
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
Nucleotide modification in RNA controls a bevy of biological processes, including RNA degradation, gene expression, and gene editing. In turn, misregulation of modified nucleotides is associated with a host of chronic diseases and disorders. However, the molecular mechanisms driving these processes remain poorly understood. To partially address this knowledge gap, we used alchemical and temperature replica exchange molecular dynamics (TREMD) simulations on an RNA duplex and an analogous hairpin to probe the structural effects of modified and/or mutant nucleotides. The simulations successfully predict the modification/mutation-induced relative free energy change for complementary duplex formation, and structural analyses highlight mechanisms driving stability changes. Furthermore, TREMD simulations for a hairpin-forming RNA with and without modification provide reliable estimations of the energy landscape. Illuminating the impact of methylated and/or mutated nucleotides on the structure-function relationship and the folding energy landscape, the simulations provide insights into modification-induced alterations to the folding mechanics of the hairpin. The results here may be biologically significant as hairpins are widespread structure motifs that play critical roles in gene expression and regulation. Specifically, the tetraloop of the probed hairpin is phylogenetically abundant, and the stem mirrors a miRNA seed region whose modification has been implicated in epilepsy pathogenesis.
Introduction
Motivated by recent discoveries regarding the fundamental role of ribonucleic acid (RNA) molecules in myriad essential functions within the cell and equipped with new experimental detection techniques, discovery of unique RNA sequences and nucleotide modifications has impressively accelerated [Citation1]. While the existence of modified nucleotides and the need for nucleotide modification to ensure stability of functional RNA structure has been long known [Citation2,Citation3], the recent improvement of sequencing and detection technologies in the field of epitranscriptomics – natural, post-transcriptional modifications to RNA – has sparked discoveries relating dynamic modification in both coding and non-coding RNAs to diverse modulation of RNA structure, cellular function, and gene expression [Citation1,Citation4,Citation5]. Although understanding the effect of nucleotide modification on RNA structure and function is relevant to advancing treatment of common medical conditions – including cancer, neurological disorders, diabetes, mitochondrial diseases [Citation6], and HIV [Citation7] – experimental determination of RNA 3D structures remains labour-intensive and difficult due to the dynamical nature of RNA molecules, the negatively charged phosphate backbone, and the difficulty of finding crystal contacts [Citation8,Citation9]. Because of new sequencing techniques, the discovery of unique RNA sequences has rapidly outpaced experimental determination of corresponding 3D structures. To bridge the gap between sequencing data and structural information, we need accurate computational structure prediction tools, which are important and necessary for advancing RNA 3D structure determination. Including the impact of nucleotide modification in calculations of RNA structure and stability is crucial to accurately compute and predict the structure, energetics, and kinetics.
Even simple modifications, such as methylation of a nucleotide, can have profound impacts on human health. For example, N6-methyladenosine (m6A) is often found in the 3ʹUTR and is known to regulate RNA localization and splicing [Citation10]. By regulating effects of the methylCpG binding protein 2 (MeCP2), suppression of m6A modification may alleviate disease symptoms in fragile X-linked mental retardation; conversely, excessive suppression of m6A is associated with autism [Citation11,Citation12]. In epilepsy-associated miRNA, m6A modification of the seed region could affect miRNA maturation and binding to mRNA targets, which may impact the severity or presence of epilepsy [Citation13,Citation14]. Endometrial cancer, hepatic cancer, breast cancer, and leukaemia tumorigenicity have been linked to upregulation or suppression of m6A, making m6A and its regulators targets for cancer detection and therapy [Citation15–19]. Similarly, ribothymidine (m5U) is elevated in breast cancer cells, and the tRNA methyltransferase 11 homolog (TRMT11) responsible for methylating guanosine to N2-methylguanosine (m2G) is associated with prostate cancer [Citation20,Citation21]. However, the underlying molecular mechanisms from which these associations arise remain poorly understood.
The growing set of known, natural modifications add to the complexity of accurate structure prediction. While determining empirical nearest neighbour energy parameters for the canonical nucleotides provided secondary structure insights and computational prediction parameters [Citation22], performing this task for all the permutations of the more than 170 known modified nucleotides would be difficult and time-consuming. A limited number of models support the incorporation of energetic parameters for modified nucleotides [Citation23,Citation24], but no current predictive model completely accounts for the impact of these modifications on RNA structure [Citation25]. Although studies have been carried out to computationally model the geometry and base pairing character of the modified RNA nucleotides [Citation26,Citation27] and an all-atom force field has been developed for 107 of the known modifications [Citation28], few predictive parameters that are validated by experimental data have been published. Information about modifications may be implicitly included in some prediction algorithms, where knowledge of structural motifs drives the energy function. However, the implicit inclusion of these modifications hampers the models in predicting the truly unmodified RNA structure because the assumption of modification is intrinsic to the models. To increase accuracy and usefulness of RNA structure prediction, we should explicitly include these naturally occurring modifications in the models.
In light of the rapid development and increasing importance of the field of epitranscriptomics, this work seeks to find relationships between nucleotide modification and functional RNA structure. RNA molecules serve to regulate a variety of cellular functions, and the functionality of an RNA is determined by its structure. While previous computational work has mainly focused on computationally determining thermodynamic parameters for unmodified duplexes with some mismatched base pairs [Citation29,Citation30], some experimental studies have extracted thermodynamic parameters for some interactions involving modified nucleotides [Citation31–35], and recent work used a combined molecular dynamics/quantum mechanics approach to successfully reproduce experimental free energy changes for base pairs with modified nucleotides [Citation36], indicating sufficient experimental progress has been made to validate computational studies of modified RNA thermodynamics. To gain insight into RNA structure and function, many experimental and computational studies focus on hairpins. Hairpin loops are a ubiquitous structural motif in RNA, have been implicated as initiators of RNA folding, and commonly engage in tertiary interactions in protein-RNA interfaces and RNA recognition sites [Citation37–39].
Here, we performed alchemical simulations on an RNA duplex and its modified and/or mutated derivatives to calculate relative free energy changes and directly compare with thermal denaturation experiments [Citation33]. Furthermore, we use structural information from alchemical simulations to support mechanisms driving modification and mutation-dependent stability changes. In the alchemical approach, a system is changed from one state of interest to another using convenient, unphysical (‘alchemical’) intermediates in order to estimate the free energy difference between the states. For instance, a nucleotide base in an RNA can be changed from the wild-type base into a modified or mutated base through a series of steps that cause wild-type base atoms to disappear and modified/mutated base atoms to appear, providing a convenient path between chemically different states. Alchemical simulations are a widely used technique for estimating free energy changes in applications ranging from drug design to structure stability studies [Citation40–43]. The optimal methodology for carrying out these simulations is a topic of vigorous, current research [Citation44,Citation45]. Due to the pervasiveness and importance of hairpins, we used temperature replica exchange molecular dynamics (TREMD) – also called parallel tempering – to explore the effect of methylation and/or mutation on the potential of mean force (PMF) for an analogous RNA hairpin and discern how these modifications affect the propensity for the RNA to form a functional hairpin. While thermodynamic experiments are vital facilitators for the understanding of RNA structure and dynamics, viewing the RNA on an atomistic level using TREMD yields structural information that is absent in thermal denaturation experiments. TREMD is an enhanced sampling method with several advantages [Citation42,Citation46], and several studies on hairpins and tetraloops have used TREMD to explore RNA folding dynamics [Citation47–59]. In comparison to other popular methods for estimating PMFs – such as umbrella sampling, steered molecular dynamics, or adaptively biased molecular dynamics – TREMD is an equilibrium method that does not require a priori knowledge or assumptions about which collective variables best identify the relevant states of the system [Citation46]. After running the TREMD simulations, we examine the PMF projected along any collective variable(s) we choose, and the PMFs are not biased from sampling being driven along any specific coordinate. Some disadvantages of TREMD are that we cannot guarantee complete sampling on a short timescale, and TREMD is more computationally demanding than non-equilibrium or collective variable-driven methods for determining PMFs. However, over long enough timescales with small enough systems, PMFs generated by TREMD are reproducible, and we can learn about the folding of the system without pre-existing coordinate biases. Both the hairpin and duplex contain a complementary 5ʹ-GAC-3ʹ helical region that mirrors the seed region of miRNA-134, and the adenosine in this miRNA is a potential methylation target that has been implied to be foundational to epilepsy pathogenesis [Citation13]. The hairpin also contains the most common tetraloop (UUCG), which forms a nucleation site for RNA folding [Citation60]. The rugged free energy landscape of this hairpin provides a wealth of information on how modifications in the loop or the stem can constrain or broaden the structural possibilities. Comparison of the PMF with thermal denaturation data shows agreement, which validates our approach. Understanding structural and dynamical changes driven by these nucleotide modifications may help discern molecular disease mechanisms and rational design of RNA nanotherapeutics [Citation61,Citation62].
MATERIALS AND METHODS
Selecting and building systems
The complementary 5ʹ-UACGACUG-3ʹ RNA duplex (see ) probed in thermal denaturation experiments was built using the Amber 18 nucleic acid builder module [Citation63,Citation64]. Experimental data are reproduced in Table S1. Single-stranded reference states were formed by deleting the unchanged strand in the duplex. The single-stranded reference state allows us to remove free energy changes that do not directly affect the free energy of duplex annealing. Hybrid structures were manually constructed from the duplex and single-stranded PDB structures. Identified as a potential site for m6A methylation, the seed region of miRNA-134 contains an analogous 5ʹ-GAC-3ʹ sequence, and modification of A to m6A may affect maturation of the miRNA and its ability to bind with proteins or target mRNA [Citation13]. Upregulation of this miRNA has been associated with epilepsy [Citation14].
Figure 1. Unmodified 2D and 3D structures. For the A) duplex and B) hairpin, analogous regions are highlighted in magenta and the experimentally probed base pair is denoted with a blue arrow. Nucleotides are numbered from 5ʹ to 3ʹ ends
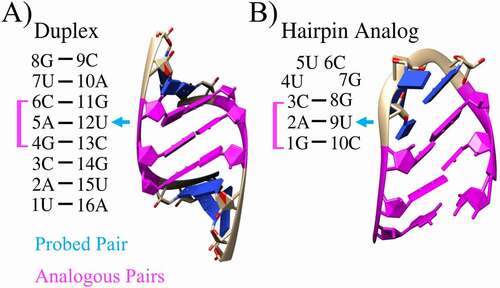
An unmodified, folded, wild-type hairpin (WT) was taken from an enzyme-activating fragment of human telomerase RNA (PDBID: 1Z31). The 10 nucleotide RNA hairpin – a tetraloop with a three base pair stem – has the sequence 5ʹ-gacUUCGguc-3ʹ, so it also contains the complementary 5ʹ-GAC-3ʹ stem probed in the duplex experiments [Citation33] and suspected to play a role in epilepsy pathogenesis via A to m6A modification [Citation13] (see ). Further, this hairpin analogue of the duplex was selected because previous studies have determined that the UUCG tetraloop with a CG closing base pair exhibits two-state behaviour with high stability, is phylogenetically abundant, and has been implicated as a nucleation site during RNA folding [Citation60].
Mutations on the hairpin and duplex were performed using UCSF Chimera [Citation65]. In accordance with modrna08 [Citation28] standards, methylation was accomplished by renaming the modified residue in the PDB file, and the geometrically correct, modified residue was subsequently included in the parameter/topology and input coordinate files built by the tleap module in Amber 18 [Citation63]. Starting from a folded 3D structure, all simulations were conducted with the recommended OL3 force field for RNA molecules and the modrna08 [Citation28] force field for modified nucleotides.
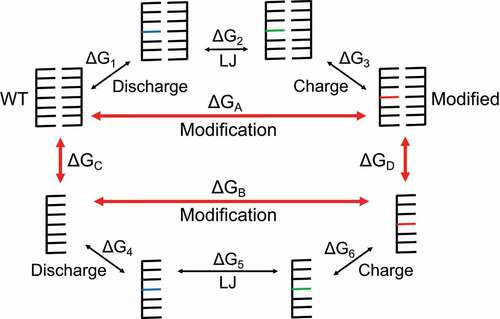
Alchemical and TREMD simulations were performed on the duplexes and hairpins using standard methods described in detail in SM Sections 2 and 3, respectively. For alchemical production runs, 11 2.4 ns simulations – corresponding to alchemical control parameter values (λ = 0.0, 0.1, …, 1.0) – were used for each of the three legs (Discharge, Lennard-Jones, Charge) in the thermodynamic cycle (see ) for both the duplexes of interest and the single-stranded reference states. Doubling the simulation time did not improve the agreement with experiment or reduce the calculated uncertainty (see Table S2), which aligns with previous findings, where longer simulations do not necessarily improve results due to bias from the initial velocities [Citation66]. For hairpin TREMD production runs, 24 replicas were simulated, each for 300 ns, with temperatures ranging from 265 to 365 K, as suggested by the predicted melting curve given by VfoldThermal [Citation67] (see Fig. S1).
Figure 2. A thermodynamic cycle formed by the states indicated by red arrows. In order to separate the electrostatic and Lennard-Jones (LJ) contributions to the free energy change, we use three main steps to go from the WT to the modified states. The first step, labelled ‘Discharge’, removes all charge from the atoms in the starting residue. The blue residues are electrically neutral WT residues. The second step, labelled ‘LJ’, replaces atoms and bonds in the WT residue with atoms and bonds from the modified residue. Because the vanishing and appearing atoms are all electrically neutral in this step, the LJ potential is the contributor to the free energy change. The green residues are electrically neutral, modified residues. The third step, labelled ‘Charge’, reintroduces charge to the modified residue to bring us to the final, modified state. The red residues represent the electrically charged, modified residues. These steps must be done for both the duplex and reference (single-stranded) states in order to calculate the relative free energy change due to modification ∆∆G
Using alchemical simulation results to estimate free energy changes
To quantify the effect of nucleotide modification on RNA energetics, we performed alchemical simulations on the RNA duplex found in experiments and its modified/mutated derivatives. In thermodynamic integration (TI) calculations, the system transitions from one end state of interest to another through unphysical (alchemical) states according to a mixed potential U(λ) in order to numerically estimate free energy change from the transformation between states, where the alchemical control parameter λ allows us to move from the starting state to the end state using a mixed potential [Citation68]. This change from one state to another is an alchemical perturbation that is accomplished via alchemical simulations of the system. In order to form a thermodynamic cycle, we use the single-stranded state as a reference state (see SM Section 4 for more details). The relative free energy due to modification ∆∆G is determined using
Practically, we break the cycle into smaller steps, where for example,
as described in . To analyse the data and validate the completeness of our sampling, the first-order polynomial (trapezoidal) integration scheme for TI (TI-Trapezoid), the cubic spline integration scheme for TI (TI-Cubic), the Bennett acceptance ratio (BAR), and MBAR energy estimators were used (see SM Section 7 for discussion and Fig. S2 for comparisons). While many estimators are available, TI and MBAR provide the most accurate results with the smallest amount of data within their distinct realms, and inconsistency between the two can indicate poor phase-space overlap or insufficient sampling [Citation69]. Estimator uncertainties were propagated by summing uncertainties of each leg of the thermodynamic cycle in quadrature (see SM Section 8). Results provided here are based on data from eight repeat runs because standard deviations provide more accurate assessments of uncertainty than the estimators and because averaged results remove bias due to initial conditions. Using alchemical analysis software [Citation70], uncorrelated, independent samples were automatically extracted from the simulation data.
Analysing TREMD trajectories
Structure clustering was performed on the basis of orthogonal reaction coordinates determined from principal component analysis, and representative frames corresponding to the centroid of each cluster were extracted for structural analysis (see SM Section 3). Additionally, the RMSD from a common, unmodified reference structure and the end-to-end distance (E2E) were determined using CPPTRAJ [Citation71] for each snapshot in each run. A Python implementation of the multistate Bennett acceptance ratio (MBAR) [Citation72] estimator was used to determine the potential of mean force (PMF) for a simulation window. The PMF projects a convoluted, multivariate free-energy landscape onto well-defined collective variables (reaction coordinates that ideally describe the dynamics of interesting aspects of the system), supplying quantitative and qualitative information to build our knowledge of the kinetics and energetic preferences of the system. Although full convergence of this system would probably require runs on the order of several μs, calculating the PMFs with increasing simulation time confirmed reasonable convergence (see Fig. S3).
For the purposes of comparing our simulated results to experimental denaturation data, we estimate the free energy of hairpin formation due to modification from the calculated PMFs (see for hairpin structure). Clustering the structures and identifying the representative frames – the frame closest to the centroid of the cluster – and their reaction coordinates allows us to verify the location of the native hairpin (NH) conformation and view long-lived alternative conformations in the energy landscape. See SM Section 10 for details.
RESULTS AND DISCUSSION
To connect thermal denaturation experiments to theory, we performed alchemical simulations on the experimentally probed, WT duplex and its modified and/or mutated variants, which enabled calculation of relative free energy changes due to denaturation. After establishing the trend between free energies from simulation data and experiment on the duplex, we used TREMD simulations of a hairpin analogue with a phylogenetically abundant tetraloop to show that the trend holds. In addition to the folding stability from the duplex simulations, the general agreement between the hairpin and duplex calculations with thermal denaturation experiments show the validity of the simulation data, so we evaluated how the hairpin folding mechanism change when perturbed by small modifications. The results of this analysis may be beneficial for understanding mechanisms driving modification-related disease and for guiding RNA nanotherapeutic design of hairpin motifs.
Comparing duplex simulation results to thermal denaturation data
Results from the alchemical simulations show that we can accurately rank the free energy change of duplex formation due to modification or mutation ∆∆G using alchemical analysis. Because we used independent simulations to modify and mutate the duplex, more errors were propagated in cases where multiple perturbations were applied. To alleviate this issue, we performed calculations for twice-perturbed systems in both alchemical directions (see Fig. S2 and Section 8 in SM). The modify-then-mutate direction first imposed the modification of the 5A/12 U (A-U) base pair to m6A-U; subsequently, the mutation of 12 U to C, G, or A was performed. In the mutate-then-modify direction, the mutation occurs first, followed by the modification. The success of these twice-perturbed results depends on the accuracy of the calculations from the singly perturbed steps (m6A-U, A-C, A-G, and A-A). Combined results from modify-then-mutate and mutate-then-modify are provided in , where the averaged uncertainties for twice-perturbed systems are on par with uncertainties for singly perturbed systems (see Table S2 and SM Section 8 for details). Provided that the paths of perturbation are exhaustively sampled, our analysis indicates that combined results from twice-perturbed systems yield uncertainties that are roughly equivalent to singly perturbed uncertainties. Given the small amount of data (<400 uncorrelated data points for each λ value), the calculated free energies from alchemical simulations were broadly successful in ranking the experimentally determined ∆∆G values, achieving a 0.93 Spearman rank correlation coefficient between our predicted ∆∆G and the experimental ranking, which shows that the ranking of the calculated free energies trends with experiment. For the unmodified mutant systems, the relative free energies estimated from the alchemical simulations appear to better match the thermal denaturation data than estimates calculated by applying the Turner rules, using the tabulated nearest neighbour Watson-Crick helices and 1 × 1 internal loop parameters [Citation73]. Some of the differences between the experimental results and Turner parameter estimates or the simulation estimates could be due to a difference between the solution conditions of the experiment. The simulations do not perfectly match the experimental solution conditions of 100 mM NaCl, 10 mM MgCl2, 10 mM Na-PIPES (pH 7.0), 5 M duplex, and the Turner parameters assume a 1.0 M NaCl solution.
Figure 3. Calculated relative free energy changes using TI-Trapezoid. Experimental [Citation33] (calculated) results and error bars are shown in black (red). The results for twice-perturbed cases are the average of the results from the modify-then-mutate and the mutate-then-modify alchemical paths. Estimates from applying the Turner rules are shown in blue [Citation73]
![Figure 3. Calculated relative free energy changes using TI-Trapezoid. Experimental [Citation33] (calculated) results and error bars are shown in black (red). The results for twice-perturbed cases are the average of the results from the modify-then-mutate and the mutate-then-modify alchemical paths. Estimates from applying the Turner rules are shown in blue [Citation73]](/cms/asset/67177dc3-0698-4a79-8d11-66b9526f68a1/krnb_a_1882179_f0003_oc.jpg)
The overlap matrices produced by alchemical analysis software [Citation70] indicate that sufficient phase-space overlap was accomplished by our λ schedule (Fig. S4). Furthermore, convergence plots indicate that we obtained reasonable convergence because time-dependent estimates of the free energy in the forward and reverse sampling directions sufficiently overlap (Fig. S5). The mutate-then-modify version of the twice-perturbed alchemical simulation produces consistently lower values of ∆∆G (Fig. S2), indicating that the agreement in magnitude with experimental values is better when the smaller perturbation is applied first, but the ranking is in slightly better agreement with experimental results when both directions are used. The improvement in ranking from using both directions is probably due to using twice as many simulations to obtain the averaged results, while the improved agreement in magnitude with experiment from using only the modify-then-mutate direction is probably due to applying the weaker perturbation first, which prevents the system from drifting too far away from the state of interest during the mutation step of the simulation. Mutating the system first causes the stronger perturbation to be applied for longer, which causes more drift away from the standard duplex structure of interest.
Determining mutation/modification-induced structural mechanisms driving duplex stability changes
Primarily a tool for free energy estimation, alchemical simulations are not optimized to identify dynamics of structure change because most of the structures are generated under an alchemical potential that is not reflective of reality. However, the structures produced under pure potentials (λ = 0 or 1) may be evaluated to check whether large, invalidating duplex deformations occurred during the simulations and to identify new interactions and preferred conformations. We found that there was not a lot of bulging or topological deformation from the mutations and modifications, even for mutations that result in purine-purine base pairs. For instance, the average heavy atom backbone RMSDs for the probed base pair from a reference structure (see SM Subsection 2.2 for more details) were 1.3 and 1.6 Å for WT conformations and mutated and/or modified conformations, respectively. The average distance between phosphate groups of the two residues remained relatively consistent across all cases (20.0 ± 0.8 Å), indicating that the small average RMSD change is likely due to small structure changes to accommodate steric strain rather than large bulging of the duplex topology at the probed site (see SM Subsection 2.2 for more details).
Specifically, the mutation of 12 U to 12A to form the A-A base pair did not result in a large physical bulge in the duplex. Instead, the 12A residue rotated to expose its sugar edge to the Watson-Crick edge of 5A. This behaviour also occurred in the modify-then-mutate 5m6A-12A structure. Because no more hydrogen bonds are disallowed in the 5m6A-12A structure than in the A-A mutated structure, the enhanced stability of this duplex in the alchemical free energy calculation relative to the 5A-12A mutant duplex is likely from the enhanced stacking strength of 5m6A with the 6 C residue. This mechanism may also explain the increased 5m6A-12 C stability in comparison to the 5A-12 C mutation. Structural details and further topology analysis can be found in Fig. S6. The small structure changes shown in the simulations did not appear to invalidate the results because we did not see any major deformations or structures that greatly deviated from the A-form helix structure, and the general trend of the results with experimental data further supports the validity of the simulations.
To determine the effect of base pairing on the conformation of the m6A residue, we examined the modified structures in both paired (duplex) and unpaired (single-stranded) states. In the single-stranded state, the methyl group prefers to point towards solution but is free to occasionally rotate into the sterically strained position. In the base-pairing duplex state, the methyl group prefers to be in a sterically strained position to facilitate the hydrogen bonding between the methylamino group and 12 U (Fig. S7). This supports the ‘spring-loaded’ mechanism proposed in previous studies [Citation33] to explain the instability caused by the methylation. The methylation makes natural formation of a base pair less probable. Similarly, once the pair is broken, the methyl group may act to push the pairing partner away and prevent reformation of the hydrogen bond.
Comparing hairpin PMFs to thermal denaturation data
To test the robustness of our hairpin TREMD simulation results, we compared the calculated ∆∆G from the 2D RMSD/E2E PMFs to thermal denaturation data [Citation33]. While the experimental system consists of RNA duplexes containing eight base pairs (5ʹ-UACGACUG- 3ʹ), the stem of our simulated hairpin (5ʹ-gacUUCGguc-3ʹ) contains the complementary 5ʹ-GAC-3ʹ sequence probed by these experiments. The hairpin also contains a tetraloop that the experimental duplexes clearly lack, and the simulations were performed with 1 M Na+ counterions and neutralizing amounts of Cl− ions, while the duplex experiments were performed with 100 mM NaCl and 10 mM MgCl2. Additionally, because the hairpin rapidly unfolds when noncanonical base pairs are introduced, there are few conformations populating the folded region of mutant PMFs after a notable amount of time. To accommodate the dearth of NH conformations at longer simulation times for noncanonical cases, we used the 5 to 300 ns window, where the NH state is still present on the PMF for all of the cases. Despite the stark differences in the systems and although the systems are not fully equilibrated at 5 ns, we can make a connection from our simulated hairpin to these experimental duplexes through the relative change in free energy of formation due to modification ∆∆G.
While the calculated ∆∆G consistently underestimates the experimental value for noncanonical base pairs, the computed free energy for m6A-U is within experimental error. This indicates that large destabilizations due to noncanonical base-pairing interactions have a weightier impact on our short-stemmed hairpin than on the corresponding experimental duplex, where the five extra base pairs help stabilize the folded helix conformation (see ). Beyond a destabilizing threshold, the NH conformation rapidly disappears from the PMF, and ∆∆G plateaus at a maximum, above which we cannot accurately determine the destabilizing effect due to modification from this probe: the three base pair stem of the hairpin becomes totally disrupted and cannot differentiate between a 2.7 kcal/mol disruption and a 5 kcal/mol disruption. However, when methylations are applied to the canonical hairpin, ∆∆G remains below the threshold set by the noncanonical cases. Interestingly, this threshold at 2.7 kcal/mol is an estimate of the absolute stability of the WT NH state because it is the maximum destabilization the hairpin can withstand: i.e., the WT NH absolute free energy of formation estimated by this hairpin study is ∆G300K ≈ −2.7 kcal/mol (see grey line in ). This prediction is in agreement with experimental thermal denaturation studies of the UUCG tetraloop, where results from several hairpins – 5ʹ-gcgUUCGcgc-3ʹ (∆G310K = −2.2 kcal/mol), 5ʹ-cgcUUCGgcg-3ʹ (∆G310K = −3.65 ± 0.41 kcal/mol), 5ʹ-ggagUUCGcucc-3ʹ (∆G310K = −2.9 ± 0.14 kcal/mol), and 5ʹ-ggacUUCGgucc-3ʹ (∆G310K = −5.07 ± 0.22 kcal/mol) – showed the enhanced stability provided by the CG closing base pair when adjacent to UUCG tetraloops [Citation60].
Figure 4. Experimental [Citation33] and calculated relative free energy changes due to modification ∆∆G. For the eight-base pair duplex, experimentally determined free energies calculated from two-state curve fits, van’t Hoff linear plots, and averages between the two-state fit and van’t Hoff fit are labelled ‘2-state fit’, ‘van’t Hoff’, and ‘Exp. average’, respectively. The values for ∆∆G calculated from the RMSD/E2E 2D PMFs of the hairpin analogue are labelled ‘Calc. from PMF’. Descriptive identifiers are provided to describe modifications and/or mutations (see ): for example, ‘m6A-U’ represents the modification of 2A (5A) to m6A in the simulated hairpin (experimental duplex), maintaining the canonical base pair with 9 U (12 U). Likewise, ‘A-C’ represents the hairpin (duplex) mutation of 9 U (12 U) to C. All hairpin (duplex) mutations were imposed on 9 U (12 U). The grey line indicates the free energy plateau, where the hairpin becomes completely destabilized. Experimental error bars are shown with respect to the experimental average. Estimates from applying the Turner rules are shown in blue
![Figure 4. Experimental [Citation33] and calculated relative free energy changes due to modification ∆∆G. For the eight-base pair duplex, experimentally determined free energies calculated from two-state curve fits, van’t Hoff linear plots, and averages between the two-state fit and van’t Hoff fit are labelled ‘2-state fit’, ‘van’t Hoff’, and ‘Exp. average’, respectively. The values for ∆∆G calculated from the RMSD/E2E 2D PMFs of the hairpin analogue are labelled ‘Calc. from PMF’. Descriptive identifiers are provided to describe modifications and/or mutations (see Fig. 1): for example, ‘m6A-U’ represents the modification of 2A (5A) to m6A in the simulated hairpin (experimental duplex), maintaining the canonical base pair with 9 U (12 U). Likewise, ‘A-C’ represents the hairpin (duplex) mutation of 9 U (12 U) to C. All hairpin (duplex) mutations were imposed on 9 U (12 U). The grey line indicates the free energy plateau, where the hairpin becomes completely destabilized. Experimental error bars are shown with respect to the experimental average. Estimates from applying the Turner rules are shown in blue](/cms/asset/53a7215f-a896-4ac0-9a9d-610375ebb6ee/krnb_a_1882179_f0004_oc.jpg)
While the trend calculated from the hairpin PMF somewhat follows the trend from the experimental duplex, the hairpin calculation reaches a plateau and fails to accurately assess free energy changes due to large disruptions. Experimentally, the five extra base pairs in the duplex lock the duplex in place, which facilitates better assessment of larger disruptive energies. In contrast, the three base pair stem of the hairpin becomes totally disrupted when mutations destabilize the hairpin beyond the free energy of formation. However, small perturbations due to methylation are below the threshold, and in the context of these small perturbations to the energy landscape, the analogy between the hairpin and duplex holds.
Investigating modification-induced hairpin structural alterations
The results from methylations with the otherwise canonically base pairing hairpin were extracted from 50 to 300 ns simulation windows for comparison to the unmodified WT. The results provided here are extracted from the 2D PMFs using RMSD and E2E as reaction coordinates (RMSD/E2E PMFs) because the physical meanings of these reaction coordinates are clear. In , the PMF for the WT, the 2m6A modification with thermal denaturation data and heavily implicated in disease, a destabilizing 5m5U modification, and a stabilizing 7m2G modification represent a summary of our findings.
Figure 5. 2D PMFs and representative frames from hairpin TREMD simulations. The representative frames and PMFs for the A) WT B) modification of 2A to m6A (2m6A), C) modification of 5 U to m5U (5m5U), D) modification of 7 G to m2G (7m2G) are shown along the end-to-end distance (E2E) and RMSD reaction coordinates. Modified carbons and ribbons in the representative frames are highlighted in green, and the figure legends colour code the energy in kcal/mol
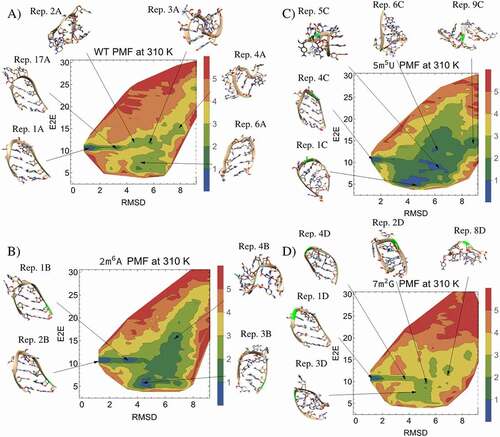
The unmodified WT RMSD/E2E PMF shows a clear preference for a folded, NH conformation with a low RMSD and an E2E distance of about 10.7 Å (see ). The apparent path of least action proceeds from the folded state through a metastable state labelled by the representative frame from cluster 17 (Rep. 17A), which is of interest because of its deepening or disappearance in the PMFs of the methylated derivatives. The representative structure from this metastable cluster indicates that unfolding of the hairpin tends to proceed from the loop, and evaluation of the representative frame indicates that disruption of the tetraloop structure is caused by disruption of the noncanonical, UG trans sugar-edge/Watson-Crick base pair between 4 U and 7 G and formation of single-stranded stacking interactions within the loop. After reaching the metastable cluster, the path of least action appears to proceed through clusters 2 and 3, respectively labelled as Rep. 2A and Rep. 3A, which are characterized by single-stranded stacking interactions. After reaching cluster 3, the path of least action diverges, leading either to a set of collapsed conformations, characterized by a low E2E distance due to terminal nucleotide stacking (Rep. 6A), or extended states, characterized by a larger E2E distance (Rep. 4A). The greatest energy barrier between the NH and random coil (RC) states is about 2.8 kcal/mol and occurs after the metastable state is reached. Swapping 9 U to form noncanonical AG, AC, or AA base pairs resulted in total disruption of the hairpin and disappearance of the NH from the energy landscape by 50 ns.
In comparison to the unmodified WT, N6-methyladenosine (m6A) methylation of nucleotide 2A (2m6A) stabilizes the metastable intermediate (Rep. 1B) between the hairpin and the RC conformations (see ). The energy barrier separating NH and RC states is lowered to about 1.7 kcal/mol, facilitating the transition. Furthermore, the 2m6A RNA PMF shows that this methylation stabilizes stacking in a collapsed conformation, where the terminal nucleotides and the methylated nucleotide together form a three-nucleotide stack (Rep. 3B). The destabilization of the folded hairpin resulting from this methylation can be visualized in the PMF, where more low energy paths can be drawn from the folded state to a high E2E distance. While methylation at this position undoubtedly destabilizes the hairpin base pair, the representative frame from cluster 2 (Rep. 2B) shows the NH conformation is a long-lived conformation. The overall destabilization is probably because this methylation stabilizes a wider range of extended and collapsed conformations through stacking, which can compete with the NH. In agreement with a previous study [Citation33], we found that to promote base-pairing in a duplex, the methyl group must rotate into a higher energy anti conformation, whereas in an unpaired context, this group prefers the lower energy syn conformation. After calculating the average dipole moment for the simulated WT 2A base atoms (|dipole| 2.21 ± 0.27 D) and m6A in both a pairing and single-stranded contexts (|dipole|
2.33 ± 0.17 D), it appears that this methylation enhances the magnitude of the dipole moment by
0.1 D and also increases the stability of the dipole moment as measured by the standard deviation of the dipole moment by
0.1 D, causing the dipole moment to remain closer to the base plane with fewer fluctuations, regardless of whether the methyl group is in the syn or anti conformation. Because most of the base atoms are constrained by the aromaticity of the purine rings, the WT 2A dipole moment fluctuations are largely driven by the orientation of the N6H2 hydrogen atoms, which can freely rotate around the nitrogen. When the amino group is methylated, the base dipole moment cannot fluctuate as freely because the methyl group is more sterically constrained, and the methyl-enhanced stability and strength of the dipole moment may contribute to the base stacking stability. The duplex destabilization and single-stranded stacking stabilization caused by m6A modification give rise to a multitude of regulatory capabilities for this modification, including mRNA stability, splicing, and translation [Citation17]. Excessive suppression and upregulation of m6A are both associated with disease [Citation11–13,Citation15–19]. This modification is the most common mRNA modification, and the relatively modest effect of m6A on the energy landscape shown here can have profound biological effects by delicately controlling pri-miRNA duplex stability and efficacy of dicer enzyme cleavage. Further, modification of mature miRNA can likely regulate the balance of available mRNA for translation by adjusting miRNA-mRNA duplex stability.
When 5 U is methylated to ribothymidine (5m5U), the hairpin structure is greatly destabilized by enhanced stability of stacking interactions with other nucleotides (see ). In the WT NH (Rep. 1A), 5 U engages in no obvious interactions with other nucleotides and is pointed towards solution on the minor groove side of the helix with the fifth carbon of the WT NH 5 U located near the phosphate backbone in the NH structure. Methylating this ring carbon pushes the nucleotide away from the backbone and forces the hairpin into a higher energy conformation, which promotes sampling of alternative structures. The greatest energy barrier between NH and RC states occurs between the NH and the metastable state and is lowered to about 1.5 kcal/mol. The minor groove has a dearth of stacking opportunities, but once m5U finds its way to the major groove, it stabilizes coil structures through stacking interactions, which naturally disrupts the NH conformation (Rep. 4 C) since the location of 5 U on the minor groove side is required to form the NH structure. Additionally, this methylation stabilizes the metastable state (Rep. 17A in shows that 5 U engages in single-stranded stacking with 6 C), which facilitates the transformation from folded hairpin to the RC conformation. The promotion of stacking interactions with other nucleotides by 5m5U that explains the tendency for this hairpin to misfold into collapsed coil conformations can be seen in Reps. 1 C, 5 C, 6 C, and 9 C in . These results indicate that methylation of 5 U in the common, stable UUCG tetraloop weakens the stability of the tetraloop and may lead to disruption of the associated stem, which could be used as a tool to facilitate RNA nanotechnology by allowing the designer to avoid formation of particular tetraloops and off-target structures. Supporting the assessment that this modification tends to destabilize the folded structure, we estimate that the relative free energy change due to 5m5U modification is ∆∆G = 0.7 kcal/mol.
Unlike most other methylations, N2-methylguanosine (m2G) modification of 7 G (7m2G) markedly stabilizes Rep. 1D, the NH conformation (see ). The energy barrier separating the NH and RC states is about 2.8 kcal/mol, similar to the WT PMF. Mechanistically, this methylation stabilizes the stacking interaction between 7 G and 8 G without greatly inhibiting base pair formation with 4 U, which partially explains the resulting PMF. In addition, many conformations that stabilize RC conformations of the WT, including the metastable conformation found in cluster 17 of (Rep. 17A), use the N2 functional group of 7 G to occupy a pocket formed by 6 C and the backbone. This methylation reduces the ability of the molecule to form the pocket in the metastable state (Rep. 4D), which results in a relative energetic destabilization of RC conformations (Reps. 2D, 3D, and 8D). In short, this methylation stabilizes the tetraloop structure without causing instabilities in the rest of the hairpin and destabilizes the metastable state and RC states, which causes it to prefer the NH structure. These results indicate that methylation of the loop-closing 7 G to m2G in this common tetraloop further stabilizes the loop and stem of the hairpin, which could also be used in RNA design. We estimate that the relative free energy change due to 7m2G modification is ∆∆G = −0.75 kcal/mol.
While the modifications discussed above provide a nice summary of our results, we also analysed other methylations in varying sequence contexts. These modifications show a broad spectrum of stabilizing and destabilizing effects with ∆∆G ranging from −0.75 to 0.72 kcal/mol. Each modification has a unique impact on the ruggedness of the PMF and on the stability of the structures in the ensemble. Most modifications have a notable impact on the energy barrier separating the NH and RC states, providing some potential insights into the kinetic changes driven by these modifications. Discussions and PMFs for other modifications can be found in Figs. S8-S16.
Conclusion
Due to the rapid development of our knowledge about the impact of RNA nucleotide modification on human health, studies of the underlying mechanics that lead to the emergence of diseases or disorders relating to nucleotide modification are increasingly in demand. In this study, we use computational modelling of structures with modified nucleotides to reproduce experimental evidence regarding the relative free energy changes that occur upon modification. First, we compared results from alchemical simulations of a duplex to thermal denaturation data, finding good agreement. Next, we established that no major deformations occur to the duplex topology after the perturbations, and we find small structural changes that point towards mechanisms that drive the free energy calculations. Then, we evaluated the effect of modification on the PMF of an analogous hairpin with a phylogenetically abundant UUCG tetraloop. Beyond a destabilizing limit, we show that the relative free energy changes extracted from hairpin PMFs reach a plateau that can be identified as an estimate of the absolute free energy of the hairpin. Furthermore, we elucidate the underlying modification-dependent mechanisms that cause alterations to the PMF for a common RNA hairpin motif. Information from this study may be useful in therapeutic design of RNA structures. The method may be used to aid in design of local features in RNA structures for RNA nanotechnology [Citation61,Citation62]. Furthermore, the results presented here may be useful in understanding duplex or tetraloop stabilization changes that occur due to RNA nucleotide modifications, which when imbalanced may give rise to neurological disease, metastatic cancer, and other chronic conditions [Citation6,Citation7,Citation11–21].
Both alchemical and TREMD simulation results indicate that we can accurately reproduce the experimental results for m6A modification, which implies that the force field parameterization for this modification is sufficient to obtain structural insights allowing for accurate study of the role of this modification in the mechanisms of epilepsy pathogenesis in silico. Here, we found that this modification destabilizes the duplex structure and stabilizes collapsed, RC conformations in a hairpin. The result here may explain the experimental finding that modification of A to m6A in the seed region of miRNA-134 and miRNA-146 would likely affect the maturation and targeting ability of the miRNA structures. Similarly, we found that other simple modifications, such as m2G and m5U, can strikingly stabilize or destabilize the hairpin structure in specific contexts, which may provide a tool for RNA nanotherapeutic design. Taking all of these context-dependent effects of modification together, this work further demonstrates the need for more experimental data of modified nucleotides in varying sequence contexts.
Supplemental Material
Download PDF (8.6 MB)Acknowledgments
This work was supported by the National Institutes of Health under Grants R01-GM117059 and R35-GM134919 to S.-J.C.; and the National Science Foundation Graduate Research Fellowship Program under Grant 1443129 to T.H.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
References
- Harcourt EM, Kietrys AM, Kool ET. Chemical and structural effects of base modifications in messenger RNA. Nature. 2017;541:339–346.
- Carell T, Brandmayr C, Hienzsch A, et al. Structure and Function of Noncanonical Nucleobases. Chem Int Ed. 2012;51:7110–7131.
- Helm M, Brule H, Degoul F, et al. The presence of modified nucleotides is required for cloverleaf folding of a human mitochondrial tRNA. Nucleic Acids Res. 1998;26:1636–1643.
- Helm M, Motorin Y. Detecting RNA modifications in the epitranscriptome: predict and validate. Nat Rev Genet. 2017;18:275–291.
- Agris PF. The importance of being modified: an unrealized code to RNA structure and function. RNA. 2015;21:552–554.
- Torres AG, Batlle E, Ribas de Pouplana L. Role of tRNA modifications in human diseases. Trends Mol Med. 2014;20:306–314.
- Lichinchi G, Gao S, Saletore Y, et al. Dynamics of the human and viral m6A RNA methylomes during HIV-1 infection of T cells. Nat Microbiol. 2016;1:16011.
- Gomez A, Toor N. Selecting New RNA Crystal Contacts. Structure. 2018;26(9):1166–1167.
- Vare VY, Eruysal ER, Narendran A, et al. Chemical and conformational diversity of modified nucleosides affects tRNA structure and function. Biomolecules. 2017;7:29.
- Zhao BS, Roundtree IA, He C. Post-transcriptional gene regulation by mRNA modifications. Nat Rev Mol Cell Biol. 2016;18:31–42.
- Cheng TL, Wang Z, Liao Q, et al. MeCP2 suppresses nuclear microRNA processing and dendritic growth by regulating the DGCR8/Drosha complex. Dev Cell. 2014;28:547–560.
- Edupuganti RR, Geiger S, Lindeboom RGH, et al. N6-methyladenosine (m6A) recruits and repels proteins to regulate mRNA homeostasis. Nat Struct Mol Biol. 2017;24:870–878.
- Rowles J, Wong M, Powers R, et al. FTO, RNA epigenetics and epilepsy. Epigenetics. 2012;7(10):1094–1097.
- Jimenez-Mateos EM, Engel T, Merino-Serrais P, et al. Silencing microRNA-134 produces neuroprotective and prolonged seizure-suppressive effects. Nat Med. 2012;18:1087–1094.
- Liu J, Eckert MA, Harada BT, et al. m6A mRNA methylation regulates AKT activity to promote the proliferation and tumorigenicity of endometrial cancer. Nat Cell Bio. 2018;20:1074–1083.
- Ma J-Z, Yang F, Zhou -C-C, et al. METTL14 suppresses the metastatic potential of hepatocellular carcinoma by modulating N6-methyladenosine-dependent primary MicroRNA processing. Hepatology. 2017;65(2):529–543.
- Chen M, Wei L, Law CT, et al. RNA N6-methyladenosine methyltransferase-like 3 promotes liver cancer progression through YTHDF2-dependent posttranscriptional silencing of SOCS2. Hepatology. 2018;67:2254–2270.
- Wu L, Wu D, Ning J, et al. High Oct4 expression: implications in the pathogenesis of neuroblastic tumours. BMC Cancer. 2019;19:1–12.
- Ianniello Z, Paiardini A, Fatica A. N6-methyladenosine (m6A): a promising new molecular target in acute myeloid Leukemia. Front Oncol. 2019;9:251.
- Willmann L, Erbes T, Halbach S, et al. Exometabolom analysis of breast cancer cell lines: metabolic signature. Sci Rep. 2015;5:13374.
- Yu YP, Ding Y, Chen Z, et al. Novel fusion transcripts associate with progressive prostate cancer. Am J Pathol. 2014;184:2840–2849.
- Andronescu M, Condon A, Turner DH, et al. In: Gorodkin J, Ruzzo WL, editors. RNA Seq. Struct. Funct. Comput. Bioinformatic Methods, Methods Mol. Biol. Vol. 1097. New York: Humana Press; 2014. p. 45–70. Chapter 3.
- Reuter JS, Mathews DH. RNAstructure: software for RNA secondary structure prediction and analysis. BMC Bioinformatics. 2010;11:129.
- Lorenz R, Bernhart SH, Honer Zu Siederdissen C, et al. ViennaRNA package 2.0. Algorithms Mol Biol. 2011;6. DOI:10.1186/1748-7188-6-26.
- Tanzer A, Hofacker IL, Lorenz R. RNA modifications in structure prediction – status quo and future challenges. Methods. 2019;156:32–39.
- Vendeix FA, Munoz AM, Agris PF. Free energy calculation of modified base-pair formation in explicit solvent: A predictive model. RNA. 2009;15:2278–2287.
- Chawla M, Oliva R, Bujnicki JM, et al. An atlas of RNA base pairs involving modified nucleobases with optimal geometries and accurate energies. Nucleic Acids Res. 2015;43:6714–6729.
- Aduri R, Psciuk BT, Saro P, et al. AMBER Force Field Parameters for the Naturally Occurring Modified Nucleosides in RNA. J Chem Theory Comput. 2007;3:1464–1475.
- Sakuraba S, Asai K, Kameda T. Predicting RNA duplex dimerization free-energy changes upon mutations using molecular dynamics simulations. J Phys Chem Lett. 2015;6:4348–4351.
- Nishida S, Sakuraba S, Asai K, et al. Estimating energy parameters for rna secondary structure predictions using both experimental and computational data. IEEE/ACM Trans Comput Biol Bioinforma. 2019;16:1645–1655.
- Chen X, Kierzek R, Turner DH. Stability and structure of RNA duplexes containing isoguanosine and isocytidine. J Am Chem Soc. 2001;123:1267–1274.
- Wright DJ, Rice JL, Yanker DM, et al. Nearest neighbor parameters for inosine·uridine pairs in RNA duplexes. Biochemistry. 2007;46:4625–4634.
- Roost C, Lynch SR, Batista PJ, et al. Structure and thermodynamics of N6-methyladenosine in RNA: a spring-loaded base modification. J Am Chem Soc. 2015;137(5):2107–2115.
- Chou F-C, Kladwang W, Kappel K, et al. Blind tests of RNA nearest-neighbor energy prediction. Proc Natl Acad Sci U S A. 2016;113:8430–8435.
- Wright DJ, Force CR, Znosko BM. Stability of RNA duplexes containing inosine·cytosine pairs. Nucleic Acids Res. 2018;46:12099–12108.
- Hopfinger MC, Kirkpatrick CC, Znosko BM. Predictions and analyses of RNA nearest neighbor parameters for modified nucleotides. Nucleic Acids Res. 2020;48:8901–8913.
- Uhlenbeck OC. Tetraloops and RNA folding. Nature. 1990;346:613–614.
- Varani G. Exceptionally Stable Nucleic Acid Hairpins. Annu Rev Biophys Biomol Struct. 1995;24:379–404.
- Deng NJ, Cieplak P. Free energy profile of RNA hairpins: a molecular dynamics simulation study. Biophys J. 2010;98:627–636.
- Seeliger D, de Groot BL. Protein thermostability calculations using alchemical free energy simulations. Biophys J. 2010;98:2309–2316.
- Procacci P. I. Dissociation free energies of drug–receptor systems via non-equilibrium alchemical simulations: a theoretical framework. Phys Chem Phys. 2016;18:14991–15004.
- Sponer J, Bussi G, Krepl M, et al. RNA structural dynamics as captured by molecular simulations: a comprehensive overview. Chem Rev. 2018;118:4177–4338.
- Lee T-S, Allen BK, Giese TJ, et al. Alchemical binding free energy calculations in AMBER20: Advances and best practices for drug discovery. Chem Inf Model. 2020; DOI:10.1021/acs.jcim.0c00613.
- Li Y, Nam K. Repulsive soft-core potentials for efficient alchemical free energy calculations. J Chem Theory Comput. 2020;16:4776–4789.
- Tanida Y, Matsuura A. Alchemical free energy calculations via metadynamics: application to the theophylline-RNA aptamer complex. J Comput Chem. 2020;41(20):1804–1819.
- Mlynsky V, Bussi G. Exploring RNA structure and dynamics through enhanced sampling simulations. Curr Opin Struct Biol. 2018;49:63–71.
- Cheng X, Cui G, Hornak V, et al. Modified replica exchange simulation methods for local structure refinement. J Phys Chem B. 2005;109:8220–8230.
- Deng N-J, Cieplak P. Molecular dynamics and free energy study of the conformational equilibria in the UUUU RNA hairpin. J Chem Theory Comput. 2007;3:1435–1450.
- Villa A, Widjajakusuma E, Stock G. Molecular dynamics simulation of the structure, dynamics, and thermostability of the RNA hairpins uCACGg and cUUCGg. J Phys Chem B. 2008;112:134–142.
- Garcia AE, Paschek D. Simulation of the pressure and temperature folding/unfolding equilibrium of a small RNA hairpin. J Am Chem Soc. 2008;130:815–817.
- Zhang Y, Zhao X, Mu Y. Conformational transition map of an RNA GCAA tetraloop explored by replica-exchange molecular dynamics simulation. J Chem Theory Comput. 2009;5:1146–1154.
- DePaul AJ, Thompson EJ, Patel SS, et al. Equilibrium conformational dynamics in an RNA tetraloop from massively parallel molecular dynamics. Nucleic Acids Res. 2010;38:4856–4867.
- Zuo G, Li W, Zhang J, et al. Folding of a small RNA hairpin based on simulation with replica exchange molecular dynamics. J Phys Chem B. 2010;114:5835–5839.
- Kuhrova P, Banas P, Best RB, et al. Computer folding of RNA tetraloops? Are we there yet? J Chem Theory Comput. 2013;9:2115–2125.
- Bottaro S, Gil-Ley A, Bussi G. RNA folding pathways in stop motion. Nucleic Acids Res. 2016;44:5883–5891.
- Kuhrova P, Best RB, Bottaro S, et al. Computer folding of RNA tetraloops: identification of key force field deficiencies. J Chem Theory Comput. 2016;12:4534–4548.
- Miner JC, Chen AA, Garcia AE. Free-energy landscape of a hyperstable RNA tetraloop. Proc Natl Acad Sci U S A. 2016;113:6665–6670. .
- Bottaro S, Bussi G, Kennedy SD, et al. Conformational ensembles of RNA oligonucleotides from integrating NMR and molecular simulations. Sci Adv. 2018;4:eaar8521.
- Cesari A, Bottaro S, Lindorff-Larsen K, et al. Fitting corrections to an RNA force field using experimental data. J Chem Theory Comput. 2019;15:3425–3431.
- Williams DJ, Hall KB. Experimental and computational studies of the G[UUCG]C RNA tetraloop11Edited by I. Tinoco. J Mol Biol. 2000;297:1045–1061.
- Hendel A, Bak RO, Clark JT, et al. Chemically modified guide RNAs enhance CRISPR-Cas genome editing in human primary cells. Nat Biotechnol. 2015;33:985–989.
- Carregal-Romero S, Fadon L, Berra E, et al. MicroRNA nanotherapeutics for lung targeting. insights into pulmonary hypertension. Int J Mol Sci. 2020;21:3253.
- Case D, Ben-Shalom IY, Brozell SR, et al. AMBER 2018. San Francisco: University of California; 2018.
- Macke TJ, Case DA. In: Leontes NB, SantaLucia J Jr., editors. Mol. Model. Nucleic Acids. Washington, DC; 1998. p. 379–393. Chapter 24. DOI: 10.1021/bk-1998-0682.ch024.
- Pettersen EF, Goddard TD, Huang CC, et al. UCSF chimera?A visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612.
- Bhati AP, Wan S, Hu Y, et al. Uncertainty quantification in alchemical free energy methods. Chem Theory Comput. 2018;14:2867–2880.
- Xu X, Zhao P, Chen S-J. Vfold: A web server for RNA structure and folding thermodynamics prediction. PLoS One. 2014;9:1–7.
- Steinbrecher T, Mobley DL, Case DA. Nonlinear scaling schemes for Lennard-Jones interactions in free energy calculations. J Chem Phys. 2007;127:214108.
- Shirts MR, Mobley DL. In: Monticelli L, Salonen E, editors. An introduction to best practices in free energy calculations. Biomol. Simulations Methods Protoc. Totowa, NJ: Humana Press; 2013. p. 271–311. DOI:10.1007/978-1-62703-017-5_11.
- Klimovich PV, Shirts MR, Mobley DL. Guidelines for the analysis of free energy calculations. J Comput Aided Mol Des. 2015;29:397–411.
- Roe DR, Cheatham TE. PTRAJ and CPPTRAJ: software for processing and analysis of molecular dynamics trajectory data. J Chem Theory Comput. 2013;9:3084–3095.
- Shirts MR, Chodera JD. Statistically optimal analysis of samples from multiple equilibrium states. J Chem Phys. 2008;129:124105.
- Mathews DH, Disney MD, Childs JL, et al. Incorporating chemical modification constraints into a dynamic programming algorithm for prediction of RNA secondary structure. Proc Natl Acad Sci USA. 2004;101:7287–7292.