inducible-dPspCas13b-BioID2/BASU/TurboID/APEX2-NES) for 48 hours, followed by selection with 1 μg/ml puromycin in growth medium for 7 days. The puromycin resistant clones were harvested by limiting dilution in cell pools. Several individual cell clones were picked, expanded and screened by immunoblotting for doxycycline-inducible expression of gene of interest. Finally, clones of interest were expanded, re-tested and stored.
Biotin labelling in live cells
For dPspCas13b-APEX2-NES transient transfection experiments, HEK293T cells were plated in 10 cm dish 18 hours prior to transfection. At 70% confluency, cells were transfected with the dPspCas13b-APEX2-NES plasmid and Psp-crRNAs. After 6 hours of transfection, the culture medium was changed. After 24 hours of transfection, biotin-phenol was added to cell culture medium to a final concentration of 500 µM for 30 min, H2O2 was then added into cell culture medium at a final concentration of 1 mM to induce biotinylation. After gently shaking the cell culture dish for 1 min, the medium was removed and cells were washed three times with PBS supplemented with quenchers (10 mM sodium azide, 10 mM sodium ascorbate and 5 mM TROLOX). Cells were scraped and transferred to 1.5 ml tubes with ice-cold PBS, spun at 3600 rpm for 5 min, flash-frozen in liquid nitrogen and stored at −80°C.
For inducibly expressing dPspCas13b-BioID2/TurboID/BASU/APEX2-NES experiments, four stable HEK293T cell lines inducibly expressing dPspCas13b- BioID2/TurboID/BASU/APEX2-NES were plated in 10 cm dish 18 hours prior to transfection. At 70% confluency, cells were transfected with 20 µg crRNA plasmid per dish. After 6 h of transfection, the culture medium was replaced with new media containing 0.1 µg/ml doxycycline. For BioID2, biotin was added to the culture medium at a final concentration of 50 µM after 15 h of transfection; for TurboID, biotin was added at a final concentration of 500 µM for 10 min before harvesting cells; for BASU, biotin was added at a final concentration of 200 µM for 2 hours before harvesting cells; for APEX2, biotin-phenol was added at a final concentration of 500 µM for 30 min and H2O2 was added at a final concentration of 1 mM for one minute before harvesting cells. All kinds of cells were harvested at 33 h after transfection. For APEX2, the medium was removed and cells were washed three times with ice-cold PBS supplemented quenchers (10 mM sodium azide, 10 mM sodium ascorbate and 5 mM TROLOX); for TurboID/BASU/BioID2, the medium was removed and cells were washed three times with ice-cold PBS. Cells were scraped and transferred to 1.5 ml tubes with ice-cold PBS, spun at 3600 rpm for 5 min, flash-frozen in liquid nitrogen and stored at −80°C.
Streptavidin magnetic beads enrichment of biotinylated proteins
Cell pellets as described above were lysed in RIPA lysis buffer (50 mM Tris, 150 mM NaCl, 0.1% SDS, 0.5% sodium deoxycholate, 1% Triton X-100, protease inhibitor cocktail [C0001, TargetMol], and 1 mM PMSF) at 4°C for 10 min. The lysates were cleared by centrifugation at 12,000 g for 10 min at 4°C. 50 µl of each lysate supernatant was reserved for detection of biotinylation activity by western blot. Streptavidin magnetic beads were washed twice with RIPA lysis buffer and then mixed with lysates supernatant together with rotation overnight at 4°C. On day 2, the beads were subsequently washed twice with 1 ml of RIPA lysis buffer, once with 1 ml of 1 M KCl, once with 1 ml of 0.1 M Na2CO3, once with 1 ml of 2 M urea in 10 mM Tris-HCl (pH 8.0), and twice with 1 ml RIPA lysis buffer. Finally, biotinylated proteins were eluted by boiling the beads in 150 µl of elution buffer (55 mM pH 8.0 Tris-HCl, 0.1% SDS, 6.66 mM DTT, 0.66 mM biotin) for 10 min and sent for mass spectrometry. All mass spectrometry data are included in Table 4.
RNA immunoprecipitation (RIP)
HEK293T cells were plated in 10 cm dish 18 h prior to transfection. At 70% confluency, cells were transfected with indicated plasmids for 24 h. Then cells were washed with ice-cold PBS, scrapped and pelleted at 2500 rpm for 5 min at 4°C. After lysed in RIPA lysis buffer containing protease inhibitor cocktail [C0001, TargetMol] and RNase inhibitor [B600478, Sangon Biotech] for 30 min at 4°C, supernatants were collected by centrifugation at 12,000 rpm for 10 min at 4°C. After saving 5% of the supernatant was saved as input, the rest of the supernatant was then incubated with 3 µg antibodies for 2 hours at 4°C with gentle rotation. Further, 30 µl protein A/G beads (20,422, Thermo Fisher) were added and incubated for 1 h at 4°C with gentle rotation. After incubation, beads were pelleted at 2,500 rpm for 30 s and washed for a total of four times: three times with lysis buffer and the last time with pre-cold PBS. Then, the co-precipitated RNAs were followed by RNA extraction and RT-qPCR.
Mass spectrometry data analysis
The raw mass spectrometry data were firstly analysed using MaxQuant and the resulting ‘proteinGroups.txt’ files were used as input for the downstream analysis. For statistical analysis, the R package DEP was applied for the analysis of protein intensity data as previous reported [Citation61]. This package’s functions include data preparation, data preparation, filtering, variance normalization and imputation of missing values, as well as statistical testing of differentially enriched proteins. The imputation type of missing values used is MinProb. The significantly enriched RBPs were determined by P-value (P-value <0.05) and fold change (fold change > 4).
Statistical analysis of bar graphs
Statistical analyses of all bar graphs were performed using Prism version 7 (GraphPad software). All data were presented as mean ± SD from data in triplicate experiments. For the comparisons, two-side Student’s t-test was applied and the statistical significance was designated as ****P < 0.0001, ***P < 0.001, **P < 0.01, *P < 0.05, or ns (P > 0.05). These have been stated in the corresponding figure legends.
Results
dRfxCas13d is not suitable for CBRPP to study RNA-protein interactions
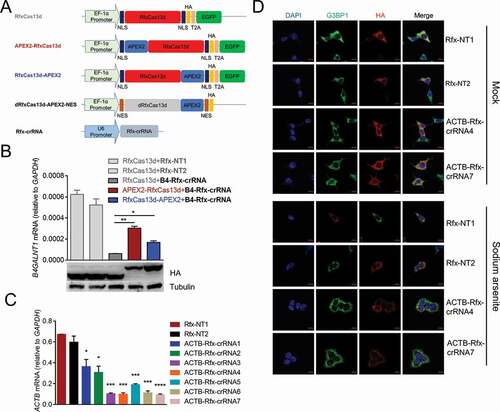
To prove the concept, we firstly selected dRfxCas13d and APEX2 for testing, because RfxCas13d is the smallest and most active one among Cas13 proteins [Citation58] and APEX2 has the fastest rate of labelling[Citation42]. To test which terminus of RfxCas13d is better to fuse with APEX2, we fused APEX2 to N-terminus or C-terminus of RfxCas13d to test whether APEX2 fusion would affect the function and expression of RfxCas13d by detecting the knockdown efficiency of RfxCas13d and western blot (). We chose B4GALNT1 mRNA as the target, since the Rfx-crRNA of B4GALNT1 had been reported effective in previous study [Citation58]. Results showed that compared with fusing APEX2 to N-terminus of RfxCas13d, fusing APEX2 to C-terminus of RfxCas13d slightly affected the knockdown efficiency of RfxCas13d, and had no effect on the expression of RfxCas13d (). Therefore, we constructed dRfxCas13d-APEX2-NES plasmid () and applied it to well-studied ACTB mRNA to test whether it would identify known RBPs of ACTB mRNA in HEK293T cells. The reason we chose HEK293T was because that it offers relatively high transfection efficiency as compared to other cell lines and is easy to handle, besides, it has been used to studying the RBPs of ACTB mRNA in several studies [Citation62–64]. Firstly, we designed seven Rfx-crRNAs targeting different regions of ACTB mRNA and validated their targeting by knockdown with an active RfxCas13d. RT-qPCR results showed that all seven ACTB Rfx-crRNAs significantly reduced ACTB mRNA level in HEK293T cells (). During cellular stress, protein synthesis is severely disrupted and bulk mRNAs are recruited to stress granules [Citation65]. Taking advantage of this phenomenon, we tracked ACTB mRNA using dRfxCas13d-APEX2-NES with optimal ACTB Rfx-crRNAs and simultaneously applied sodium arsenite to induce cellular stress, resulting in an accumulation of mRNAs, including ACTB mRNAs, into stress granules, which makes the positional relationship between dRfxCas13d-APEX2-NES and ACTB mRNA more visible. Results showed that dRfxCas13d-APEX2-NES colocalized with the stress granule marker G3BP1 regardless of co-transfection with ACTB Rfx-crRNAs or non-targeting Rfx-crRNAs (). This indicated that dRfxCas13d-APEX2-NES may non-specifically accumulate with ACTB mRNA. We also constructed dRfxCas13d-APEX2-NLS plasmid and designed the Rfx-crRNAs targeting NEAT1 to study paraspeckles. We found that the localization of dRfxCas13d-APEX2-NLS had no difference between the non-targeting Rfx-crRNA group and the NEAT1 targeting Rfx-crRNA group (data not shown), which is consistent with results of the Chen group [Citation66]. These data suggested that dRfxCas13d is not suitable for CBRPP to study RNA–protein interactions.
Figure 2. dRfxCas13d is not suitable for CBRPP to study RNA-protein interactions
Transient transfection of dPspCas13b-APEX2-NES to identify the RBPs of ACTB mRNA is not effective
Recent study showed that dPspCas13b is the most efficient dCas13 protein to label RNA [Citation66], so we replaced dRfxCas13d with dPspCas13b and added a flexible linker 3x(GGGGS) between dPspCas13b and APEX2 to avoid mutual influence (). Since PspCas13b and RfxCas13d cannot share the crRNAs, we redesigned four ACTB Psp-crRNAs and validated their targeting. Results showed that all four ACTB Psp-crRNAs significantly reduced ACTB mRNA level in HEK293T cells, and that the knockdown efficiency and expression level were comparable between PspCas13b and PspCas13b-APEX2, indicating fusing APEX2 to C-terminus of PspCas13b does not influence the function and expression of PspCas13b (). Therefore, we constructed dPspCas13b-APEX2-NES plasmid and applied it to track ACTB mRNA with optimal ACTB Psp-crRNAs. In addition, sodium arsenite was applied to induce the formation of stress granules. Results showed that dPspCas13b-APEX2-NES colocalized with the stress granule marker G3BP1 co-transfected with ACTB Psp-crRNAs, but not non-targeting Psp-crRNAs (). Besides, RIP-qPCR assays showed that pulldown of dPspCas13b-APEX2-NES with ACTB Psp-crRNAs resulted in significant enrichment of ACTB mRNA compared to controls using non-targeting Psp-crRNAs (). These data indicated that dPspCas13b-APEX2-NES, under the guidance of ACTB Psp-crRNAs, can specifically track ACTB mRNA in cells. Furthermore, co-transfection of dPspCas13b-APEX2-NES and ACTB Psp-crRNAs in HEK293T cells did not affect the mRNA and protein level of ACTB (), suggesting this system did not affect the stability and translation of ACTB mRNA. Then, we transiently transfected dPspCas13b-APEX2-NES and ACTB Psp-crRNAs into HEK293T cells to performed a 1-min proximity labelling reaction, followed by streptavidin beads enrichment of biotinylated proteins and liquid chromatography-tandem mass spectrometry (LC-MS/MS) (). Western blot results showed that biotinylation occurred (). MS results showed that two proteins were significantly enriched in the ACTB Psp-crRNAs group (transfected with ACTB Psp-crRNA 3 and 4 together) relative to the non-targeting Psp-crRNAs group (transfected with non-targeting Psp-crRNA 1 and 2 together), but the RBPs of ACTB mRNA was not included (). These data suggested that transient transfection of dPspCas13b-APEX2-NES to identify the RBPs of ACTB mRNA is not effective.
Figure 3. Transient transfection of dPspCas13b-APEX2-NES to identify the RBPs of ACTB mRNA
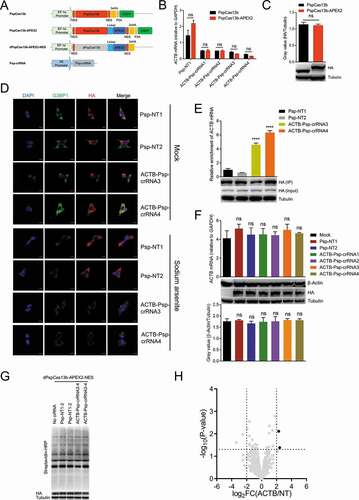
We speculated that such unsatisfactory results may be due to the high expression of dPspCas13b-APEX2-NES or the properties of APEX2 itself. If the protein expression level of dPspCas13b-APEX2-NES is too high, or the copy number of dPspCas13b-APEX2-NES proteins exceeds that of the target RNAs, some redundant dPspCas13b-APEX2-NES proteins cannot be directed to the target RNAs with the guidance of specific crRNAs, so background proteins would be labelled, resulting in low signal-to-noise ratio. Besides, it’s known that APEX2-based labelling is often specific to low-abundance amino acids such as tyrosine [Citation67,Citation68], so it is possible that labelling will not occur if surface-exposed tyrosine is not available.
Inducible expression of dPspCas13b-BioID2-NES successfully identifies the RBPs of ACTB mRNA
For further optimization, we next used other three PBL enzymes (BioID2, TurboID and BASU) to compare with APEX2 for testing which enzyme is optimal, and simultaneously leveraged the Tet-On 3 G inducible expression system to keep the expression of fusion proteins at a low level in HEK293T cells (Supplementary ). Firstly, we constructed PspCas13b-BioID2/TurboID/BASU and proved that like APEX2, fusing BioID2, TurboID or BASU to C-terminus of PspCas13b does not influence the function and expression of PspCas13b by detecting the knockdown efficiency of PspCas13b and western blot (). Then, Inducible-dPspCas13b-BioID2/TurboID/BASU/APEX2-NES (dPspCas13b-BioID2/TurboID/BASU/APEX2-NES under the control of TRE3G promoter) were constructed and stably transfected into HEK293T cells stably expressing Tet-On 3 G transactivator protein, to generate four cell lines inducibly expressing dPspCas13b-BioID2/TurboID/BASU/APEX2-NES (). The optimal biotin concentration and labelling time for each PBL enzyme was adopted to induce biotinylation based on previous research [Citation32,Citation39,Citation40,Citation42,Citation69]. For BioID2, 50 µM biotin for 18 h was used; for TurboID, 500 µM biotin for 10 minutes was used; for APEX2, 500 µM biotin-phenol for 30 min followed by a 1-min exposure to 1 mM H2O2 was used; for BASU, 200 µM biotin for 2 h was used. Western blot results showed that all four cell lines can be induced by doxycycline in a dose-dependent manner, and that BioID2, TurboID and APEX2 had biotinylation activity but not BASU (). 0.1 µg/ml doxycycline was adopted for subsequent experiments, since protein expression and biotinylation activity were low but detectable under this condition. Subsequently, we used dPspCas13b-BioID2/TurboID/Apex2-NES inducibly expressing cell lines to identify the proteins interacting with ACTB mRNA (). Western blot results showed that biotinylation occurred (). We analysed MS data obtained from these three cell lines, and found that in the cell lines inducibly expressing dPspCas13b-TurboID2/APEX2-NES, several proteins were significantly enriched in the ACTB Psp-crRNAs group (transfected with ACTB Psp-crRNA 3 and 4 together) relative to the non-targeting Psp-crRNAs group (transfected with non-targeting Psp-crRNA 1 and 2 together), but the RBPs of ACTB mRNA was not included (Supplementary ). However, in the cell line expressing dPspCas13b-BioID2-NES, several known RBPs of ACTB mRNA (marked in red dots) were significantly enriched in the ACTB Psp-crRNAs group relative to the non-targeting Psp-crRNAs group (). Among these proteins, IGF2BP1, also known as zipcode-binding protein 1 (ZBP1), interacts with the ‘zipcode’ in the 3ʹUTR of ACTB mRNA via HNRNPK homology (KH) domains to regulate the localization and translation of ACTB mRNA [Citation70]. Of note, the ‘zipcode’ of ACTB mRNA is exactly between the protospacer sequences of two ACTB Psp-crRNAs. HNRNPA1, HNRNPC and HNRNPA2B1 are common RBPs that are involved not only in processing heterogeneous nuclear RNAs into mRNAs, but also mRNA stability and translational regulation [Citation71]. DHX9 was reported to interact with the 3ʹUTR of ACTB mRNA [Citation72]. NPM1 was reported to be associated in a sequence-independent manner with the region ~10 nucleotides upstream of the poly(A) signal (AAUAAA) [Citation73]. To test whether unknown RBPs of ACTB mRNA were enriched by this method, we performed RIP-qPCR to confirm several new RBPs of ACTB mRNA from the significantly enriched proteins (HNRNPA1 as positive control). These data indicated that inducible expression of dPspCas13b-BioID2-NES successfully identify the RBPs of ACTB mRNA.
Figure 4. Identifying the RBPs of ACTB mRNA in cells inducibly expressing dPspCas13b-BioID2/TurboID/APEX2-NES
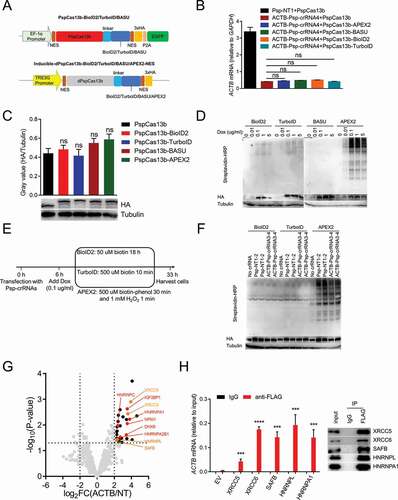
Unlike TurboID or APEX2, BioID2 used in CBRPP generates a history of RNA–protein interactions over time, which can capture some transient RNA-protein interactions, such as those occur during various stages of the cell cycle. Besides, the results obtained using BioID2 in CBRPP represent the accumulation of biotinylated proteins over time. The proteins that interact with the target RNA are labelled and accumulated during this time, and those background proteins that occasionally appear near the target RNA without mutual interaction may be labelled but not accumulated, which results in high signal-to-noise ratio. Therefore, inducible expression of dPspCas13b-BioID2 is recommended to study the RBPs of the target RNA.
Identifying the RBPs of NORAD using CBRPP
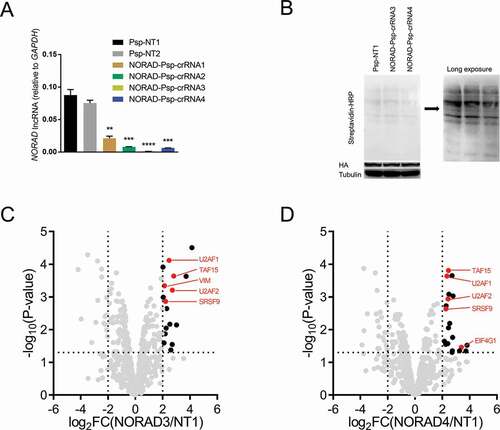
To determine whether CBRPP can identify RBPs of long non-coding RNAs (lncRNAs), we applied CBRPP to study the RBPs of NORAD, a broadly expressed and highly conserved mammalian lncRNA [Citation74–76]. NORAD has been called the ‘defender of the genome’ due to its key role in maintaining genome integrity [Citation74]. Besides, several studies indicated that the RBPs of NORAD are important for the function of NORAD [Citation75–77]. Therefore, identifying the RBPs of NORAD in vivo is meaningful. To this end, we designed four Psp-crRNAs targeting NORAD and validated their targeting (). Two optimal NORAD Psp-crRNAs (3 and 4) were selected and, respectively, transfected into HEK293T cells inducibly expressing dPspcas13b-BioID2-NES to study the RBPs of NORAD. Western blot results showed that biotinylation occurred (). MS results showed that, relative to the non-targeting Psp-crRNA 1 group, five known RBPs of NORAD were significantly enriched in the NORAD Psp-crRNA 3 group (U2AF1, U2AF2, TAF15, SRSF9, VIM) and the NORAD Psp-crRNA 4 group (U2AF1, U2AF2, TAF15, SRSF9, EIF4G1) respectively. Of these enriched RBPs of NORAD, four proteins (U2AF1, U2AF2, TAF15, SRSF9) were enriched in both groups (). These data suggested that CBRPP can be used not only to study RBPs of mRNAs but also RBPs of lncRNAs.
Figure 5. Identifying the RBPs of NORAD using CBRPP
Discussion and conclusion
Here we proposed a new RNA-centric method named CBRPP by combining dCas13 with proximity-based labelling. With some optimizations, we finally determined that inducible expression of dPspCas13b-BioID2-NES is most suitable for studying RNA–protein interactions. In the presence of specific Psp-crRNAs, the dPspCas13b-BioID2-NES fusion protein is directed to the target RNA, then BioID2 in the chimera biotinylates nearby proteins of the target RNA. With the strong interaction between biotin and streptavidin, biotinylated proteins can be easily enriched and identified.
Compared with previous RNA-centric methods, CBRPP has several advantages. First, CBRPP does not require pre-labelling of the target RNA [Citation17], MS2 insertion in advance [Citation31], or designing antisense probes [Citation22,Citation24,Citation78–80] to purify RNA-protein complexes. In dPspCas13b-BioID2-NES positive cells, only crRNAs are required. Second, using CBRPP, RBPs labelling is done in a living cell state without manipulating RNA-protein complexes in vitro, so it almost preserves the natural structure of the target RNA, while avoiding the possible disruption of RNA–protein interactions and RNA degradation. Third, CBRPP can capture weak and transient RNA–protein interactions, taking advantage of proximity-based labelling[Citation36].
As with any technology, CBRPP has its limitations. Since proximity-based labelling is in a distance-dependent manner, proteins identified by CBRPP may be not the RBPs of the target RNA but merely proximate proteins. Therefore, it is necessary to confirm the interactions between the target RNA and candidate proteins identified by CBRPP with RIP or CLIP. Due to the large size of dPspCas13b-BioID2-NES, its binding to the target RNA may affect the binding of the original interacted protein at this site. In addition, the long labelling time required for BioID2 prevents CBRPP from isolated analysis of RNA–protein interactions that occur over short period of time.
According to our experience, there are three crucial factors for the success of CBRPP. First, it is necessary to find potent crRNAs for analysis, and testing multiple crRNAs at the same time is recommended. Second, the expression level of dPspCas13b-BioID2-NES should be controlled at a low level in case the copy number of fusion proteins exceeds that of the target RNAs, resulting in low signal-to-noise ratio. Third, setting up an appropriate control group is necessary. In our experiments, an appropriate control group can be compared with the experimental group to exclude background proteins identified in the experimental group as far as possible.
In summary, in this study we developed an effective RNA-centric method to identify proteins associated with an endogenous RNA of interest in native cellular context without pre-editing of the target RNA, cross-linking or RNA-protein complexes manipulation in vitro. Although we have only studied ACTB mRNA and NORAD lncRNA using CBRPP, in principle CBRPP can also be used to study other RNA types. Besides, for long lncRNAs, taking Xist as an example, by designing different crRNAs target different regions of Xist, CBRPP can not only study the RBPs of Xist, but also the RBPs at a certain position of Xist [Citation21,Citation22]. Furthermore, CBRPP is suitable for studying the mechanism of diseases caused by abnormal RNA–protein interactions. For example, frontotemporal dementia (FTD) and amyotrophic lateral sclerosis (ALS) have been found to develop from the repeated GGGGCC sequence transcribed from an intron in the C90RF72 gene [Citation81]. These toxic RNA sequences consist of a varying number of repeats and have been observed to construct intermolecular hairpin structure which slow their decay and allow them the functionality to interact with proteins inducing irregular splicing [Citation82]. Therefore, identifying RBPs of these toxic RNAs by CBRPP is meaningful for understanding how these toxic RNAs disrupt splicing and cause diseases.
Author Contributions
Y.L. and F.Y. conceived this project. Y.L. analyzed the data and wrote the paper. Y.L., SD.L., L.C., H.D. and F.Y. revised the paper. Y.L., SD.L., L.C. and YJ.L. performed most experiments. SJ.L. contributed to imaging.
Supplemental Material
Download Zip (1.5 MB)Acknowledgments
This work was supported by the National Natural Science Foundation of China (31570891) and the National Key Research and Development Program of China (Grant #2016YFA0500302).
Supplementary material
Supplemental data for this article can be accessed here.
Disclosure statement
The authors declare no competing interests.
Additional information
Funding
Related Research Data
References
- Lee SR, Lykke-Andersen J. Emerging roles for ribonucleoprotein modification and remodeling in controlling RNA fate. Trends Cell Biol. 2013;23(10):504–510.
- Muller-McNicoll M, Neugebauer KM. How cells get the message: dynamic assembly and function of mRNA-protein complexes. Nat Rev Genet. 2013;14(4):275–287.
- Gerstberger S, Hafner M, Tuschl T. A census of human RNA-binding proteins. Nat Rev Genet. 2014;15(12):829–845.
- Di Liegro CM, Schiera G, Di Liegro I. Regulation of mRNA transport, localization and translation in the nervous system of mammals (Review). Int J Mol Med. 2014;33(4):747–762.
- Bugaut A, Balasubramanian S. 5ʹ-UTR RNA G-quadruplexes: translation regulation and targeting. Nucleic Acids Res. 2012;40(11):4727–4741.
- Ma W, Mayr C, Membraneless Organelle A. Associated with the endoplasmic reticulum enables 3ʹUTR-mediated protein-protein interactions. Cell. 2018;175(6):1492–506 e19.
- Jain A, Vale RD. RNA phase transitions in repeat expansion disorders. Nature. 2017;546(7657):243–247.
- Baltz AG, Munschauer M, Schwanhausser B, et al. The mRNA-bound proteome and its global occupancy profile on protein-coding transcripts. Mol Cell. 2012;46(5):674–690.
- Castello A, Fischer B, Eichelbaum K, et al. Insights into RNA biology from an atlas of mammalian mRNA-binding proteins. Cell. 2012;149(6):1393–1406.
- Yum K, Wang ET, Kalsotra A. Myotonic dystrophy: disease repeat range, penetrance, age of onset, and relationship between repeat size and phenotypes. Curr Opin Genet Dev. 2017;44:30–37.
- Chau A, Kalsotra A. Developmental insights into the pathology of and therapeutic strategies for DM1: back to the basics. Dev Dyn. 2015;244(3):377–390.
- Ramanathan M, Porter DF, Khavari PA. Methods to study RNA-protein interactions. Nat Methods. 2019;16(3):225–234.
- Licatalosi DD, Mele A, Fak JJ, et al. HITS-CLIP yields genome-wide insights into brain alternative RNA processing. Nature. 2008;456(7221):464–469.
- Nicholson CO, Friedersdorf M, Keene JD. Quantifying RNA binding sites transcriptome-wide using DO-RIP-seq. RNA. 2017;23(1):32–46.
- McMahon Aoife C, Rahman R, Jin H, et al. TRIBE: hijacking an RNA-editing enzyme to identify cell-specific targets of RNA-Binding proteins. Cell. 2016;165(3):742–753.
- Lapointe CP, Wilinski D, Saunders HA, et al. Protein-RNA networks revealed through covalent RNA marks. Nat Methods. 2015;12(12):1163–1170.
- Zheng X, Cho S, Moon H, et al. Detecting RNA-protein interaction using end-labeled biotinylated RNA oligonucleotides and immunoblotting. Methods Mol Biol. 2016;1421:35–44.
- Leppek K, Stoecklin G. An optimized streptavidin-binding RNA aptamer for purification of ribonucleoprotein complexes identifies novel ARE-binding proteins. Nucleic Acids Res. 2014;42(2):e13.
- Lee HY, Haurwitz RE, Apffel A, et al. RNA-protein analysis using a conditional CRISPR nuclease. Proc Natl Acad Sci U S A. 2013;110(14):5416–5421.
- Engreitz JM, Pandya-Jones A, McDonel P, et al. The Xist lncRNA exploits three-dimensional genome architecture to spread across the X chromosome. Science. 2013;341(6147):1237973.
- Chu C, Zhang QC, da Rocha ST, et al. Systematic discovery of Xist RNA binding proteins. Cell. 2015;161(2):404–416.
- Simon MD, Wang CI, Kharchenko PV, et al. The genomic binding sites of a noncoding RNA. Proc Natl Acad Sci U S A. 2011;108(51):20497–20502.
- Zeng F, Peritz T, Kannanayakal TJ, et al. A protocol for PAIR: PNA-assisted identification of RNA binding proteins in living cells. Nat Protoc. 2006;1(2):920–927.
- Matia-Gonzalez AM, Iadevaia V, Gerber AP. A versatile tandem RNA isolation procedure to capture in vivo formed mRNA-protein complexes. Methods. 2017;118-119:93–100.
- Tsai BP, Wang X, Huang L, et al. Quantitative profiling of in vivo-assembled vivo-assembled RNA-protein complexes using a novel integrated proteomic approach. Mol Cell Proteomics. 2011;10(4):M110 007385.
- Sugimoto Y, Konig J, Hussain S, et al. Analysis of CLIP and iCLIP methods for nucleotide-resolution studies of protein-RNA interactions. Genome Biol. 2012;13(8):R67.
- Kim B, Kim VN. fCLIP-seq for transcriptomic footprinting of dsRNA-binding proteins: lessons from DROSHA. Methods. 2019;152:3–11.
- Meisenheimer KM, Koch TH. Photocross-linking of nucleic acids to associated proteins. Crit Rev Biochem Mol Biol. 1997;32(2):101–140.
- Hoffman EA, Frey BL, Smith LM, et al. Formaldehyde crosslinking: a tool for the study of chromatin complexes. J Biol Chem. 2015;290(44):26404–26411.
- Li X, Song J, Yi C. Genome-wide mapping of cellular protein-RNA interactions enabled by chemical crosslinking. Genomics Proteomics Bioinformatics. 2014;12(2):72–78.
- Mukherjee J, Hermesh O, Eliscovich C, et al. beta-Actin mRNA interactome mapping by proximity biotinylation. Proc Natl Acad Sci U S A. 2019;116(26):12863–12872.
- Ramanathan M, Majzoub K, Rao DS, et al. RNA-protein interaction detection in living cells. Nat Methods. 2018;15(3):207–212.
- Zhang W, Xie M, Shu MD, et al. A proximity-dependent assay for specific RNA-protein interactions in intact cells. RNA. 2016;22(11):1785–1792.
- Jung J, Lifland AW, Alonas EJ, et al. Characterization of mRNA-cytoskeleton interactions in situ using FMTRIP and proximity ligation. PLoS One. 2013;8(9):e74598.
- Zurla C, Jung J, Blanchard EL, et al. A novel method to quantify RNA-protein interactions in situ using FMTRIP and proximity ligation. Methods Mol Biol. 2017;1468:155–170.
- Kim DI, Roux KJ. Filling the void: proximity-based labeling of proteins in living cells. Trends Cell Biol. 2016;26(11):804–817.
- Trinkle-Mulcahy L. Recent advances in proximity-based labeling methods for interactome mapping. F1000Res. 2019;8:8.
- Roux KJ, Kim DI, Raida M, et al. A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J Cell Biol. 2012;196(6):801–810.
- Kim DI, Jensen SC, Noble KA, et al. An improved smaller biotin ligase for BioID proximity labeling. Mol Biol Cell. 2016;27(8):1188–1196.
- Branon TC, Bosch JA, Sanchez AD, et al. Efficient proximity labeling in living cells and organisms with TurboID. Nat Biotechnol. 2018;36(9):880–887.
- Rhee HW, Zou P, Udeshi ND, et al. Proteomic mapping of mitochondria in living cells via spatially restricted enzymatic tagging. Science. 2013;339(6125):1328–1331.
- Lam SS, Martell JD, Kamer KJ, et al. Directed evolution of APEX2 for electron microscopy and proximity labeling. Nat Methods. 2015;12(1):51–54.
- Gupta GD, Coyaud E, Goncalves J, et al. A dynamic protein interaction landscape of the human centrosome-cilium interface. Cell. 2015;163(6):1484–1499.
- Huang A, Tang Y, Shi X, et al. Proximity labeling proteomics reveals critical regulators for inner nuclear membrane protein degradation in plants. Nat Commun. 2020;11(1):3284.
- Uezu A, Kanak DJ, Bradshaw TW, et al. Identification of an elaborate complex mediating postsynaptic inhibition. Science. 2016;353(6304):1123–1129.
- Opitz N, Schmitt K, Hofer-Pretz V, et al. Capturing the Asc1p/Receptor Asc1p/Receptor for activated activate C kinase kinas 1 (RACK1) microenvironment at the head region of the 40S ribosome with quantitative bioid in yeast. Mol Cell Proteomics. 2017;16(12):2199–2218.
- Chen AL, Kim EW, Toh JY, et al. Novel components of the Toxoplasma inner membrane complex revealed by BioID. mBio. 2015;6(1):e02357–14.
- Martell JD, Deerinck TJ, Sancak Y, et al. Engineered ascorbate peroxidase as a genetically encoded reporter for electron microscopy. Nat Biotechnol. 2012;30(11):1143–1148.
- Hung V, Zou P, Rhee HW, et al. Proteomic mapping of the human mitochondrial intermembrane space in live cells via ratiometric APEX tagging. Mol Cell. 2014;55(2):332–341.
- Knott GJ, Doudna JA. CRISPR-Cas guides the future of genetic engineering. Science. 2018;361(6405):866–869.
- Shalem O, Sanjana NE, Hartenian E, et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. 2014;343(6166):84–87.
- Fellmann C, Gowen BG, Lin PC, et al. Cornerstones of CRISPR-Cas in drug discovery and therapy. Nat Rev Drug Discov. 2017;16(2):89–100.
- Kim VN. RNA-targeting CRISPR comes of age. Nat Biotechnol. 2018;36(1):44–45.
- Terns MP. CRISPR-based technologies: impact of RNA-targeting systems. Mol Cell. 2018;72(3):404–412.
- Abudayyeh OO, Gootenberg JS, Konermann S, et al. C2c2 is a single-component programmable RNA-guided RNA-targeting CRISPR effector. Science. 2016;353(6299):aaf5573.
- Abudayyeh OO, Gootenberg JS, Essletzbichler P, et al. RNA targeting with CRISPR-Cas13. Nature. 2017;550(7675):280–284.
- Cox DBT, Gootenberg JS, Abudayyeh OO, et al. RNA editing with CRISPR-Cas13. Science. 2017;358(6366):1019–1027.
- Konermann S, Lotfy P, Brideau NJ, et al. Transcriptome Engineering with RNA-Targeting Type VI-D CRISPR Effectors. Cell. 2018;173(3):665–76 e14.
- Myers SA, Wright J, Peckner R, et al. Discovery of proteins associated with a predefined genomic locus via dCas9-APEX-mediated proximity labeling. Nat Methods. 2018;15(6):437–439.
- Gao XD, Tu LC, Mir A, et al. C-BERST: defining subnuclear proteomic landscapes at genomic elements with dCas9-APEX2. Nat Methods. 2018;15(6):433–436.
- Zhang X, Smits AH, van Tilburg GB, et al. Proteome-wide identification of ubiquitin interactions using UbIA-MS. Nat Protoc. 2018;13(3):530–550.
- Farina KL, Huttelmaier S, Musunuru K, et al. Two ZBP1 KH domains facilitate beta-actin mRNA localization, granule formation, and cytoskeletal attachment. J Cell Biol. 2003;160(1):77–87.
- Eliseeva I, Vasilieva M, Ovchinnikov LP. Translation of human beta-actin mRNA is regulated by mtor pathway. Genes (Basel). 2019;10(2):10.
- Haimovich G, Ecker CM, Dunagin MC, et al. Intercellular mRNA trafficking via membrane nanotube-like extensions in mammalian cells. Proc Natl Acad Sci U S A. 2017;114(46):E9873–E82.
- Wolozin B, Ivanov P. Stress granules and neurodegeneration. Nat Rev Neurosci. 2019;20(11):649–666.
- Yang LZ, Wang Y, Li SQ, et al. Dynamic Imaging of RNA in living cells by CRISPR-Cas13 systems. Mol Cell. 2019;76(6):981–97 e7.
- Echols N, Harrison P, Balasubramanian S, et al. Comprehensive analysis of amino acid and nucleotide composition in eukaryotic genomes, comparing genes and pseudogenes. Nucleic Acids Res. 2002;30(11):2515–2523.
- Tourasse NJ, Li WH. Selective constraints, amino acid composition, and the rate of protein evolution. Mol Biol Evol. 2000;17(4):656–664.
- Lu M, Wei W. Proximity labeling to detect RNA-protein interactions in live cells. FEBS Open Bio. 2019;9(11):1860–1868.
- Chao JA, Patskovsky Y, Patel V, et al. ZBP1 recognition of beta-actin zipcode induces RNA looping. Genes Dev. 2010;24(2):148–158.
- Geuens T, Bouhy D, Timmerman V. The hnRNP family: insights into their role in health and disease. Hum Genet. 2016;135(8):851–867.
- Eliscovich C, Shenoy SM, Singer RH. Imaging mRNA and protein interactions within neurons. Proc Natl Acad Sci U S A. 2017;114(10):E1875–E84.
- Palaniswamy V, Moraes KC, Wilusz CJ, et al. Nucleophosmin is selectively deposited on mRNA during polyadenylation. Nat Struct Mol Biol. 2006;13(5):429–435.
- Ventura A. NORAD: defender of the Genome. Trends Genet. 2016;32(7):390–392.
- Munschauer M, Nguyen CT, Sirokman K, et al. The NORAD lncRNA assembles a topoisomerase complex critical for genome stability. Nature. 2018;561(7721):132–136.
- Lee S, Kopp F, Chang TC, et al. Noncoding RNA NORAD regulates genomic stability by sequestering PUMILIO proteins. Cell. 2016;164(1–2):69–80.
- Elguindy MM, Kopp F, Goodarzi M, et al. PUMILIO, but not RBMX, binding is required for regulation of genomic stability by noncoding RNA NORAD. Elife. 2019;8:e48625.
- Chu C, Qu K, Zhong FL, et al. Genomic maps of long noncoding RNA occupancy reveal principles of RNA-chromatin interactions. Mol Cell. 2011;44(4):667–678.
- McHugh CA, Guttman M. RAP-MS: A method to identify proteins that interact directly with a specific RNA molecule in cells. Methods Mol Biol. 2018;1649:473–488.
- West JA, Davis CP, Sunwoo H, et al. The long noncoding RNAs NEAT1 and MALAT1 bind active chromatin sites. Mol Cell. 2014;55(5):791–802.
- Osborne RJ, Thornton CA. RNA-dominant diseases. Hum Mol Genet. 2006;15(No 2):R162–9. 15 Spec
- Orr HT. Toxic RNA as a driver of disease in a common form of ALS and dementia. Proc Natl Acad Sci U S A. 2013;110(19):7533–7534.