
ABSTRACT
CRISPR-Cas systems are prokaryotic adaptive immune systems that recognize and cleave nucleic acid targets using small RNAs called CRISPR RNAs (crRNAs) to guide Cas protein(s). There is increasing evidence for the broader endogenous roles of these systems. The CRISPR-Cas9 system of Francisella novicida also represses endogenous transcription using a non-canonical small RNA (scaRNA). We examined whether the crRNAs of the native F. novicida CRISPR-Cas systems, Cas12a and Cas9, can guide transcriptional repression. Both systems repressed mRNA transcript levels when crRNA-target complementarity was limited, and led to target cleavage with extended complementarity. Using these parameters we engineered the CRISPR array of Cas12a to guide the transcriptional repression of a new and endogenous target. Since the majority of crRNA targets remain unidentified, this work suggests that a re-analysis of crRNAs for endogenous targets with limited complementarity could reveal new, diverse regulatory roles for CRISPR-Cas systems in prokaryotic biology.
Introduction
CRISPR (clustered, regularly interspaced, short, palindromic repeats) – Cas (CRISPR-associated) systems are widespread in prokaryotes, where their best described role is as adaptive immune systems that protect against foreign genetic elements [Citation1,Citation2]. They are highly diverse and can be divided into two classes, characterized by the nature of the effector protein(s) used for nucleic acid targeting; Class 1 systems have a multi-protein effector complex, while Class 2 systems have a single protein effector [Citation3]. Each class is further subdivided into types and subtypes that are continually expanding as more distant variants of these systems are discovered [Citation3].
In particular, the Class II systems, especially type II (containing CRISPR-Cas9) and type V (containing CRISPR-Cas12a/Cpf1) systems, have garnered significant research attention for their ability to be repurposed for molecular biological and therapeutic genome engineering applications [Citation4,Citation5]. Both Type II and V systems cleave foreign nucleic acid targets using their effectors in complex with a small RNA, CRISPR RNA (crRNA) [Citation6–8]. Each crRNA contains a repeat which is bound by the effector protein and a spacer that binds to complementary sequences on foreign nucleic acid targets, guiding the effector proteins to cleave the target (called the protospacer) and protect the cell [Citation6–8]. Within these systems, the biochemical variabilities between subtypes and orthologous effectors are being continually uncovered and often harnessed for genome engineering [Citation4,Citation9,Citation10]. Further characterization of CRISPR-Cas systems in their native genetic contexts and the bacterial hosts in which they are maintained will likely continue to uncover unique features of different systems [Citation11–14].
The pathogen Francisella novicida encodes a type II-B CRISPR-Cas9 system (FnoCas9) and a type V-A CRISPR-Cas12a (FnoCas12a) system, both of which are capable of directing DNA cleavage [Citation8,Citation15–17]. The CRISPR arrays from each CRISPR-Cas system are transcribed and processed into individual crRNAs; the CRISPR-Cas9 array (CRISPR1) has 13 spacers and the Cas12a array (CRISPR2) has 9 spacers [Citation8,Citation15,Citation18]. Both F. novicida systems contain spacers against the same putative prophage, suggesting that they have the ability to defend against a similar threat [Citation15]. The CRISPR arrays of both systems recognize and restrict protospacers corresponding to spacers in the CRISPR arrays, and can be engineered within F. novicida to restrict new targets through modification of the spacers in the genome [Citation17]. While the Cas12a crRNA independently directs the protein to its target, Cas9 interacts with the crRNA as an RNA duplex with tracrRNA (trans-activating crRNA), a second small RNA transcribed from the CRISPR-Cas9 locus [Citation19].
To avoid lethal autoimmunity-like cleavage of the CRISPR array in the chromosome by their own crRNAs, these two systems (as well as others) utilize a protospacer adjacent motif (PAM) [Citation6,Citation8,Citation17,Citation20,Citation21]. A PAM is a short nucleotide sequence directly adjacent to the protospacer that is required for crRNA:protospacer interaction and canonically allows for differentiation of self and non-self sequences [Citation22–25]. For example, a PAM must be present in foreign DNA sequences to be targeted, but no PAMs are present in the endogenous CRISPR arrays, precluding self-targeting. It has been suggested that if the PAM is present adjacent to a self-target, other elements of the CRISPR-Cas system would have to be mutated to prevent cell death caused by cytotoxic self-targeting and cleavage of the bacterial genome [Citation20,Citation23,Citation26,Citation27].
In addition to crRNA-directed cleavage, FnoCas9 represses the expression of endogenous genes, promoting virulence by facilitating evasion of the host immune system [Citation28–30]. Cas9 has been shown in other pathogens to regulate traits that are important for virulence, such as attachment to host cells and intracellular survival [Citation31–36]. In Group B Streptococcus, Cas9 appears to regulate a significant portion of the genome; cas9 mutants are attenuated in vivo and have altered protein abundance of multiple virulence determinants [Citation35,Citation36]. We recently interrogated the mechanism for endogenous gene repression by FnoCas9 and developed an updated model [Citation29]. We found that FnoCas9 can act as a transcriptional repressor, using tracrRNA and an additional CRISPR-associated RNA, scaRNA, to guide FnoCas9 to bind but not cleave endogenous DNA targets located near transcriptional start sites (TSSs) [Citation29]. Repression by scaRNA requires a PAM, and the limited scaRNA:DNA target complementarity facilitates the cleavage-independent binding of FnoCas9 to the DNA that leads to transcriptional repression. This provided the first example that a PAM-dependent self-targeting event by a cleavage-capable Cas9 can be stably maintained in the bacterium while also modulating physiology, and recalls early and continued discussions of the biological importance of self-targeting RNAs that are associated with CRISPR-Cas systems [Citation20,Citation37–39]. Interestingly, computational predictions of scaRNAs in organisms with type II CRISPR-Cas systems suggest a correlation between the presence of these small RNAs and increased strain virulence [Citation33].
We hypothesized that endogenous crRNAs could direct FnoCas9 and FnoCas12a to regulate transcription in addition to DNA cleavage, though a mechanism similar to scaRNA. For other orthologs of Cas9, imperfect binding of crRNAs to target DNA can inhibit cleavage without blocking all interaction with the DNA target, although the role of these non-dsDNA cleavage activities in bacterial physiology has yet to be determined [Citation6,Citation40–42]. Utilizing this principle, Cas9 has been engineered for use in non-native cell types to alter transcription using reduced crRNA:target DNA complementarity while retaining cleavage capability of the effector [Citation41,Citation43,Citation44]. Most recently, these principles were applied to an engineered Acidaminococcus sp. BV3L6 Cas12a (AsCas12a), in which multiplexed crRNAs were used to coordinate cleavage and transcriptional activation in mammalian cells [Citation45].
These crRNA-directed advancements in Cas9 and Cas12a engineering and the natural ability of FnoCas9 to direct transcriptional repression using scaRNA with incomplete complementarity to the DNA, highlight the possibility that transcriptional regulation through incomplete RNA:DNA complementarity is a native capability of many CRISPR-Cas systems, in particular Cas9 and Cas12a. We utilized the model system F. novicida to investigate whether altering crRNA target:homology could turn the native FnoCas9 and FnoCas12a CRISPR-Cas systems into transcriptional regulators using crRNA. Notably, this bacterium naturally encodes both systems and it is unknown whether either endogenous catalytically active system is able to repress transcription with crRNAs.
We first tested crRNA-mediated transcriptional repression using the native CRISPR-Cas12a system of F. novicida, which unlike the CRISPR-Cas9 system, does not have a known scaRNA. Like the CRISPR-Cas9 system, however, Cas12a is catalytically active in vitro and within the bacterium, as directed by its native crRNAs [Citation8,Citation17]. We observed that small target modifications that reduced the amount of complementarity to crRNA spacers were sufficient for transitioning the activity of FnoCas12a from cleavage to transcriptional repression, providing evidence that FnoCas12a could contribute to bacterial physiology beyond DNA defense. Based on this finding, we engineered the FnoCas12a CRISPR array by replacing the existing array with a spacer for an endogenous target, successfully harnessing the transcriptional regulatory activity of this protein to repress the levels of a native transcript within F. novicida. We subsequently demonstrated that the endogenous FnoCas9 effector displayed similar ability to repress transcription with reduced crRNA:DNA target complementarity, which is supported by engineering and structural studies [Citation40–42,Citation44]. The ability of partially complementary crRNAs to regulate endogenous targets may represent a widespread function of the CRISPR-Cas12a and CRISPR-Cas9 machineries, and may be a general activity of other CRISPR-Cas systems as well. Thus, in addition to demonstrating that the functional role of CRISPR-Cas systems can be rationally engineered by modifications to crRNAs, these data suggest that a broad re-analysis of known crRNAs for partial complementarity to endogenous targets could reveal new regulatory functions of CRISPR-Cas systems in prokaryotic biology.
Results
We previously demonstrated that scaRNA mediates FnoCas9 binding to the F. novicida chromosome and repression of transcription, using 15 bases of complementarity between its tail and the target DNA [Citation29]. When the complementarity between scaRNA and the chromosome was artificially extended to 20 bases, scaRNA instead directed lethal cleavage of the chromosome by FnoCas9 [Citation29]. scaRNA-mediated repression requires the DNA target to be adjacent to a PAM and located within the promoter or directly downstream of the transcriptional start site (TSS) of the gene targeted for repression [Citation29]. It was unclear if FnoCas12a crRNAs could act similarly to scaRNA and direct these two distinct functions in F. novicida. To determine whether Cas12a crRNAs with reduced complementarity to a DNA target could repress transcription without inducing cleavage, and given the absence of available phages for F. novicida, we developed a panel of FnoCas12a target plasmids with complementarity to spacer 5 in CRISPR2 (cas12a CRISPR array) ( A).
Figure 1. crRNA complementarity to target DNA shifts Cas12a from transcriptional repression to DNA cleavage. A) Schematic of the cas12a locus in F. novicida. B) Diagram of the pBav-derived vectors (3654 bp) used in , and C)) Sequence alignment of the PAM + Cas12a protospacer sequences with varying bases of consecutive complementarity to the spacer 5 of CRISPR2, used in B-C. The FnoCas12a PAM sequence (5ʹ-TTTN-3ʹ) is underlined. Protospacer sequences were inserted directly downstream of the TSS and directly upstream of the gfp start. D) Transformation of WT F. novicida and Δcas12a with plasmids with 9 bp, 11 bp, 15 bp, and full complementarity (in consecutive bases) to the Cas12a spacer between the promoter and gfp. Results are presented as transformation efficiency relative to a plasmid with no complementarity to the crRNA (n = 3, error bars represent s.e., **p ≤ 0.005). E-G) Relative gfp transcript levels were measured by qRT-PCR from constructs containing sequences with different lengths of consecutive complementarity to the Cas12a spacer (0, 9, 11, 15 bp) between a synthetic promoter and gfp, and normalized to the transcript levels of the kanamycin resistance cassette on the plasmid. E) Results are presented as % gfp transcript from each plasmid in WT relative to Δcas12a. F-G) Relative gfp transcript level from each plasmid in F) WT and G) Δcas12a (n = 6, error bars represent s.e., *p ≤ 0.05; **p ≤ 0.005; ***p ≤ 0.001)
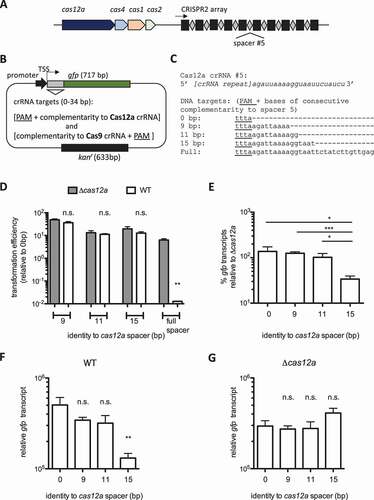
Figure 2. Cas12a can be engineered to repress endogenous targets. A) Model of Cas9 in complex with tracrRNA and scaRNA binding to the 1104 and 1101 5ʹ UTRs on the F. novicida chromosome with the scaRNA tail, which results in repression of 1104–1101 transcription. The base-pairing interactions between scaRNA and each UTR are depicted. B) Schematic of the strategy used to engineer the cas12a CRISPR array for endogenous repression. C) Model of Cas12a interacting with the 1104 5ʹ UTR with the engineered crRNA to repress 1104–1102 transcription. The predicted base-pairing interactions between the engineered crRNA and each 5ʹ UTR are depicted. The PAM is underlined in the 11045' UTR and absent in the 1101 5' UTR. D-G) Relative levels of (D) 1104, (E) 1103, (F) 1102, and (G) 1101 transcripts were measured by qRT-PCR in WT, Δcas9, Δcas12a, WT + engineered cas12a crRNA, Δcas9 + engineered cas12a crRNA, and Δcas9Δcas12a + engineered cas12a crRNA strains. Significance is determined relative to WT unless otherwise noted (n = 6, error bars represent s.e., *p ≤ 0.05; **p ≤ 0.005; ***p ≤ 0.001)
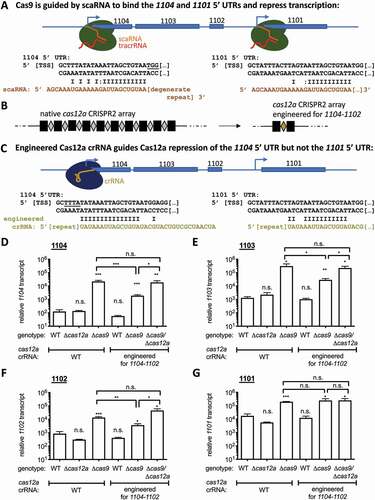
Figure 3. Reduced crRNA:target complementarity shifts Cas9 function from DNA cleavage to transcriptional repression. A) Schematic of the cas9 locus in F. novicida. B) Sequence alignment of the Cas9 protospacer target and PAM (5ʹ-NGG-3ʹ; underlined) inserted into the vector (Figure 1B and S1) with varying bases of consecutive complementarity to the Cas9 crRNA, used in C-D. C) Transformation of WT F. novicida and Δcas9 with the plasmids containing constructs from (B) with 8 bp, 11 bp, 15 bp, and full complementarity (consecutive bases) to the Cas9 spacer between the promoter and gfp. Results are presented as transformation efficiency relative to a plasmid with no complementarity to the crRNA (n = 5–6, error bars represent s.e., **p ≤ 0.005). D) Relative gfp transcript levels were measured by qRT-PCR from constructs containing targets with varying lengths of consecutive complementarity to the Cas9 crRNA: 0, 8 (n = 3), 11, or 15 bp. Results are presented as % relative gfp transcript in WT relative to that in Δcas9 for each plasmid (n = 6, error bars represent s.e., *p ≤ 0.05; **p ≤ 0.005)
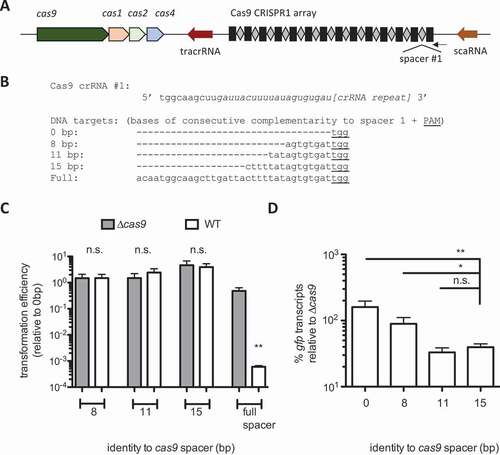
Spacer 5 in CRISPR2 corresponds to a protospacer in a putative prophage that is only found in strains of Francisella that do not have spacers against it [Citation15]. This spacer was selected based on previous work on the endogenous function of the F. novicida CRISPR-Cas12a system that demonstrated efficient transformation inhibition of target plasmids containing the protospacer for spacer 5 (CRISPR2), restricting transformation to almost the limit of detection for the assay. It is important to note that the expression level of each crRNA can affect activity since there is inherent competition for the effector protein, as observed between scaRNA and crRNAs for FnoCas9 [Citation29].
We designed the target plasmids based on the constructs used previously to determine the mechanism of scaRNA-mediated transcriptional repression [Citation29]. In that study, target sequences with varying levels of complementarity to scaRNA, and adjacent to an FnoCas9 PAM, were inserted between a promoter and gfp sequence on the plasmids. gfp transcript levels were then measured by qRT-PCR (quantitative reverse transcriptase PCR) to assess the relative ability of Cas9 to repress transcription from each construct [Citation29]. Herein, we utilized the same synthetic constitutive promoter (p146) to drive the expression of gfp. Rather than complementarity to scaRNA and inclusion of a Cas9 PAM, these constructs contained an FnoCas12a PAM (5ʹ-TTTN-3ʹ) immediately upstream of 0, 9, 11, 15, or 29 bases of uninterrupted complementarity to CRISPR2 spacer 5, inserted between the TSS and gfp (, Figure S2A) [Citation17,Citation46]. The 29 base pair construct represents full complementarity to the native CRISPR2 spacer 5 sequence.
We first determined which of the plasmids were targeted for cleavage by FnoCas12a by measuring the level of transformation inhibition (, S2A). There was high transformation efficiency for plasmids containing 0, 9, 11, or 15 bases of consecutive complementarity to the crRNA, and no differences in transformation between strains with and without cas12a (). However, transformation efficiency with the construct containing the full protospacer was strongly inhibited in WT relative to Δcas12a (). To remove any variability from strain differences in transformation efficiency, we normalized the transformation efficiency of WT to a Δcas12a strain for each plasmid, and found that transformation was only restricted with the full spacer target plasmid (Figure S3A). For this assay the full protospacer was used as a measure of the DNA targeting efficiency of the crRNA at its natural processed length, which is the greatest extent of crRNA:DNA target complementarity that can occur naturally. Twenty bases of crRNA complementarity is the standard protospacer length used to direct Cas9 and Cas12a cleavage. We observed no difference in targeting efficiency between the 20 bp and full protospacer targets, indicating that FnoCas12a has maximal targeting efficiency with the naturally processed crRNA (Figure S3B).
Following transformation of each plasmid into WT and Δcas12a strains, the relative gfp levels were measured by qRT-PCR. gfp transcript levels from the plasmid with 15 bases of complementarity to the crRNA were significantly lower in WT compared to Δcas12a, indicating Cas12a-dependent repression (). In contrast, no repression of gfp was observed from plasmids with 0, 9, or 11 bases of complementarity to the FnoCas12a crRNA in WT relative to Δcas12a (). In some instances, there were additional bases of predicted complementarity between the crRNA and protospacer outside the regions of consecutive complementarity that could influence binding affinity of the crRNA to the DNA. These are depicted in Figure S2 but do not alter the conclusion that extent of complementarity shifts effector function between repression and cleavage. The impact of base pairing and PAM presence on repression in these assays is summarized in Figure S6. Collectively, these results indicate that catalytically active FnoCas12a can both repress transcription and direct target cleavage, and that this functional differentiation can be determined by the extent of complementarity between the target and the crRNA.
We further validated the functionality of Cas12a-mediated transcriptional repression by engineering FnoCas12a to repress an endogenous operon. FnoCas9 uses scaRNA to repress four genes encoded on two mRNA transcripts (1104–1102 and 1101) () [Citation29]. We replaced the endogenous CRISPR2 array with a new spacer sequence with adjoining Cas12a repeats, placing the engineered spacer in the same location in the chromosome and under the same promoter as the native array (). This strategy has been used to engineer Cas12a to restrict new DNA targets in F. novicida at least as efficiently as restriction by FnoCas12a guided by CRISPR2 spacer 5, suggesting that engineering the crRNA by this methodology results in effective transcription and processing [Citation17]. Herein, we engineered the FnoCas12a CRISPR array to have partial complementarity to the 5ʹ untranslated region (UTR) of the 1104–1102 open reading frames (ORF), which are transcribed on a single mRNA (). To investigate the specificity of FnoCas12a-mediated targeting, we designed the engineered crRNA so that it could not bind the 1101 5ʹ UTR, unlike scaRNA which naturally targets both regions (). Despite an 11 bp stretch of complementarity between the engineered Cas12a spacer and the 5ʹ 1101 UTR, there is no adjacent PAM, which is required for effector binding and activity (). As a control for baseline transcript level, we measured the level of each transcript in the native FnoCas9 regulon in WT, Δcas12a, and Δcas9 strains, observing that the levels of 1104, 1103, 1102 and 1101 were not altered in the Δcas12a mutant compared to WT, but were expressed at higher levels in the Δcas9 strain (). This confirmed the requirement of FnoCas9 for the repression of these genes and further demonstrated that FnoCas12a is not naturally involved in the transcriptional regulation of these Cas9 targets (). We then measured the levels of 1104, 1103, 1102, and 1101 in a WT strain with the engineered FnoCas12a CRISPR array, as a control for baseline transcript level. This strain did not exhibit a difference in 1104–1102 transcript levels compared to a Δcas12a mutant or WT strain with an unmodified CRISPR array, likely because the 5ʹ UTR is already being efficiently bound and transcription repressed by scaRNA in complex with Cas9, both of which are intact in this strain (). These results indicate that directing another CRISPR effector to compete for the scaRNA binding site is not an efficient way to improve the level of transcriptional repression of the Cas9 regulon in F. novicida. Compared to a Δcas9 strain that is unable to repress these genes naturally, the Δcas9 strain with the engineered Cas12a crRNA had reduced levels of the 1104–1102 transcript (). This suggested that Cas12a actively repressed these genes in the presence of the engineered crRNA. To determine if the transcriptional repression in the engineered strain was Cas12a-dependent, we compared the transcript levels of 1104–1102 in the Cas12a-engineered strain (Δcas9, intact Cas12a) to an engineered strain without Cas9 and Cas12a (Δcas9/Δcas12a). We found that the 1104–1102 transcript level was similar to that of the Δcas9 strain when cas12a was deleted from the strain with an engineered CRISPR2 array (). This result demonstrates that Cas12a can be engineered to repress transcription through replacement of the spacers in its CRISPR array (). The level of 1104–1102 repression by the engineered Cas12a crRNA was not as robust as endogenous repression of the same transcript by scaRNA, despite a similar level of complementarity between the RNAs and the DNA targets and similar proximity to the TSS ( and Figure S4A-E). This is also observed in the dominance of the Cas9-mediated repression in a strain with intact scaRNA and Cas12a engineered to repress the 1104–1102 transcript as well (, S4E). This suggests that Cas9 is guided by scaRNA to have a stronger or more stable interaction with the 1104 5ʹ UTR than Cas12a guided by the engineered crRNA. Conversely, when the CRISPR arrays of both systems were previously engineered for the same exogenous plasmid DNA target, they exhibited similar levels of plasmid restriction. Taken together, these results suggest that Cas12a may be less effective than Cas9 in transcriptional repression. As an additional control, 1101 transcript level was unaltered between the Δcas9 and Δcas12a/Δcas9 engineered strains, consistent with a specific effect of FnoCas12a on 1104–1102 when guided by the engineered crRNA ().
To address any Cas9-independent contribution of scaRNA to the repression of the 1104 transcript in strains with the Cas12a locus engineered to target the 5ʹ UTR, we compared the levels of 1104 and 1101 between a scaRNA mutant, a scaRNA mutant with the intact Cas12a system engineered to target only 1104–1102 (not 1101), and a cas9/cas12a double mutant. The Δcas9/Δcas12a strain has scaRNA and all of the crRNAs from both loci intact, while the scaRNA mutant has all of the effectors and crRNAs intact, but is missing the small RNA we previously demonstrated to work with Cas9 to repress 1104–1101. Finally, we included a strain missing scaRNA and with intact Cas9, with Cas12a engineered to repress 1104–1102 but not 1101. In this assay 1104 was used to represent the levels of the 1104–1102 transcript, since 1103 and 1102 are encoded on the same transcript as 1104 and exhibited the same changes in RNA level between the different strains (). Between the ΔscaRNA and Δcas9/Δcas12a strains, there was no difference in the levels of 1104 or 1101, indicating that scaRNA does not contribute to the repression of 1104 or 1101 endogenously without the effector proteins. In a ΔscaRNA strain with Cas12 intact and the CRISPR2 array engineered to target 1104, but not 1101, Cas12a reduced the 1104 but not 1101 transcript level (Figure S4A-B). This indicates that scaRNA does not contribute to the repression by engineered Cas12a, and that a gene specific to the natural regulon of scaRNA (1101) is not repressed in the engineered Cas12a strain in the absence of cas9 and scaRNA. We also demonstrate that the levels of 1104 transcripts between a ΔscaRNA and Δcas9 strain with Cas12a engineered to target 1104–1102 are the same. Similarly, between ΔscaRNA and Δcas9 strains with a WT cas12a CRISPR array, the levels of 1104 are the same (Figure S4C-E). The similar phenotypes of the scaRNA and cas9 mutants are consistent with previous work indicating that detectable levels of scaRNA in the cell are dependent on the presence of Cas9, and we therefore expect the cas9 mutant to behave like a cas9/scaRNA double mutant. Although we cannot rule out the possibility that the Cas9 system could have some influence on the transcriptional repression observed with the reprogrammed Cas12a, such repression would be through a mechanism that has yet to be demonstrated [Citation29]. Taken together, these results show that Cas12a can repress endogenous transcription and highlight the functional flexibility of the F. novicida CRISPR-Cas systems.
We next sought to investigate whether this RNA-determined functional shift of FnoCas12a from cleavage to transcriptional repression could be a broader function of crRNAs of other systems by extrapolating to the crRNAs of FnoCas9. To test the ability of crRNAs to direct Cas9 to repress transcription, we again used a panel of plasmids containing a construct with the p146 promoter driving the expression of gfp. A sequence was inserted between the transcriptional start site and gfp that contained an FnoCas9 PAM (5ʹ-NGG-3ʹ) immediately downstream of 0, 8, 11, 15, or 34 bases of consecutive complementarity to the first spacer in the FnoCas9 array (spacer 1 in CRISPR1) (, , S2B). The number of complementary bases in these constructs was selected based on what was used previously to measure transcriptional repression by FnoCas9 and scaRNA [Citation29]. The 34 base construct corresponds to the full native spacer sequence. Spacer 1 in CRISPR1, one of the most highly expressed spacers of the cas9 locus, has been shown to restrict transformation at a similar efficiency as gnoCas12a is able to restrict transformation with the protospacer corresponding to CRISPR2 spacer 5 [Citation17]. Therefore, for the purposes of interrogating native capabilities of the CRISPR-Cas9 system and making functional comparisons to Cas12a, the spacers used from each system were selected based on activity level rather than RNA expression level relative to the other spacers in each array.
To confirm that full, but not partial (15 consecutive bases, or 16 bases over a 20 bp stretch adjacent to the PAM), complementarity between the crRNA and DNA target leads to cleavage, we measured the ability of WT and Δcas9 strains to inhibit transformation with each of the plasmids (, , S2B). The plasmid encoding the full protospacer reflects the cleavage activity with the crRNA as it is naturally processed. Plasmids with 0, 8, 11, or 15 consecutive bases of complementarity to the crRNA, directly adjacent to the repeat, were transformed at the same efficiency into WT F. novicida and Δcas9, indicating the lack of plasmid restriction ( and S4A). Conversely, transformation with the plasmid harbouring the full (34 bp) sequence complementary to the FnoCas9 crRNA spacer resulted in significantly reduced levels of transformation in WT compared to a Δcas9 mutant ( and S4A).
Next, we quantified gfp transcript levels from plasmids with 0, 8, 11, or 15 bp consecutive complementarity to the crRNA and adjacent to a PAM, all of which transformed with high efficiency into WT and Δcas9 strains. For plasmids containing 11 and 15 bases of consecutive complementarity to crRNA, gfp transcripts were lower in WT as compared to Δcas9, indicating that FnoCas9 significantly repressed the mRNA (). In contrast, FnoCas9 did not similarly repress gfp from plasmids with either 0 or 8 bp (10 bpover a 20 bp sequence) of complementarity (, S2B). The gfp transcript levels from each plasmid in WT were normalized to the levels in a Δcas9 strain in each condition to remove plasmid specific variability. gfp levels were significantly reduced with 11 bp (13 bp over a 20 bp sequence) and 15 bp (16 bp over a 20bp sequence) of complementarity, while there was not a significant or consistent trend in gfp levels from strains with these plasmids in the absence of cas9 (Figure S5B-C). These data are consistent with the level of complementarity required for FnoCas9-scaRNA-mediated gfp repression from a plasmid (8 bases are insufficient while 11 bases are sufficient) [Citation29]. In addition, this highlights a difference with FnoCas12a, where 11 bases of complementarity was insufficient to repress gfp levels from the plasmid construct (). Future work would be necessary to elucidate the exact number of base pairs that are required for repression. Together, the data indicate that an F. novicida crRNA can direct FnoCas9 to repress transcription from a target plasmid. These results indicate that similar to scaRNA, the ability of FnoCas9 to repress transcription or cleave DNA is determined by the extent of complementarity between the target and the guiding crRNA.
Discussion
In this study we explored whether modulating the complementarity between a crRNA from each of the two native CRISPR-Cas systems of F. novicida and their DNA targets could facilitate transcriptional repression. Both the FnoCas12a and FnoCas9 systems could efficiently repress transcription without cleaving the DNA when the target sequence contained 15 consecutive bases of complementarity to the crRNA with no more than 16 bases of total interaction over a 20 base pair stretch (, , S2B), but cleaved when there was full complementarity of the crRNA spacer to the target (, ). We harnessed this functionality to, for the first time, engineer cleavage-capable Cas12a to repress transcription ().
We observed that when reduced complementarity between a crRNA and DNA target resulted in a loss of cleavage by Cas12a, the effector gained the ability to repress transcription, and this crRNA-directed shift between DNA restriction and transcriptional repression could be extrapolated to a different CRISPR-Cas system, Cas9. These data are supported by a recent study demonstrating the ability of an engineered AsCas12a to exhibit crRNA-directed transcriptional activation in eukaryotic cells [Citation45]. This study also highlighted the importance of ortholog-specific differences in activity and mechanism for these applications, as Lachnospiraceae bacterium ND2006 Cas12a (LbCas12a) was a significantly less efficient transcriptional activator in this system [Citation45]. This demonstrates that the functional diversity amongst orthologous effector proteins, something that has been frequently observed for Cas9 functions, also is observed for Cas12a non-cleavage activities. Furthermore, Cas12a, unlike Cas9, is able to process its own crRNAs without the assistance of cellular RNases. This makes the system highly versatile as it is able to process crRNAs that could guide multiple effector functions [Citation45,Citation47,Citation48]. Our data highlight the unexplored ability of cleavage-capable Cas12a systems, and specifically FnoCas12a, to act as regulators of transcription, a feature that may be important for the natural functions of these systems ().
For prokaryotes, one benefit of modulating base-pair complementarity for repressing transcription with Cas9 or Cas12a is that they are able to retain the protective function of the system in targeting and cleaving invading DNA while gaining a mode of transcriptional control. The alternative, which is use of a full-length crRNA by a catalytically inactive effector, would require the cell to produce a second large protein with reduced functional versatility. In the case of Cas9, its ability to bind DNA with partially complementary crRNAs is supported by in vitro, structural, and heterologous expression studies demonstrating that Cas9 crRNAs with partial target complementarity prevent cleavage of DNA [Citation8,Citation40,Citation41]. Coordinated double-strand cleavage by Cas9 involves a conformational shift into a cleavage-activated state that depends on sufficient base pairing between the crRNA and DNA target [Citation40]. However, binding can still occur when there is insufficient crRNA:target complementarity to direct Cas9 cleavage. The similar ability of both Cas12a and Cas9 to interfere with transcriptional repression and restrict foreign DNA suggests that crRNA-directed functional flexibility could be a feature that is conserved in other systems as well.
There are likely to be other examples of these effectors mediating endogenous repression via reduced crRNA:DNA target complementarity, but we do not think that is the primary function of the crRNAs in F. novicida. Using the parameters of [Citation1] zero mismatches with the protospacer within the first 10 bases of the crRNA directly following the repeat [Citation2], a PAM, and [Citation3] location within 100 bases of the start of a CDS (coding sequence), we did not identify endogenous crRNA targets for CRISPR1 or CRISPR2 in F. novicida. These parameters can be used to predict protospacers, because when scaRNA was searched across the F. novicida U112 genome, the only sequences that met the above criteria were the 1104 and 1101 5ʹ UTRs that are both bound and repressed by scaRNA. Importantly, this does not rule out the existence of other alternative CRISPR-Cas functions of these systems in F. novicida that occur via mechanisms that have yet to be uncovered.
Beyond F. novicida, it is likely that there are unidentified endogenous or exogenous crRNA targets that are repressed by CRISPR-Cas systems using a partial crRNA complementarity mechanism. These repressive functions could arise in a variety of ways, including simply based on probability. We previously demonstrated that just 11 consecutive bases of scaRNA:protospacer complementarity are needed to guide FnoCas9 binding and transcriptional repression, which is similar to what we observe for FnoCas9 with crRNAs herein. While the ability to repress expression involves numerous factors such as proximity to a TSS, promoter strength, and flexibility of PAM preference, based on complementarity alone and assuming a ~ 2 bp PAM, Cas9 would randomly encounter a sequence of DNA that it could bind at a frequency of ~1 in 70Mb, or ~1 in 20 bacterial genomes. Therefore, it is not impossible that Cas9 could bind DNA by random chance in a way that confers a fitness advantage to the bacterium. Another likely pathway for the evolution of self-targeting spacers with partial complementarity and capable of transcriptional repression is the acquisition of a self-targeting spacer or self-targeted sequence into the chromosome. In this scenario, the bacteria that could survive would have mutations in the PAM, the CRISPR system, or the spacer:protospacer interaction. Thus, a lethal or near-lethal self-targeting event could select for crRNAs with endogenous binding capabilities while maintaining the cleavage capability of the system if the mutation that enabled survival was in the spacer:protospacer interaction. These represent two non-exhaustive possibilities for the evolution of self-regulating spacers, and highlight the potential for transcriptional regulation and endogenous DNA binding to be more common functions of CRISPR-Cas systems than has been appreciated.
In other Cas9 and Cas12a systems, we anticipate that there are other mechanisms of non-canonical crRNA-driven activity, some of which have likely already been identified. For example, multiple orthologs of Cas9 cleave ssRNA with a ~ 20 bp crRNA:target RNA interaction. Although the biological importance of this mechanism is not fully understood, it is reasonable to consider that an RNA degradation mechanism could be playing a role in gene regulation in some systems [Citation49–51].
Other CRISPR-Cas systems, including those that use multi-protein effector complexes, may utilize crRNAs for endogenous regulatory functions. A systematic analysis of Type I-E spacer sequences in an E. coli strain found that self-targeting spacers were more likely to target the host than foreign nucleic acids [Citation52]. Most interestingly, the bioinformatic predictions of the crRNA binding sites suggest that they regulate endogenous transcription by binding in a PAM-dependent manner to transcriptionally active regions of the genome [Citation52], which is in appearance similar to what we describe here. Experimentally, work has suggested that extending the crRNA spacer in the type I-E system could enhance gene silencing when targeted upstream of a CDS while retaining DNA degradation, and suggests that PAM-distal mutations to the target disrupt both functions to a similar extent [Citation53].
Furthermore, there are multiple examples of crRNAs influencing bacterial physiology. In the type I-E CRISPR-Cas system of Pseudomonas aeruginosa, a crRNA directs degradation of the mRNA for lasR, a transcriptional regulator which helps the pathogen evade the mammalian immune system [Citation54]. The type I-F CRISPR-Cas system of P. aeruginosa is involved in regulating motility through partial complementarity of a crRNA to a prophage [Citation55].
Interestingly, self-targeting spacers were identified early in the study of CRISPR-Cas systems, and recent analyses suggest they may be found in about 7% of the genomes in the CRISPRminer database [Citation20,Citation38,Citation56]. It is likely some, if not many, of these spacers have activities that benefit the host, such as gene regulation [Citation37,Citation38]. CRISPR-Cas systems are highly diverse which is likely reflected in the existence of multiple mechanisms of crRNA-based gene regulation, and we propose that one of these is through partial crRNA complementarity to target DNA, especially for type II and type V systems as we have demonstrated. It remains unknown how many self-targeting spacers may exist with reduced complementarity to endogenous targets and which could result in transcriptional regulation, or if they would have been identified in previous efforts to predict protospacers. For example, one study searched for targets of 363,460 unique spacers and found that predicted protospacers could only be assigned to 7% of the spacers queried, using a search criteria of 95% coverage and identity between spacer and predicted target [Citation57]. Many of these targets were likely unidentified because their exogenous targets have yet to be sequenced, but it is also possible that some interact with known targets via reduced complementarity and therefore would not have been captured by the search parameters.
Advances in our ability to survey and identify CRISPR arrays and orphan CRISPR elements, as well as in interrogating and identifying PAMs and PAM flexibilities across systems, will continue to enable more robust investigations of self-targeting by CRISPR-Cas systems [Citation58–62]. It will be important to determine the precise mismatch and energetic parameters that distinguish between cleavage and binding for each system within the context of interest, in order to comprehensively search for regulatory targets. The ideal free energy models will also incorporate diverse and non-trivial parameters that preclude consecutive and non-consecutive complementarity between the protospacer and the crRNA. These include promoter strength and proximity to the crRNA binding site, location of the binding interaction, supercoiling of the target, the energetics of heteroduplex formation and PAM recognition, and flexibility or diversity of the PAM in different strains [Citation29,Citation63,Citation64]. Importantly, as we have shown, regulatory functions of CRISPR-Cas systems can co-exist with the canonical adaptive immune functions for multiple CRISPR-Cas systems. We hypothesize that as self-targeting spacers are investigated further on an individual and systematic basis, many more roles for CRISPR-Cas systems in diverse aspects of prokaryotic physiology will be revealed.
Materials and methods
Strains and growth conditions
Francisella novicida strains were grown at 37°C shaking. Tryptic Soy Broth (VWR international) with 0.2% cysteine (BD biosciences) supplementation was used as the growth media for liquid cultures. For colony-based assays strains were grown on Typtic Soy Agar (VWR international) plates with 0.1% cysteine at 37°C. For selection of mutants and maintenance of plasmids, media was supplemented with kanamycin sulphate (30 μg/ml, Fisher Scientific). For removal of the kanamycin resistance cassette, the system utilized tetracycline selection, in which media was supplemented with 15 μg/ml tetracycline (Alfa Aesar) and the strains were grown at 30°C during the unmarking.
Francisella novicida mutant generation
All strains used in experiments were derived from Francisella novicida U112 (‘WT’) (Appendix Table S1). Δcas12a and Δcas9/Δcas12a deletion mutants and the engineered crRNA array were made using allelic exchange with the primers listed in Appendix Table S2. The engineered crRNA was made by replacing the Cas12a CRISPR array with a repeat-spacer-repeat in the same location, with the spacer targeting the 1104 5ʹ UTR. Linear allelic exchange products were used as the substrates for the mutations, the 500–1000 bp regions of homology for recombination into specific sites of the genome flanking the new inserted sequence (where necessary) and a selection cassette. For mutant selection a kanamycin selectable marker was used, containing Flp recombinase target sites (FRT) between the flanking sequences to enable unmarking [Citation65]. Fragments for construction of the allelic exchange products were amplified from genomic DNA (isolated with Qiaquick Tissue Extraction Kit) and sewn together using overlapping PCR as described previously [Citation29]. Phusion high-fidelity DNA polymerase (New England Biolabs) was used for PCR amplification and the Qiaquick gel extraction kit (Qiagen) was used to purify DNA from PCR reactions. Chemically competent F. novicida was transformed with the allelic exchange substrates [Citation65] and recombinants were selected for on kanamycin and confirmed by amplification and sequencing (Genewiz) using ‘ampli’ and ‘seq’ primers (Appendix Table S2) [Citation29]. The selection cassette was removed a vector containing the Flp recombinase in trans, using temperature and tetracycline selection as described previously (Appendix Table S1) [Citation66]. The Δcas9 strain was described in previous work (Appendix Table S1) [Citation28,Citation30,Citation67].
Plasmid construction
Plasmids (Appendix Table S1) were constructed in the broad host range vector pBAV1K-T5-GFP (pBAV) using the primers listed in Appendix Table S2[Citation68]. In the plasmids used in the transformation and Cas9-/Cas12a-dependent transcriptional repression assays, the promoter and RBS that drive gfp expression in WT pBAV were replaced with a synthetic constitutive promoter ‘p146’ followed by varying amounts of homology to the Cas9 or Cas12a crRNAs and respective PAMs, in front of gfp [Citation29,Citation46]. As per the functioning of each system, the Cas9 PAM was placed directly after the target sequence (5ʹ-NGG-3ʹ) while the Cas12a PAM was inserted before the target sequence directly after the TSS (5ʹ-TTTN-3ʹ) (, ). Plasmids were constructed, amplified and isolated from competent E. coli (NEB 5-alpha) as described previously [Citation29].
Transformation assays
Chemically competent F. novicida was made by 10-fold concentration of log-phase cultures (OD600 nm of 0.8–1.0) in 4°C chemical transformation buffer (CTB) as described previously [Citation29,Citation65]. As described in Ratner et al., 2019, competent cells were mixed with DNA and incubated at 37°C shaking (20 min), followed by a 2 hour recovery in 1 ml of liquid growth media at 37°C shaking and plating on selective agar plates overnight (37°C) [Citation29]. For transformations with plasmid vectors, the same amount of plasmid was transformed into WT and either the Δcas9 and Δcas12a strains (~500 ng per plasmid). Transformants were enumerated for each strain and compared between WT and either Δcas9 or Δcas12a strains to determine the % transformation relative to the CRISPR-effector protein-deficient strain [Citation29]. As described in [Citation29], Cas9 and Cas12a repression of gfp transcript levels was measured by isolating transformants from each strain and for each plasmid, for growth in liquid growth media with kanamycin supplementation. At OD600 nm of 0.8–1.0, RNA was isolated from the transformants and used to measure gfp transcript level in each strain (see qRT-PCR methods).
Quantitative real-time PCR
RNA extraction and qRT-PCR were conducted from cultures at OD600 nm of 0.8–1.0 as described in [Citation29]. RNA was extracted using TRI-reagent and a Direct-zol RNA MiniPrep Kit (Zymo Research) followed by Turbo DNaseI (Ambion Biosciences) [Citation29]. Power Sybr Green RNA-to-CT one-step kit (Applied Biosystems) was used for measurement of transcript levels by qRT-PCR (Primers in Appendix Table S2 [Citation29]. To determine 2-ΔΔct, CT values for 1104–1101 were normalized to DNA helicase II (uvrD, FTN_1594) and plasmid gfp transcript levels were normalized to the kanamycin resistance cassette [Citation28,Citation29]. For the plasmid assays, the results are presented as % transcript in WT relative to the uninterrupted mRNA levels in the respective CRISPR-effector deficient strain (Δcas9 or Δcas12a strains).
Statistical analysis
Unpaired, two-tailed, Student’s t-tests were used to determine significance.
Disclosure of interest
The authors have filed a provisional patent application related to this work and declare no other competing interests.
Supplemental Material
Download Zip (427.3 KB)Acknowledgments
We thank the members of the Weiss lab for their feedback and support on this project.
Supplementary material
Supplemental data for this article can be accessed here.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- Barrangou R, Fremaux C, Deveau H, et al. CRISPR provides acquired resistance against viruses in prokaryotes. Science. 2007;315(5819):1709–1712.
- Marraffini LA, Sontheimer EJ. CRISPR interference limits horizontal gene transfer in staphylococci by targeting DNA. Science. 2008;322(5909):1843–1845.
- Koonin EV, Makarova KS, Zhang F Diversity, classification and evolution of CRISPR-Cas systems. Curr Opin Microbiol. 2017;37:67–78.
- Makarova KS, Wolf YI, Iranzo J, et al. Evolutionary classification of CRISPR–Cas systems: a burst of class 2 and derived variants. Nature Rev Microbiol. 2020;18(2):67–83.
- Ratner HK, Weiss DS. Overview of CRISPR-Cas9 Biology. In: Doudna JAMP, editor. CRISPR-Cas: A laboratory manual. Cold Spring Harbor, New York: Cold Spring Harbor Press; 2016. (12):1025–1038.
- Jinek M, Chylinski K, Fonfara I, et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337(6096):816–821.
- Gasiunas G, Barrangou R, Horvath P, et al. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc Natl Acad Sci U S A. 2012;109(39):E2579–86.
- Zetsche B, Gootenberg JS, Abudayyeh OO, et al. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell. 2015;163(3):759–771.
- Shmakov S, Smargon A, Scott D, et al. Diversity and evolution of class 2 CRISPR-Cas systems. Nat Rev Microbiol. 2017;15(3):169–182.
- Yan WX, Hunnewell P, Alfonse LE, et al. Functionally diverse type V CRISPR-Cas systems. Science. 2019;363(6422):88–91.
- Yao R, Liu D, Jia X, et al. Cas9/Cas12a biotechnology and application in bacteria. Synth Syst Biotechnol. 2018;3(3):135–149.
- Wu WY, Lebbink JHG, Kanaar R, van der Oost J. et al., Genome editing by natural and engineered CRISPR-associated nucleases. Nat Chem Biol. 2018;14(7):642–651.
- Swarts DC, Jinek M. Cas9 versus Cas12a/Cpf1: structure-function comparisons and implications for genome editing. Wiley Interdiscip Rev RNA 2018:e1481. 9 5
- Liao C, Slotkowski RA, Achmedov T, et al. The Francisella novicida Cas12a is sensitive to the structure downstream of the terminal repeat in CRISPR arrays. RNA Biol. 2019;16(4):404–412.
- Schunder E, Rydzewski K, Grunow R, et al. First indication for a functional CRISPR/Cas system in Francisella tularensis. Int J Med Microbiol. 2013;303(2):51–60.
- Fonfara I, Le Rhun A, Chylinski K, et al. Phylogeny of Cas9 determines functional exchangeability of dual-RNA and Cas9 among orthologous type II CRISPR-Cas systems. Nucleic Acids Res. 2014;42(4):2577–2590.
- Ratner HK, Weiss DS. Francisella novicida CRISPR-Cas systems can functionally complement each other in DNA defense while providing target flexibility. J Bacteriol. 2020. 202 12 10.1128/JB.00670-19
- Chylinski K, Le Rhun A, Charpentier E. The tracrRNA and Cas9 families of type II CRISPR-Cas immunity systems. RNA Biol. 2013;10(5):726–737.
- Deltcheva E, Chylinski K, Sharma CM, et al. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature. 2011;471(7340):602–607.
- Stern A, Keren L, Wurtzel O, et al. Self-targeting by CRISPR: gene regulation or autoimmunity? Trends Genet. 2010;26(8):335–340.
- Sapranauskas R, Gasiunas G, Fremaux C, et al. The Streptococcus thermophilus CRISPR/Cas system provides immunity in Escherichia coli. Nucleic Acids Res. 2011;39(21):9275–9282.
- Deveau H, Barrangou R, Garneau JE, et al. Phage response to CRISPR-encoded resistance in Streptococcus thermophilus. J Bacteriol. 2008;190(4):1390–1400.
- Mojica FJ, Diez-Villasenor C, Garcia-Martinez J, et al. Short motif sequences determine the targets of the prokaryotic CRISPR defence system. Microbiology. 2009;155(Pt 3):733–740.
- Westra ER, van Erp PB, Kunne T, et al. CRISPR immunity relies on the consecutive binding and degradation of negatively supercoiled invader DNA by Cascade and Cas3. Mol Cell. 2012;46(5):595–605.
- Shah SA, Erdmann S, Mojica FJM, et al. Protospacer recognition motifs: mixed identities and functional diversity. RNA Biol. 2013;10(5):891–899.
- Marraffini LA, Sontheimer EJ. Self versus non-self discrimination during CRISPR RNA-directed immunity. Nature. 2010;463(7280):568–571.
- Vercoe RB, Chang JT, Dy RL, et al. Cytotoxic chromosomal targeting by CRISPR/Cas systems can reshape bacterial genomes and expel or remodel pathogenicity islands. PLoS Genet. 2013;9(4):e1003454.
- Sampson TR, Saroj SD, Llewellyn AC, et al. A CRISPR/Cas system mediates bacterial innate immune evasion and virulence. Nature. 2013;497(7448):254–257.
- Ratner HK, Escalera-Maurer A, Le Rhun A, et al. Catalytically active Cas9 mediates transcriptional interference to facilitate bacterial virulence. Mol Cell. 2019;75(3):498–510.e5.
- Jones CL, Sampson TR, Nakaya HI, et al. Repression of bacterial lipoprotein production by Francisella novicida facilitates evasion of innate immune recognition. Cell Microbiol. 2012;14(10):1531–1543.
- Ma K, Cao Q, Luo S, et al. Cas9 enhances bacterial virulence by repressing the regR transcriptional regulator in streptococcusagalactiae. Infect Immun 2018;86(3). 3
- Louwen R, Horst-Kreft D, de Boer AG, et al. A novel link between Campylobacter jejuni bacteriophage defence, virulence and Guillain-Barre syndrome. Eur J Clin Microbiol Infect Dis. 2013;32(2):207–226.
- Guzina J, Chen WH, Stankovic T, et al. In silico analysis suggests common appearance of scaRNAs in type II systems and their association with bacterial virulence. Front Genet 2018;9:474.
- Ratner HK, Sampson TR, Weiss DS. I can see CRISPR now, even when phage are gone: a view on alternative CRISPR-Cas functions from the prokaryotic envelope. Curr Opin Infect Dis. 2015;28(3):267–274.
- Spencer BL, Deng L, Patras KA, et al. Cas9 contributes to group B streptococcal colonization and disease. Front Microbiol 2019;10:1930.
- Gao NJ, Al-Bassam MM, Poudel S, et al. Functional and proteomic analysis of Streptococcus pyogenes virulence upon loss of its native Cas9 nuclease. Front Microbiol 2019;10(1967).
- Bikard D, Marraffini LA Control of gene expression by CRISPR-Cas systems. F1000prime Reports. 2013;5:47.
- Wimmer F, Beisel CL CRISPR-Cas systems and the paradox of self-targeting spacers. Front Microbiol 2020;10:3078.
- Heussler GE, O’Toole GA. Friendly fire: biological functions and consequences of chromosomal targeting by CRISPR-Cas systems. J Bacteriol. 2016;198(10):1481–1486.
- Sternberg SH, LaFrance B, Kaplan M, et al. Conformational control of DNA target cleavage by CRISPR-Cas9. Nature. 2015;527(7576):110–113.
- Bikard D, Jiang W, Samai P, et al. Programmable repression and activation of bacterial gene expression using an engineered CRISPR-Cas system. Nucleic Acids Res. 2013;41(15):7429–7437.
- Fu Y, Sander JD, Reyon D, et al. Cas nuclease specificity using truncated guide RNAs. Nat Biotechnol. 2014;32(3):279–284.
- Kiani S, Chavez A, Tuttle M, et al. Cas9 gRNA engineering for genome editing, activation and repression. Nat Methods. 2015;12(11):1051–1054.
- Wu X, Scott DA, Kriz AJ, et al. Genome-wide binding of the CRISPR endonuclease Cas9 in mammalian cells. Nat Biotechnol. 2014;32(7):670–676.
- Breinig M, Schweitzer AY, Herianto AM, et al. Multiplexed orthogonal genome editing and transcriptional activation by Cas12a. Nat Methods. 2019;16(1):51–54.
- McWhinnie RL, Nano FE. Synthetic promoters functional in Francisella novicida and Escherichia coli. Appl Environ Microbiol. 2014;80(1):226–234.
- Fonfara I, Richter H, Bratovič M, et al. The CRISPR-associated DNA-cleaving enzyme Cpf1 also processes precursor CRISPR RNA. Nature. 2016;532(7600):517–521.
- Zetsche B, Heidenreich M, Mohanraju P, et al. Multiplex gene editing by CRISPR-Cpf1 using a single crRNA array. Nat Biotechnol. 2017;35(1):31–34.
- Dugar G, Leenay RT, Eisenbart SK, et al. CRISPR RNA-dependent binding and cleavage of endogenous RNAs by the Campylobacter jejuni Cas9. Mol Cell. 2018;69(5):893–905.e7.
- Strutt SC, Torrez RM, Kaya E, et al. RNA-dependent RNA targeting by CRISPR-Cas9. Elife. 2018;7. 10.7554/eLife.32724
- Rousseau BA, Hou Z, Gramelspacher MJ, et al. Programmable RNA cleavage and recognition by a natural CRISPR-Cas9 system from Neisseria meningitidis. Mol Cell. 2018;69(5):906–14.e4.
- Bozic B, Repac J, Djordjevic M. Endogenous gene regulation as a predicted main function of type I-E CRISPR/Cas system in E. Coli. Molecules. 2019;24(4).
- Luo ML, Jackson RN, Denny SR, et al. The CRISPR RNA-guided surveillance complex in Escherichia coli accommodates extended RNA spacers. Nucleic Acids Res. 2016;44(15):7385–7394.
- Li R, Fang L, Tan S, et al. Type I CRISPR-Cas targets endogenous genes and regulates virulence to evade mammalian host immunity. Cell Res. 2016;26(12):1273–1287.
- Heussler GE, Cady KC, Koeppen K, et al. Clustered regularly interspaced short palindromic repeat-dependent, biofilm-specific death of Pseudomonas aeruginosa mediated by increased expression of phage-related genes. MBio. 2015;6(3):e00129–15.
- Zhang F, Zhao S, Ren C, et al. CRISPRminer is a knowledge base for exploring CRISPR-Cas systems in microbe and phage interactions. Commun Biol. 2018;1(1):180.
- Shmakov SA, Sitnik V, Makarova KS, et al. The CRISPR spacer space is dominated by sequences from species-specific mobilomes. mBio. 2017;8(5):e01397–17.
- Almendros C, Guzmán NM, García-Martínez J, et al. Anti-cas spacers in orphan CRISPR4 arrays prevent uptake of active CRISPR–Cas I-F systems. Nat Microbiol. 2016;1(8):16081.
- Milicevic O, Repac J, Bozic B, et al. A simple criterion for inferring CRISPR array direction. Front Microbiol 2019;10(2054).
- Biswas A, Staals RHJ, Morales SE, et al. CRISPRDetect: A flexible algorithm to define CRISPR arrays. BMC Genomics. 2016;17(1):356.
- Couvin D, Bernheim A, Toffano-Nioche C, et al. CRISPRCasFinder, an update of CRISRFinder, includes a portable version, enhanced performance and integrates search for Cas proteins. Nucleic Acids Res. 2018;46(W1):W246–w51.
- Leenay Ryan T, Maksimchuk Kenneth R, Slotkowski Rebecca A, et al. Identifying and visualizing functional PAM diversity across CRISPR-Cas systems. Mol Cell. 2016;62(1):137–147.
- Farasat I, Salis HM. A biophysical Model of CRISPR/Cas9 activity for rational design of genome editing and gene regulation. PLoS Comput Biol. 2016;12(1):e1004724.
- Vigouroux A, Oldewurtel E, Cui L, et al. Tuning dCas9’s ability to block transcription enables robust, noiseless knockdown of bacterial genes. Mol Syst Biol. 2018;14(3):e7899.
- Llewellyn AC, Jones CL, Napier BA, et al. Macrophage replication screen identifies a novel Francisella hydroperoxide resistance protein involved in virulence. PLoS ONE. 2011;6(9):e24201.
- Gallagher LA, McKevitt M, Ramage ER, et al. Genetic dissection of the Francisella novicida restriction barrier. J Bacteriol. 2008;190(23):7830–7837.
- Weiss DS, Brotcke A, Henry T, et al. In vivo negative selection screen identifies genes required for Francisella virulence. Proc Natl Acad Sci U S A. 2007;104(14):6037–6042.
- Bryksin AV, Matsumura I, Mokrousov I. Rational design of a plasmid origin that replicates efficiently in both gram-positive and gram-negative bacteria. PLoS One. 2010;5(10):e13244
#x2005;
Appendix Table 1. Strains and plasmids used in this study plasmids used and the supporting genotype and reference information
Appendix Table 2. RT-PCR and Cloning Primers. Sequences and purpose of primers used in this study