
ABSTRACT
Pseudomonas aeruginosa harbours two redundant RNA-binding proteins RsmA/RsmN (RsmA/N), which play a critical role in balancing acute and chronic infections. However, in vivo binding sites on target transcripts and the overall impact on the physiology remains unclear. In this study, we applied in vivo UV crosslinking immunoprecipitation followed by RNA-sequencing (UV CLIP-seq) to detect RsmA/N-binding sites at single-nucleotide resolution and mapped more than 500 binding sites to approximately 400 genes directly bound by RsmA/N in P. aeruginosa. This also verified the ANGGA sequence in apical loops skewed towards 5ʹUTRs as a consensus motif for RsmA/N binding. Genetic analysis combined with CLIP-seq results suggested previously unrecognized RsmA/N targets involved in LPS modification. Moreover, the RsmA/N-titrating RNAs RsmY/RsmZ may be positively regulated by the RsmA/N-mediated translational repression of their upstream regulators, thus providing a possible mechanistic explanation for homoeostasis of the Rsm system. Thus, our study provides a detailed view of RsmA/N-RNA interactions and a resource for further investigation of the pleiotropic effects of RsmA/N on gene expression in P. aeruginosa.
Introduction
The gram-negative bacterium Pseudomonas aeruginosa is an opportunistic pathogen infecting humans that thrives in diverse environments. It can cause serious biofilm-associated infections in burn wounds, indwelling devices, and the lungs of immunocompromised cystic fibrosis patients [Citation1]. The infection processes are governed via complex post-transcriptional regulatory mechanisms stimulated by certain environmental stresses such as nutrient starvation and antibiotic exposure [Citation2]. Such post-transcriptional control networks are typically composed of globally acting RNA-binding proteins (RBPs) together with small non-coding RNAs (sRNAs).
Host factor protein for the replication of phage qβ (Hfq) is one of the most well-known RBPs that acts as an RNA chaperone and helps sRNAs basepair with target mRNAs for repression or activation of the translations [Citation3]. Similar to Escherichia coli or Salmonella, Hfq contributes to varied phenotypes such as quorum sensing, virulence, and antibiotic resistance in P. aeruginosa [Citation4–6]. Likewise, the CsrA family, originally discovered as a carbon storage regulator in E. coli, also plays a global role in post-transcriptional control in many bacteria [Citation7]. Unlike Hfq, the CsrA family functions by repressing translation directly by competing with the ribosome rather than acting as a matchmaker between sRNA and mRNA in Gram-negative bacteria. CsrA family proteins bind to target RNAs via a stem-loop structure comprising an ANGGA motif. Conversely, translational suppression is inhibited by at least three sRNAs, namely, CrsB, CrsC, and McaS, each containing multiple GGA motifs in E. coli [Citation8,Citation9].
P. aeruginosa has two CsrA homologues RsmA and RsmN (also known as RsmF) and four redundant sRNAs RsmV, RsmW, RsmY, and RsmZ that function as antagonists of RsmA and RsmN [Citation10–12]. RsmA and RsmN (RsmA/N) are structurally distinct with respect to the position of an α helix constituent; RsmA is composed of five consecutive β sheets and a C-terminal α helix whereas RsmN consists of a uniquely inserted α helix between the β2 and β3 sheets [Citation13,Citation14]. RsmA/N-titrating sRNAs, RsmY/RsmZ (RsmY/Z), are regulated by GacA/GacS (GacAS) two-component system. When the transmembrane histidine protein kinase (HPK) GacS is activated by certain environmental signals, it phosphorylates its cognate response regulator GacA [Citation15]. Upon phosphorylation, GacA promotes the transcription of RsmY/Z [Citation16]. While RsmW is activated in the stationary phase or by high temperature [Citation11], the level of RsmV expression is relatively low during the whole course of the growth, independent of GacAS activity [Citation12]. Both RsmA/N bind to some, but not all, common regulatory targets via a conserved arginine residue. Most of the known RsmA/N targets such as genes encoding type VI secretion systems (T6SS) and exopolysaccharide biosynthesis are subject to direct translational repression [Citation17,Citation18]. In contrast, the genes encoding motility and type III secretion systems (T3SS) are positively regulated by RsmA/N [Citation15,Citation19], thus modulating the transition between acute and chronic infections in P. aeruginosa.
These relationships between RsmA/N and target genes have been extensively investigated by phenotypic assays combined with genetic analysis in vitro. However, the complexity of in vivo RNA-based regulation makes attaining a comprehensive understanding of the mode of actions of RsmA/N difficult. To address this, recent studies have identified target transcripts, that are bound by members of the CsrA/Rsm family via co-immunoprecipitation approaches [Citation20,Citation21]. Such methods can identify major RNA ligands. However, they do not provide sufficient information with respect to the RBPs-binding sites within each transcript. Herein, we performed in vivo UV crosslinking immunoprecipitation followed by RNA-seq (UV CLIP-seq) with RsmA/N. In this method, the complex of RsmA/N and bound RNAs are UV cross-linked and an additional RNA trimming step is performed to identify their regulatory targets at a single-nucleotide resolution in vivo. Our approach found more than 500 potential-binding sites genome-wide, which enabled us to gain new insights into the biological functions and the similarities and differences between the two redundant Rsm RBPs in P. aeruginosa.
Materials and methods
Strains and growth conditions
Strains used herein are enlisted in Table S3. All experiments were performed using P. aeruginosa PAO1 or its derived strains. Each strain was cultured at 37°C in Luria-Bertani (LB) medium. Samples were collected at the OD600 values indicated in the figures. Where indicated, the appropriate AHLs were added at the following final concentrations: C4-HSL (Cayman Chemicals) at 10 μM and 3OC12-HSL (RTI International) at 2 μM. Antibiotics and arabinose were used as needed at concentrations listed as follows. For P. aeruginosa, gentamicin, tetracycline, and arabinose at concentrations 50 μg/ml, 80 μg/ml, and 0.1%, respectively, were used. Similarly, for E. coli, gentamicin and tetracycline at concentrations 10 μg/ml and 20 μg/ml, respectively, were used.
Strain construction
P. aeruginosa PAO1 chromosomal mutants were constructed based on the conjugative transfer of appropriate plasmids and homologous recombination between chromosomes and plasmids as described previously [Citation22]. The detail is provided in Supplementary Information. Plasmids and oligonucleotides used herein are enlisted in Table S3.
UV crosslinking, immunoprecipitation, and RNA purification
Bacterial cultures with volume 200 ml of three replicates were maintained up to an OD600 of 2.0 in LB medium. For RsmN expression from the multicopy plasmid, 0.1% arabinose was added at an OD600 of 0.8. UV cross-linking and immunoprecipitation were performed in accordance with the previously published protocol with minor modifications [Citation23]. Briefly, half of the culture at an OD600 of 2.0 from each condition was irradiated at 800 mJ of UV light at 254 nm. After UV cross-linking, the sample was centrifuged at 3,600 rpm for 30 min at 4°C along with the non-cross-linked control samples.
Cell pellets were lysed in FastPrep 24 (MP-Biomedicals) at 6 m/s for 1 min with 1 ml of 0.1-mm glass beads and 800 μl NP-T buffer (50 mM NaH2PO4, 300 mM NaCl, 0.05% Tween20, pH 8.0). NP-T supplemented with 8 M urea was added to each supernatant at an equal volume and incubated for 5 min at 65°C with shaking at 900 rpm. Anti-FLAG magnetic beads were washed three times with 500 μl NP-T buffer, resuspended in 125 μl NP-T buffer, and treated with a 10-fold diluted suspension of urea-treated samples. After 1 h of incubation at 4°C, samples were washed twice with 500 μl high-salt buffer (50 mM NaH2PO4, 1 M NaCl, 0.05% Tween20, pH 8.0), followed by two washes with 500 μl NP-T buffer. Beads were resuspended in benzonase mix [500 units benzonase nuclease (E1014, Sigma-Aldrich) in NP-T buffer with 1 mM MgCl2] and incubated for 10 min at 37°C with shaking at 900 rpm. After one wash with high-salt buffer and two washes with CIP buffer (100 mM NaCl, 50 mM Tris-HCl, pH 7.4, 10 mM MgCl2), beads were resuspended in 200 μl CIP mix (20 units of calf intestinal alkaline phosphatase (M0290, NEB) in CIP buffer) and incubated for 30 min at 37°C with shaking at 800 rpm.
Here, radioactive isotope (RI) labelling was performed only in the preliminary investigation to check whether UV crosslinking did enrich RNA-protein complex (). After one wash with high-salt buffer and two washes with PNK buffer (50 mM Tris-HCl, pH 7.4, 10 mM MgCl2, 0.1 mM spermidine), beads were resuspended in 200 μl PNK buffer and separated into 20 μl and 180 μl volumes to be used subsequently for western blotting and RI labelling, respectively. For RI labelling, beads were resuspended in 100 μl PNK mix [10 units of T4 polynucleotide kinase (EK0032, ThermoFisher Scientific), 10 μCi γ-32P-ATP in PNK buffer]) and incubated for 30 min at 37°C, following the addition of 1 μl 10 mM non-radioactive ATP and incubation for 5 min at 37°C. After two washes with NP-T buffer, beads were resuspended in 25 μl 2xProtein loading buffer and incubated for 5 min at 95°C. Supernatants were transferred to fresh tubes. The elution was repeated twice. For western blotting, 20 μl separated beads were washed twice with 100 μl NP-T buffer and resuspended in 10 μl 2xProtein loading buffer and incubated for 5 min at 95°C. Supernatants were transferred to fresh tubes. The elution was repeated twice.
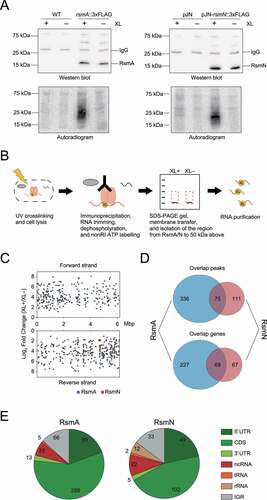
Figure 1. Overview of RsmA/N CLIP-seq. (A) Representative figures of western blotting (top) and autoradiogram (bottom) of the CLIP-enriched RsmA/N-RNA complexes. XL+: crosslinking, XL-: non-crosslinking. (B) Graphical summary of nonRI CLIP-seq approach. (C) The distribution of RsmA/N binding sites across the P. aeruginosa genome. Peaks from RsmA and RsmN CLIP-seq are highlighted in blue and red, respectively. (D) Overlapping peaks (top) and genes (bottom) between RsmA and RsmN bindings. Two peaks from both RBPs wherein both the start and stop positions are within 40 nt were considered as the same peak. (E) Classification of RsmA/N peaks into RNA classes (5ʹ UTR, CDS, 3ʹ UTR, sRNA, tRNA, rRNA, and Intergenic peaks). Note that a peak may be classified into multiple classes based on its position
In the CLIP experiments for RNA purification and cDNA preparation, RI labelling was not used (). After CIP reaction, beads were resuspended in 100 μl PNK mix [10 units of T4 polynucleotide kinase (EK0032, ThermoFisher Scientific), 1 μl 10 mM non-radioactive ATP in PNK buffer] and incubated for 30 min at 37°C, followed by addition of 1 μl 10 mM non-radioactive ATP and incubation for 5 min at 37°C. After two washes with NP-T buffer, beads were resuspended in 30 μl 2xProtein loading buffer and incubated for 5 min at 95°C. Supernatants were transferred to fresh tubes. The elution was repeated twice.
Aliquots of 55 μl were loaded and separated via SDS-polyacrylamide gel electrophoresis (12% resolving gel) at 30 mA while moving through the stacking gel, which was then increased to 40 mA. After RNA electrophoresis, the RNA was transferred from the gel to the Protran membrane (#10,600,016, GE Healthcare). The membrane was placed on a clean glass surface and cut from the prestained protein markers to 50 kDa above them. Each membrane piece was cut into smaller pieces and placed in 2 ml fresh tubes with 400 μl PK solution [1.3 mg/ml Proteinase K (EO0491, ThermoFisher Scientific), and 10 units of RNase inhibitor (#10,777,019, Invitrogen) in 2xPK buffer (100 mM Tris-HCl, pH 7.9, 10 mM EDTA, 1% SDS)], followed by incubation for 1 h at 37°C with shaking at 1,000 rpm. The samples were subsequently incubated with 100 μl PK buffer containing 9 M urea for 1 h at 37°C with shaking at 1,000 rpm. Thereafter, 450 μl of supernatants from proteinase K-treated membranes were mixed with an equal volume of phenol:chloroform:isoamyl alcohol in phase-lock gel tubes and incubated for 5 min at 30°C with shaking at 1,000 rpm. Each mixture was centrifuged for 12 min at 13,000 rpm at 4°C, and 400 μl of the aqueous phase was precipitated with three volumes of ice-cold ethanol, 1/30 volume of 3 M NaOAc (pH 5.2), and 1 μl of GlycoBlue (AM9515, Invitrogen) for 2 h at −20°C. The precipitated pellet was washed with 80% ethanol, briefly dried for 5 min at 20°C, and resuspended in 11 μl of sterilized water.
cDNA library preparation and sequencing
A cDNA library of the CLIP samples was prepared using the NEBNext Multiplex Small RNA Library Prep Set for Illumina (#E7300, NEB) in accordance with the manufacturer’s instructions. RT primer and both 3ʹ/5ʹ SR adapters were diluted 10-fold with nuclease-free water before use. cDNA was converted from 2.5 μl of each purified RNA. Reverse-transcribed cDNAs were amplified by 20 cycles PCR. PCR products were concentrated to 10 μl using the MinElute PCR Purification kit and separated by 6% polyacrylamide gel with 7 M urea. Bands between 130 bp and 250 bp were cut and purified from the gel in accordance with the manufacturer’s instructions. Purified cDNAs of 5 μl volume were amplified by six cycles PCR and concentrated to 10 μl using the MinElute PCR Purification kit. Final cDNA libraries were quantified using the Qubit dsDNA assay (Q32854, Invitrogen) and Agilent 2100 Bioanalyzer DNA HS (Agilent). Twelve cDNA libraries from CLIP were pooled on an Illumina HiSeq2500 and sequenced in paired-end mode (2 × 50 cycles).
Sequence processing, mapping, normalization, and peak calling
To assure high sequence quality, read 1 (R1) and read 2 (R2) files containing the Illumina paired-end reads in FASTQ format were quality and adapter-trimmed by Cutadapt [Citation24] version 1.15/1.16 using a cut-off Phred score of 20. Reads without any remaining bases were discarded (command-line parameters: -q 20 -m 1 -a AGATCGGAAGAGCACACGTCTGAACTCCAGTCAC -A GATCGTCGGACTGTAGAACTCTGAACGTGTAGATCTCGGTGGTCGCCGTATCATT). To eliminate putative PCR duplicates, paired-end reads were collapsed using FastUniq [Citation25] After trimming, we applied the pipeline READemption [Citation26] version 0.4.5 to align all reads longer than 11 nt to the P. aeruginosa PAO1 chromosome (NCBI accession no. NC_002516.2) reference genome using segemehl [Citation27] version 0.2.0 with accuracy cut-off of 80%.
Read counts per position were analysed using reads that mapped uniquely to single genomic position. The core of positions present in cross-linked and non-cross-linked library pairs was isolated, as described previously [Citation28]. Positions with low read counts were filtered from both cross-linked and non-crosslinked libraries using 10 standard deviations from 0 as a threshold (Supplementary Figure 7A). The distribution of ratios between the paired cross-linked and non-crosslinked samples presented a clear bimodal distribution, with modes corresponding to background (high counts in both libraries) and cross-link-enriched positions, which were separated by k-means clustering. Size factors were then calculated from the background positions present across all replicates against a geometric mean pseudo-reference, as introduced in DESeq [Citation29].
We applied PEAKachu v0.1.0 (https://github.com/tbischler/PEAKachu) for peak calling similarly to as previously described protocol [Citation28]. First, BAM files for the respective pairs of cross-linked and non-cross-linked libraries were used to run in paired-end (-P) and paired-replicates (-r) mode. The maximum fragment size (-M) was set to 50 and annotations generated as GFF format were used to map overlapping features to the called peaks. Normalization was performed in the ‘manual’ mode using previously determined size factors (see above). Other parameters were set to default. Second, the boundary of initial peaks was set through block definition computed by the blockbuster algorithm [Citation30] based on pooled read alignments from all cross-linked libraries using default parameters. Third, the PEAKachu tool ran the DESeq2 package [Citation31] to analyse the significance of peak enrichment in the cross-linked libraries relative to the non-crosslinked libraries with parameter values as follows: mad-multiplier (-m) 1.0, fold change (-f) 1.0, and adjusted p-value (-Q) 0.05. Finally, PEAKachu was used for each replicon and strand to generate normalized coverage plots to facilitate data visualization.
RsmA/N-Enrichment index calculation
We applied READemption to map reads on P. aeruginosa PAO1 chromosome (NCBI accession no. NC_002516.2) reference genome, assess nucleotide-wise coverage, and count the number of reads overlapping with each of the annotation entries on the sense strand [Citation26]. Raw data for total RNA-seq were retrieved from NCBI’s Gene Expression Omnibus (GEO, http://www.ncbi.nlm.nih.gov/geo) under accession number GSE136112 [Citation23]. These RNA-seq data were generated using total RNA extracted from cross-linked samples grown to OD600 = 2.0. The resulting read counts were subjected to differential expression analysis of cross-linked RsmA/N-binding RNAs vs. total RNAs via DESeq2 [Citation31]. Genes with log2 Fold Change (cross-linked RsmA or RsmN-binding RNAs/total RNAs) of 1.807 or greater (RNAs 3.5-fold more abundant in the RsmA/N-bound fractions than in the total RNA) were selected as RsmA/N targets as with the case of previously performed RsmN RIP-seq (Supplementary Table 2) [Citation20].
Analysis of sequence and structural motifs
The sequences of peaks were used to perform MEME sequence motif analysis [Citation32]. Minimum and maximum motif widths were set at 6 and 50, respectively, while other parameters were set to default. Structural motifs of the sequences of peak regions extended by additional 10 nt upstream and downstream were analysed using CMfinder 0.2.1 [Citation33]. The minimum length of single stem-loop candidates was set to 20, while other parameters were set to default. Each analysed motif was visualized using R2R [Citation34].
Statistical and other analysis
Descriptive statistical analyses for peak and gene overlapping between RsmA and RsmN, peak distributions across the P. aeruginosa genome, and peak classification among RNA classes were performed using Microsoft Excel. Meta-gene analyses across detected mRNAs or the P. aeruginosa genome were performed with in-house script using Python3. Genes identified via CLIP-seq analysis were functionally characterized using the PseudoCAP annotation (http://www.pseudomonas.com). Gene Ontology enrichment analysis was performed for mRNAs identified via CLIP-seq analysis using DAVID (https://david.ncifcrf.gov/summary.jsp). Default parameters and databases were used for the analysis.
Translational fusion assay
P. aeruginosa wild type and ΔrsmA/N strains carrying the sfGFP translational fusions were grown overnight in 1 ml LB medium with 80 μg/ml tetracycline at 37°C. An aliquot with 2 μl volume was resuspended in 200 μl LB medium with 80 μg/ml tetracycline and transferred to black polystyrene 96-well microplates with a clear, flat bottom (Corning). The medium was additionally supplemented with gentamicin with concentration 50 μg/ml and 0.1% arabinose for rsmA/N and SPA0079 expression from multicopy plasmids. Fluorescence polarization (FP476/510) was measured with SpectraMax GeminiXS (Molecular Devices) every 15 min for 6 h at 37°C with agitation for 1 min before fluorescence measurement. The fluorescent cut-off was set to 495 nm. Thereafter, the final optical density (OD) of each culture was obtained by GeneQuant 1300 (Biochrom). GFP activity was expressed in arbitrary units as FP476/510/OD600. The detail of the construction of translational fusion plasmid is provided in Supplementary Information.
qRT-PCR
Total RNA was extracted by the hot phenol extraction method followed by DNase treatment. cDNAs from 1 μg DNase-treated purified RNA were obtained using PrimeScriptTM RT Master Mix (#RR036A, TAKARA) following the manufacturer’s instruction. qRT-PCR was performed for triplicates. Reactions were performed in a total volume of 20 μl containing 10 μl of TB Green Fast qPCR Mix (#RR430A, TAKARA), 0.4 μl of ROX reference dye II, 0.8 μl of each primer (10 μM), 7 μl of RNase-free water and 1 μl of template DNA. PCR and fluorescence measurements were performed using the viiA7 (Applied Biosystems) with the following program; preheating at 95°C for 30 s, followed by 40 cycles of denaturation at 95°C for 5 s and annealing at 60°C for 20 s. The gene rpoD was used as an internal standard [Citation35]. Relative gene expression was calculated using the ΔΔCt method [Citation36].
5ʹ and 3ʹ rapid amplification of cDNA ends (RACE)
5ʹ and 3ʹ RACE assays were performed as previously described with some modifications [Citation37]. For 5ʹ RACE assay, 15 μg of DNA-depleted RNA was incubated with 12.5 units of RNA 5ʹ pyrophosphohydrolase (RppH) (#M0356, NEB) in a 50 μl reaction for 1 h at 37°C. The same volume of RNA without the RppH reaction was prepared to be used as a negative control. After incubation, RppH-reacted and control RNAs were purified and 1.25 μM 5ʹRACE adaptor (5ʹ–GAU AUG CGC GAA UUC CUG UAG AAC GAA CAC UAG AAG AAA–3ʹ) was mixed. The samples were denatured at 95°C for 5 min. The adaptor was ligated using T4 RNA Ligase 1 overnight at 16°C. The adaptor-ligated RNA was purified, annealed with gene-specific primer and reverse-transcribed using AffinityScript Multiple Temperature Reverse Transcriptase (#600,107, Agilent) with following conditions; 42°C for 20 min, 55°C for 20 min, and 70°C for 15 min in a 20 μl reaction. One-micro-litre of reverse-transcribed cDNA was amplified by nested PCR with a total volume of 50 μl containing 0.4 μM of 5ʹadaptor-specific primer, 0.4 μM of 1st round gene-specific primer, 200 μM of each dNTP, 1x PCR buffer, and 1.25 units of TaKaRa Taq HS (#R007A, TAKARA). Second PCR was performed with a total volume of 50 μl containing 1 μl of PCR product obtained after the first round, 0.4 μM of 5ʹadaptor-specific primer, 0.4 μM of 2nd round gene-specific primer, 200 μM of each dNTP, 1x PCR buffer, and 1.25 units of TaKaRa Taq HS (#R007A, TAKARA). Following conditions were used for both PCRs: preheating at 95°C for 3 min, followed by 30 cycles of denaturation at 94°C for 30 s, annealing at 56°C for 30 s, extension at 72°C for 30 s, and then final extension at 72°C for 7 min.
For 3ʹ RACE assay, 15 μg of DNA-depleted RNA was incubated with 25 units of calf intestinal alkaline phosphatase (CIP) (#M0290, NEB) in a 50 μl reaction volume for 1 h at 37°C. After incubation, RNA reacted with CIP was purified and 1.25 μM 3ʹRACE adaptor (5ʹ-phosphate-UUC ACU GUU CUU AGC GGC CGC AUG CUC-idT −3ʹ) was mixed. The samples were denatured at 95°C for 5 min. The adaptor was ligated overnight using T4 RNA Ligase 1 at 17°C. The adaptor-ligated RNA was purified, annealed with gene-specific primer and reverse-transcribed using AffinityScript Multiple Temperature Reverse Transcriptase (#600,107, Agilent) with following conditions; 42°C for 20 min, 55°C for 20 min and 70°C for 15 min in a 20 μl reaction. Reverse-transcribed cDNA of 1 μl was amplified by PCR in a total volume of 50 μl containing 0.4 μM of 3ʹadaptor-specific primer, 0.4 μM of gene-specific primer, 200 μM of each dNTP, 1x PCR buffer, and 1.25 units of TaKaRa Taq HS (#R007A, TAKARA). The PCR was conducted as follows: preheating at 95°C for 3 min, followed by 30 cycles of denaturation at 94°C for 30 s, annealing at 56°C for 30 s, extension at 72°C for 30 s, and then final extension at 72°C for 7 min.
PCR products were separated on 4% NuSieve GTG agarose gel, the band of interest was cut, gel-eluted, and cloned into a BamHI/HindIII digested pUC19. Bacterial colonies obtained after the transformation were screened by colony PCR. The PCR fragments were purified by QIAquick PCR purification kit and sequenced with an ABI Genetic analyser 3500 (Applied Biosystems).
Northern blotting
Purified RNA of 10 μg was denatured at 95°C for 5 min in gel loading buffer II (#AM8547, Invitrogen) and separated by 6% polyacrylamide gel with 7 M urea at 300 V for 2 h. Separated RNA was electro-transferred from the gel to Hybond-N+ membranes (GE Healthcare) at 50 V for 1 h at 4°C and the membranes were UV-crosslinked (120 mJ/cm2). Northern blotting was performed with the Roche DIG system. DNA probes for 5S rRNA and SPA0079 were amplified with PCR using primers described in Table S3 and PCR DIG Probe Synthesis Kit (#11,636,090,910, Roche). The reaction was performed with a total volume of 50 μl containing 0.2 μM of forward primer, 0.2 μM of reverse primer, 200 μM of dATP, dCTP, and dGTP, 130 μM of dTTP, 70 μM of DIG-11-dUTP, 1x PCR buffer, and 2.625 units of Enzyme mix, Expand High Fidelity (#1,732,641, Roche). The PCR conditions were set as follows: preheating at 95°C for 2 min, followed by 30 cycles of denaturation at 95°C for 30 s, annealing at 60°C for 30 s, extension at 72°C for 40 s and then final extension at 72°C for 7 min.
UV-cross-linked membranes were prehybridized with 10 ml of DIG EasyHyb for 30 min at 50°C. Thereafter, DIG-labelled DNA probes were hybridized overnight at 50°C in 15 ml of DIG EasyHyb. Membranes were washed every 15 min in 5× Saline Sodium Citrate (SSC)/0.1% SDS, 1× SSC/0.1% SDS and 0.5× SSC/0.1% SDS buffers at 50°C followed by washing with maleic acid buffer (0.1 M maleic acid, 0.15 M NaCl, 0.3% Tween-20 pH 7.5) for 5 min at 37°C. Thereafter, membranes were blocked with blocking solution (#11,585,762,001, Roche) for 45 min at 37°C, and probed with 75 mU/ml Anti-Digoxigenin-AP (#11,093,274,001, Roche) in blocking solution for 45 min at 37°C. Membranes were then washed again in maleic acid wash buffer in two 15-min steps and equilibrated with detection buffer (0.1 M Tris–HCl, 0.1 M NaCl, pH 9.5). Signals were visualized by CDP-star (#12,041,677,001, Roche) with Fusion Fx Imaging System (Vilber-Lourmat).
Western blotting
One-hundred bacterial cultures at OD600 = 2.0 were centrifuged at 10,000 rpm for 2 min at 22°C and resuspended in 100 μl 1xProtein loading buffer, followed by incubation for 5 min at 95°C. Aliquots of 5 μl were separated on 10% TGX gel (Bio-Rad) and subsequently transferred onto a polyvinylidene difluoride (PVDF) membrane using a Transblot Turbo Transfer System (Bio-Rad).
For the CLIP procedure, 5 μl aliquots of heat-denatured immunoprecipitated RNA-protein complex were loaded and separated via SDS-polyacrylamide gel electrophoresis (12% resolving gel) at 30 mA per gel while moving through the stacking gel, which was then increased to 40 mA. After electrophoresis, proteins were electro-transferred onto a PVDF membrane.
Transferred membranes were blocked in 1xTBS-T buffer (20 mM Tris, 150 mM NaCl, 0.1% Tween20, pH 7.6) with 10% skim milk for 45 min. Thereafter, the membrane was probed overnight at 4°C with monoclonal α-FLAG (#31,430, ThermoFisher Scientific; 1:10,000) antibody diluted in 1× TBS-T buffer containing 3% bovine serum albumin, washed three times for 5 min each in 1× TBS-T buffer, probed for 1 h with anti-mouse-HRP-antibody (F1804, Sigma-Aldrich; 1:50,000) diluted in 1× TBS-T buffer containing 3% bovine serum albumin, and washed three times for 5 min each in 1× TBS-T buffer. Chemiluminescent signals were detected using Immobilon Western Chemiluminescent HRP substrate (#WBKLS0500, Millipore) and measured with Fusion Fx Imaging System (Vilber-Lourmat).
Results
Transcriptome-wide mapping of RsmA/N-binding sites
In order to identify RsmA/N-binding sites through the P. aeruginosa transcriptome, we used a chromosomally tagged rsmA::3×FLAG strain and plasmid-inducible rsmN::3×FLAG strain (Supplementary Discussion 1). Both were grown to early stationary phase (OD600 = 2.0) and UV irradiated to induce covalent bonds between RNAs and the corresponding proteins in vivo. Autoradiography and western blot analyses indicated that UV crosslinking and co-immunoprecipitation with anti-FLAG antibodies along with stringent washing enriched RsmA-RNA and RsmN-RNA complexes (). It should be noted that we omitted RNA radiolabelling steps and cut membranes from the regions consisting of RsmA/N bands up to the region 50 kDa above for RNA purification ().
Purified RNAs from cross-linked and non-cross-linked samples in triplicates were reverse transcribed into cDNA and subjected to Illumina sequencing. By performing a comparative analysis of called peaks from RsmA/N-binding regions between cross-linked and non-crosslinked libraries using DESeq2, we detected 411 and 186 of binding sites for RsmA and RsmN with significant enrichment (false discovery rate (FDR) <0.05 and minimum expression = 1.0) throughout the P. aeruginosa transcriptome, respectively (Supplementary Table 1, ). We identified 75 overlapping peaks between RsmA and RsmN, with overlapping peaks defined as the peaks that exhibited both the start and stop positions within 40 nt of each other, thus constituting 69 overlapping genes (). Since this CLIP-seq was carried out in conditions similar to the previously performed Hfq CLIP-seq [Citation23], the genes that overlapped between RsmA/N and Hfq were also investigated. A total of 25 genes including the genes encoding outer membrane proteins were common among all three RBPs (Supplementary Figure 2A). Gebhardt et al. recently performed chromatin immunoprecipitation with cells grown in the presence or absence of rifampicin followed by high-throughput DNA sequencing (ChIPPAR-seq) for P. aeruginosa RsmA to capture a subset of nascent transcripts [Citation21]. When compared with the list of transcripts that are regulated co-transcriptionally by RsmA, approximately 25% of RsmA-binding genes were also detected in our CLIP-seq (Supplementary Figure 2B). In addition, Romero et al. mapped more than 500 transcripts directly bound by RsmN using RNA immunoprecipitation and sequencing (RIP-seq) [Citation20]. We observed that a limited number of RsmN-binding genes were detected in both our CLIP-seq and the RIP-seq (Supplementary Figure 2C). To compare our CLIP-seq data with RsmN RIP-seq more precisely, we additionally calculated an enrichment index by comparing read counts for RsmN-binding RNAs in our CLIP-seq to total RNA expression (Supplementary Table 2). We set the same cut-off value as with above-mentioned RsmN RIP-seq (see Materials and methods). As a result, approximately 17% of RsmN-enriched genes were also detected in our CLIP-seq. Calculating the enrichment index for RsmA-binding RNAs as well, we eventually found 757 and 890 enriched genes for RsmA and RsmN, respectively, with 538 overlapping genes (hereSupplementary Table 2).
Finally, significant peaks were classified on the basis of RNA classes using previously generated UTR annotation lists [Citation23,Citation38,Citation39] and manually curated P. aeruginosa sRNA lists [Citation40–42]. Most of the peaks were mapped within mRNAs, in which more than 40% of the binding sites in CDSes overlapped with the first quarter of the CDSes (, Supplementary Figure 3A and B). Interestingly, approximately half of ncRNAs bound by RsmA were annotated as cis-antisense RNAs (asRNAs) (Supplementary Figure 3C), suggesting that certain post-transcriptional regulatory mechanisms involving RsmA and asRNAs are still unrecognized (Supplementary Discussion 2). Taken together, RsmA/N CLIP-seq analysis identified hundreds of binding sites associated with each RBP.
Similarities and differences in sequence and structural motifs bound by RsmA/N
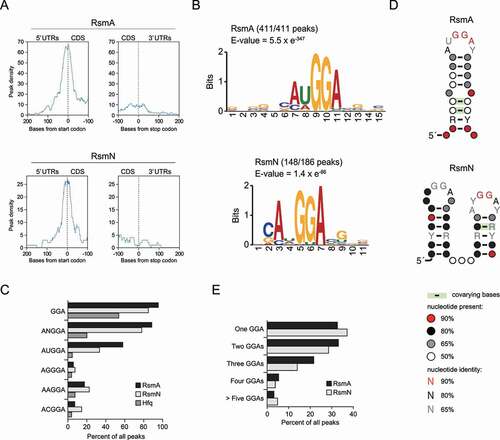
RsmA/N bind to conserved GGA sequences within or close to Shine-Dalgarno sequences of target mRNAs [Citation19]. More precisely, parallel systematic evolution of ligands by exponential enrichment (SELEX) studies for RsmA/N have identified a common consensus motif CANGGAYG positioned in a hexaloop region of the stem-loop structure [Citation43]. To understand the consensus sequence bound by RsmA/N and its position in the target transcripts in vivo, the peak density of RsmA/N peaks across all detected mRNAs was determined via meta-gene analysis using start or stop codons as reference points. Strong peak densities were observed around start codons but not around stop codons, showing that P. aeruginosa RsmA/N preferentially bind to the 5ʹ UTRs of mRNAs (). This result is consistent with CsrA family protein being translational repressor competing with ribosomes at Shine-Dalgarno sequence [Citation44]. Next, sequence motifs for RsmA/N-bindings were determined using all detected RsmA/N-binding sequences by MEME motif analysis. Top-ranked motifs of both RsmA/N contained the ANGGA sequence (). All RsmA-binding sites exhibited the motif. In fact, more than 80% of detected RsmA/N peaks demonstrated at least one minimal GGA motif, and the second nucleotide of the ANGGA sequence for RsmA binding showed a preference for uracil (). Unlike RsmA, the second nucleotide position of the ANGGA sequence for RsmN binding is more tolerant, accepting any nucleotides except for guanine (–C). In order to understand whether the detected motifs were likely to be present in the apical loop, all detected RsmA/N-binding sequences were subjected to CMfinder structural motif analysis. The top-ranked structural motifs from CMfinder were predicted as highly conserved stem-loop with the GGA sequence in the loop regions (). Interestingly, the top-ranked structural motif for RsmN binding demonstrated two stem-loops with GGA sequences adjacent to each other, consistent with the previous report showing that RsmN requires two binding sites of the known target tssA1 [Citation43]. Therefore, the number of GGA sequences per peak was investigated to evaluate whether RsmN requires two adjacent GGA sequences for high-affinity binding. Although we expected that RsmN might tend to bind peaks corresponding to more than two GGA motifs, no significant difference was observed between RsmA and RsmN (). Altogether, RsmA/N CLIP-seq demonstrates ANGGA in loop regions as a general recognition sequence/structural motif, which is in clear accordance with previously published works.
Figure 2. Consensus motif for P. aeruginosa RsmA/N bindings. (A) Meta-gene analysis for RsmA/N bindings along mRNAs with start and stop codons as the reference points. (B) MEME sequence motif analysis for all RsmA (411) and RsmN (186) peaks. The numbers indicate the peaks containing predicted sequence motifs. (C) Percentage of peaks with indicated sequences. (D) CMfinder structural motif analysis of all RsmA (411) and RsmN (186) peak sequences extended with 10 nt upstream and downstream. Top-ranked structural motifs are shown. (E) Percentage of peaks with the indicated number of GGA sequence per peak sequence extended with 10 nt upstream and downstream
RsmA/N CLIP-seq expands global-binding targets in P. aeruginosa
When the small non-coding RNAs RsmY/Z are highly expressed, RsmA/N are titrated away from target RNAs playing an important role in the regulation of virulence factors associated with acute and chronic infections. The results of RsmA/N CLIP-seq conducted in this study demonstrate that the read coverage of RsmY/Z was highest among all other genes (, Supplementary Table 1). RsmY/Z exhibits multiple GGA motif sites for RsmA/N binding and high coverage positions of RsmA/N binding corresponded to their GGA motifs although the comparison of cross-linked and non-cross-linked of RsmA CLIP-seq did not show a significant enrichment of RsmY/Z (). To assess the interaction with RsmY/Z by an alternative approach, we calculated an enrichment index defined as ratio between CLIP-enriched RNA and total RNA. The RNA-seq data was previously generated using total RNA extracted from cross-linked samples grown to OD600 = 2.0 [Citation23]. RsmY and RsmZ exhibit high RsmA-enrichment index of 8.49 and 9.02, indicating they are 3.59 × 102 and 5.19 × 102 more abundant in RsmA-bound samples, respectively (Supplementary Table 2). Unlike RsmA/N, Hfq predominantly associated with their Rho-independent terminators () [Citation23,Citation45]. Our data also show that RsmA/N indeed bind to well-known target mRNAs. For examples, high CLIP-seq coverages were found at the 5ʹUTR of the gene tssA1 encoding structural component of the Hcp secretion island-I-encoded T6SS and 5ʹUTR of the gene mucA encoding anti-sigma factor (). The RsmA/N-binding sites detected on these mRNAs fold into hairpins with GGA motifs (). Collectively, the data suggest that RsmA/N CLIP-seq was able to capture the bona fide RsmA/N-binding sites.
Figure 3. RsmA/N CLIP-seq captures previously known RsmA/N-binding sites. (A) Read coverage from RsmA/N at the sRNAs RsmY/Z loci. Vertical axis indicates each read count. XL+: crosslinking, XL-: non-crosslinking. (B) Secondary structures of sRNAs RsmY/Z predicted by Mfold [Citation54]. Red and shaded letters indicate RsmA/N and Hfq binding peaks, respectively. Bold letters indicate GGA sequence as a common binding motif of RsmA/N. (C) Read coverage at the tssA1 and mucA loci from RsmA/N CLIP-seq are indicated. (D) Secondary structures of RsmA/N binding sites at the tssA1 and mucA were predicted using Mfold [Citation54]. Red letters indicate GGA sequence as a common binding motif of RsmA/N
![Figure 3. RsmA/N CLIP-seq captures previously known RsmA/N-binding sites. (A) Read coverage from RsmA/N at the sRNAs RsmY/Z loci. Vertical axis indicates each read count. XL+: crosslinking, XL-: non-crosslinking. (B) Secondary structures of sRNAs RsmY/Z predicted by Mfold [Citation54]. Red and shaded letters indicate RsmA/N and Hfq binding peaks, respectively. Bold letters indicate GGA sequence as a common binding motif of RsmA/N. (C) Read coverage at the tssA1 and mucA loci from RsmA/N CLIP-seq are indicated. (D) Secondary structures of RsmA/N binding sites at the tssA1 and mucA were predicted using Mfold [Citation54]. Red letters indicate GGA sequence as a common binding motif of RsmA/N](/cms/asset/2d27130d-c498-4227-a357-e25e3c91f45d/krnb_a_1917184_f0003_oc.jpg)
To determine the biological processes in which RsmA/N-binding RNAs were enriched, DAVID enrichment analysis was performed for mRNAs containing peaks, with a modified Fisher’s exact p-value threshold <0.1 [Citation46]. Genes associated with cell wall organization, polysaccharide biosynthetic process, O-antigen biosynthetic process, and lipopolysaccharide (LPS) biosynthetic process were enriched (). Among the enriched pathways, high peak density was detected within the wbp gene cluster involved in O5 O-antigen biosynthesis (). Since the category represents an unknown role for RsmA/N, we decided to explore them in more detail.
Figure 4. The wbp gene cluster is post-transcriptionally regulated by RsmA/N. (A) DAVID enrichment analysis of RsmA/N peaks. The results of biological process are presented. (B) RsmA/N peak density distribution along the P. aeruginosa chromosome in bins of 2 × 104 basepairs. (C) Read coverage at the wbp gene cluster from RsmA/N CLIP-seq. TSS annotations (black arrows) were derived from a previous study [Citation39]. Vertical axis indicates each read count. RsmA and RsmN-binding peaks with significant enrichment (FDR < 0.05) are indicated as red and blue bars, respectively. The genes shown in red were used for the following translational fusion assay. (D) Super-folder GFP translational fusion assay for the indicated genes between wild type and rsmAN mutant. Welch’s t-test results are indicated: N.S., not significant; *, p-value < 0.05; **, p-value < 0.01; ***, p-value < 0.001. The reported values with the standard error represent the average of three independent experiments
![Figure 4. The wbp gene cluster is post-transcriptionally regulated by RsmA/N. (A) DAVID enrichment analysis of RsmA/N peaks. The results of biological process are presented. (B) RsmA/N peak density distribution along the P. aeruginosa chromosome in bins of 2 × 104 basepairs. (C) Read coverage at the wbp gene cluster from RsmA/N CLIP-seq. TSS annotations (black arrows) were derived from a previous study [Citation39]. Vertical axis indicates each read count. RsmA and RsmN-binding peaks with significant enrichment (FDR < 0.05) are indicated as red and blue bars, respectively. The genes shown in red were used for the following translational fusion assay. (D) Super-folder GFP translational fusion assay for the indicated genes between wild type and rsmAN mutant. Welch’s t-test results are indicated: N.S., not significant; *, p-value < 0.05; **, p-value < 0.01; ***, p-value < 0.001. The reported values with the standard error represent the average of three independent experiments](/cms/asset/8178331d-a40d-4ec5-84f1-945a30876da0/krnb_a_1917184_f0004_oc.jpg)
The wbp gene cluster consists of three genes responsible for the assembly of the O-antigen (wzx, wzy, and wzz), 12 genes involved in the biosynthesis assembly of the nucleotide sugars of the O unit (wbpABCDEGHIJKLM), three insertion genes encoding putative transposase and integrase (PA3142–PA3144) between wbpL and wbpM, and two non-LPS-related genes (hisF2 and hisH2) between wbpG and wzx [Citation47]. Significant peaks indicating RsmA binding were observed in wzz, wbpA, wbpB, hisH2, hisF2, wbpG, wbpH, wbpL, and wbpM genes whereas those indicating RsmN binding were observed in wbpA, wbpE, wbpI, and wbpM genes (). We verified the function with respect to RsmA/N-mediated gene regulation against six genes of the cluster using super-folder GFP (sfGFP) translational fusion assay (Supplementary Discussion 3). Of them, four (wzz, wbpA, wbpE, and wbpG) were significantly upregulated upon rsmA/N deletion, suggesting that RsmA/N post-transcriptionally repress the translation of each gene (). Altogether, our RsmA/N CLIP-seq results found new RsmA/N regulatory targets associated with LPS modification.
Possible homoeostatic regulation of RsmY/Z expressions by an RsmA/N-controlled feedback loop
The expression of RsmA/N-titrating sRNAs RsmY/Z is activated by GacAS two-component system [Citation16]. The GacAS system is controlled by three additional HPKs: RetS, PA1611, and LadS [Citation48,Citation49]. In addition to these complex regulatory pathways, RsmY and RsmZ RNA levels are independently affected by the histidine phosphotransfer protein HptB and endoribonuclease CafA (RNase G), respectively [Citation50,Citation51]. Expression of hptB is regulated by additional membrane-binding proteins SagS and ErcS [Citation50], whereas the cafA expression is under the control of another two-component system BfiR/BfiS [Citation51]. Among those proteins involved in the homoeostasis of RsmY/Z expression, we observed RsmA/N-binding peaks in the sagS, gacS, ladS, hptB, and cafA genes (). Since HptB is the decision hub for RsmY transcription via the phosphorylation of additional regulatory components HsbA/HsbD and flagellar gene expression via FlgM sequestration [Citation52], we focused on RsmA/N-mediated hptB gene regulation. We also investigated whether RsmA/N directly regulated cafA gene expression since CafA specifically affects the level of RsmZ RNA with its endoribonucleolytic activity.
Figure 5. RsmA/N-controlled feedback loop for RsmY/Z sRNAs expressions. (A) Graphical summary of RsmA/N bindings with GacAS regulatory network. Orange and purple ovals are membrane-bound histidine kinases and response regulators in two-component systems, respectively. Asterisks indicate that RsmA/N peaks are associated with the genes. (B) Read coverage at the cafA and hptB loci from RsmA/N CLIP-seq. Vertical axis indicates each read count. RsmN-binding peaks with significant enrichment (FDR < 0.05) are indicated as red bars. (C) Super-folder GFP translational fusion assay for the indicated genes between wild type and rsmAN mutant. (D) Super-folder GFP translational fusion assay for the indicated genes in rsmAN mutant with indicated plasmids. For exogenous RsmA/N expressions, 0.1% arabinose was added. (E) Relative expressions of RsmY/Z in PAO1 wild type and ΔrsmA/N. RNA was extracted from cultures at OD600 = 2.0 and RsmY/Z abundances were quantified by qRT-PCR. Welch’s t-test results are indicated: *, p-value < 0.05; **, p-value < 0.01; ***, p-value < 0.001 (C and E) and One-way ANOVA and Tukey’s HSD test results are indicated: **, p-value < 0.01; N.S., not significant (D). The reported values with the standard error represent the average of three independent experiments
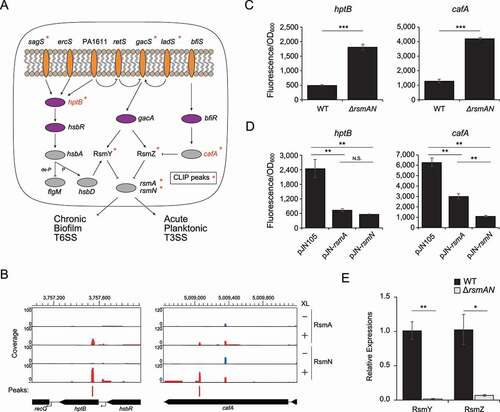
We observed RsmA/N-binding peaks in both hptB and cafA coding sequences, though only RsmN binding was statistically significant (). Therefore, we performed the sfGFP translational fusion assay to understand the RsmA/N associated gene regulation of hptB and cafA. Both were significantly upregulated in an rsmA/N deletion strain (). In order to investigate whether both RsmA and RsmN individually repress hptB and cafA translation, RsmA and RsmN were exogenously expressed from multicopy plasmid pJN105 in ΔrsmA/N background and GFP fluorescence was measured. As expected, each was capable of repressing translation (). If hptB expression is negatively regulated by RsmA/N, downstream HptB-dependent secretion and biofilm anti-sigma factor HsbA should be phosphorylated and diguanylate cyclase HsbD should activate RsmY expression. Additionally, if cafA expression is negatively regulated by RsmA/N, degradation of RsmZ by CafA should be alleviated. To confirm these hypotheses, RsmY/Z abundance between the wild type and ΔrsmAN strains was quantified by qRT-PCR. As expected, RsmY/Z levels were significantly reduced in the ΔrsmAN strain (), which is consistent with a previous report that shows low RsmY/Z abundance in an rsmA mutant [Citation48,Citation53]. Altogether, this suggests that RsmY/Z expression is balanced by RsmA/N-controlled feedback loop.
RsmA/N bind to multiple sRNAs other than RsmY/Z in P. aeruginosa
Besides RsmY/Z, we observed high read coverage and significant enrichment in cross-linked samples for several sRNAs. For example, SPA0035 sRNA, a transcript from the minus strand of the intergenic region between ada and PA2119, was significantly bound to RsmA/N, but not to Hfq (Supplementary Figure 4A, left) [Citation23]. As another example, the PAO1-specific asRNA SPA0066 transcribed from the opposite strand of PA3993 was also significantly bound to RsmA/N, but not to Hfq (Supplementary Figure 4A, right) [Citation23]. It should be noted that SPA0066 overlapped in the antisense orientation of the 5ʹ UTR of a gene encoding a putative transposase of the IS116/IS110/IS902 family. This transposase family exists in six genomic positions in PAO1, all of which demonstrate identical asRNAs (SPA0004–8 and SPA0066). The SPA0079 sRNA, a transcript from the minus strand of the intergenic region comprising 1,011 nt between PA2763 and PA2764, was significantly bound to RsmA/N, as well as to Hfq () [Citation23]. The majority of these sRNAs carry GGA motifs and many corresponding peaks fold into hairpins ( and Supplementary Figure 4B). Considering the significant binding to both Hfq and RsmA/N, we decided to investigate the characteristic of SPA0079 as an RsmA/N-binding sRNA in next section.
Figure 6. SPA0079 does not titrate RsmA/N under our conditions (A) Read coverage at the SPA0079 locus from RsmA/N CLIP-seq. Vertical axis indicates each read count. XL+: crosslinking, XL-: non-crosslinking. (B) Secondary structure of SPA0079 in P. aeruginosa PAO1 predicted by Mfold [Citation54]. Red letters indicate AUGGA sequence as a common binding motif of RsmA/N. (C) Super-folder GFP translational fusion assay for cafA gene between pJNS105 control vector and pJNS-SPA0079 overexpression vector. For exogenous SPA0079 expression, 0.1% arabinose was added. The reported values represent the average of three independent experiments and the standard error
![Figure 6. SPA0079 does not titrate RsmA/N under our conditions (A) Read coverage at the SPA0079 locus from RsmA/N CLIP-seq. Vertical axis indicates each read count. XL+: crosslinking, XL-: non-crosslinking. (B) Secondary structure of SPA0079 in P. aeruginosa PAO1 predicted by Mfold [Citation54]. Red letters indicate AUGGA sequence as a common binding motif of RsmA/N. (C) Super-folder GFP translational fusion assay for cafA gene between pJNS105 control vector and pJNS-SPA0079 overexpression vector. For exogenous SPA0079 expression, 0.1% arabinose was added. The reported values represent the average of three independent experiments and the standard error](/cms/asset/937a2ab2-733e-4f5e-b3e7-c65f8e5af7f6/krnb_a_1917184_f0006_oc.jpg)
SPA0079 sRNA does not sequester RsmA/N away from target mRNAs under our conditions
We first determined the SPA0079 boundaries by 5ʹ/3ʹ RACE analysis. RNA pyrophosphohydrolase RppH treatment followed by PCR amplification selectively enriched 5ʹ terminus with triphosphate ends of SPA0079 (the band indicated by a red arrow in Supplementary Figure 5B). The 3ʹ terminus of SPA0079 corresponded to the canonical U repeats of the rho-independent terminator ( and Supplementary Figure 5A). The SPA0079 secondary structure predicted by Mfold [Citation54] exhibited only a single stem-loop, having a strong resemblance to a rho-independent transcription terminator (). The expression of SPA0079 in the wild type and the deletion mutant were measured throughout the course of growth (Supplementary Figure 5C). The maximal level of the SPA0079 expression was reached in the late stationary phase at the OD600 of 6.0 in the wild type (Supplementary Figure 5C, lanes 1–5). In contrast, SPA0079 sRNA was undetectable in the ΔSPA0079 mutant strain (Supplementary Figure 5C, lane 6).
A previous study has suggested that two quorum sensing (QS) pathways LasI/LasR and RhlI/RhlR might be related to the expression of SPA0079 [Citation55]. Furthermore, the DNA sequence of the upstream of −35 promoter motif is similar to the consensus motif of LasR/RhlR binding (Supplementary Figure 5A). To investigate QS-dependent SPA0079 expression, northern blotting analysis was performed with a ΔlasIΔrhlI double mutant strain with or without the exogenous supplementation of two acyl homoserine lactones (AHLs) 3OC12-HSL and C4-HSL. SPA0079 displayed strict dependence on QS as it accumulated within 1 h after AHLs were added (Supplementary Figure 5D). Additionally, SPA0079 was not detectable in a ΔlasI strain, whereas it was detected moderately in a ΔrhlI strain (Supplementary Figure 5E). Taken together with the published report [Citation55], these results suggest that the expression of SPA0079 is controlled by the QS systems. Another previous report showed that Hfq stabilizes RsmY by blocking cleavage by RNase E [Citation45]. We investigated whether Hfq might affect SPA0079. Northern blotting showed no difference in the SPA0079 levels in a Δhfq mutant compared to its levels in wild type and ΔrsmA/N mutant (Supplementary Figure 5F).
To investigate whether SPA0079 indeed titrates RsmA/N away from the mRNA target, the activity of sfGFP fused with 5ʹ UTR and CDS of cafA was assayed with or without SPA0079 overexpression. Although we speculated that a cafA::sfGFP fusion would be activated by the induction of SPA0079, no clear difference was observed in GFP activity in the SPA0079 overexpression state compared to that in the ΔrsmA/N mutant strain (). Considering that the level of SPA0079 expression reaches maximum in the late stationary phase (Supplementary Figure 5C), and that RsmY/Z predominantly bind to RsmA/N under the conditions in which CLIP-seq was performed, it may be that SPA0079 sRNA competes or complements RsmY/Z and also target the mRNAs of RsmA/N under specific conditions not captured in our assay. Future studies are required to determine whether or not SPA0079 is a functional RsmA/N antagonist.
Discussion
Two conserved and redundant RBPs RsmA/N play a critical role in balancing acute and chronic infections in P. aeruginosa. So far, the interactions of RsmA/N with target RNAs have been extensively explored in vitro. However, it is prudent to understand the mode of actions in the complex transcriptional networks in vivo. In this study, we performed RsmA/N CLIP-seq analysis and demonstrated more than 500 genome-wide RsmA/N-binding sites. Many genes identified in this study could be attractive targets for further elucidating the regulatory mechanisms of RsmA/N in P. aeruginosa.
When compared with previously published data, RsmA/N-binding genes demonstrated in this study were relatively few in number (Supplementary Figure 2). This variation may be attributed to the differences in the experimental setup. RsmA ChIPPAR-seq captured nascent transcript at mid-log phase and significant-binding positions are defined as enrichment in the minus rifampicin library compared with the plus rifampicin library [Citation21]. RsmN RIP-seq was previously performed during the late stationary phase and significant-binding genes were defined using an enrichment index generated by comparison of co-immunoprecipitation library with total RNA [Citation20]. In addition, the small overlap of detected genes with previous reports perhaps derives from the limitations of bioinformatics analysis (Supplementary Discussion 4). Nevertheless, numerous known targets of RsmA/N were observed from our CLIP-seq (). As examples, we showed high CLIP-seq coverages observed at the 5ʹUTRs of the mRNAs tssA1 and mucA and sRNAs RsmY/Z, leading to the identification of their RsmA/N-binding sites in vivo ().
The advantage of CLIP-seq is to detect protein-binding sites on the transcript at a single-nucleotide resolution. This approach has demonstrated sequence/structural-binding motifs of the CsrA family in E. coli and Salmonella [Citation44,Citation56]. Herein, we investigated the similarities and differences between P. aeruginosa RsmA and RsmN on the basis of sequence/structural motifs and a typical distribution of the detected peaks (). The results of the peak distribution among mRNAs were similar between the two RBPs, indicating that RsmA/N peaks are highly enriched at only 5ʹ UTRs, which is consistent with the previous reports [Citation20,Citation57] (–D). In addition, sequence and structural motif analyses demonstrated that the stem-loop structure with the ANGGA sequence in its loop region is characteristic of RsmA/N peaks. Moreover, approximately half of RsmA/N-binding transcripts identified in this study have two or more core GGA motifs adjacent to RsmA/N-binding sites. Because RsmA/N exists as homodimer with two identical-binding sites, our data suggest that single transcripts simultaneously interact with both binding sites through multiple GGA motifs in vivo. However, it remains unclear whether such bridging of single transcripts to each site is necessary for RsmA/N function. It should be noted that mixed CsrA heterodimers with RNA-binding-defective monomers have comparable RNA-binding affinity to WT CsrA for target transcripts containing two GGA motifs but shows weak activity for in vitro repression of the translation of target transcripts [Citation58].
Although all of the RsmA peaks detected in this study include the ANGGA motif, RsmN peaks seem to have a more permissive binding motif (see the number of detected peaks in ). From the molecular perspective, the RsmA homologue RsmE in Pseudomonas fluorescens CHA0 needs the looped out nucleotide base N in ANGGA motif and baseparing in the stem for rigid binding through Leu55 to Ala57 in the C-terminal α helix as well as the correct stacking [Citation59]. In contrast, the α helix in RsmN is located within the internalized β2 and β3 sheets, constituting the hydrophobic core that acts as a potential new RNA-binding site [Citation14]. Thus, RsmN may bind with a more flexible loop motif independent of C-terminal α helix even without obvious ANGGA sequence. The structural motif of RsmN peaks consisted of two tandem stem-loop structures with GGA motifs (). Consistent with this, an in vitro SELEX study and mutational analysis have demonstrated long RsmA/N targets with two consensus GGA motifs, and found that two tandem GGA sites are required only for RsmN binding [Citation43]. Overall, our CLIP-seq analysis combined with previous reports suggested a unique two-sidedness of RsmN binding; tandem stem-loop structures with two GGAs or flexible sequences without obvious ANGGA sequence.
Further genetic analysis has elucidated new regulatory targets of RsmA/N involved in O5 O-antigen biosynthesis (). The three genes wzx, wzy, and wzz are responsible for the assembly of the O-antigen and other genes wbpABCDEGHIJKLM are involved in the biosynthesis assembly of the nucleotide sugars of the O unit [Citation47]. Since we observed RsmA bindings to both wzz and wzz2 encoding O-antigen chain length regulators and validated the RsmA/N-mediated translational repression of wzz (, Supplementary Table 1), active post-transcriptional regulation through RsmA/N would lead to an LPS-rough phenotype, which is often observed in P. aeruginosa isolates from chronic pulmonary infections [Citation60,Citation61]. In a recent study, low levels of wzz2 expression led to the conversion to mucoid phenotype with alginate production, and a transcriptional factor AmrZ negatively regulated the expression [Citation62]. Under the same AlgU regulon with AmrZ, AlgR activates alginate production and in turn, directly stimulates RsmA expression [Citation63]. This might accelerate modulation of O-antigen length and eventually biofilm formation [Citation64]. Translational repression of genes involved in O-antigen biosynthesis through RsmA/N is also consistent with the findings demonstrating that the absence of B-band O-antigen is correlated with the increase of T3SS-mediated cytotoxicity that is activated by RsmA/N directly [Citation65]. Taking into account previous ChIP-seq analysis showing that T3SS master regulator ExsA binds to promoter sequences of wbpA and wbpH [Citation66], the wbp gene cluster is likely to be under the both transcriptional and translational regulation mediated by RsmA/N.
Free RsmA/N and the level of the titrating sRNAs are controlled through homoeostatic regulation. Although it was previously shown that RsmA exerts a positive effect on RsmY and RsmZ transcription in P. aeruginosa and Pseudomonas protegens [Citation48,Citation53,Citation67,Citation68], the mechanisms underlying the positive feedback loop remain unclear. In this study, hptB encoding the histidine phosphotransfer protein and cafA encoding the ribonuclease G were found as potential candidate RsmA/N regulatory targets (). If RsmA/N repress the translation of hptB, RsmY expression would be stimulated via the phosphorylation of HsbA and subsequent activation of HsbD [Citation50]. In addition to RsmY, if CafA expression is repressed in an RsmA/N-dependent manner, RsmZ would escape from degradation owing to the endoribonucleolytic activity [Citation51], thus constituting a three-repressor feedback loop to maintain RsmY/Z levels. However, we still cannot rule out the possibility that RsmA/N binding to RsmY/Z simply confers protection from degradation. Future studies should be tested to determine whether deletion of hptB and cafA abolishes the effects of RsmA/N on the abundance of RsmY/Z. Meanwhile, since RsmY/Z expression increases throughout the growth period [Citation16], other factors might be involved in the homoeostatic regulations of RsmY/Z. For example, polynucleotide phosphorylase PNPase regulates the stabilities of RsmY/Z [Citation69]. Our CLIP-seq data has identified RsmA binding to the gene encoding PNPase (Supplementary Table 1), which implies that RsmA may affect RsmY/Z through the availability of PNPase.
Finally, we discovered 41 RsmA/N-binding sRNAs besides known RsmA/N-titrating sRNAs (Supplementary Table 1). Among them, SPA0079 was highly enriched in both Hfq and RsmA/N CLIP-seq, motivating us to further investigate its properties. The level of SPA0079 expression reached its maximum in late stationary phase and was undetectable in ΔlasIΔrhlI mutant, whereas exogenous supplementation of two AHLs complimented the expression ( and Supplementary Figure 5), strongly supporting the fact that SPA0079 is regulated by two QS pathways [Citation55]. Although we expected that SPA0079 would alleviate RsmA/N-mediated translational repression, no clear activation of translational fusion activity was observed with SPA0079 sRNA overexpression (). Given that RsmY/Z predominantly bind to RsmA/N under the conditions in which CLIP-seq was performed and the effect of SPA0079 is negligible, future studies should be performed using ΔrsmY/Z mutant to determine whether or not the SPA0079 sRNA is indeed an RsmA/N antagonist. A recent paper shows that sRNA179 (also annotated as SPA0034) expression stimulates RsmY transcription [Citation70]. Additionally, the expression of sRNA179 is activated by QS similar to SPA0079 [Citation55]. Considering these observations, QS might suppress the RsmA/N regulatory system through two different post-transcriptional pathways, highlighting the complexity of Rsm regulatory systems.
Supplemental Material
Download Zip (5.5 MB)Acknowledgments
We thank Akiko Yokota and Tetsushi Suyama for their assistance with the registration of this study in AIST. We also thank Thorsten Bischler for his help with CLIP-seq analysis and Jörg Vogel for his helpful comments on this study.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
Raw sequence data are available in the DDBJ Sequenced Read Archive (https://www.ddbj.nig.ac.jp/dra/index-e.html) under the accession number DRA010307.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Mulcahy LR, Isabella VM, Lewis K. Pseudomonas aeruginosa biofilms in disease. Microb Ecol. 2014 July;68(1):1–12.
- Holmqvist E, Wagner EGH. Impact of bacterial sRNAs in stress responses. Biochem Soc Trans. 2017 November;45(6):03.
- Chao Y, Vogel J. The role of Hfq in bacterial pathogens. Curr Opin Microbiol. 2010 February;13(1):24–33.
- Sonnleitner E, Hagens S, Rosenau F, et al. Reduced virulence of a hfq mutant of Pseudomonas aeruginosa O1. Microb Pathog. 2003;35(5):217–228.
- Sonnleitner E, Schuster M, Sorger-Domenigg T, et al. Hfq-dependent alterations of the transcriptome profile and effects on quorum sensing in Pseudomonas aeruginosa. Mol Microbiol. 2006 March;59(5):1542–1558.
- Pusic P, Sonnleitner E, Krennmayr B, et al. Harnessing metabolic regulation to increase Hfq-dependent antibiotic susceptibility in Pseudomonas aeruginosa. Front Microbiol. 2018;9:9.
- Romeo T, Gong M, Liu MY, et al. Identification and molecular characterization of csrA, a pleiotropic gene from Escherichia coli that affects glycogen biosynthesis, gluconeogenesis, cell size, and surface properties. J Bacteriol. 1993 August;175(15):4744–4755.
- Babitzke P, Romeo T. CsrB sRNA family: sequestration of RNA-binding regulatory proteins. Curr Opin Microbiol. 2007 April;10(2):156–163.
- Holmqvist E, Vogel J. A small RNA serving both the Hfq and CsrA regulons. Genes Dev. 2013 May 15;27(10):1073–1078.
- Kay E, Humair B, Denervaud V, et al. Two GacA-dependent small RNAs modulate the quorum-sensing response in Pseudomonas aeruginosa. J Bacteriol. 2006 August;188(16):6026–6033.
- Miller CL, Romero M, Karna SL, et al. RsmW, Pseudomonas aeruginosa small non-coding RsmA-binding RNA upregulated in biofilm versus planktonic growth conditions. BMC Microbiol. 2016 July 19;16(1):155.
- Janssen KH, Diaz MR, Gode CJ, et al. RsmV, a small noncoding regulatory RNA in Pseudomonas aeruginosa that sequesters RsmA and RsmF from target mRNAs. J Bacteriol. 2018 August 15;200:16.
- Marden JN, Diaz MR, Walton WG, et al. An unusual CsrA family member operates in series with RsmA to amplify posttranscriptional responses in Pseudomonas aeruginosa. Proc Natl Acad Sci U S A. 2013 September 10;110(37):15055–15060.
- Morris ER, Hall G, Li C, et al. Structural rearrangement in an RsmA/CsrA ortholog of Pseudomonas aeruginosa creates a dimeric RNA-binding protein, RsmN. Structure. 2013 September 3;21(9):1659–1671.
- Goodman AL, Merighi M, Hyodo M, et al. Direct interaction between sensor kinase proteins mediates acute and chronic disease phenotypes in a bacterial pathogen. Genes Dev. 2009 January 15;23(2):249–259.
- Brencic A, McFarland KA, McManus HR, et al. The GacS/GacA signal transduction system of Pseudomonas aeruginosa acts exclusively through its control over the transcription of the RsmY and RsmZ regulatory small RNAs. Mol Microbiol. 2009 August;73(3):434–445.
- Irie Y, Starkey M, Edwards AN, et al. Pseudomonas aeruginosa biofilm matrix polysaccharide Psl is regulated transcriptionally by RpoS and post-transcriptionally by RsmA. Mol Microbiol. 2010 October;78(1):158–172.
- Allsopp LP, Wood TE, Howard SA, et al. RsmA and AmrZ orchestrate the assembly of all three type VI secretion systems in Pseudomonas aeruginosa. Proc Natl Acad Sci U S A. 2017 July 18;114(29):7707–7712.
- Heurlier K, Williams F, Heeb S, et al. Positive control of swarming, rhamnolipid synthesis, and lipase production by the posttranscriptional RsmA/RsmZ system in Pseudomonas aeruginosa PAO1. J Bacteriol. 2004 May;186(10):2936–2945.
- Romero M, Silistre H, Lovelock L, et al. Genome-wide mapping of the RNA targets of the Pseudomonas aeruginosa riboregulatory protein RsmN. Nucleic Acids Res. 2018 April;46(13):30.
- Gebhardt MJ, Kambara TK, Ramsey KM, et al. Widespread targeting of nascent transcripts by RsmA in Pseudomonas aeruginosa. Proc Natl Acad Sci U S A. 2020 April;117(19):24.
- Hmelo LR, Borlee BR, Almblad H, et al. Precision-engineering the Pseudomonas aeruginosa genome with two-step allelic exchange. Nat Protoc. 2015 November;10(11):1820–1841.
- Chihara K, Bischler T, Barquist L, et al. Conditional Hfq association with small noncoding RNAs in Pseudomonas aeruginosa revealed through comparative UV cross-linking immunoprecipitation followed by high-throughput sequencing. mSystems. 2019;4(6):e00590–19.
- Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnetjournal. 2011;17:1.
- Xu H, Luo X, Qian J, et al. FastUniq: a fast de novo duplicates removal tool for paired short reads. PLoS One. 2012;7(12):e52249.
- Forstner KU, Vogel J, Sharma CM. READemption-a tool for the computational analysis of deep-sequencing-based transcriptome data. Bioinformatics. 2014 December 1;30(23):3421–3423.
- Hoffmann S, Otto C, Doose G, et al. A multi-split mapping algorithm for circular RNA, splicing, trans-splicing and fusion detection. Genome Biol. 2014 February 10;15(2):R34.
- Holmqvist E, Li L, Bischler T, et al. Global maps of ProQ binding In Vivo reveal target recognition via RNA structure and stability control at mRNA 3ʹ Ends. Mol Cell. 2018 June 7;70(5):971–982 e6.
- Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11(10):R106.
- Langenberger D, Bermudez-Santana C, Hertel J, et al. Evidence for human microRNA-offset RNAs in small RNA sequencing data. Bioinformatics. 2009 September 15;25(18):2298–2301.
- Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550.
- Bailey TL, Johnson J, Grant CE, et al. The MEME Suite. Nucleic Acids Res. 2015 July 1;43(W1):W39–49.
- Yao Z, Weinberg Z, Ruzzo WL. CMfinder–a covariance model based RNA motif finding algorithm. Bioinformatics. 2006 February 15;22(4):445–452.
- Weinberg Z, Breaker RR. R2R–software to speed the depiction of aesthetic consensus RNA secondary structures. BMC Bioinformatics. 2011 January 4;12(1):3.
- Beaudoin T, Zhang L, Hinz AJ, et al. The biofilm-specific antibiotic resistance gene ndvB is important for expression of ethanol oxidation genes in Pseudomonas aeruginosa biofilms. J Bacteriol. 2012 June;194(12):3128–3136.
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001 December;25(4):402–408.
- Argaman L, Hershberg R, Vogel J, et al. Novel small RNA-encoding genes in the intergenic regions of Escherichia coli. Curr Biol. 2001 June 26;11(12):941–950.
- Kingsford CL, Ayanbule K, Salzberg SL. Rapid, accurate, computational discovery of Rho-independent transcription terminators illuminates their relationship to DNA uptake. Genome Biol. 2007;8(2):R22.
- Gill EE, Chan LS, Winsor GL, et al. High-throughput detection of RNA processing in bacteria. BMC Genomics. 2018 March 27;19(1):223.
- Ferrara S, Brugnoli M, De Bonis A, et al. Comparative profiling of Pseudomonas aeruginosa strains reveals differential expression of novel unique and conserved small RNAs. PLoS One. 2012;7(5):e36553.
- Gomez-Lozano M, Marvig RL, Molin S, et al. Genome-wide identification of novel small RNAs in Pseudomonas aeruginosa. Environ Microbiol. 2012 August;14(8):2006–2016.
- Gomez-Lozano M, Marvig RL, Tulstrup MV, et al. Expression of antisense small RNAs in response to stress in Pseudomonas aeruginosa. BMC Genomics. 2014 September;11(15):783.
- Schulmeyer KH, Diaz MR, Bair TB, et al. Primary and secondary sequence structure requirements for recognition and discrimination of target RNAs by Pseudomonas aeruginosa RsmA and RsmF. J Bacteriol. 2016 September 15;198(18):2458–2469.
- Holmqvist E, Wright PR, Li L, et al. Global RNA recognition patterns of post-transcriptional regulators Hfq and CsrA revealed by UV crosslinking in vivo. Embo J. 2016 May 02;35(9):991–1011.
- Sorger-Domenigg T, Sonnleitner E, Kaberdin VR, et al. Distinct and overlapping binding sites of Pseudomonas aeruginosa Hfq and RsmA proteins on the non-coding RNA RsmY. Biochem Biophys Res Commun. 2007 January 19;352(3):769–773.
- Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44–57.
- Rocchetta HL, Burrows LL, Lam JS. Genetics of O-antigen biosynthesis in Pseudomonas aeruginosa. Microbiol Mol Biol Rev. 1999 September;63(3):523–553.
- Ventre I, Goodman AL, Vallet-Gely I, et al. Multiple sensors control reciprocal expression of Pseudomonas aeruginosa regulatory RNA and virulence genes. Proc Natl Acad Sci U S A. 2006 January 3;103(1):171–176.
- Goodman AL, Kulasekara B, Rietsch A, et al. A signaling network reciprocally regulates genes associated with acute infection and chronic persistence in Pseudomonas aeruginosa. Dev Cell. 2004 November;7(5):745–754.
- Bordi C, Lamy MC, Ventre I, et al. Regulatory RNAs and the HptB/RetS signalling pathways fine-tune Pseudomonas aeruginosa pathogenesis. Mol Microbiol. 2010 June;76(6):1427–1443.
- Petrova OE, Sauer K. The novel two-component regulatory system BfiSR regulates biofilm development by controlling the small RNA rsmZ through CafA. J Bacteriol. 2010 October;192(20):5275–5288.
- Bouillet S, Ba M, Houot L, et al. Connected partner-switches control the life style of Pseudomonas aeruginosa through RpoS regulation. Sci Rep. 2019 April 24;9(1):6496.
- Perez-Martinez I, Haas D. Azithromycin inhibits expression of the GacA-dependent small RNAs RsmY and RsmZ in Pseudomonas aeruginosa. Antimicrob Agents Chemother. 2011 July;55(7):3399–3405.
- Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31(13):3406–3415.
- Thomason MK, Voichek M, Dar D, et al. A rhlI 5′ UTR-derived sRNA regulates RhlR-dependent quorum sensing in Pseudomonas aeruginosa. mBio. 2019;10(5):5.
- Potts AH, Vakulskas CA, Pannuri A, et al. Global role of the bacterial post-transcriptional regulator CsrA revealed by integrated transcriptomics. Nat Commun. 2017 November 17;8(1):1596.
- Pessi G, Williams F, Hindle Z, et al. The global posttranscriptional regulator RsmA modulates production of virulence determinants and N-acylhomoserine lactones in Pseudomonas aeruginosa. J Bacteriol. 2001 November;183(22):6676–6683.
- Mercante J, Edwards AN, Dubey AK, et al. Molecular geometry of CsrA (RsmA) binding to RNA and its implications for regulated expression. J Mol Biol. 2009 September 18;392(2):511–528.
- Duss O, Michel E, Diarra Dit Konte N, et al. Molecular basis for the wide range of affinity found in Csr/Rsm protein-RNA recognition. Nucleic Acids Res. 2014 April;42(8):5332–5346.
- Burrows LL, Chow D, Lam JS. Pseudomonas aeruginosa B-band O-antigen chain length is modulated by Wzz (Ro1). J Bacteriol. 1997 March;179(5):1482–1489.
- Deretic V, Schurr MJ, Yu H. Pseudomonas aeruginosa, mucoidy and the chronic infection phenotype in cystic fibrosis. Trends Microbiol. 1995;3(9):351–356.
- Cross AR, Goldberg JB. Remodeling of O antigen in mucoid Pseudomonas aeruginosa via transcriptional repression of wzz2. mBio. 2019 February 19;10(1):1.
- Stacey SD, Williams DA, Pritchett CL. The Pseudomonas aeruginosa two-component regulator AlgR directly activates rsmA expression in a phosphorylation independent manner. J Bacteriol. 2017 March;199(18):20.
- Ghafoor A, Hay ID, Rehm BH. Role of exopolysaccharides in Pseudomonas aeruginosa biofilm formation and architecture. Appl Environ Microb. 2011 August;77(15):5238–5246.
- Augustin DK, Song Y, Baek MS, et al. Presence or absence of lipopolysaccharide O antigens affects type III secretion by Pseudomonas aeruginosa. J Bacteriol. 2007 March;189(6):2203–2209.
- Huang H, Shao X, Xie Y, et al. An integrated genomic regulatory network of virulence-related transcriptional factors in Pseudomonas aeruginosa. Nat Commun. 2019 July 3;10(1):2931.
- Intile PJ, Diaz MR, Urbanowski ML, et al. The AlgZR two-component system recalibrates the RsmAYZ posttranscriptional regulatory system to inhibit expression of the Pseudomonas aeruginosa type III secretion system. J Bacteriol. 2014 January;196(2):357–366.
- Wang Z, Huang X, Liu Y, et al. GacS/GacA activates pyoluteorin biosynthesis through Gac/Rsm-RsmE cascade and RsmA/RsmE-driven feedback loop in Pseudomonas protegens H78. Mol Microbiol. 2017 September;105(6):968–985.
- Chen R, Weng Y, Zhu F, et al. Polynucleotide Phosphorylase regulates multiple virulence factors and the stabilities of small RNAs RsmY/Z in Pseudomonas aeruginosa. Front Microbiol. 2016;7:247.
- Janssen KH, Corley JM, Djapgne L, et al. Hfq and sRNA 179 Inhibit Expression of the Pseudomonas aeruginosa cAMP-Vfr and Type III secretion regulons. mBio. 2020 June 16;11(3):3.