
ABSTRACT
The ability to precisely alter the genome holds immense potential for molecular biology, medicine and biotechnology. The development of the Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) into a genomic editing tool has vastly simplified genome engineering. Here, we explored the use of chemically synthesized chimeric oligonucleotides encoding a target-specific crRNA (CRISPR RNA) fused to a single-stranded DNA repair template for RNP-mediated precision genome editing. By generating three clinically relevant oncogenic driver mutations, two non-stop extension mutations, an FGFRi resistance mutation and a single nucleotide change, we demonstrate the ability of chimeric oligos to form RNPs and direct Cas9 to effectively induce genome editing. Further, we demonstrate that the polarity of the chimeric oligos is crucial: only chimeric oligos with the single-stranded DNA repair template fused to the 3’-end of the crRNA are functional for accurate editing, while templates fused to the 5’-end are ineffective. We also find that chimeras can perform editing with both symmetric and asymmetric single-stranded DNA repair templates. Depending on the target locus, the editing efficiency using chimeric RNPs is similar to or less than the efficiency of editing using the bipartite standard RNPs. Our results indicate that chimeric RNPs comprising RNA-DNA oligos formed from fusing the crRNA and DNA repair templates can successfully induce precise edits. While chimeric RNPs do not display an advantage over standard RNPs, they nonetheless represent a viable approach for one-molecule precision genome editing.
KEYWORDS:
Introduction
Precise, targeted alteration of DNA has been a longstanding goal in research. The discovery of the Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) [Citation1–3] and its subsequent development into a genomic editing tool [Citation4–9] has ushered in the modern era of genome editing by enabling both targeted gene disruption and precision genome engineering. The type II CRISPR system from Streptococcus pyogenes which uses the hallmark Cas9 protein as a molecular scissor is the best characterized and most widely used CRISPR editing tool. For the delivery of CRISPR/Cas9 into human cells, plasmids and viral vectors are commonly used vehicles. While the latter is plagued by several safety concerns such as genome integration, prolonged Cas9 expression and immune responses, the former is often silenced in primary cells or can also have immunotoxic effects. Preformed Cas9 ribonucleoprotein complexes (RNPs), which avoid these disadvantages, have emerged as a viable and highly effective alternative to vector mediated gene editing [Citation10–16].
RNPs are composed of a CRISPR RNA (crRNA) and trans-activating CRISPR RNA (tracrRNA) duplex complexed with the Cas9 protein [Citation4]. The crRNA is composed of a 20 nucleotide target-specific region at the 5’-end followed by a tracrRNA-binding region at the 3’-end. The crRNA and tracrRNA can also be engineered as a single-guide RNA (sgRNA) [Citation4]. The crRNA- or sgRNA-dictated sequence-specific recognition of the target site along with the presence of the NGG protospacer adjacent motif (PAM) leads to Cas9 mediated cleavage resulting in a double-stranded break (DSB) [Citation4,Citation17]. Such DSBs can be repaired by the Non-homologous end joining pathway (NHEJ) [Citation18] or the Homology directed repair pathway (HDR) [Citation19]. The NHEJ pathway is error-prone leading to the generation of an uncontrolled mixture of insertions and deletions (indels), which could be used for gene disruption. The HDR pathway can accurately repair DSBs in the presence of a donor DNA repair template. However, the low frequency of HDR in most cells along with challenges involved in donor DNA template delivery have greatly limited the relevance of HDR. Multiple strategies to improve the efficiency of HDR have been developed, including cell synchronization, transient blocking of the NHEJ pathway [Citation20,Citation21] and covalent attachment of single-stranded DNA repair templates (ssDNA) to the Cas9 protein to improve the spatial and temporal colocalization of the repair template [Citation22–25].
Here, we explore the use of RNA-DNA chimeric oligonucleotides (chimeras) comprising a target-specific crRNA fused to a ssDNA repair template for precision genome editing. Thus, chimeras in the RNP complex with the Cas9 protein can perform the dual function of crRNA mediated direction of Cas9 and ssDNA repair template mediated precision repair. They represent a viable precision genome editing technique, which may decrease the complexity of the editing reagent and physically link the locus-guiding crRNA to the mutation-defining repair template.
Materials and methods
Cell culture
Human embryonic kidney (HEK) 293 T cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM; Corning and Thermo-Fisher) supplemented with 10% (vol/vol) foetal bovine serum (FBS; Thermo-Fisher) in a humidified 5% CO2 incubator at 37°C.
Oligonucleotide synthesis
The chimeric oligonucleotides (chimeras) were synthesized by Integrated DNA Technologies (IDT) using the RNA ultramer platform. A total of 16 chimeric oligonucleotides were designed. Twelve chimeric oligonucleotides were designed to create three oncogenic driver mutations: the G35A and A183C mutations in the KRAS gene and the G374T mutation in the TP53 gene. Additional four chimeric oligonucleotides were designed to create the nonstop extension mutations T1657C in the SMAD4 gene [Citation26] and T502C in the CDKN2A gene, a resistance mutation A1645C to the FGFR inhibitor Pemigatinib in the FGFR2 gene [Citation27] and a G87C mutation in the RNF2 gene, respectively. Each chimeric oligonucleotide was an RNA-DNA hybrid comprising a target binding crRNA region and either a symmetric or an asymmetric ssDNA repair template. The crRNA sequence in the chimeric oligonucleotide was also used as a separate Alt-R CRISPR-Cas9 crRNA (crRNA) along with an ALT-R CRISPR-Cas9 tracrRNA (tracrRNA) from IDT. Symmetric ssDNA repair templates for each of the three targets were also used as single-stranded oligodeoxynucleotides (ssODNs) from IDT. The details and sequences of the chimeric oligonucleotides, crRNAs and repair templates can be found in Supplementary Tables 1 and 2, respectively.
Chimeric oligonucleotide and RNA preparation
The chimeric oligonucleotides, crRNAs and tracrRNA were resuspended in nuclease-free duplex buffer (IDT) to a final concentration of 200 μM. The tracrRNA was mixed with either the chimeric oligonucleotide or the crRNA in equimolar concentrations to a final duplex concentration of 100 μM. The mixture was heated to 95°C for 5 min and allowed to cool to room temperature on the bench top.
RNP assembly
To prepare RNPs, 104 pmol of the HiFi-Cas9 v3 protein (IDT) was incubated with either 120 pmol of the chimeric oligo-tracRNA duplex or 120 pmol of the crRNA-tracrRNA duplex in 1x PBS to a final volume of 5 μL per reaction. The mixture was incubated at room temperature for 20 min.
Nucleofection
The chimeric RNPs and the standard RNPs were delivered into cells by nucleofection according to previously described methods [Citation10,Citation11,Citation28] using the SF Cell Line 4D-NucleofectorTM X Kit (Lonza) and the Amaxa 4D-Nucleofector device (Lonza). In brief, HEK293T cells grown to 70% confluency were washed with 1x PBS, trypsinized and counted. The total number of cells needed were transferred to a sterile 15 mL tube and centrifuged at 100 g for 5 min at room temperature. The supernatant was removed and the pellet was washed with 5 mL of 1x PBS followed by centrifugation at 100 g for 5 min at room temperature. The cells were resuspended in 20 μL of supplemented nucleofector solution SF (Lonza) per 3.5 × 105 cells. For each reaction with chimeric RNPs, 20 μL of resuspended cells was mixed with 5 μL of the pre-formed chimeric RNP and 1 μL of electroporation enhancer (IDT). For each reaction with the standard RNPs, 20 μL of resuspended cells was mixed with 5 μL of the standard RNP, 1 μL of electroporation enhancer (IDT) and 120 pmol of the respective ssODN. The final mixture for both chimeric RNPs and standard RNPs was pipetted up and down two times and carefully transferred to separate wells of a 16-well Nucleocuvette™ strip. The Nucleocuvette™ strip was placed into the 4D-Nucleofector™ X Unit and the cells were nucleofected using the CM130 program. The Nucleocuvette™ strip was removed from the device and 80 μL of pre-warmed culture media was added to each of the nucleofected wells in the Nucleocuvette™ strip. The cells were resuspended by gentle pipetting and transferred to a prepared 48 well plate containing 180 μL culture media and 1 μM HDR enhancer v3 (IDT). The cells were incubated in a humidified 5% CO2 incubator at 37°C.
Genomic DNA isolation
96 h post nucleofection, the cells were harvested by trypsinization followed by centrifugation at 100 g for 5 min and the genomic DNA was isolated using DNeasy Blood & Tissue Kit (Qiagen) according to the manufacturer’s recommendation.
PCR and sanger sequencing
50 ng of genomic DNA was used for PCR amplification using the Q5 DNA polymerase (NEB) and primer pairs spanning the target sequences for KRAS G35A, KRAS A183C, TP53 G374T, SMAD4 T1657C, FGFR2 A1645C, CDKN2A T502C and RNF2 G87C. A touchdown PCR with the following thermal cycle program was used: initial denaturation at 98°C for 30 s followed by 30 cycles of 98°C for 10 s, 69°C for 20 s (−0.5°C per cycle in each subsequent cycle) with an extension at 72°C for 3 min. The annealing temperature was subsequently set to the annealing temperature of the specific primer pair for another 29 cycles with a final extension for 7 min at 72°C. Following PCR, the amplicons were loaded on a 1% agarose-SYBR safe gel and visualized. The correct bands were excised from the gel and eluted using the GeneJET gel extraction kit (Thermo Fisher Scientific) according to the manufacturer’s recommendation. The primer sequences can be found in Supplementary Table 3. The PCR amplicons were then sent for Sanger sequencing.
ICE analysis of gene editing rates
Editing rates using Sanger sequencing chromatograms were analysed using Synthego’s ICE tool available at ice.synthego.com. The Sanger sequencing files for the edited samples and unedited controls were uploaded into ICE along with the crRNA and ssODN sequences. The tool compared the sequence traces in the edited and unedited Sanger files and generated an overview of the rate of indels and accurate editing (preprint: Hsiau et al., 2019).
Next Generation DNA sequencing
Genomic sites of interest were amplified from genomic DNA samples and sequenced on an Illumina MiSeq. In brief, target-specific amplification primers (Supplementary Table 3) were used for a first round of PCR (PCR 1) to amplify the genomic region of interest. In addition to the target-binding site, the forward and reverse primers also comprised a 15 nucleotide scaffold sequence to serve as a primer binding site for PCR 2. This was done to facilitate multiplexing within biological replicates and allow PCR 2 to be carried out with the same primer pairs. PCR 1 reactions of 50 μl were performed with 0.5 μM of each forward and reverse primer, 50 ng of genomic DNA and 0.5 μl of Q5 DNA polymerase. A touchdown PCR with the following thermal cycle program was used: initial denaturation at 98°C for 30 s followed by 30 cycles of 98°C for 10 s, 69°C for 20 s (−0.5°C per cycle in each subsequent cycle) with an extension at 72°C for 3 min. The annealing temperature was subsequently set to the annealing temperature of the specific primer pair for another 29 cycles with a final extension for 7 min at 72°C. A second PCR reaction was carried out using the product of the first PCR reaction as a template and degenerate forward primers that included the Illumina adapter sequence and reverse primers with unique indices. Specifically, a 50 μl PCR 2 reaction for each target contained 0.5 μM of each degenerate forward primer and reverse Illumina indexing primer, 10 ng of the purified PCR 1 reaction, and 25 μl NEBNext® Ultra™ II Q5® Master Mix (NEB). The PCR 2 reactions were carried out as follows: 98°C for 2 min, followed by 15 cycles of 98°C for 10 s, 65°C for 75 s, with a final 65°C extension for 2 min. The PCR 2 products were evaluated by electrophoresis in a 1.0% agarose-SYBR-safe gel. PCR 2 products were purified by gel extraction using a GeneJET gel extraction kit (ThermoFisher Scientific) and eluted with 50 μl water. The PCR 2 products were then further concentrated using AMPure XP beads (Beckman Coulter) and eluted in a final volume of 13 μl. DNA concentration was measured by fluorometric quantification using a Qubit (ThermoFisher Scientific). The clean and concentrated samples from the PCR 2 reaction were pooled and sequenced on an Illumina MiSeq instrument according to the manufacturer’s protocols. Sequencing reads were demultiplexed using Galaxy [Citation29,Citation30] and the alignment of amplicons to a reference sequence was done using CRISPResso2 [Citation31]. Editing efficiencies of chimeric and standard RNPs were calculated as the percentage of (number of reads with the desired edit that do not contain indels)/(number of total reads).
Results
Structure of the chimeric oligonucleotides
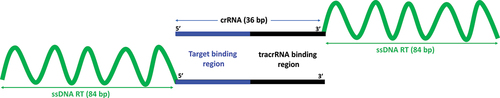
Each chimeric oligonucleotide is designed and synthesized as a single 120 bp oligonucleotide. Each chimera consists of 36 RNA nucleotides pertaining to the target-specific crRNA and 84 DNA nucleotides pertaining to the target- and mutation-specific single-stranded DNA repair template (ssDNA RT). For each of the three oncogenic driver mutations KRAS G35A, KRAS A183C and TP53 G374T, a 3’-end and a 5’-end chimeric oligonucleotide with a repair template containing the desired point mutation flanked by homology arms of equal length (symmetric repair template) is designed. In the 3’-chimeric oligonucleotide, the ssDNA repair template is fused to the 3’-end of the crRNA sequence (), whereas in the 5’-chimeric oligonucleotide, the ssDNA repair template is fused to the 5’-end of the crRNA sequence (). Each ssDNA repair template sequence of the chimeric oligonucleotides contains one of the desired point mutations KRAS G35A, KRAS A183C and TP53 G374T, respectively. Since previous studies have shown that the asymmetric ssDNA repair templates induce higher editing rates [Citation32,Citation33], we additionally designed 3’-end chimeric oligonucleotides with asymmetric repair templates for these three targets.
Figure 1. Schematic showing the structure of 3’-end chimeric oligonucleotides (upper panel) and 5’-end chimeric oligonucleotides (lower panel).
Polarity of the chimeric oligonucleotides is crucial for genome editing
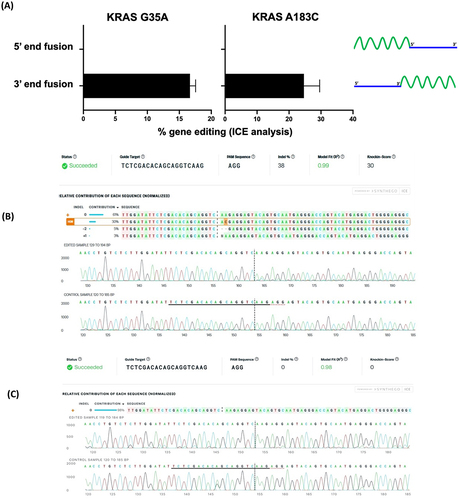
To analyze the effect of polarity of the chimeric oligonucleotides, we perform editing to create the KRAS G35A, KRAS A183C and TP53 G374T mutations. From Sanger sequencing and ICE analysis of the cells edited using chimeric oligonucleotides, we find that for the two KRAS mutations, only the 3’-end chimeras are active for genome editing, while the 5’-end chimeras appear unable to trigger even cleavage at the target locus according to the ICE analysis (, C). In the case of TP53, neither the 3’- nor the 5’-end chimeric oligos is active for gene editing. This indicates that chimeric oligonucleotides can direct Cas9 for successful gene editing, in a locus-specific manner and only when the ssDNA repair template is fused to the 3’-end of the crRNA.
Figure 2. Gene editing rates as determined by ICE analysis for (A) 3’-end oligonucleotides (chimera 1 for KRAS G35A and chimera 2 for KRAS 183C) with symmetric ssDNA RTs and 5’-end chimeric oligonucleotides (chimera 3 for KRAS G35A and chimera 4 for KRAS 183C) with symmetric ssDNA RT. Data for TP53 G374T is not shown since no editing is obtained. Data is obtained from three independent experiments. Error bars show mean ± SEM. Representative ICE analysis output is shown for (B) a 3’-end chimeric oligonucleotide (chimera 2) and (C) a 5’-end chimeric oligonucleotide (chimera 4).
Chimeras with symmetric or asymmetric design of the repair template induce gene editing
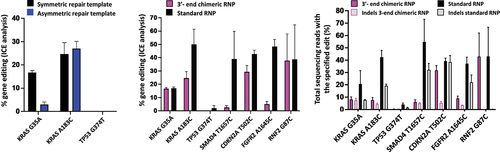
We analyze the effect of repair template symmetry within the chimeras for the two KRAS mutations and the TP53 mutation. For each of the KRAS mutations, the chimeras with symmetric ssDNA repair templates (chimera 1 for KRAS G35A and chimera 2 for KRAS A183C, respectively) and the chimeras with asymmetric ssDNA repair templates (chimera 5 for KRAS G35A and chimera 6 for KRAS A183C respectively) are active for gene editing (). For KRAS A183C, chimera 6 with the asymmetric ssDNA repair template shows higher editing, but for KRAS G35A, chimera 1 with the symmetric ssDNA repair template shows higher editing. At the TP53 locus, as seen with the symmetric repair template chimera, the asymmetric repair template containing chimera is also unable to induce successful DNA cleavage.
Figure 3. Gene editing rates as determined by ICE analysis (A) for editing with 3’-end chimeric RNPs with symmetric or asymmetric repair templates and (B) for the comparison of editing rates between 3-‘end chimeric RNPs with symmetric repair template and standard RNPs with co-delivered symmetric ssODN and (C) gene editing rates as determined by NGS. For comparison, the data using the chimeras with symmetric DNA repair template for KRAS G35A, KRAS A183C and TP53 G374T in (A) and (B) is the same data as the data depicted in . Data is obtained from three independent experiments. Error bars show mean ± SEM.
Comparable or lower editing efficiency of 3’-end chimeric RNPs compared to standard RNP
Based on the results of the polarity of chimeric oligonucleotides and the symmetry of repair templates, we have designed four additional chimeric oligonucleotides for SMAD4, CDKN2A, FGFR2 and RNF2 genes as 3’-end chimeras with a symmetric repair template. We compare the editing rates between the active 3’-end chimeric oligonucleotide RNPs and the standard RNPs using ICE analysis ( B) and next generation sequencing (NGS) ( C). From the ICE analyses ( B), the editing rates for the KRAS G35A and RNF2 G87C mutations are comparable between the chimeric RNP and the standard RNP, while for the KRAS A183C, SMAD4 T1657C, CDKN2A T502C and FGFR2 A1645C mutations, the chimeric RNPs show lower editing rates than the standard RNPs. For TP53 G374T, the standard RNP shows the lowest editing efficiency among all RNPs, while the chimeric RNP does not induce detectable cleavage as determined by ICE analysis. The NGS analysis ( C) shows comparable rates of editing between 3’-end chimeric RNP and standard RNP for CDKN2A T502C and RNF2 G87C mutations. Chimeric RNPs show lower editing rates as determined from NGS for the oncogenic driver mutations in KRAS and the SMAD4 and FGFR2 mutations. For the TP53 mutation, similar to ICE analysis, NGS also reveals that the rate of editing with the standard RNP is the lowest among all the targets while chimeric RNPs do not induce any editing. The NGS analysis further reveals a trend towards lower indel rates of the chimeric RNPs for KRAS A183C, SMAD4 T1657C, CDKN2A T502C and FGFR2 A1645C, but in three out of these four cases, this is also associated with a lower editing efficiency generating correct point mutations.
Discussion
In summary, our study shows that chimeric oligonucleotides, which fuse the crRNA and a single-stranded donor repair template, can perform the dual functions of sequence-specific recruitment of Cas9 to the target site and mutation-specific precision genomic repair. We demonstrate that the polarity of the chimeric oligonucleotide is a critical factor and only 3’-end chimeras which have the ssDNA repair template fused to the 3’-end of the crRNA can perform successful genome editing. Chimeric RNPs can successfully create two oncogenic driver mutations in the KRAS gene, a non-stop extension mutation each in the SMAD4 and CDKN2A genes, a Pemigatinib resistance mutation in the FGFR2 gene and a single nucleotide change in the RNF2 gene at a rate either comparable to or lower than the rates mediated by standard RNPs which use a crRNA-tracrRNA duplex plus a separate co-delivered ssODN. For TP53 G374T, the chimeric RNPs yield no detectable cleavage as determined with both ICE analysis and next generation sequencing. However, since the efficiency of the standard RNP is the lowest at this locus, it indicates that the TP53 locus may be more recalcitrant to editing. Further, for the 5’-end fusion chimeras, the ICE analyses show no detectable cleavage or indels which indicates that such chimeras may either be unable to complex into RNPs with Cas9 or as a result of their molecular structure be unable to direct sequence-specific targeting of Cas9, both of which might be reflected in the apparent lack of cleavage observed for the 5'-end fusion chimeras.
Chimeric RNPs deliver all the components of genome editing simultaneously into the cells which allows for the spatial and temporal co-localization of the ssDNA repair template with the rest of the editing machinery. This could be expected to show higher editing rates than standard RNPs where the ssDNA repair template is co-delivered. Thus, it is surprising to find that chimeric oligonucleotides do not lead to a significant increase in editing rates and rather showed lower editing rates than a standard RNP. This difference could be due to reduced mobility of the ssDNA repair template within the chimera in contrast to the untethered co-delivered ssODN and could perhaps be solved by introducing linker sequences between the crRNA and ssDNA repair templates in the chimeric oligonucleotide. While current synthesis pipelines do not allow for longer chimeric oligonucleotides due to length constraints, it would nevertheless be possible to generate such chimeras using other techniques such as click chemistry. Additionally, similar to work done with crRNAs, chemical modifications of chimeric oligos to improve stability and activity could potentially increase the efficiency of chimeras for precise editing. Chimeric crRNA-templates could have two potential applications in the future – at least for selected loci and mutations: 1) they could reduce the complexity of in vivo and therapeutic applications by merging two molecules into only one which needs to be delivered and tested – and 2) they physically link the target-defining crRNA and the mutation-defining repair template and could thus be envisioned to be used in pooled approaches combining different chimeras for gene editing.
In conclusion, we demonstrate the use of dual functional 3’-end chimeric oligonucleotides as a viable strategy for precision gene editing.
Supplemental Material
Download MS Excel (15.7 KB)Disclosure statement
S. Diederichs is coowner of siTOOLs Biotech, Martinsried, Germany, without relation to this work. No potential conflict of interest was reported by the other authors.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
References
- Ishino Y, Shinagawa H, Makino K, et al. Nucleotide sequence of the iap gene, responsible for alkaline phosphatase isozyme conversion in Escherichia coli, and identification of the gene product. J Bacteriol. 1987;169:5429–5433.
- Mojica FJM, Juez G, Rodriguez-Valera F. Transcription at different salinities of Haloferax mediterranei sequences adjacent to partially modified PstI sites. Mol Microbiol. 1993;9:613–621.
- van Soolingen D, de Haas PE, Hermans PW, et al. Comparison of various repetitive DNA elements as genetic markers for strain differentiation and epidemiology of Mycobacterium tuberculosis. J Clin Microbiol. 1993;31:1987–1995.
- Jinek M, Chylinski K, Fonfara I, et al. A Programmable Dual-RNA–Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science. 2012;337(6096):816–821.
- Cho SW, Kim S, Kim JM, et al. Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat Biotechnol. 2013;31:230–232.
- Cong L, Ran FA, Cox D, et al. Multiplex Genome Engineering Using CRISPR/Cas Systems. Science. 2013;339(6121):819–823.
- Mali P, Yang L, Esvelt KM, et al. RNA-Guided Human Genome Engineering via Cas9. Science. 2013;339(6121):823–826.
- Doudna JA, Charpentier E. The new frontier of genome engineering with CRISPR-Cas9. Science. 2014;346(6213):1258096.
- Ran FA, Hsu PD, Wright J, et al. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013;8(11):2281–2308.
- Kim S, Kim D, Cho SW, et al. Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Res. 2014;24:1012–1019.
- Ramakrishna S, Kwaku Dad A-B, Beloor J, et al. Gene disruption by cell-penetrating peptide-mediated delivery of Cas9 protein and guide RNA. Genome Res. 2014;24:1020–1027.
- Choi JG, Dang Y, Abraham S, et al. Lentivirus pre-packed with Cas9 protein for safer gene editing. Gene Ther. 2016;23:627–633.
- Montagna C, Petris G, Casini A, et al. VSV-G-Enveloped Vesicles for Traceless Delivery of CRISPR-Cas9. Mol Ther Nucleic Acids. 2018;12:453–462.
- Mangeot PE, Risson V, Fusil F, et al. Genome editing in primary cells and in vivo using viral-derived Nanoblades loaded with Cas9-sgRNA ribonucleoproteins. Nat Commun. 2019;10(1):45.
- Campbell LA, Coke LM, Richie CT, et al. Gesicle-Mediated Delivery of CRISPR/Cas9 Ribonucleoprotein Complex for Inactivating the HIV Provirus. Mol Ther. 2019;27(1):151–163.
- Gee P, Lung MSY, Okuzaki Y, et al. Extracellular nanovesicles for packaging of CRISPR-Cas9 protein and sgRNA to induce therapeutic exon skipping. Nat Commun. 2020;11(1):1334.
- Jiang F, Doudna JA. CRISPR–Cas9 Structures and Mechanisms. Annu Rev Biophys. 2017;46:505–529.
- Lieber MR. The Mechanism of Double-Strand DNA Break Repair by the Nonhomologous DNA End-Joining Pathway. Annu Rev Biochem. 2010;79:181–211.
- San Filippo J, Sung P, Klein H. Mechanism of Eukaryotic Homologous Recombination. Annu Rev Biochem. 2008;77:229–257.
- Liu M, Rehman S, Tang X, et al. Methodologies for Improving HDR Efficiency. Front Genet. 2019;9:691.
- Bischoff N, Wimberger S, Maresca M, et al. Improving Precise CRISPR Genome Editing by Small Molecules: is there a Magic Potion? Cells. 2020;9:1318.
- Ruff P, Koh KD, Keskin H, et al. Aptamer-guided gene targeting in yeast and human cells. Nucleic Acids Res. 2014;42:e61–e61.
- Carlson-Stevermer J, Abdeen AA, Kohlenberg L, et al. Assembly of CRISPR ribonucleoproteins with biotinylated oligonucleotides via an RNA aptamer for precise gene editing. Nat Commun. 2017;8:1711.
- Savic N, Ringnalda FC, Lindsay H, et al. Covalent linkage of the DNA repair template to the CRISPR-Cas9 nuclease enhances homology-directed repair. eLife. 2018;7:e33761.
- Aird EJ, Lovendahl KN, St.Martin A, et al. Increasing Cas9-mediated homology-directed repair efficiency through covalent tethering of DNA repair template. Commun Biol. 2018;1:54.
- Dhamija S, Yang CM, Seiler J, et al. A pan-cancer analysis reveals nonstop extension mutations causing SMAD4 tumour suppressor degradation. Nat Cell Biol. 2020;22(8):999–1010.
- Krook MA, Bonneville R, Chen H-Z, et al. Tumor heterogeneity and acquired drug resistance in FGFR2-fusion-positive cholangiocarcinoma through rapid research autopsy. Cold Spring Harb Mol Case Stud. 2019;5:a004002.
- Zuris JA, Thompson DB, Shu Y, et al. Cationic lipid-mediated delivery of proteins enables efficient protein-based genome editing in vitro and in vivo. Nat Biotechnol. 2015;33:73–80.
- Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011;17:10.
- Gordon A. FASTQ/A short-reads pre-processing tools. 2010.
- Clement K, Rees H, Canver MC, et al. CRISPResso2 provides accurate and rapid genome editing sequence analysis. Nat Biotechnol. 2019;37(3):224–226.
- Richardson CD, Ray GJ, DeWitt MA, et al. Enhancing homology-directed genome editing by catalytically active and inactive CRISPR-Cas9 using asymmetric donor DNA. Nat Biotechnol. 2016;34:339–344.
- Renaud J-B, Boix C, Charpentier M, et al. Improved Genome Editing Efficiency and Flexibility Using Modified Oligonucleotides with TALEN and CRISPR-Cas9 Nucleases. Cell Rep. 2016;14(9):2263–2272.