
ABSTRACT
A persisting obstacle in human immunology is that blood-derived leukocytes are notoriously difficult to manipulate at the RNA level. Therefore, our knowledge about immune-regulatory RNA-networks is largely based on tumour cell-line and rodent knockout models, which do not fully mimic human leukocyte biology. Here, we exploit straightforward cell penetrating peptide (CPP) chemistry to enable efficient loss-of-function phenotyping of regulatory RNAs in primary human blood-derived cells. The classical CPP octaarginine (R8) enabled antisense peptide-nucleic-acid (PNA) oligomer delivery into nearly 100% of human blood-derived macrophages without apparent cytotoxicity even up to micromolar concentrations. In a proof-of-principle experiment, we successfully de-repressed the global microRNA-155 regulome in primary human macrophages using a PNA-R8 oligomer, which phenocopies a CRISPR-Cas9 induced gene knockout. Interestingly, although it is often believed that fairly high concentrations (μM) are needed to achieve antisense activity, our PNA-R8 was effective at 200 nM. RNA-seq characterized microRNA-155 as a broad-acting riboregulator, feedback restraining a late myeloid differentiation-induced pro-inflammatory network, comprising MyD88-signalling and ubiquitin-proteasome components. Our results highlight the important role of the microRNA machinery in fine-control of blood-derived human phagocyte immunity and open the door for further studies on regulatory RNAs in difficult-to-transfect primary human immune cells.
INTRODUCTION
Despite the undeniable success of antibiotics and vaccines, infectious and inflammatory diseases remain a major cause of death, according to the World Health Organization (WHO)[Citation1]. Particularly problematic are imbalances in the production of soluble inflammation mediators, which account for the majority of death cases due to organ failure, e.g. in sepsis, community acquired pneumonia or COVID-19 [Citation2,Citation3]. Mammalian immune cells possess a myriad of control mechanisms to counteract such inflammation-associated pathologies upon pathogen sensing [Citation4]. The rapid detection of pathogens by the innate immune system is achieved through evolutionarily conserved pattern recognition receptors (PRRs), which sense foreign signatures of viruses and bacteria. A prototypic example is the Toll-like receptor 4 (TLR4), which senses bacterial cell wall lipopolysaccharides (LPS). Upon activation of TLR4, a signalling cascade depending on the signalling adapters myeloid differentiation primary response gene 88 (MyD88) and TAK1-binding protein 2 (TAB2) culminates in the nuclear translocation of the inflammation master transcription factor nuclear factor-kB (NFκB) [Citation4]. To prevent from false-positive NFκB activation and uncontrolled inflammation, the MyD88 signalling cascade is feedback controlled at multiple levels. Riboregulators of the microRNA and long non-coding RNA (lncRNA) class play an important role in this process [Citation5,Citation6]. MicroRNA-155 and microRNA-146a, for instance, are induced following toll-like receptor 4 (TLR4-activation and feedback inhibit TLR4-signalling by base-pairing and interfering with the messengers of key TLR signal transduction components, such as MyD88, TAB2, TNF receptor-associated factor 6 (TRAF6) or interleukin-1 receptor-associated kinase 1 (IRAK1) [Citation7–9]. Under certain conditions, however, microRNA-155 may also promote inflammatory gene expression through different mRNA targets, therefore its overall contribution to inflammation remains controversial [Citation10,Citation11]. In addition, our present knowledge about the roles of non-coding RNAs such as microRNA-155 in immune cells is based on small animal and tumour cell models, which do not fully mimic human leukocyte immunobiology [Citation12,Citation13]. The investigation of the roles of non-coding RNAs such as microRNA-155 in primary human innate immune cells is presently hampered by the resistance of these cells to transduction and limited viability under nucleic acid transfection conditions [Citation14–16].
The most straightforward possibility for targeted RNA inhibition without relying on transfection is the use of cell-permeable RNA antisense-inhibitors; however, the intrinsic impossibility of achieving an effective cellular uptake remains an efficacy-limiting factor of commonly used inhibitors [Citation17,Citation18]. Such inhibitors are typically composed of stable oligomers of nucleic acid mimics, such as 2ʹ-O-methyl RNA, peptide nucleic acid (PNA) or phosphorodiamidate morpholino (PMO) [Citation17]. Conjugation to nanoparticles or cell-penetrating peptides (CPPs) [Citation19] has turned antisense drugs into powerful tools, which are even arriving in the clinics (e.g. SRP-5051) [Citation20,Citation21]. Examples of CPPs are protein transduction domains like human immunodeficiency virus (HIV) Tat protein or Penetratin, or synthetic peptides, whose sequences often are highly cationic, amphipathic or hydrophobic. CPP-coupled oligomers have been successfully tested in animal models, e.g. as antivirals [Citation22] or as exon-skipping agents for treatment of Duchenne muscular dystrophy [Citation23]. Despite the advances already made and the few precedents of CPPs as vectors targeting mammalian leukocytes, CPPs that confer efficient delivery of antisense oligomers to primary human blood-derived leukocytes to our knowledge have remained unexplored. The obvious lack of knowledge as to effective RNA inhibition strategies in leukocytes has prevented from dissecting the contributions of regulatory RNAs to primary human blood cell immunity. Closing this gap might help harnessing the therapeutic potential of small and long regulatory RNAs controlling immune-signalling to protect from inflammation-induced pathologies.
Here, we make use of classic CPP chemistry to enable efficient antisense-inhibition and phenotyping of non-coding RNAs in primary human immune-cells. Comparing well-established CPPs, we found octaarginine (R8) to confer the best delivery of antisense PNA oligomers into peripheral blood cells. PNA-R8 was delivered into nearly 100% of blood-derived macrophages at submicromolar concentration (200 nM), with no apparent signs of cytotoxicity. To demonstrate its utility for RNA loss-of-function phenotyping in primary human immune cells, we directed the PNA-R8 chemistry against microRNA-155. RNA-seq revealed global microRNA-155 target de-repression upon blood-derived macrophage treatment with 200 nM of a PNA-R8 blocking the mature microRNA seed, thereby confirming sequence-specific PNA-R8 activity. Gratifyingly, comparison of the primary cell data to results obtained in parallel with a CRISPR-Cas9 induced microRNA-155 knockout cell line revealed an agreement in the core-set of target mRNAs under negative influence by the microRNA. Besides the signalling adaptors MyD88 and TAB2, this included a broader network of interlinked TLR- and ubiquitination-pathway members, induced during myeloid immune cell differentiation from haematopoietic stem cells. These results suggest microRNA-155 as an imortant fine-tuner, restraining rather than promoting inflammation in human myeloid immune cells outside of the bone-marrow niche to prevent from pro-inflammatory immune pathologies. We believe that our study will help closing the gap between small animal and human primary cell models in immunological research.
Results
PNA-design and PNA-delivery into human leukocytes by various CPPs
Presently, regulatory RNA circuits in primary human myeloid immune cells remain difficult to study, due to the general fragility of these cell types under nucleic acid transfection conditions. To overcome this problem, we initially tested the ability of antisense PNAs coupled to standard CPPs to derepress and thus expose the regulome of immune-associated non-coding RNAs in human blood-derived cells. As a target, we chose microRNA-155, a well-characterized non-coding RNA in peripheral blood mononuclear cells (PBMCs), which regulates their inflammatory activity through a defined short seed-sequence motif [Citation7,Citation24]. In a first step, we determined the ability of classic CPPs to deliver anti-miR-155 or scrambled PNAs coupled to 5/6-carboxyfluorescein (FAM) into primary human immune cells by fluorescence activated cell sorting (FACS) analysis ( and ). Among the chosen CPPs were the polyarginine CPP R8, derived from protein transduction domains [Citation25], and the second generation analogue (secR8). In the latter CPP, 6-aminohexanoic acid (Ahx) spacers are added to lower the charge density, thus improving uptake and endosomal release due to enhanced flexibility [Citation26]. In 2008 the Gait lab introduced a new class of CPPs, referred to as peptide nucleic acid internalization peptides (Pips). They extended the lead secR8 CPP by a central hydrophobic core motif [Citation27]. These Pip CPPs exhibit more efficient internalization in vitro and in vivo [Citation28–30]. From the available Pip CPPs, for this project we selected Pip6a, which consists of arginine-rich Ahx and β-alanine (B) spaced flanking regions and a five amino acid hydrophobic core motif (-YQFLI-), because it displayed the highest activity experimentally [Citation28,Citation31]. For the PNA probe design, we built on previously published work with a trilysine peptide (K3-) and pH-low insertion peptide (pHLIP)-coupled anti-miR-155 PNAs, administered to mice [Citation12,Citation13]. To compensate for a base-exchange in the human compared to the murine microRNA-155 orthologue, we adapted the anti-miR-155 PNA-sequence accordingly (Fig. S1A). Furthermore, we shortened the inhibitor sequence from 23 to 18 PNA bases, in favour of reduced synthesis cost and effort, while expecting to retain the specific blockage of the majority of microRNA-155 seed and accessory bases (Fig. S1A) [Citation32].
Figure 1. Structure of PNA-CPP probes. Left to right: fluorophore (‘label’), PNA-sequence and cell penetrating peptide (‘peptide’) are specified. FAM = 5/6-fluorescein, B = β-alanine, X = 6-aminohexanoic acid (Ahx), scr = scrambled. – = none
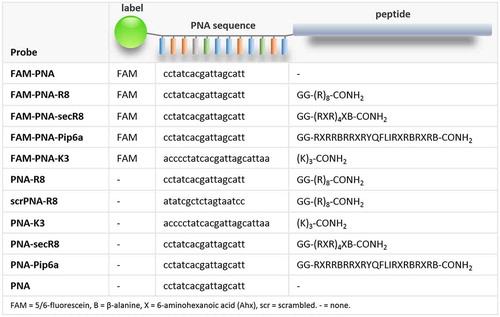
Figure 2. PNA delivery into human PBMCs via different peptide moieties. A) Anti-miR-155 PNAs were coupled to CPPs and FAM, and their delivery into human peripheral blood-derived mononuclear cells (PBMCs) was studied. Cells were counterstained with CD14 (monocyte/macrophage), CD66b (granulocyte), CD19 (B-cell) and CD4 (T-cell) specific antibodies. B) FACS analysis of human PBMCs. Top left: Forward-/Sideward-Scatter plot of untreated PBMCs. Bottom left: qRT-PCR analysis (Row Z-scores, colour-coded according to legend) of indicated marker expression in flow-sorted cell populations, using CD14-, CD66b-, CD19- or CD4-specific antibodies. Middle and right: Same cells as top left, but counterstained with subpopulation markers introduced above. Predominant area occupied by the respective subpopulations is indicated. Cells positive for the indicated markers are highlighted in blue. C) Left: total percentage of FAM-positive PBMCs upon challenge with indicated FAM-PNA-constructs (200 nM) or DMSO. Right: same as left, but for myeloid (CD14+/CD66b+) and lymphoid (CD19+/CD14+) subpopulations. D) Representative scatter plots of PBMCs treated with FAM-labelled control or PNA-R8. FAM-positive populations highlighted in green. Differences between groups of samples were evaluated by 1way-ANOVA with multiple comparisons (* = P-value ≤ 0.05, ** = P-value ≤ 0.01). Error bars indicate standard deviations, based on ≥ 3 independent experimental replicates
To determine the efficacy of FAM-PNA-CPP delivery into human blood cell populations, PBMCs were counter-stained with fluorophore-labelled T- and B-lymphocyte (CD4+ and CD19+), monocyte/macrophage (CD14+) and granulocyte (CD66b+) marker antibodies, enabling discrimination of lymphoid and myeloid PBMC subpopulations ( and B). Antibody-specificity was verified by quantitative real time polymerase chain reaction (qRT-PCR) analysis of CD14, CD66b, CD19 and CD4 mRNA expression in FACS-purified cell-populations stained with the respective antibodies (). PBMC treatment with the different FAM-PNA-CPP conjugates at a concentration of 200 nM shifted the cells into the FAM (‘green’) channel with varying efficacy. Interestingly, all types of FAM-PNA-CPPs preferentially accumulated in myeloid cells and were only poorly delivered into lymphoid cells ( and D). Of note, all CPPs outcompeted the K3 peptide, previously used for systemic PNA-delivery in mice [Citation12]. Among all tested CPPs, R8 displayed the best delivery into human PBMCs, with ~70% of the myeloid cell population shifting into the FAM-positive gate at 200 nM.
PNA-CPP toxicity and delivery into blood-derived macrophages
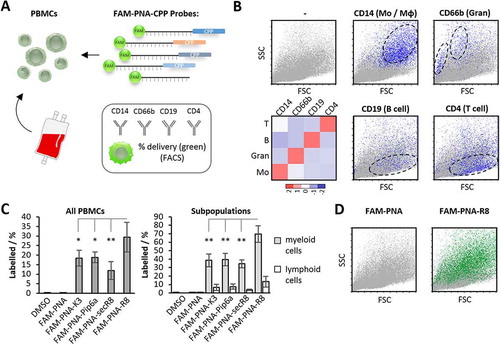
Inflamed tissue releases soluble factors, which attract PBMCs from the bloodstream. Upon diapedesis from the vasculature into inflamed tissue, monocytes maturate into macrophages, which orchestrate local innate immunity [Citation33]. To account for this major arm of myeloid leukocyte driven immunity and explore the inhibitory potential of PNAs in myeloid cells in more detail, we studied the delivery of the FAM-PNA-CPP conjugates into blood-derived macrophages by FACS, confocal microscopy, and bioactivity assays (). Similar to our results with whole PBMCs, the PNA-R8 construct outperformed the other FAM-PNA-CPPs in FACS analysis, with respect to both macrophage fluorescence intensity and percentage of labelled cells ( and C). At 200 nM, nearly all cells (> 98%) were positive for the FAM-PNA-R8 – even at a reduced concentration of just 20 nM > 60% of the cells were still FAM-positive. In stark contrast, the labelling efficiency for the SecR8, K3 and Pip6a conjugates was only 32%, 12% and 8%, respectively (). Of note, none of the tested PNA constructs evoked relevant signs of cytotoxicity in macrophages at 200 nM (Fig. S1B). To narrow down the tolerable range of PNA-R8 we recorded its cytotoxicity at higher concentrations. Considerable toxicity was observed starting from 10 µM (). This is 50-fold above the 200 nM concentration used for subsequent functional inhibitor experiments. Antisense-mediated inhibition of intracellular RNA targets requires that the PNA-CPP is not trapped in the plasma membrane or endosomal compartments. We therefore studied the subcellular distribution of FAM-labelled PNA-R8 supplemented into blood-derived macrophage cultures by standard and confocal fluorescence microscopy. The FAM-PNA-R8 signal was evenly dispersed throughout the cytoplasm and nucleus of DAPI-counterstained cells, suggesting successful intracellular delivery without endosomal trapping ( and B, Fig. S1C). We further observed that the uptake of FAM-PNA-R8 was temperature and largely energy-independent (Fig. S1D). Although the exact mechanism is unclear passive uptake of R8 was previously reported [Citation34]. Taken together, among several classic CPPs, R8 enables most efficient PNA delivery into blood-derived myeloid immune cells at a concentration fully compatible with cell viability.
Figure 3. PNA delivery into human blood-derived macrophages. A) Anti-miR-155 PNAs, coupled to CPPs and to FAM, were applied to CD14+ monocyte derived macrophages to study their intracellular delivery and cytotoxicity. B) Geo-median fluorescence intensity (Geo-MFI) fold-change of macrophages after treatment with the indicated FAM-PNA constructs and concentrations, compared to background fluorescence. C) Same as B), but with percentage of labelled macrophages. D) Cytotoxicity of PNA-R8 and scrPNA-R8 as well as R8 towards macrophages at indicated concentrations. Viability was assessed after 72 h employing the resazurin cell viability assay. Error bars indicate standard deviations, based on ≥ 3 independent experimental replicates
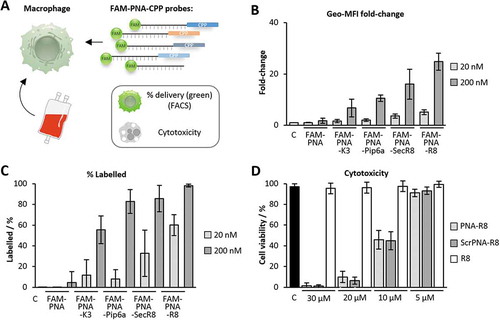
Figure 4. Fluorescence microscopy of PNA-treated blood-derived macrophages. A) Representative FACS analysis of macrophages treated with 200 nM anti-miR-155 PNA not coupled to R8 (top) or coupled to R8 (bottom). Dashed line separates background from FAM-fluorescence. B) Representative fluorescence microscopy pictures showing macrophages left untreated or treated for 24 h with 200 nM FAM-labelled, R8-CPP coupled anti-miR-155 PNA (FAM-PNA-R8). Nuclei counterstained with DAPI. DIC = differential interference contrast images. Scale-bar: 20 µm
Determination of the microRNA-155 targetome for functional PNA-CPP tests
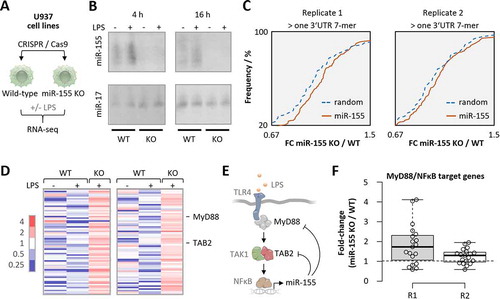
After achieving efficient PNA delivery into blood-derived human phagocytes at submicromolar concentration, we sought to assess the target RNA-inhibition potential of R8-coupled short PNAs in these cells. To this end, the inhibition of microRNA-155 by anti-miR-155-R8 compared to K3-coupled PNA and scrambled PNA-R8 treatment of blood-derived macrophages was determined by RNA-seq analysis of global microRNA-155 target mRNA expression. We hypothesized that in these experiments the PNA-R8 would outperform the activity of a K3 linked PNA, given the superior delivery of PNA-R8 into human blood-derived cells at 20 nM (60% compared to 12%) and 200 nM (98% compared to 56%; ). In preparation for these investigations, we determined the full spectrum of microRNA-155 controlled messenger RNAs by unbiased RNA-seq profiling of microRNA-155 knockout and wild-type human U937 macrophage cell lines, generated using CRISPR-Cas9 (). Of note, primary human macrophages were largely resistant to CRISPR-vector transfection and sensitive to lipofection, underlining that this approach is not applicable to primary human phagocytes (Fig. S2A; Fig. S1B). In CRISPR-edited U937 cells, loss of base-line and TLR-induced microRNA-155 expression was confirmed by Northern Blot and RNA-seq analysis of control- and 4 h Salmonella lipopolysaccharide (LPS)-challenged knockout compared to wild-type cells (, Fig. S2B). RNA-seq data analysis revealed the expected de-repression of mRNAs with > 1 microRNA-155 3ʹ UTR 7-mer seed match in LPS-stimulated knockout compared to wild-type cells (). Reactome-pathway analysis confirmed an enrichment of TLR-signalling associated terms among mRNAs with a de-repression ≥ 1.5-fold and ≥ 1 microRNA-155 3ʹ UTR seed match (Fig. S2C), in line with the presumed function of microRNA-155 in innate immune sensing [Citation7]. Among these de-repressed mRNAs were the major, experimentally determined targets of microRNA-155 in the pro-inflammatory TLR4-MyD88-NFκB immune-signalling pathway, TAB2 and MyD88 ( and E) [Citation7,Citation9,Citation35]. Conversely, expression of mRNAs known to be under positive control of the TLR-MyD88-NFκB signalling axis was elevated in microRNA-155 knockout compared to wild-type cells (). Thus, RNA-seq analysis of microRNA-155 deficient cells confirmed a role of this microRNA in TLR-MyD88 immune signalling and provided a reference set of mRNAs controlled by microRNA-155, for subsequent PNA inhibitor tests in primary human blood-derived cells.
Figure 5. RNA-seq analysis of microRNA-155 knockout U937 macrophages. A) Schematic representation of experimental setup. PMA-differentiated U937 cells were transfected with control or microRNA-155 KO CRISPR constructs and clonal populations were raised, treated with 100 ng/mL LPS for 4 h or left untreated and subjected to RNA-seq analysis. B) Northern blot analysis of microRNA-155 and −17 expression in 4 h and 16 h LPS- or control-treated wild-type (WT) and knockout (KO) U937 macrophages. C) Cumulative density fraction analysis of microRNA-155 7-mer target RNA expression compared to random 7-mer RNAs, in two RNA-seq replicates of microRNA-155 KO versus WT cells. D) Heatmap representation of the fold-changes (over base-mean) of microRNA-155 3ʹ UTR target-site containing mRNAs elevated ≥ 1.5 fold in microRNA-155 KO compared to WT U937 macrophages. Two RNA-seq replicates. MyD88 and TAB2 mRNA are indicated. E) Schematic representation of the TLR4 signalling pathway with microRNA-155 feedback through MyD88 and TAB2 targeting. F) MyD88/NFκB target mRNA expression in microRNA-155 knockout compared to wild-type U937 macrophages. Two RNA-seq replicates shown
Global microRNA-155 target de-repression exclusively upon PNA-R8 treatment
To determine the target inhibition potential of R8- compared to K3-coupled PNA antisense oligomers, the above introduced microRNA-155 inhibitors () and scrambled controls were re-synthesized without FAM-label, followed by supplementation into blood-derived macrophage cultures at 200 nM. 24 h upon PNA addition, macrophages were activated with LPS for an additional 4 h and subjected to RNA-seq analysis (). Confirming successful immune-activation, macrophages readily induced the expression of inflammation mediator interleukin 1β (IL1β) upon LPS-stimulation (). Treatment with the microRNA-155-targeting PNA-R8 but not with the scrambled PNA-R8 globally lifted the expression of mRNAs identified in the CRISPR-edited KO cell lines as microRNA-155-supressed transcripts (, Fig. S2D). In contrast, the K3 coupled inhibitor did not significantly impact the expression of microRNA-155-controlled mRNAs (). Therefore, only our PNA-R8, which, of note has a shorter PNA sequence than previous inhibitors [Citation12,Citation13,Citation36], yields a selective functional response to target repression at low concentration. Overlay of the obtained transcriptome datasets from microRNA-155 knockout and PNA-inhibitor treated cells confirmed MyD88 and TAB2 mRNAs as microRNA-155 targets elevated in all replicates from both experiments and revealed the expected over-representation of pattern recognition receptor (PRR)-NFκB and PRR-mitogen activated protein kinase (MAPK) signalling pathways among these mRNAs (, E and F). In contrast to microRNA-155 knockout and PNA-R8 inhibitor treatment, PNA-K3 inhibitor treatment did not significantly impact MyD88 and TAB2 mRNA expression (). Network analysis of mRNAs derepressed upon PNA-R8 treatment suggested that in primary human phagocytes microRNA-155 controls a tightly interlinked network of factors involved in NFκB-dependent innate immunity e.g. as TLR-signalling molecules or ubiquitin-proteasome dependent regulators (). Importantly, the mRNAs establishing this network were co-induced with microRNA-155 during terminal myeloid haematopoiesis, as determined by comparison of the transcriptome profiles of human haematopoietic stem cells (HSCs), common myeloid precursors (CMPs) and peripheral monocytes (Mono, ). No such co-induction was observed for mRNAs not derepressed upon microRNA-155 inhibition (). This suggests that human microRNA-155 acts as a specific negative feedback regulator of a broad innate immune signalling network established late during myeloid lineage differentiation, when mature immune cells exit the bone marrow niche.
Figure 6. RNA-seq analysis of blood-derived macrophages treated with microRNA-155 inhibitors. A) Schematic representation of experimental setup. Blood-derived macrophages were treated with K3- and R8-coupled PNAs (200 nM) and subjected to LPS-treatment (100 ng/mL for 4 h) and RNA-seq analysis. B) qRT-PCR analysis of IL1β expression in LPS-treated macrophages compared to mock-treatment. C) Comparison of the expression of KO-confirmed microRNA-155 targets (from ) in blood-derived macrophages treated with indicated PNA inhibitors. Two RNA-seq replicates are shown, with fold-changes compared to base-mean. D) Heatmap showing RNA-seq-determined mRNA fold-changes ≥ 1.5-fold in microRNA-155 KO and ≥ 1.2-fold in R8 inhibitor-treated cells. Fold-changes colour-coded as indicated to the left. MyD88 and TAB2 mRNAs highlighted. E) Enriched KEGG pathways and corresponding P-values associated with the mRNAs from D). F) RNA-seq determined fold-changes (compared to base-mean) of MyD88 and TAB2 expression in microRNA-155 KO and PNA inhibitor treated macrophages. Differences between groups of samples were evaluated by 1way-ANOVA with multiple comparisons. P-values ≤ 0.05 are shown
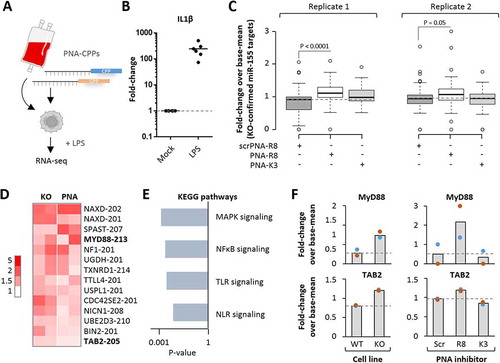
Figure 7. Verification and comparison of microRNA-155 inhibitory activity of PNA-CPPs. A) Induced network plot with proteins encoded by microRNA-155-supressed mRNAs from Fig. 6D (green) and their connecting interactors (light blue). Physiological inputs of this network in the form of TLR4-agonists and cellular ubiquitin and output in the form of NFκB-activation are shown in red. B) Left: schematic illustration of haematopoietic stem cell (HSC) differentiation to common myeloid precursors (CMP) and monocytes (Mono) in the bone marrow niche. Right top: base-mean fold-changes of microRNA-155 targeted mRNAs from A) in HSC, CMP and Mono (significance in Mono vs HSC and Mono vs CMP, respectively). Right middle: same as top, but with mRNAs not supressed by microRNA-155. Right bottom: Base-mean fold-changes (colour-coded) of microRNA-155 host-gene (HG) expression in HSC, CMP and Mono. C) qRT-PCR analysis of MyD88, TAB2 and IL1β mRNA expression in primary human blood-derived macrophages, treated with 200 nM of the indicated PNAs or left untreated (solvent control) for 24 h. D) Same as C, but showing results for all anti-miR-155 PNA constructs, unconjugated, or conjugated to R8 (scrambled (scr) and anti-miR-155), secR8, Pip6a, K3. Heatmap shows colour-coded Row Z-scores of three experimental replicates per condition. Differences between groups of samples were evaluated by 1way-ANOVA with multiple comparisons (* = P-value ≤ 0.05, ** = P-value ≤ 0.01). Error bars indicate standard deviations, based on ≥ 3 independent experimental replicates
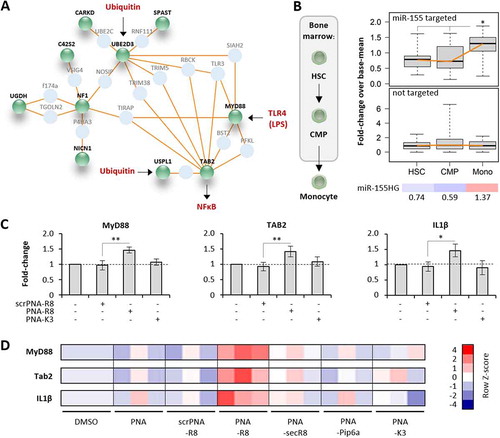
Validation of the RNA-seq data by qRT-PCR confirmed elevated expression of innate immune signalling components MyD88 and TAB2 upon R8 but not K3 microRNA-155 PNA inhibitor treatment of blood-derived macrophages (). R8 inhibitor treatment also increased expression of the messenger of MyD88- and TAB2-dependent inflammation mediator IL1β (). In line with the FACS-based PNA delivery results ( and C), qRT-PCR additionally proved the R8 inhibitor to outcompete secR8 and Pip6a CPP coupled inhibitors, which did not confer significant de-repression of microRNA-155 target mRNAs or of IL1β mRNA (). Of note, Northern blot analysis suggested the R8-inhibitor to antagonize microRNA-155 through steric inhibition rather than degradation (Fig. S2E). Thus, treatment with 200 nM R8-, but not secR8-, Pip6a- or K3-coupled antisense PNA-inhibitor recapitulates molecular loss-of-function phenotypes observed in microRNA-155 deficient cells, proving target-specific steric blocking activity of PNA-R8 in primary human blood-derived cells at non-toxic, submicromolar concentrations.
Taken together, our study reveals preferred PNA-CPP entry into myeloid rather than lymphoid human blood cells and determines octaarginine as a CPP enabling passive PNA delivery and RNA-inhibition in myeloid immune cells at submicromolar concentration. Our experiments with a microRNA antisense PNA-R8 inhibitor reveal microRNA-155 to feedback-control a broad pro-inflammatory immune-signalling network induced during late myelopoiesis, when human phagocytes become committed to anti-pathogen defence. We therefore suggest the PNA-R8 design as a preferential antisense inhibitor constellation for interference in the RNA circuits of primary human myeloid immune cells, which had remained difficult to manipulate and are frequently involved in leading human diseases.
Discussion
The intricate RNA circuits of primary human myeloid immune cells have remained difficult to dissect. In addition to their short life span, these cells are highly resistant to viral transduction and vector DNA take-up [Citation16] (Fig. S2A), precluding e.g. CIRSPR-Cas9- or small hairpin RNA (shRNA)-approaches. As an alternative, antisense oligomer and small interfering RNA (siRNA) transfection has been used to interfere in the RNA-circuits of human blood-derived cells [Citation37,Citation38], however, limited viability under lipofection conditions may severely affect the obtained results (Fig. S2A, Fig. S1B). Published insights into the RNA-circuits controlling immune-cell activation have therefore largely been obtained using small animal and tumour cell models, which, however, do not fully mimic human immune cell behaviour [Citation6,Citation39,Citation40]. Self-delivering antisense oligomers, traversing the plasma-membrane without a need for transfection constitute a feasible alternative. Previous studies, however, have indicated that functional oligomer delivery is particularly difficult to achieve for the leukocyte compartment. In a mouse strain expressing an aberrantly spliced green fluorescent protein (GFP)-transgene, for instance, restoration of GFP expression by a splice-switching PNA-K4 was observed in several tissues. Even at micromolar concentration, however, no PNA activity, was detected in the bone-marrow, as the major leukocyte reservoir [Citation41]. Attempts to target the bone marrow niche through a homing peptide failed to achieve a gain in splice-switching PNA activity. Cholesterol-conjugation on the other hand, increased splice-switching but failed to improve delivery [Citation42]. This suggests that besides improvement of delivery, antisense-oligomer activity needs to be closely examined before effective blood-cell targeting conjugations can be achieved. Surprisingly, simple CPP conjugation for efficient human myeloid leukocyte RNA targeting had been missing from the available literature, although these may fulfil both criteria.
By using an antisense PNA inhibitor coupled to the classic CPP R8, our present study succeeds in the derepression and functional characterization of the targetome of immune-regulatory RNA microRNA-155 in primary human blood-derived phagocytes for the first time, to our knowledge. Encouragingly, our research shows that PNA-R8 can block target RNA activity in human macrophages already at submicromolar concentrations (200 nM), predicting less toxic effects compared to other oligonucleotide compounds requiring micromolar concentrations, such as the PNA-K3 construct [Citation12]. Furthermore, we achieved more efficient antisense inhibition although we shortened the inhibitor sequence compared to earlier studies with PNAs targeting microRNA-155 [Citation12,Citation13,Citation36], reducing synthesis cost and effort. In mammalian myeloid cells, microRNA-155 both positively and negatively regulates the phagocyte inflammatory response to infectious agents and soluble immune-mediators [Citation10,Citation24]. Our present study, using efficient PNA inhibitors, shows that microRNA-155 primarily targets components of the pro-inflammatory macrophage MyD88-NFκB signalling network to restrain inflammation, rather than serving as a positive regulator, as previously speculated [Citation43]. In addition to core MyD88-pathway components such as MyD88 and TAB2, our results suggest the regulome of microRNA-155 in human macrophages to entail a broader network of regulators, controlling pro-inflammatory immune responses at multiple levels. Examples are regulatory components of the ubiquitin-proteasome system, such as ubiquitin-conjugating enzyme E2 D3 (UBE2D3) or ubiquitin specific peptidase like 1 (USPL1), known to contribute to TLR-based immune signalling [Citation44,Citation45]. Intriguingly, this complex network of microRNA-155 controlled mRNAs seems to be co-induced with the microRNA during late differentiation of myeloid cells from pluripotent bone marrow precursors. This suggests that the ability to mount an inflammatory response and to contain this response through riboregulators is tightly interlinked and co-acquired when immune cells emerge from the bone marrow niche, to counteract pathological inflammatory conditions, such as a systemic inflammatory response syndrome (SIRS) [Citation46].
Besides the modulation of pro-inflammatory immune signalling, microRNA-155 constitutes a host- and virulence-factor of Epstein-Barr virus (EBV) and Kaposi’s sarcoma-associated herpesvirus (KSHV), respectively [Citation47,Citation48]. Recently, microRNA-155 was even speculated to represent a potential host factor of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) [Citation49]. Furthermore, microRNA-155 has been assigned oncogenic properties [Citation50], thus, antisense-targeting of microRNA-155 might have therapeutic potential beyond modulation of NFκB-triggered immunity. In fact, studies with pHLIP peptide and cyclic peptide coupled microRNA-155 antagonists, administered to mice and cell lines, suggest a value of such compounds as cancer therapeutics [Citation13,Citation36]. Of note, however, pHLIP specifically targets cancer cell environments and pHLIP and cyclic peptide constructs were applied at concentrations ≥ 1 µM, where microRNA-155 sequestration induces cytotoxicity [Citation13,Citation36]. Furthermore, pHLIP and cyclic peptide constructs are less straight forward to synthesize than the peptides tested in the present work and their utility for targeting of the human innate immune system remains unknown. In our present study, we demonstrate RNA inhibition with more easy-to-synthesize peptides at submicromolar concentration, backed up by mechanistic insights into PNA activity in myeloid immune cells by global RNA-seq analysis. We believe that besides the ability to deliver antisense-chemistry into immune cells at submicromolar concentration, the ease-of-synthesis of the here studied peptides could be a significant advantage in the context of prospective basic research and clinical applications. MicroRNA-dependent re-writing of dendritic cell signalling pathways, for instance was suggested as a strategy to improve cancer vaccination approaches [Citation51]. Whereas PNA-R8 enables highly efficient interference in myeloid immune cell RNA-circuits, it should be noted that the ability of R8 to deliver antisense oligomers into lymphoid cells is limited (). In these cells, however, microRNA mimic and inhibitor transfection approaches seem feasible [Citation52], suggesting that further approaches, such as nebulized nanoparticle mediated pulmonary delivery [Citation53] could be employed for therapeutic targeting at the site of disease.
Furthermore, in addition to microRNAs, lncRNAs are emerging as important regulators of human myeloid inflammatory responses to infectious agents [Citation6]. Macrophage interferon-regulatory lncRNA 1 (MaIL1) for instance is essential for TLR-based activation of type I interferon (IFN-I) expression in human macrophages [Citation38]. Several other lncRNAs, such as p50-associated COX-2 extragenic RNA (PACER) or cardiac apoptosis-related lncRNA (CARLR) were reported to fulfil critical functions in cellular defence against infectious agents and pro-inflammatory immune-activation of myeloid cells [Citation5,Citation6]. Similar to the immune-modulatory microRNAs, however, the available studies are largely based on cell-line and small animal models. Analogous PNA-R8 compounds could enable the intervention in lncRNA and also mRNA circuits of primary human phagocytes without compromising at cell viability. Since lncRNAs and mRNAs, unlike microRNAs, cannot be blocked full-length with PNA probes, different targeting mechanisms would be required. The well-documented splice-switching activity of antisense-oligomers could be employed for this purpose [Citation18]. As a result, the dissection of the complex small and long regulatory and protein-coding RNA circuits controlling human phagocyte immunity could substantially improve our understanding of the pathomechanisms contributing to misguided inflammatory responses and uncontrolled colonization by infectious agents – major threats to human health. Our study demonstrates that appropriate CPP-PNA conjugates can serve as tools for the dissection of the regulatory RNA circuits of primary myeloid immune cells, as exemplified here by decryption of the regulon of microRNA-155 in human blood-derived macrophages using PNA-R8.
Materials and Methods
Cell culture
U937 monocytes (ATCC, CRL-1593.2) were grown in RPMI 1640 medium (Gibco, Thermo Fisher, Cat# 31870025) with 10% foetal calf serum (FCS, Biochrom, Cat# S 0613) 1% penicillin/streptomycin solution (Gibco, Thermo Fisher, Cat# 15140122).
Blood buffy coats were obtained from healthy donors (deidentified prior to use) from the Centre for Transfusion Medicine and Haemotherapy in Giessen, Germany. All donors consent to their blood samples being used for scientific purposes.
Human peripheral blood derived mononuclear cells (PBMCs) were isolated from buffy coats by density gradient centrifugation (Lymphoprep medium, Stem Cell Technologies, Cat# 07851, used according to the manufacturer’s instructions). Monocytes were isolated from PBMCs by MACS purification with anti-CD14 beads (Miltenyi Biotec, Cat# 130–050-201). PBMCs and blood-derived monocytes were cultivated in X–Vivo 15 medium (Lonza Bioscience, Cat# BEBP02-061Q), supplemented with 5% FCS and optionally with 15 ng/mL recombinant human GM-CSF (PreproTech, Cat# 300–03) for macrophage generation. All cells were cultured at 37°C in a humidified chamber in the presence of 5% CO2.
Cell based assays
U937 cells were differentiated into macrophage-like cells by addition of 20 nM phorbol 12-myristate 13-acetate (PMA, Sigma Aldrich, Cat# P1585) for 72 hours. Blood-derived macrophages and U937 macrophages were differentiated in 12-well plates, at a density of 4 × 105 cells/mL. For immune-activation, cells were stimulated with 1 µg/mL Salmonella enterica serovar typhimurium lipopolysaccharide (LPS, Sigma Aldrich, Cat# L6143) for the indicated durations. Cells were pre-treated with the indicated amounts of PNA oligonucleotide or DMSO (Sigma Aldrich, Cat# D8418) as a solvent control for 24 h in X–Vivo 15 medium, supplemented with 2.5% FCS, prior to further stimulations and subsequent sample collection.
CRISPR-Cas9
MicroRNA-155 knockout cells were generated as described previously [Citation24]. Briefly, a synthetic DNA segment (Integrated DNA Technologies) was introduced into the pX458 CRISPR vector [Citation54] introducing a second guideRNA expression cassette. The two introduced guideRNAs were directed against the transcriptional start-site of the microRNA-155 host gene [Citation24]. MicroRNA-155 and empty control CRISPR constructs were transfected into U937 monocytes using Lipofectamine 3000 (Invitrogen, Thermo Fisher, Cat# L3000008) according to the manufacturer’s protocol. Transfected cells were recognized by GFP-expression and single-cell sorted into 96-well plates using an, Aria III cell sorter (BD) with 100 µm nozzle. 96-well plates were pre-filled with RPMI 1640 complete medium, containing 100 µg/mL Normocin (InvivoGen, Cat# ant-nr-1). Upon single cell clonal expansion knockout cell populations were detected by genomic DNA isolation (Nucleospin tissue kit; Macherey Nagel, Cat# 740,952.50) and PCR with primers flanking the excised DNA-element [Citation24]. For investigation of primary human macrophage viability under transfection conditions, empty pX458 plamid DNA (no guideRNA insert) was transfected as described above and cells were stained with Annexin V-APC (BD Pharmingen, Cat# 550,474) and Propidium Iodide (BD Pharmingen, Cat# 556,463) for viability analysis.
Real-time PCR
Real-time PCR was performed using the High-Capacity cDNA Reverse Transcription kit (Applied Biosystems, Thermo Fisher, Cat# 4368813) and the Luna Universal qPCR Master Mix (NEB, Cat# M3003) according to the manufacturer’s instructions. Threshold cycle (CT) values and melt-curves were recorded on a Quantstudio3 device (Applied Biosystems). Fold-changes in expression, with U6 snRNA as a reference RNA, were calculated based on CT values using the 2−ΔΔCT method [Citation55]. Real-time PCR primers (Metabion GmbH) are listed in Table S1.
RNA purification
RNA was purified by TRIzol (Invitrogen, Thermo Fisher, Cat# 15596026)/chloroform extraction (Sigma Aldrich, Cat# 132950). To remove DNA contaminants prior to real-time PCR or RNA-seq analysis, the RNA was incubated with DNaseI (Thermo Scientific, Thermo Fisher, Cat# EN0521) and RNase Inhibitor (Promega, Cat# N2511) for 30 min at 37°C, followed by extraction with PCI (Sigma Aldrich, Cat# X985.1). Extracted RNA was precipitated using 3 volumes of 30:1 ethanol/5 M sodium acetate and Glycoblue (Invitrogen, Thermo Fisher, Cat# AM9516) as a precipitation aid, followed by one wash with 70% ethanol.
Northern Blot
For Northern blot analysis of microRNA expression, 60 µg of RNA per lane in 15 µl Gel loading buffer II (Invitrogen, Thermo Fisher, Cat# AM8546G) were denatured for 5 min at 95°C prior to size separation on 15% PAA (Carl Roth, Cat# 3030.1), 8 M Urea (Carl Roth, Cat# X999) TBE gels. Size-separated RNA was transferred in a wet-blot tank onto a positively charged nylon membrane (Roche, Cat# 11417240001) at 50 V, 50 min in TBE buffer. The membrane was subsequently UV cross-linked at 120 mJ/cm² using a HL2000 Hybrilinker (UVP). For RNA detection, the blots were hybridized with DNA probes (Table S1, Metabion GmbH), 3ʹ DIG-labelled using the 2nd Generation DIG-Tailing Kit (Roche, Cat# 3353583910). Probes were hybridized over night at 42°C in DIG EasyHyb Buffer (Roche, Cat# 11603558001). For all subsequent steps, including blot development, the DIG Wash and Block Buffer Set (Roche, Cat# 11585762001) and CDP-STAR (Roche, Cat# 11685627001) were used according to the manufacturer’s instructions. Chemiluminescence signals were recorded using an Chemostar Imager (Intas).
Flow cytometry
For analysis of PNA entry into human leukocytes, cells were treated with 20 nM or 200 nM FAM-labelled constructs or DMSO as a solvent control for 24 h in X–Vivo 15 medium, supplemented with 2.5% FCS, prior to PBS (Capricorn Scientific, Cat# PBS-1A) washes and collection of macrophages by scraping in PBS, containing 0.5% FCS. Cells were transferred on ice and subjected to FACS analysis within the next 60 min. For visualization of monocytes, granulocytes, B cells and T cells, PBMCs were stained with anti-CD14-APC (Biolegend, Cat# 367117), anti-CD66b-APC (eBioscience, Cat# 17–0666-61), anti-CD19-APC (Biolegend, Cat# 392503) or anti-CD4-PE (eBioscience, Cat# 12–0048-41) antibodies in PBS, 0.1% FCS for 30 min on ice, followed by one wash with PBS. Antibody specificity was validated by cell-sorting (Aria III device) and real-time PCR analysis of the expression of the respective markers. FACS data were recorded using a Guava easyCyte flow cytometer (Millipore). Exported FCS 3.0 files were imported into Flowing Software (http://flowingsoftware.btk.fi/) for further analysis. Intact cells were first gated in the forward-/side-scatter plot and FAM-positive, PE-positive or APC-positive cells were detected in the respective fluorescence channels, gated and quantified.
Cytotoxicity assay
For toxicity analysis, cell viability was assessed by resazurin cell viability assay [Citation56]. After incubation with PNA constructs or controls at various concentrations for 72 h, 20 μL of a 1 mM solution of resazurin (Alfa Aesar, Cat# B21187.06) in PBS was added to each well and mixed appropriated. After addition, the plate was shaken for 10 s using an Infinite M200 Pro (TECAN) plate reader, fluorescence was measured using the plate reader with the following settings: fluorescence bottom read, excitation wavelength: 560 nm, excitation band width: 9 nm, emission wavelength: 590 nm, emission bandwidth: 20 nm, 25 readings per well in 30 min intervals until saturation in the untreated cells was reached. The slope of each individual well was calculated and percentage of cell survival in the untreated control was set as 100%. Relative viability = (experimental slope – background slope)/(slope of untreated control-background slope) × 100%. Values were obtained in triplicates per sample/control and in ≥ 3 independent experimental replicates.
Uptake mechanism
To investigate the PNA-R8 uptake mechanism blood-derived monocytes were seeded in 24-well plates and differentiated into macrophages as described above. Under reduced serum conditions (X–Vivo 15 medium, supplemented with 2.5% FCS) cells were optionally pre-treated with 10 mM NaN3/2dG (ACROS Organics, Cat# 190381000 and Cat# 111980050, 200 mM stock in H2O) for ATP depletion for 30 min [Citation57]. For 4°C conditions another plate was incubated on ice and ice-cold solutions were used. Cells were then treated with 200 nM FAM-PNA-R8 for 2 h, washed twice with PBS supplemented with 0.5% FCS before FACS analysis as describes above.
Microscopy
Blood-derived monocytes were seeded on coverslips and differentiated into macrophages as described above. Subsequently, they were treated with the indicated amounts of FAM-labelled PNA oligonucleotide or DMSO as a solvent control for 24 h in X–Vivo 15 medium, supplemented with 2.5% FCS. Cells were then washed once with PBS, followed by fixation with 4% PFA (Carl Roth, Cat# 0335) for 10 min at room temperature and washed twice with PBS. Cells were then stained with DAPI (ATT Bioquest, Cat# 17510) at a dilution of 1:2000 in ultrapure H2O for 10 min. Cells were washed twice with PBS and then mounted on a glass slide with Mowiol 4–88 (Carl Roth, Cat# 0713.1) containing 25 mg/ml of DABCO (Carl Roth, Cat# 0718.1). Slides were dried overnight at room temperature in the dark and further stored at 4°C. For bright field images were acquired via Zeiss Plan-Apochromat 100x/1.45 Oil objective. Fluorescent images were taken with ET-YFP and ET-DAPI filter cubes (Chroma, USA). Fluorescence images shown in Fig. S1C were acquired on a Zeiss Axio Observer.Z1 microscope (Zeiss, Germany) equipped with a pco.edge sCMOS camera. Images were evaluated with ImageJ software [Citation58].
RNA-seq analysis
RNA-seq libraries were generated using the TruSeq stranded mRNA library kit (Illumina, Cat# 20040532) according to the manufacturer’s instructions. Libraries were analysed by single-ends sequencing with 50 bp read length using a NextSeq500 system. Upon FastQC analysis and quality trimming reads were mapped to the GENCODE GRCh38 human reference annotation using the CLC Genomics Workbench (Qiagen) with standard settings (mismatch cost = 2; insertion cost = 3; deletion cost = 3; length fraction = 0.8; similarity fraction = 0.8). For subsequent expression analysis, datasets were filtered for genes with RPKM (reads per kilobase of transcript, per million mapped reads) values ≥ 0.5 under at least one condition in both analysed experimental replicates. Cumulative density plot analysis was performed in Excel (Microsoft Office). Heat-maps were generated using Java TreeView [Citation59] and KEGG and Reactome pathway analysis (overrepresentation analysis) and induced network analysis was done using ConsensusPathDB [Citation60]. All RNA-seq data produced in this study are deposited and publically available in the NCBI GEO database (accession no. GSE152740). RNA-seq raw datasets for haematopoietic stem cells (EGAD00001000939, 8 replicates), common myeloid precursors (EGAD00001000907, 3 replicates) and CD16- monocytes (EGAD0000100928, 3 replicates) were generated by and obtained through the Blueprint Consortium [Citation61].
Solid phase synthesis and compound characterization
The peptide nucleic acids (PNAs) were synthesized manually according the procedures of Fmoc based solid-phase peptide synthesis. The synthesis was carried out using commercially available Fmoc/Bhoc PNA monomers (PNA Bio, Cat# FMA-1001; Cat# FMG-1001; Cat# FMC-1001; Cat# FMT-1001) and Fmoc amino acids (Iris Biotech) as described below. After cleavage and purification, the probes were analysed by analytical RP-HPLC and high-resolution electrospray ionization (HRMS ESI) mass spectrometry. Detailed compound characterizations are provided in the supplementary information file.
Mass spectrometry
High resolution electrospray ionization mass spectra (HRMS ESI) were acquired with a LTQ-FT Ultra mass spectrometer (Thermo Fischer Scientific).
UV–vis spectroscopy
Concentration determinations and yield quantification of PNA compounds were performed on a Tecan (Switzerland) Spark 20 M multimode microplate reader at room temperature by applying the corresponded molar extinction coefficient of the measured compound with Lambert-Beer law:
A = ε ∙ c ∙ l
where A = absorbance; ε = molar extinction coefficient; c = concentration; l = path length. All measurements were performed with 800 µL sample in a 1400 μL quartz cuvette (Hellma Analytics (104 F-QS)) with a pathlength of 1 cm.
The concentrations of the stock solutions of PNA in ultrapure water were determined by measuring the UV absorbance at 260 nm and utilizing the molar extinction coefficient obtained by applying the following equation [Citation62]:
ε260 nm = [(8.8 x nt) + (7.3 ∙ nc) + (11.7 ∙ ng) + (15.4 ∙ na)] ∙ 0.9 ∙ 103 M−1cm−1
Where n represents the frequency of occurrence of the bases in the conjugate.
High-performance liquid chromatography
For preparative purposes a Varian (USA) ProStar Preparative HPLC System with a preparative Juptier 10 u C18 300 Å column (10 μm, 250 × 10 mm; Phenomenex) with a flow rate of 8 mL/min was used. An UV detector at wavelength of 220 nm and 260 nm was used for signal detection. The eluents were ultrapure H2O with addition of 0.1% TFA (A) and MeCN with addition of 0.1% TFA (B).
For analytical purposes, an Agilent 1260 Infinity II HPLC-System (Agilent Technologies) with an eclipse XDB-C18 column (5 μm, 4.6 × 150 mm, Agilent) or with a EC 125/2 Nucleodur 100-C18 ec column (Macherey Nagel) was used. For the gradients an isocratic regime during the first 5 min, followed by a linear gradient of B as stated below with a flow rate of 1.00 mL/min (Agilent column) or 0.20 mL/min (MN column) were used. An UV detector at wavelength of 220 nm and 260 nm was used for signal detection. The eluents were ultrapure H2O with addition of 0.05% TFA (A) and MeCN with the addition of 0.03% TFA (B).
gradient 1: 2% to 30% of solvent B
gradient 2: 5% to 75% of solvent B
Solid-phase synthesis
General protocol
The synthesis was performed in a 2 mL polypropylene reactor with plunger and frit (25 μm poresize, Multi Syn Tech GmbH). TentaGel® S RAM resin (0.19 mmol/g, RAPP Polymere, Cat# R28023) was used as solid support. The amounts of reagents of the following synthesis protocol correspond to 2.5–5 μmol scale:
Swelling
The resin was swollen in 2 mL DMF for 30 min.
Deprotection of the temporal Fmoc group
The resin was treated twice with 500 µL piperidine (20% in DMF, v%) for 3 min each and subsequently washed with DMF (5 × 1.5 mL), CH2Cl2 (5 × 1.5 mL) and DMF (5 × 1.5 mL).
Coupling of Fmoc-amino acids
The Fmoc-amino acid (4.00 equiv), Oxyma (4.00 equiv) and DIC (4.00 equiv) were dissolved in DMF (0.20 M) and pre-activated for 3 min. The mixture was then added to the resin. After 45 min, the resin was washed with DMF (5 × 1.5 mL), CH2Cl2 (5 × 1.5 mL) and DMF (5 × 1.5 mL).
Coupling of Fmoc-PNA monomer
The Fmoc-PNA monomers (4.00 equiv), PyBOP (4.00 equiv) and NMM (8.00 equiv) were dissolved in DMF (0.20 M) and pre-activated for 3 min. The mixture was then added to the resin. After 45 min, the resin was washed with DMF (5 × 1.5 mL), CH2Cl2 (5 × 1.5 mL) and DMF (5 × 1.5 mL).
Capping
The resin was treated with 500 µL of a solution of 2,6-lutidine/Ac2O/DMF (6:5:89, v/v/v) for 5 min. The resin was washed with DMF (5 × 1.5 mL), CH2Cl2 (5 × 1.5 mL) and DMF (5 × 1.5 mL). After the last coupling, the resin was washed with (5 × 1.5 mL), CH2Cl2 (10 × 1.5 mL) and finally dried under reduced pressure.
Cleavage
The dried resin was treated with a solution of TFA/TiPS/DCM/H2O (90:2.5:2.5:5, v/v/v/v, 2.0 mL for 5 μmol scale and 1 mL for 2.5 μmol). After 150 min, the resin was washed with TFA (5 × 500 µL) and the combined eluates were added to dry ice-cold Et2O (1 mL of Et2O for each 100 μL of cleavage mixture). Centrifugation (4000 rpm, 5 min, 4°C) yielded a precipitate, which was washed with cold Et2O. The residue was dried under reduced pressure.
Purification
The crude product was dissolved in ultrapure H2O. The purification was performed as stated in the experimental procedures (High-performance liquid chromatography, for preparative purposes). Afterwards fractions containing the product were freeze-dried.
Characterization
Products were identified via analytical RP-HPLC-MS on an Agilent 1260 Infinity II LC system. The HPLC traces of all purified compounds can be found in Fig. S3-S13 in the supplemental information file.
H-cctatcacgattagcatt-GG-R8-CONH2 (PNA-R8) was obtained as a white solid (718 nmol, 14.4%). tR = 12.8 min (gradient 2, Fig. S3). Formula: C245H345N133O65. HRMS-ESI+ (m/z): [M + 8H]8+ calcd.: 775.1054; found: 775.1050.
H-atatcgctctagtaatcc-GG-R8-CONH2 (scrPNA-R8) was obtained as a white solid (764 nmol, 15.3%). tR = 12.7 min (gradient 2, Fig. S4). Formula: C245H345N133O65. HRMS-ESI+ (m/z): [M + 8H]8+ calcd.: 775.1054; found: 775.1052.
FAM-cctatcacgattagcatt-GG-R8-CONH2 (FAM-PNA-R8) was obtained as a pale-yellow solid (483 nmol, 9.66%). tR = 16.1 min (gradient 2, Fig. S5). Formula: C266H355N133O71. HRMS-ESI+ (m/z): [M + 8H]8+ calcd.: 819.8614; found: 819.8606.
H-cctatcacgattagcatt-GG-(RXR)4XB-CONH2 (PNA-secR8) was obtained as a white solid (5.6 nmol, 0.22%). tR = 15.2 min (gradient 2, Fig. S6). Formula: C278H405N139O71. HRMS-ESI+ (m/z): [M + 10 H]10+ calcd.: 683.9316; found, 683.9315.
FAM-cctatcacgattagcatt-GG-(RXR)4XB-CONH2 (FAM-PNA-secR8) was obtained as a white solid (4.9 nmol, 0.20%). tR = 16.4 min (gradient 2, Fig. S7). Formula: C299H415N139O77. HRMS-ESI+ (m/z): [M + 10 H]10+ calcd.: 719.8366; found: 719.8362.
H-cctatcacgattagcatt-GG-RXRRBRRXRYQFLIRXRBRXRB-CONH2 (PNA-Pip6a) was obtained as a white solid (486 nmol, 9.72%). tR = 13.1 min (gradient 2, Fig. S8). Formula: C325 H476N154O81. HRMS-ESI+ (m/z): [M + 7H]7+ cald 1120.4067; found, 1120.4079.
FAM-cctatcacgattagcatt-GG-RXRRBRRXRYQFLIRXRBRXRB-CONH2 (FAM-PNA-Pip6a) was obtained as a pale yellow solid (178 nmol, 7.12%). tR = 13.9 min (gradient 2, Fig. S9). Formula: C346H486N154O87. HRMS-ESI+ (m/z): [M + 9 H]9+ calcd.: 911.4344; found: 911.4346
H-cctatcacgattagcat-GG-K(FAM)-CONH2 (FAM-PNA) was obtained as a pale-yellow solid (175 nmol, 7.00%). tR = 24.7 min (gradient 1, Fig. S10). Formula: C213H257N99O60. HRMS-ESI+ (m/z): [M + 6 H]6+ calcd.: 861.1762; found: 861.1757.
H2N-K-acccctatcacgattagcattaa-KKK-CONH2 (PNA-K3) was obtained as a white solid (126 nmol, 2.52%). tR = 16.2 min (gradient 2, Fig. S11). Formula: C270H356N138O71. HRMS-ESI+ (m/z): [M + 7H]7+ calcd.: 953.8439; found: 953.8444.
FAM-K-acccctatcacgattagcattaa-KKK-CONH2 (FAM-PNA-K3) was obtained as a pale-yellow solid (258 nmol, 5.16%). tR = 17.1 min (gradient 2, Fig. S12). Formula: C291H366N138O77. HRMS-ESI+ (m/z): [M + 8 H]8+ calcd.: 879.1199; found: 879.1207.
H-cctatcacgattagcatt-GG-CONH2 (PNA) was obtained as a white solid (283 nmol, 11.3%) tR = 24.1 min (gradient 1, Fig. S13). Formula: C207H265N103O60. HRMS-ESI+ (m/z): [M + 6 H]6+ calcd.: 860.1890; found: 860.1917.
Author Contributions
GL and MF synthesized all compounds. GL, HJ, MF, and LNS performed experiments. AN and TS constructed and sequenced RNA-seq libraries. LNS performed bioinformatics analysis. BS provided funding and proof-read the manuscript. GL, LNS and OV designed experiments and wrote the manuscript. LNS and OV supervised the study and provided funding.
Data Availability
All RNA-seq datasets generated in this study have been deposited in the NCBI GEO repository and can be accessed through the GEO series number GSE152740.
Supplementary information is supplied as separate file
Supplemental Material
Download PDF (557.9 KB)Acknowledgments
We would like to thank Kerstin Hoffmann for assisting in cell isolation. This work was supported by the DFG SFB-TR84 “Innate Immunity of the Lung” (to L.N.S. / project C10 and B.S. / project C01), the DFG TRR81 “Chromatin Changes in Differentiation and Malignancies” (TRR81/3 Z04) (to O.V.), Philipps-Universität Marburg Forschungsförderfonds (project: development of novel inhibition strategies by targeting the human long non-coding RNA complement of the inflammatory response, to L.N.S. and O.V.), Bundesministerium für Bildung und Forschung (ERACoSysMed2 – SysMed-COPD – FKZ 031L0140; e:Med CAPSYS – FKZ 01ZX1604E; both to B.S.).
This study makes use of data generated by the Blueprint Consortium. A full list of the investigators who contributed to the generation of the data is available from www.blueprint-epigenome.eu. Funding for the project was provided by the European Union’s Seventh Framework Programme (FP7/2007-2013) under grant agreement no 282510 – BLUEPRINT.
Disclosure statement
The authors declare no competing interests.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
References
- Furman D, Campisi J, Verdin E, et al. Chronic inflammation in the etiology of disease across the life span. Nat Med. 2019;25(12):1822–1832.
- Jose RJ, Manuel ACOVID-19. COVID-19 cytokine storm: the interplay between inflammation and coagulation. Lancet Respir Med. 2020;8(6):e46–e7.
- Kellum JA, Kong L, Fink MP, et al. Understanding the inflammatory cytokine response in pneumonia and sepsis: results of the genetic and inflammatory markers of sepsis (GenIMS) study. Arch Internal Med. 2007;167:1655–1663.
- Leifer CA, Medvedev AE. Molecular mechanisms of regulation of toll-like receptor signaling. J Leukoc Biol. 2016;100:927–941.
- Schulte LN, Bertrams W, Stielow C, et al. ncRNAs in inflammatory and infectious diseases. Methods Mol Biol. 2019;1912:3–32.
- Walther K, Schulte LN. Role of long non-coding RNAs in innate immunity and inflammation. RNA Biol. 2020; Nov 19;1–17. doi:10.1080/15476286.2020.1845505
- Schulte LN, Westermann AJ, Vogel J. Differential activation and functional specialization of miR-146 and miR-155 in innate immune sensing. Nucleic Acids Res. 2013;41:542–553.
- Taganov KD, Boldin MP, Chang KJ, et al. NF-kappa B-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proceedings of the National Academy of Sciences of the United States of America 2006; 103:12481–12486.
- Tang B, Xiao B, Liu Z, et al. Identification of MyD88 as a novel target of miR-155, involved in negative regulation of Helicobacter pylori-induced inflammation. FEBS Lett. 2010;584:1481–1486.
- O’Connell RM, Chaudhuri AA, Rao DS, et al. Inositol phosphatase SHIP1 is a primary target of miR-155. Proceedings of the National Academy of Sciences of the United States of America 2009; 106:7113–7118.
- Mehta A, Baltimore D. MicroRNAs as regulatory elements in immune system logic. Nat Rev Immunol. 2016;16:279–294.
- Fabani MM, Abreu-Goodger C, Williams D, et al. Efficient inhibition of miR-155 function in vivo by peptide nucleic acids. Nucleic Acids Res. 2010;38(13):4466–4475.
- Cheng CJ, Bahal R, Babar IA, et al. MicroRNA silencing for cancer therapy targeted to the tumour microenvironment. Nature. 2015;518(7537):107–110.
- Dong SXM, Caballero R, Ali H, et al. Transfection of hard-to-transfect primary human macrophages with Bax siRNA to reverse resveratrol-induced apoptosis. RNA Biol. 2020;17(6):755–764.
- Keller AA, Maess MB, Schnoor M, et al. Transfecting macrophages. Macrophages: Methods and Protocols 2018;1784:187–195.
- Miyakawa K, Matsunaga S, Yokoyama M, et al. PIM kinases facilitate lentiviral evasion from SAMHD1 restriction via Vpx phosphorylation. Nat Commun. 2019;10(1):1844.
- Shen X, Corey DR. Chemistry, mechanism and clinical status of antisense oligonucleotides and duplex RNAs. Nucleic Acids Res. 2018;46(4):1584–1600.
- Godfrey C, Desviat LR, Smedsrod B, et al. Delivery is key: lessons learnt from developing splice-switching antisense therapies. EMBO Mol Med. 2017;9(5):545–557.
- Lehto T, Ezzat K, Wood MJA, et al. Peptides for nucleic acid delivery. Adv Drug Deliv Rev. 2016;106:172–182.
- Shieh PB. Emerging strategies in the treatment of duchenne muscular dystrophy. Neurotherapeutics. 2018;15(4):840–848.
- Roberts TC, Langer R, Wood MJA. Advances in oligonucleotide drug delivery. Nat Rev Drug Discov. 2020;19:673–694.
- Nan Y, Zhang YJ. Antisense phosphorodiamidate morpholino oligomers as novel antiviral compounds. Front Microbiol. 2018;9:750.
- Tsoumpra MK, Fukumoto S, Matsumoto T, et al. Peptide-conjugate antisense based splice-correction for Duchenne muscular dystrophy and other neuromuscular diseases. EBioMedicine. 2019;45:630–645.
- Janga H, Aznaourova M, Boldt F, et al. Cas9-mediated excision of proximal DNaseI/H3K4me3 signatures confers robust silencing of microRNA and long non-coding RNA genes. PloS One. 2018;13(2):e0193066.
- Futaki S, Suzuki T, Ohashi W, et al. Arginine-rich peptides. An abundant source of membrane-permeable peptides having potential as carriers for intracellular protein delivery. J Biol Chem. 2001;276(8):5836–5840.
- Rothbard JB, Kreider E, VanDeusen CL, et al. Arginine-rich molecular transporters for drug delivery: role of backbone spacing in cellular uptake. J Med Chem. 2002;45(17):3612–3618.
- Ivanova GD, Arzumanov A, Abes R, et al. Improved cell-penetrating peptide-PNA conjugates for splicing redirection in HeLa cells and exon skipping in mdx mouse muscle. Nucleic Acids Res. 2008;36(20):6418–6428.
- Hammond SM, Hazell G, Shabanpoor F, et al. Systemic peptide-mediated oligonucleotide therapy improves long-term survival in spinal muscular atrophy. Proceedings of the National Academy of Sciences of the United States of America 2016; 113:10962–10967.
- Yin H, Saleh AF, Betts C, et al. Pip5 transduction peptides direct high efficiency oligonucleotide-mediated dystrophin exon skipping in heart and phenotypic correction in mdx mice. Mol Ther. 2011;19(7):1295–1303.
- Gait MJ, Arzumanov AA, McClorey G, et al. Cell-Penetrating Peptide Conjugates of Steric Blocking Oligonucleotides as Therapeutics for Neuromuscular Diseases from a Historical Perspective to Current Prospects of Treatment. Nucleic Acid Ther. 2019;29(1):1–12.
- Betts C, Saleh AF, Arzumanov AA, et al. Pip6-PMO, A New Generation of Peptide-oligonucleotide Conjugates With Improved Cardiac Exon Skipping Activity for DMD Treatment. Mol Ther Nucleic Acids. 2012;1:e38.
- Silverman AP, Kool ET. Detecting RNA and DNA with templated chemical reactions. Chem Rev. 2006;106:3775–3789.
- Gerhardt T, Ley K. Monocyte trafficking across the vessel wall. Cardiovasc Res. 2015;107(3):321–330.
- Hirose H, Takeuchi T, Osakada H, et al. Transient focal membrane deformation induced by arginine-rich peptides leads to their direct penetration into cells. Mol Ther. 2012;20:984–993.
- Ceppi M, Pereira PM, Dunand-Sauthier I, et al. MicroRNA-155 modulates the interleukin-1 signaling pathway in activated human monocyte-derived dendritic cells. Proceedings of the National Academy of Sciences of the United States of America 2009; 106:2735–2740.
- Soudah T, Khawaled S, Aqeilan RI, et al. AntimiR-155 cyclic peptide-PNA conjugate: synthesis, cellular uptake, and biological activity. ACS Omega. 2019;4:13954–13961.
- Iiott NE, Heward JA, Roux B, et al. Long non-coding RNAs and enhancer RNAs regulate the lipopolysaccharide-induced inflammatory response in human monocytes.. Nat Commun. 2014;5:5.
- Aznaourova M, Janga H, Sefried S, et al. Noncoding RNA MaIL1 is an integral component of the TLR4-TRIF pathway. Proceedings of the National Academy of Sciences of the United States of America 2020; 117:9042–9053.
- Zschaler J, Schlorke D, Arnhold J. Differences in innate immune response between man and mouse. Crit Rev Immunol. 2014;34(5):433–454.
- Mestas J, Hughes CCW. Of mice and not men: differences between mouse and human immunology. J Iimmunol. 2004;172(5):2731–2738.
- Sazani P, Gemignani F, Kang SH, et al. Systemically delivered antisense oligomers upregulate gene expression in mouse tissues. Nat Biotechnol. 2002;20(12):1228–1233.
- Halloy F, Iyer PS, Cwiek P, et al. Wildner-Verhey van Wijk N, et al. Delivery of oligonucleotides to bone marrow to modulate ferrochelatase splicing in a mouse model of erythropoietic protoporphyria. Nucleic Acids Research 2020;48:4658–4671.
- Bayraktar R, Bertilaccio MTS, Calin GA. The Interaction Between Two Worlds: microRNAs and Toll-Like Receptors. Front Immunol. 2019;10:1053.
- Shi Y, Yuan B, Zhu W, et al. Ube2D3 and Ube2N are essential for RIG-I-mediated MAVS aggregation in antiviral innate immunity. Nat Commun. 2017;8(1):15138.
- Gewurz BE, Towfic F, Mar JC, et al. Genome-wide siRNA screen for mediators of NF-kappaB activation. Proceedings of the National Academy of Sciences of the United States of America 2012; 109:2467–2472.
- Balk RA. Systemic inflammatory response syndrome (SIRS): where did it come from and is it still relevant today? Virulence. 2014;5(1):20–26.
- Gottwein E, Mukherjee N, Sachse C, et al. A viral microRNA functions as an orthologue of cellular miR-155. Nature. 2007;450(7172):1096–1099.
- Yin Q, McBride J, Fewell C, et al. MicroRNA-155 is an epstein-barr virus-induced gene that modulates epstein-barr virus-regulated gene expression pathways. J Virol. 2008;82(11):5295–5306.
- Wyler E, Mösbauer K, Franke V, et al. Bulk and single-cell gene expression profiling of SARS-CoV-2 infected human cell lines identifies molecular targets for therapeutic intervention. BioRxiv. 2020. https://doi.org/10.1101/2020.05.05.079194
- O’Connell RM, Rao DS, Chaudhuri AA, et al. Sustained expression of microRNA-155 in hematopoietic stem cells causes a myeloproliferative disorder. J Exp Med. 2008;205(3):585–594.
- Lai X, Dreyer FS, Cantone M, et al. Network- and systems-based re-engineering of dendritic cells with non-coding RNAs for cancer immunotherapy. Theranostics. 2021;11(3):1412–1428.
- Hargreaves BKV, Roberts SE, Derfalvi B, et al. Highly efficient serum-free manipulation of miRNA in human NK cells without loss of viability or phenotypic alterations is accomplished with TransIT-TKO.. PloS One. 2020;15(4):15.
- Vencken S, Foged C, Ramsey JM, et al. Nebulised lipid-polymer hybrid nanoparticles for the delivery of a therapeutic anti-inflammatory microRNA to bronchial epithelial cells.. ERJ Open Res. 2019;5(2):5.
- Ran FA, Hsu PD, Wright J, et al. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013;8:2281–2308.
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(T)(-Delta Delta C) method. Methods. 2001;25:402–408.
- O’Brien J, Wilson I, Orton T, et al. Investigation of the alamar blue (resazurin) fluorescent dye for the assessment of mammalian cell cytotoxicity. Eur J Biochem. 2000;267:5421–5426.
- Qian ZQ, Martyna A, Hard RL, et al. Discovery and mechanism of highly efficient cyclic cell-penetrating peptides. Biochemistry. 2016;55:2601–2612.
- Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–675.
- Saldanha AJ. Java Treeview–extensible visualization of microarray data. Bioinformatics. 2004;20:3246–3248.
- Kamburov A, Wierling C, Lehrach H, et al. ConsensusPathDB–a database for integrating human functional interaction networks. Nucleic Acids Res. 2009;37:D623–8.
- Adams D, Altucci L, Antonarakis SE, et al. BLUEPRINT to decode the epigenetic signature written in blood. Nat Biotechnol. 2012;30:224–226.
- Vazquez O, Seitz O. Cytotoxic peptide-PNA conjugates obtained by RNA-programmed peptidyl transfer with turnover. Chem Sci. 2014;5:2850–2854.