
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
RNA methylation, especially 6-methyladenosine (m6A)-modified RNAs, plays a specific role in DNA damage response (DDR). Here, we also observe that RNA modified at 8-methyladenosine (m8A) is recruited to UVA-damaged chromatin immediately after microirradiation. Interestingly, the level of m8A RNA at genomic lesions was reduced after inhibition of histone deacetylases and DNA methyltransferases. It appears in later phases of DNA damage response, accompanied by active DNA demethylation. Also, PARP inhibitor (PARPi), Olaparib, prevented adenosine methylation at microirradiated chromatin. PARPi abrogated not only m6A and m8A RNA positivity at genomic lesions, but also XRCC1, the factor of base excision repair (BER), did not recognize lesions in DNA. To this effect, Olaparib enhanced the genome-wide level of γH2AX. This histone modification interacted with m8A RNAs to a similar extent as m8A RNAs with DNA. Pronounced interaction properties we did not observe for m6A RNAs and DNA; however, m6A RNA interacted with XRCC1 with the highest efficiency, especially in microirradiated cells. Together, we show that the recruitment of m6A RNA and m8A RNA to DNA lesions is PARP dependent. We suggest that modified RNAs likely play a role in the BER mechanism accompanied by active DNA demethylation. In this process, γH2AX stabilizes m6A/m8A-positive RNA-DNA hybrid loops via its interaction with m8A RNAs. R-loops could represent basic three-stranded structures recognized by PARP-dependent non-canonical m6A/m8A-mediated DNA repair pathway.
Introduction
Cells developed sophisticated mechanisms for maintaining genome integrity. Genome injury leads to single-strand breaks (SSBs) or more deleterious double-strand breaks (DSBs) in DNA. In a simplistic view, three repair mechanisms recognize single-strand damage in DNA. Base excision repair (BER) eliminates damaged bases by generating SSBs via the function of DNA glycosylases. The next step of DNA repair is mediated by DNA polymerase β (polβ). This enzyme is recruited to DNA lesions by PARP1 (poly[ADP-ribose] polymerase 1), but subsequently, the XRCC1 protein is responsible for PARP1 release from SSBsCitation1. During long-patch BER, together with PCNA, pol δ and pol ε mediate DNA synthesis Citation2. After that, DNA ligases are responsible for the final step of the BER mechanism. For example, DNA ligase IIIα, in parallel with XRCC1, catalyzes the so-called short-patch BER in human cells [Citation3]. On the other hand, DNA ligase I plays a role in long-patch BER [Citation4]. The second mechanism, nucleotide excision repair (NER), recognizes UV-induced DNA lesions. Such molecular lesions are mainly cyclobutane pyrimidine dimers (CPDs) or 6–4 photoproducts. In this case, the DNA polymerase uses an undamaged DNA strand as a template for synthesizing the damaged strand. NER is distinguished as Global Genomic NER or Transcription Coupled NER. It is well known that many proteins function in the NER pathway, including XPA, XPB, XPC, XPD, XPE, XPF, and XPG, CSA, CSB, RASD23A, RAD23B or others [Citation5]. The third SSB repair mechanism is mismatch repair (MMR) which recognizes mismatches in bases or insertion/deletion mispairs that appear during DNA replication and recombination Citation6.
The most deleterious are so-called double-strand breaks (DSBs), in which incorrect repair leads to tumorigenesis. To avoid pathological processes caused by non-physiological repair of DSBs, the following two canonical and mechanistically distinct repair pathways are initiated: Non-homologous end joining (NHEJ), active mainly in the G1 phase of the cell cycle, and less erroneous homologous recombination repair (HRR) that needs nascent chromatids and, thus, the entry of the cells to the S phase of the cell cycle. It is well known that the HRR also proceeds in the G2 phase of the cell cycle [Citation7]. In the S/G2 phases, the NHEJ repair mechanism can also work, but only when the HRR process fails in pathological states Citation8. In a physiological condition, 53BP1 (p53-binding protein 1) and its interacting partner RIF1, proteins of NHEJ, form a barrier inhibiting DNA-end resection that is, on the other hand, activated by the BRCA1 protein interacting with CtIP in a phospho-dependent manner [Citation9,Citation10]. From the view of NHEJ repair machinery, Ku proteins (Ku70 and Ku80) are essential factors that recognize asymmetric DSBs and recruit DNA protein kinase catalytic subunits (DNA-PKcs). This enzyme phosphorylates the Artemis protein having a 5’-3’ exonuclease activity. This process leads to the formation of blunt DSBs. After that, DNA ligase joins two strands of DNA that lost original genetic information. On the other hand, more sequentially precise HRR needs a 3’ overhang and subsequent synthesis of a complementary strand according to the sister chromatid template or following chromatid of the homologous chromosome. As an early step of HRR is considered the binding of the MRN (Mre11/Rad50/NBS1) protein complex to asymmetric double-strand break sites containing 3’ overhangs that are generated by nucleolytic degradation of the 5’ strands. In this case, also RPA covers 3’ overhangs to protect DNA against nucleases. Such single-strand DNA nucleofilaments are recognized by the RAD51 protein that recruits the BRCA2 protein. This protein complex replaces RPA. In this DNA repair process, RAD51 helps to search for homologous DNA and mediates the formation of so-called D-loops. This step needs a homologous DNA template. In this case, non-cross-over HRR or cross-over HRR can be recognized [Citation11].
DNA repair machinery has its epigenetic regulations. It is generally accepted that epigenetic adaptations of DNA repair systems involve the function of phosphorylated histones H2AX (γH2AX). Also, histone H4 lysine 20 methylation (H4K20me2/me3) or H3K79 methylation, as prominent binding partners of the 53BP1 protein [Citation12,Citation13], represent crucial repair factors at DSB sites in mammalian cells [Citation14–18]. From the epigenetic point of view, ionizing radiation causes changes in H3K36 methylation and H3K9me3, which represent additional epigenetic factors of DDR [Citation19–24]. It is known that H3K9me3 at damaged chromatin is linked to the activation of histone acetyltransferase TIP60, furthermore mediating specific histone acetylation at DNA lesions [Citation23,Citation24]. Also, H2AXK119 ubiquitination and H4K16 acetylation were observed at damaged chromatin. Similarly, the NuRD complex, consisting of histone deacetylases 1 and 2 (HDAC1 and HDAC2), is recruited to the damaged genome. This epigenetic event causes deacetylation of H3K56 [Citation25]. We additionally observed H3K9 deacetylation at microirradiated chromatin. This phenomenon was likely mediated by HDAC1 that significantly accumulated at DSB sites [Citation26].
It was recently published that histone signature and co-transcriptionally modified RNAs participate in DNA damage response. It was shown that a novel non-canonical repair pathway is mediated via RNAs methylated on N6 -adenosine (m6A) [Citation27,Citation28]. It is well known that this RNA modification is regulated by specific ‘writers’, so-called methyltransferases METTL3, METTL14, and METTL16, and recognized by ‘readers’ like the YTHDC1 protein. As fundamental m6A ‘erasers’ are considered FTO (fat mass and obesity-associated protein) and ALKBH5 proteins (summarized by [Citation29]). Recently, we additionally showed that mainly METTL16 is recruited to UVA-microirradiated chromatin in later steps of DDR, while the levels of METTL3 and METTL14 proteins remain stable at DNA lesions [Citation27]. Xiang et al. (2017) [Citation28] published that N6-methyladenosinepresent on RNA appears at DNA lesions immediately after local laser microirradiation, and we documented that this DDR-related event is additionally accompanied by depletion of 2,2,7-methylguanosine (m3G/TMG) in RNA [Citation27]. Significantly, in parallel with the described changes in epitranscriptome, UV-irradiation additionally decreases the global cellular level of N1-methyladenosine (m1A) in RNAs [Citation27]. In this case, surprisingly, when m6A RNAs disappeared from DNA lesions, the level of FTO remained stable [Citation27]. The above-mentioned experimental data documents that the function of m6A-specific ‘writers’ and ‘erasers’ is essential for the regulation of not only gene expression but also for DNA damage repair. For instance, it was shown that in the absence of METTL3, there is a delay in the repair of UV-induced cyclobutane pyrimidine dimers (CPDs), and cells are more sensitive to UV light [Citation28,Citation30]. It is also well known that DNA polymerases participate in such DNA damage responses. In this case, DNA polymerase κ (Pol κ), playing a role in nucleotide excision repair (NER), works in parallel with the catalytic activity of METTL3 recruited to damaged chromatin. Xiang et al. (2017) [Citation28] showed that exogenous overexpression of DNA Pol κ rescues the defect of CPD elimination in METTL-depleted cells. This observation suggests that the fast recruitment of DNA Pol κ to the damage site is potentially due to m6A RNA deposition at UV-irradiated genomic regions [Citation30]. Xiang et al. (2017) [Citation28] suggested that METTL3/METTL14 complex (but not METTL3/WTAP complex) in parallel with FTO (but not ALKBH5) serves as the ‘writer’ and the ‘eraser’ of the m6A in RNA accumulating at UV-damaged chromatin. Zhang et al. (2020) [Citation31] showed that ATM-mediated phosphorylation at serine 43 could activate METTL3, and such phosphorylated protein accumulates at DNA lesions, where it acts as methyltransferase mediating the m6A in RNA. Also, m6A ‘reader’, the YTHDC1 protein, is recruited to damaged sites [Citation31]. These all observations indicate the existence of a non-canonical m6A-mediated DNA repair pathway. It was revealed that this non-canonical process is dependent on the PARP [Poly (ADP-ribose) polymerase] and is specific for UV-irradiated chromatin but not for ionizing-radiation induced foci (IRIF) [Citation28,Citation29,Citation32–37]. Zhang (2017) [Citation37] suggested that the m6A mediated DNA repair pathway is independent of the phosphorylation of histone H2AX and is activated at damage sites with hybrid DNA-RNA loops. It was published that such RNA-loops appear in the genome because of DNA damage at transcription sites, and some studies showed their link to the Transcription-Coupled Nucleotide Excision Repair mechanism (TC-NER) [Citation38].
We were inspired by the above-mentioned studies showing novel and RNA-dependent DNA repair principles. To continue with the study of the role of RNA modifications in DNA damage response, we addressed whether RNA methylated at 8-adenosine (m8A RNA) can recognize UV-induced DNA lesions. This RNA modification is known to be catalysed by the SAM (S-Adenosyl methionine)-dependent methyltransferase Cfr [Citation39]. The studies were performed in bacteria, especially in Escherichia coli, in which the Cfr methyltransferase is responsible for the resistance to five different classes of antibiotics. It was described that the Cfr-mediated modification was determined on nucleotide A2503 of 23S rRNA, and also Cfr can catalyse methylation leading to the formation of 2,8-dimethyladenosine. However, the mutation of single conserved cysteine residues in the SAM motif CxxxCxxC of Cfr abolishes its activity [Citation39].
Here, we show m8A RNA positivity at DNA lesions experimentally induced in human cells. The local high positivity of m8A RNA at UV-damaged chromatin depended on the PARP function. It was shown by other authors that PARP inhibitors trap the PARP1 and PARP2 enzymes in damaged DNA. Such PARP-DNA complexes are more likely to be cytotoxic than unrepaired SSBs induced by PARP inhibitors themselves [Citation1,Citation40]. Since the presence of PARP1 at damaged chromatin suppresses the activities of BER factors, including APE1, Polβ, and LigIIIα, it seems likely that PARP1 function is preferentially linked to the BER mechanism [Citation41,Citation42]. However, an indirect role of PARP1 in BER was shown by Strom et al. (2011) [Citation43], suggesting that indirect regulation of the DNA repair process is via auto PARylation of PARP1 and PARP2 or PARylation of various chromatin-related proteins that facilitate the recruitment of DNA repair factors [Citation44]. From this view, we show the PARP-dependent appearance of m8A RNAs at DNA lesions, and we also discuss the existence of an RNA-dependent non-canonical DNA repair pathway involving both m6A RNAs [Citation45] and RNAs methylated on 8-adenosine.
Results
The 8-methyladenosine (m8A)-modified RNA is recruited to UVA-microirradiated chromatin
We analysed whether m6A RNA [Citation26–28] and m8A RNA are recruited to UV-induced DNA lesions. As the first step, we studied the distribution properties of m6A RNA and m8A RNA in the nucleoplasm. We observed a lower level of both RNA modifications in the nucleoplasm of non-irradiated cells, while the density of m6A RNAs and m8A RNAs, measured in the whole cell nuclei, was higher after laser microirradiation. The highest m6A RNA and m8A RNA levels were found in microirradiated genomic region. These data implied that modified RNA is not recruited from the surrounding genome to damaged sites, but RNA is directly methylated at DNA lesions (). After initiation experiments, we studied the recruitment kinetics of m8A RNAs to DNA lesions. We observed that m8A RNAs recognize micro-irradiated chromatin immediately after local laser exposure. However, m8A RNAs’ positivity weakens 15–30 minutes after microirradiation (). As the next step, we analysed if distinct inhibitors of epigenetic processes and DNA repair can change the recruitment properties of 8A RNAs to DNA lesions. We studied the effect of Suv420H1 and Suv420H2 inhibitor A196 (A196iCitation[46]) affectioning H4K20me2/me3, a prominent 53BP1 binding partner playing a role in the NHEJ repair mechanism [Citation18,Citation47] (). In the case of histone signature, replication must be considered from the view of the dilution of the histone marks, including H4K20me2/me3. As a result, a reduced level of the 53BP1 protein, as an H4K20me3 binding partner, at DNA lesions is expected in the S/G2 phases of the cell cycle. However, this was not the case for the 53BP1 protein because we recently showed a comparable level of 53BP1 at microirradiated chromatin of cells studied in the G1, S, and G2 cell cycle phases [Citation48]. These data show that a subset of old and likely modified histones is deposited into newly-synthesized chromatin after replication. Alternatively, as discussed by Budhavapu et al. [Citation49] rather, histone-modifying enzymes, but not the parental histones, represent epigenetic factors that remain associated with the DNA through replication. In this case, these enzymes can re-establish the epigenetic information on the newly assembled chromatin. Interestingly, Svobodova Kovarikova et al. [Citation18] showed a reduced level of H4K20me3 at UV-induced DNA lesions in the S/G2 cell cycle phases, so we might expect a reduced level of 53BP1 at microirradiated chromatin in G2 cells, but this was not that case [Citation48]. Moreover, the 53BP1 protein can bind to γH2AX, which is also a 53BP1 interacting partner; thus, not only H4K20me2/me3 but also γH2AX can be a key factor in this DNA repair mechanism (summarized in [Citation50]).
Figure 1. Recruitment of m6A RNA and m8A RNA to locally irradiated chromatin. (A) Localization of m6A RNA and m8A RNA in the nucleoplasm of non-irradiated and microirradiated HeLa cells. (B) Quantification of the density of m6A RNA and m8A RNA in the whole cell nuclei of non-irradiated and microirradiated cells. (C) m8A RNA was studied at DNA lesions of (a) non-treated cells, (b) after the treatment of Suv20h1/2 inhibitor A196 (A196i) affectioning H4K20me2/me3, after inhibition of RNA polymerases I and II (c) Actinomycin D and (D) α-amanitin. (E) We performed ATM depletion and (F) inhibition of DNA methyltransferase by 5-aza deoxycytidine. Inhibitors of histone deacetylases (HDACs) (G), suberoylanilide hydroxamic acid (Vorinostat; syn. SAHA), and (H) Trichostatin A (TSA) were studied. (I) PARP was inhibited by Olaparib. Regions of interest (ROI) are shown in white frames. Scale bars show 5 µm. (D, E) Quantification of m8A RNA at locally microirradiated chromatin in non-treated cells and after different treatments is shown. (F) Western blots show effects of selected treatments from panel C on the level of H4K20me2/me3, H3K9ac, and γH2AX. Data were normalized to the total protein levels and quantification is shown in panel (G).
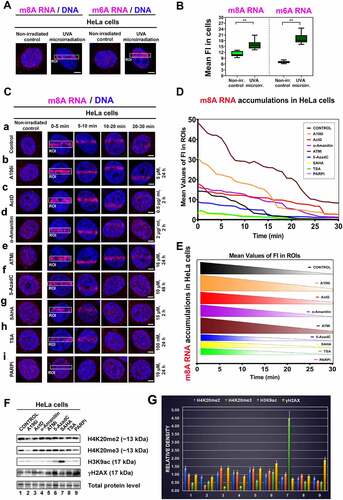
We also addressed how the following compounds affect m8A RNA positivity at DNA lesions. We analysed the effect of inhibitors of RNA polymerases I and II (Actinomycin D and α-amanitin). We performed ATM inhibition (ATMi), which could affect DDR processes. We also studied the effect of inhibition of DNA methyltransferase by 5-aza deoxycytidine or inhibition of histone deacetylases (HDACs) by Trichostatin A (TSA) or suberoylanilide hydroxamic acid (Vorinostat; syn. SAHA) (-). Moreover, we inhibited PARP with Olaparib for 1 h and 24 hours (, 1-F). In the suggested experiments, quantification analyses showed that ATMi and A196i strengthen the accumulation of m8A RNA at locally micro-irradiated chromatin (,). At the same time, HDAC inhibitors, TSA, and SAHA (inducing hyperacetylation) reduced the level of m8A RNAs at locally induced DNA lesions (,). Significantly, PARP inhibitor Olaparib completely abrogated m8A RNA positivity at DNA lesions (). Recruitment kinetics of m8A RNA to DNA lesions also changed over time. The most pronounced DDR-related accumulation of m8A RNA was 0–5 min after micro-irradiation. The significantly reduced m8A RNA signal at DNA lesion was 15–30 min post-irradiation, especially in 5-AzaC, TSA treated the cells, or after SAHA treatment (). We quantified the density of m8A RNA in the microirradiated region in time, and the treatment-dependent differences in this parameter are shown in . Also, we studied the effect of selected inhibitors on histone signature, and we confirmed that A196i reduces the level of H4K20me2, and especially H4K20me3. SAHA caused H3K9 hyperacetylation, and both 5-AzaC and PARPi increased the level of γH2AX; thus, these compounds induced DNA damage (. Taken together, these experiments imply that especially PARP inhibitor Olaparib and HDAC inhibitors reduce the level of m8A RNA at DNA lesions.
A distinction in the density of m6A RNA and m8A RNA at DNA lesions is only in the G1 phase of the cell cycle, and m6A RNA positivity at damaged chromatin is METTL3-dependent
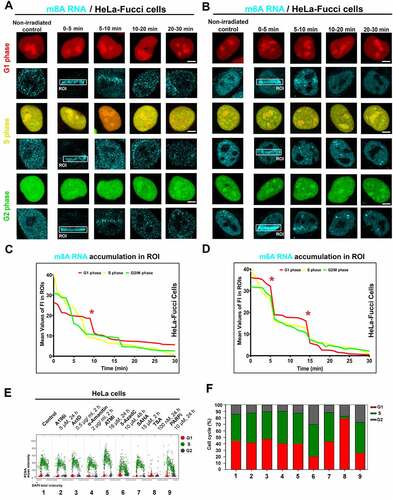
To the phenomenon mentioned above, we additionally observed that the recruitment of m8A RNA to DNA lesions is stable during the interphase of the cell cycle. It is documented in HeLa cells stably expressing the so-called FUCCI system. It is possible to recognize cells in the G1 phase expressing RFP-tagged cdt1 and G2 phase cells expressing GFP-tagged geminin. The S-phase can be identified according to the weak expression of both markers, see . Density of m8A RNA in irradiated chromatin of FUCCI cells was quantified in , showing a maximum intensity peak 10 minutes after microirradiation. Similarly, m6A RNA recognized locally induced DNA lesions in all cell cycle phases (). Notably, a distinction in the density of m6A RNA and m8A RNA recruitment to DNA lesions was only in the G1 phase of the cell cycle. In detail, m6A RNAs had a maximum peak of 5 and 15 minutes at DNA lesions, while the highest level of m8A in RNA was in UV-damaged chromatin at 10 minutes postirradiation (). We also studied the effect of selected inhibitors on the cell cycle profile. By the use of quantitative microscopy, we observed that especially 5-AzaC, TSA, and PARPi change the cell cycle profile (). As mentioned above, the positivity of modified RNAs in DNA lesions was in all cell cycle phases studied (); thus, the changes in the cell cycle could not influence the fact that modified RNAs recognize UV-damaged chromatin irrespectively of the cell cycle.
Figure 2. Accumulation of m8A RNA and m6A RNA to DNA lesion during interphase. Using the FUCCI cellular system expressing RFP-tagged cdt1 in the G1 phase and GFP-tagged geminin in the G2 phase of the cell cycle, we observed an accumulation of both (A) m8A RNA and (B) m6A RNA to microirradiated chromatin in G1, S, and G2 phases of the cell cycle. Quantification of (C) m8A RNA and (D) m6A RNA fluorescence intensity in irradiated regions of G1, S, and G2 cells.
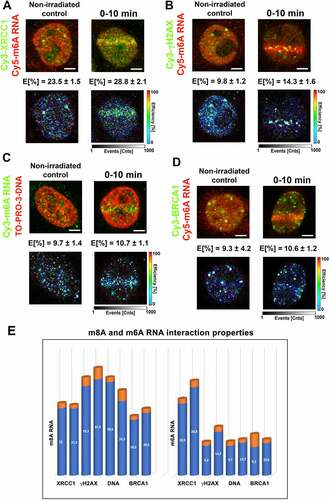
In parallel with the kinetics of m8A RNA at locally induced DNA lesions after PARP inhibition ( Ci, D, E), we have also studied the kinetics of m6A RNA at DNA lesions after PARP inhibition. We came to similar results for both m6A RNA and m8A RNA, meaning that the PARP inhibitor (Olaparib) abolished the positivity of both m6A RNA and m8A RNA at DNA lesions ( Ai , and 3A, B). In this case, we suggest that PARPi-induced changes in the cell cycle profile could not influence our conclusion on PARP-dependent and cell cycle-independent recruitment of modified RNAs to UV-irradiated chromatin ().
Figure 3. The level of m6A RNA in microirradiated regions and the effect of METTL3 inhibitor. (A) m6A RNA accumulated in microirradiated chromatin 0–5 min after laser exposure. (A, B) Quantification by ImageJ software shows that PARP inhibitor Olaparib reduced m6A RNA positivity at DNA lesions. (C-E) METTL3 inhibitors, STM2457 (see ref[Citation51].), reduced the level of m6A RNA at microirradiated chromatin. Regions of interest (ROI) are shown by rectangles or, in later phases of DDR, by arrows for better visibility. Statistically, significant differences are shown at p ≤ 0.01. Scale bars show 5 µm.
![Figure 3. The level of m6A RNA in microirradiated regions and the effect of METTL3 inhibitor. (A) m6A RNA accumulated in microirradiated chromatin 0–5 min after laser exposure. (A, B) Quantification by ImageJ software shows that PARP inhibitor Olaparib reduced m6A RNA positivity at DNA lesions. (C-E) METTL3 inhibitors, STM2457 (see ref[Citation51].), reduced the level of m6A RNA at microirradiated chromatin. Regions of interest (ROI) are shown by rectangles or, in later phases of DDR, by arrows for better visibility. Statistically, significant differences are shown at p ≤ 0.01. Scale bars show 5 µm.](/cms/asset/6bfdce0c-87d3-4113-898b-bd1240c47621/krnb_a_2139109_f0003_oc.jpg)
In the next step, we continued to address whether the appearance of m6A RNA at UV-damaged chromatin is mediated via METTL3 methyltransferase. Recently, we showed that the level of METLL3 and METTL14 at DNA lesions is stable, which can be sufficient for DDR-related RNA methylation on 6-adenosine [Citation27]. From this view, we additionally tested selective METTL3 inhibitor (STM2457) [Citation51], and we observed a reduced level of m6A RNAs at DNA lesions induced in STM2457-treated cells. The DDR-specific inhibitory effect caused by the STM2457 compound was dependent on its concentration. In this case, we detected the most significant inhibitory effect for 2 µM concentration of STM2457 at 5–20 minutes after microirradiation (). For this type of analysis, we tested the concentration and time of the treatment as recommended in Yankovska et al. (2021) [Citation51].
PARP inhibitor increased γH2AX positivity in the whole cell nuclei, did not change the level of CPDs at locally induced DNA lesions, and abolished the positivity of XRCC1 and APE1 to microirradiated chromatin
We found that the PARP inhibitor, Olaparib, has no potential to change the levels of NER factors, including XPC and CPDs at DNA lesions. In contrast, Olaparib potentiates γH2AX positivity in the whole cell nuclei (, Ba-c). PARPi also increased the number of γH2AX positive foci per cell (). Olaparib prevents the accumulation of XRCC1 and APE1, factors of BER mechanism, to UVA-damaged chromatin (). In detail, the level of XRCC1 was stable before and after microirradiation (Fig. 4Ea). At the same time, APE1 density in the irradiated genome was reduced 0–5 min postirradiation in Olaparib-treated cells (). The number of XRCC1-positive DNA repair foci was not affected by Olaparib treatment, but the number of APE1-positive repair foci increased after this treatment (Fig. 4Eb, Fb) we calculated repair foci larger than 1 µm).
Figure 4. PARP inhibitor Olaparib abolished XRCC1 and APE1 recruitment to microirradiated chromatin, but no changes were observed in NER factors. (A) PAPRi did not change the levels of XPC and CPDs at DNA lesions, while Olaparib potentiates γH2AX positivity in the whole cell nuclei. Panel (B) shows quantification of fluorescence intensity of (a) γH2AX, (b) XPC, (c) CPDs in microirradiated chromatin. (C) PARPi causes an increase of γH2AX positive foci per cell. (D) PARPi prevents the recruitment of XRCC1 and APE1 to UVA-damaged chromatin but does not change the number of (E) XRCC1 positive foci. (F) The number of APE1 positive foci increased after PARP inhibition. Data are shown as the mean ± SE (standard error), and statistically significant differences are shown at p ≤ 0.01. (G) Treatment with an alkylating agent, Melphalan, increased the number of XRCC1-positive foci, but γH2AX positivity and m8A RNA level remained stable after Melphalan treatment.
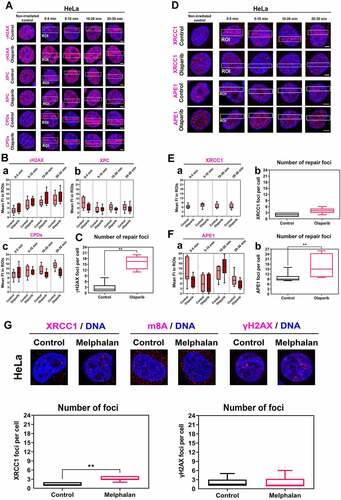
We also studied the effect of an alkylating agent potentially inducing genomic lesions recognized by the BER mechanism. For this experiment, we used Melphalan, a clinically used cytostatic for multiple myeloma treatment [Citation52], and we observed that Melphalan increases the number of XRCC1-positive DNA repair foci, but no changes were found when we compared m8A RNA nuclear level and γH2AX-positive foci in non-treated and Melphalan-treated cells ().
PARP inhibitor reduced the level of PARP1 in the cell nucleus and increased the number of 53BP1- and MDC1-positive DNA repair foci
Because PARP inhibitor Olaparib abrogated positivity of m8A RNA, XRCC1, and APE1 at DNA lesions (), we have additionally analysed if PARPi affects factors of non-homologous end joining (NHEJ) and homologous recombination repair. We also tested whether RNAse H treatment affects the level of m8A RNA at microirradiated chromatin. In this case, in Olaparib treated cells, m8A RNA was barely detectable, similarly to in the cells affected by RNAse H (). An inhibitor of ATM did not prevent m8A RNA accumulation at microirradiated chromatin (). In non-treated and Olaparib treated cells, we also calculated the number of DNA repair foci, positive on 53BP1, phosphorylated form of 53BP1, RIF1, BRCA1, RAD51, and MDC1 proteins ( Ba-c, Ba’-c’, Ca-c, Ca’-c’). After Olaparib treatment, the number of DNA repair foci, positive on 53BP1pS1778, RIF1, BRCA1, and RAD51 was not changed ( Bb’, c’; Ca’, b’). An exception was the increased number of 53BP1- and MDC1-positive repair foci, which appeared after Olaparib treatment ( Ba’, Cc’).
Figure 5. PARP inhibitor affects specific DNA repair foci and erases PARP1 positivity in microirradiated chromatin. (A) One hour after the cell treatment by PARPi, there was a low m8A RNA positivity at DNA lesions, and RNase H reduced the m8A RNA abundance in the whole genome. ATMi did not change the m8A RNA level in microirradiated chromatin. (B-C) PARPi significantly changed the number of (Ba, a’) 53BP1-, and (Cc, c’) MDC1-positive foci but did not change the number of (Bb, b’) 53BP1pS1778-, (Bc, c’) RIF1-, (Ca, a’) BRCA, (Cb, b’) RAD51-positive foci. Data are shown as the mean ± SE (standard error). Statistically, significant differences are shown at p ≤ 0.01. (D) PARP inhibitor reduced the level of PARP1 in both the whole genome and microirradiated genomic regions (ROI).
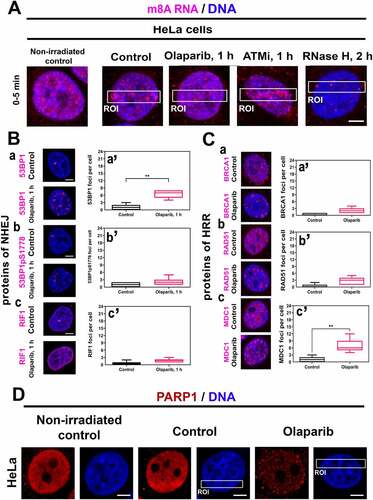
In addition to the above-mentioned observations, we verified if the PARP1 protein is recruited to UV-induced DNA lesions. We found an increased PARP1 level after microirradiation, and significantly, PARPi completely reduced PARP1 positivity in the whole genome, and in these cells, PARP1 did not recruit to microirradiated chromatin ().
m8A RNA interacted with DNA and γH2AX, while the highest FLIM-FRET efficiency was observed for m6A RNA and the XRCC1 protein
Using FLIM-FRET analysis, we studied a degree of interaction between m8A RNA (m6A RNA) and XRCC1, a protein that, similarly to m8A RNA or m6A RNA, was recruited to DNA lesions in microirradiated cells. Significantly, the treatment by PARP inhibitor abolished the accumulation of m8A RNA, m6A RNA, and XRCC1 in the microirradiated genome (Fig. 1Ai; 3A, B; 4D). When studying interaction properties, in the case of m8A RNA and XRCC1, we have observed the FLIM-FRET efficiency on the so-called cut-off level that we established at 20% [Citation53] (). However, we found a significant FLIM-FRET efficiency when we analysed m8A RNA interaction with γH2AX or m8A RNA binding to DNA. In these cases, FLIM-FRET efficiency was approximately 30%. Significantly, UV-microirradiation did not change the protein-m8A RNA or m8A RNA-DNA interaction properties ().
Figure 6. m8A RNA interacts with γH2AX and DNA. FLIM-FRET analysis showed a weak interaction between (A) m8A RNA and the XRCC1 protein but a significant interaction between (B) m8A RNA and γH2AX or (C) m8A RNA and DNA. (D) The FLIM-FRET efficiency for m8A RNA and BRCA1 was on the cut-off level, which is ~ 20%.
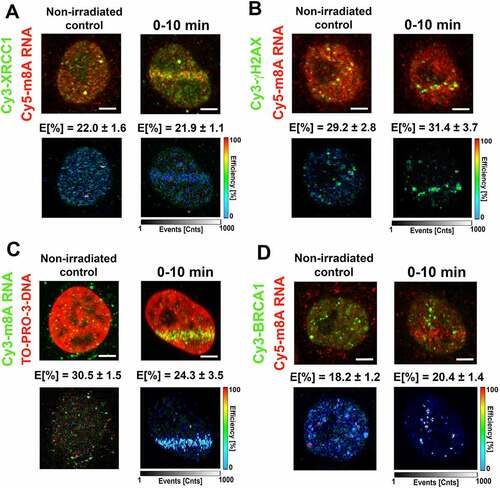
Surprisingly, in the case of m6A RNA, we have observed the highest FLIM-FRET efficiency for m6A RNA and XRCC1 (~28% in microirradiated cells); however, no interaction we found for m6A RNA and γH2AX; m6A RNA and DNA, or m6A RNA and BRCA1 (). For comparison of interaction properties studied here, see in .
Figure 7. m6A RNA did not interact with γH2AX, DNA, and BRCA1. FLIM-FRET analysis showed a strong interaction between (A) m6A RNA and XRCC1, but no interaction was shown when analysing (B) m6A RNA and γH2AX or (C) m6A RNA and DNA or (D) m6A RNA and the BRCA1 protein. (E) Comparison of interaction properties in the following interaction partners: m8A RNA-XRCC1; m8A RNA-γH2AX, m8A RNA- DNA, and m8A RNA-BRCA1 and m6A RNA-XRCC1; m6A RNA-γH2AX, m6A RNA- DNA, and m6A RNA-BRCA1.
In the case of m8A RNA (m6A RNA) and its interaction with the BRCA1 protein, FLIM-FRET efficiency was ~20% (~10%) (). For the explanation, we tested an interaction between m6A RNA (or m8A RNA) with BRCA1 due to the observation published by Zhang et al. (2020) [Citation31] or San Martin Alonso and Noordermeer (2021) [Citation54], showing that the BRCA1 protein is recruited to m6A-modified R-loops, associated with DNA breaks. According to these authors, this type of RNA modification is essential for recognizing R-loops by the BRCA1 protein. To this point, we have to mention that the kinetics of m6A /m8A RNAs and the BRCA1 protein at DNA lesions is distinct: methylated RNAs appear at damaged chromatin 0–10 min after microirradiation (Fig. 1Ca, 3A, B), while BRCA1 recruits to the lesions 20 min post-irradiation [Citation55]. Summarizing FLIM-FRET data in , it is evident that the highest degree of interaction is between m8A RNA and γH2AX or DNA, and also m6A RNA significantly interacted with XRCC1.
Together, our data support the existence of m6A/m8A RNA-DNA hybrid loops. In such hybrid structures, DNA interacts with RNA via 8-methyladenosine, and loops are likely stabilized by the interaction between m8A RNA and γH2AX (). Moreover, both m6A RNA and m8A RNA interact with the XRCC1 protein, a member of the BER mechanism.
The level of 5-hydroxymethylation (5hmC) and 5-carboxymethylation (5caC) was increased at locally microirradiated chromatin
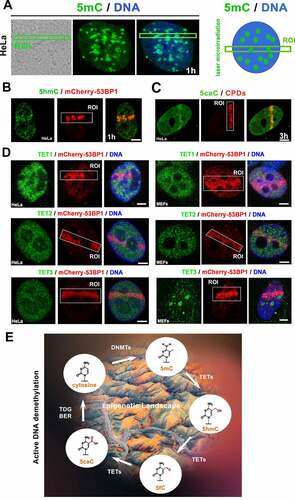
In cells exposed to microirradiation, we observed that the level of 5-methyl cytosine (5mC) is relatively high but stable (), but the density of 5hmC (5-hydroxymethyl cytosine) was increased in microirradiated chromatin (). Most significantly, the level of 5-caC (5-carboxymethylation in DNA) increased in the microirradiated genomic region (; note: density of 5hmC at microirradiated ROI is weaker than the level of 5caC, see ). Interestingly to this observation, when the cell populations were exposed to UV light, the level of Ten-Eleven Translocation enzymes 1–3 (TET1, TET2, and TET3, responsible for the active DNA demethylation) was relatively stable (). The stable level of these DNA demethylating proteins could be sufficient for the active DNA demethylation () appearing in the microirradiated genome as the later epigenetic event of DDR ().
Figure 8. The active DNA demethylation in microirradiated chromatin. (A) nuclear distribution pattern of 5mC was studied in the microirradiated genome. (B) The level of 5hmC at locally induced DNA lesion is shown, similarly to (C) the level of 5caC. (D) TET1, TET2, and TET3 protein levels were studied in DNA lesions induced by UV laser in HeLa and MEFs cells. Scale bars show 4 µm. (E) Illustration of DNA demethylation processes.
Discussion
Based on recently published data, it is evident that not only DNA but also RNAs must cope with genome injury. From this view, a fundamental observation of Xiang et al. (2017) [Citation28] showed that m6A modified RNA and RNA-metabolizing proteins contribute to UV-induced DNA damage response. Also, Yu et al. (2021) [Citation56] showed that the METTL3-METTL14 complex, similarly to m6A RNA nuclear reader YTHDC1, contributes to DNA repair. We observed the same for m8A RNAs, which appeared at DNA lesions in a PARP-dependent manner (Ci, D, E). From this view, Zhang et al. (2017) [Citation31] suggested the existence of a new PARP- and METTL3/METTL14-dependent non-canonical m6A RNA-mediated DNA repair pathway recognizing UV damaged chromatin. However, Caron et al. (2019) [Citation57] showed that PARP1 regulates DNA end resection of DSBs. These authors revealed that PARP1 protects DNA ends from nucleolytic degradation, but when PARP1 was depleted, it induced DNA hyper-resection due to weak DNA strand protection by the Ku80 factor. After PARP inhibition, the function of RIF1 and 53BP1, the proteins of the NHEJ, was abolished at DSB sites. Together, the above-mentioned observations show that m6A RNA and m8A RNA, in parallel with METTL3/METTL14 methyltransferases, are required for the repair of damaged chromatin [Citation27,Citation28]. However, the exact repair pathway, mediated via modified RNAs, is not fully elucidated.
In this case, an essential structural phenomenon represents hybrid DNA-RNA loops in which m6A RNA is considered as a template for homologous recombination repair (HRR) [Citation58,Citation59]. Also, m6A RNA readers, like YTHDF2 or YTHDC1, can recognize m6A RNA in R-loops, and these DDR-related conformation changes in chromatin could be responsible for the efficient DNA repair. Several authors suggested that R-loops at DNA lesions act positively and negatively, especially in homologous recombination repair [Citation29, Citation32–36, Citation60,Citation611]. Interestingly, R-loops in the vicinity of DSB sites can recruit factors of HRR, including RAD52 and BRCA1, in turn counteracting the anti-resection activity of the Shieldin complex, consisting of C20orf196 (also known as SHLD1), FAM35A (SHLD2), CTC-534A2.2 (SHLD3) and REV7 proteins recognizing DSB sites and working in 53BP1- and RIF1-mediated NHEJ repair pathway [Citation62]. Recently, Abakir et al. (2020) [Citation63] showed that m6A RNA is responsible for R-loops stability. Depletion of YTHDF2 caused an increased incidence of DSB sites. On the other hand, our data imply that m6A and m8A RNAs are involved in the repair of DNA lesions, primarily via a BER mechanism. We showed that the BER scaffold protein, XRCC1, in parallel with m6A and m8A RNAs, recruits to UV-induced DNA lesions in a PARP-dependent manner ( 3 3-C, , 4D). PARP-dependence of XRCC1 function has also been reported by, for example, Adamowicz et al. (2021) [Citation64]. Also, the role of XRCC1 and PARP in the direct SSB repair was shown by Strom et al. (2010) [Citation43]. In addition to these observations, we show the most pronounced interaction between the XRCC1 protein and m8A RNA, or XRCC1 and m6A RNA, in which FLIM-FRET efficiency was strengthened after UV microirradiation (). From this view, it is essential to mention that UV light induces CPDs that are recognized by NER factors [Citation65]. Therefore, the exact role of XRCC1 and modified RNAs is not clear.
It must also be considered that the formation of hybrid RNA-DNA loops needs end-resection in S-phase. Thus, this process preferentially should proceed in the S phases of the cell cycle, but we have observed both m6A RNA and m8A RNA positivity at DNA lesions induced in the G1, S, and G2 phases of the cell cycle (). Due to the functioning of modified RNAs during all phases of interphase and m6A/m8A RNA interaction with XRCC1, our results imply that m6A and m8A-modified RNAs rather contribute to the BER repair pathway.
In the literature, there is information about the appearance of R-loops in the RNAP II (RNA polymerase II) pause sites close to transcription termination sites [Citation66,Citation67]. Therefore, according to these data, the formation of methylated R-loops should be linked to the Transcription-Coupled Nucleotide Excision Repair (TC-NER) mechanism. In this case, an accumulation of the phosphorylated form of RNAP II is expected at UV-induced DNA lesions. However, we did not observe such a phenomenon [Citation68]. Moreover, NER factors like CPDs and XPC were not affected by PARP inhibition ()(). Together, in our experimental system, it seems likely that preferentially non-coding RNAs, but not mRNA, are components of R-loops in the irradiated genome. As Michelini et al. (2017) [Citation69] or Vagbøa and Geir Slupphaug (2020) [Citation59] showed, damage-induced long non-coding RNAs (dilncRNAs) or miRNAs can participate in DDR. Also, we must consider that these RNAs can be damaged by radiation; thus, their modifications could also be markers of RNA injury.
Here, we also find that the active DNA demethylation proceeds at DNA lesions as a later step of DDR. Especially, a high level of (5caC) was observed in damaged chromatin (). This observation fits well with the data published by Zhu (2009) [Citation70], demonstrating in Arabidopsis thaliana that a subfamily of DNA glycosylases promotes DNA demethylation through a base excision repair mechanism. Our observation suggests that active DNA demethylation also works in human cells via a base excision repair process activated by DNA damage. This nuclear event is mediated by TET enzymes () oxidizing 5-methylcytosines (5mC) to 5-hydroxymethylcytosine (5hmC), formyl cytosine (5fC), and 5-carboxylcytosine (5caC) in a Fe(II) and alpha-ketoglutarate (α-KG)-dependent manner [Citation71]. Here, we observe an identical density of TET proteins before and after microirradiation, which did not exclude the possible functioning of these demethylating enzymes in the DDR mechanism studied ().
Together, we showed and confirmed that the appearance of m8A RNA and m6A RNA at DNA lesions is PARP dependent (Ci, 3A, B). Due to a pronounced interaction between modified RNAs and XRCC1 and active DNA demethylation at the irradiated genome, we suggest that RNA modifications play a role in the BER mechanism, or alternatively, there is an existence of a non-canonical RNA-mediated DNA repair pathway, as suggested by Zhang et al. (2017) [Citation45]. We also consider that R-loops are formed at DNA damage sites [Citation72], showing that modified RNAs on m6A and m8A are essential for stabilizing DNA lesions. We also consider that UV-irradiation induces not only SSBs but also DSBs as the secondary structures, or UV irradiation can induce various types of DNA lesions. Thus, we observed a significant interaction between a DSB marker γH2AX and m8A RNAs (). Suggested mechanism here shows a reciprocal link between epigenetic and epitranscriptomic events regulating DNA damage repair.
Materials and methods
Cell cultivation and treatment
The human cervix adenocarcinoma (HeLa) cell line (ATCC® CCL-2TM, ATCC, UK) was cultivated in EMEM (Eagle’s Minimum Essential Medium, Merck, Germany) supplemented with 10% foetal calf serum (FCS) and the appropriate antibiotics. Mouse embryonic fibroblasts (MEFs) are a gift from Prof. Thomas Jenuwein, Max Planck Institute of Immunology and Epigenetics, Freiburg, Germany). MEF cells were cultivated in DMEM (Dulbecco’s modified Eagle’s medium, Merck, Germany) supplemented with 10% FCS, β-mercaptoethanol (#31350-010, Thermo Fisher Scientific, USA), nonessential amino acids (100× NEAA; #1140-035, Thermo Fisher Scientific, USA), sodium pyruvate (#11360-039, Thermo Fisher Scientific, USA), 1.5 g of NaHCO3 (#S5761, Merck, Germany), and appropriate antibiotics at 37°C in a humidified atmosphere containing 5% CO2 (also see [Citation73]).
To study the cell cycle-dependent recruitment of proteins to DNA lesions, we used HeLa-FUCCI cells expressing RFP-Cdt1 in the G1 phase and GFP-geminin in the S/G2/M phases previously been described in detail [Citation74] (Life Technologies; http://www.lifetechnologies.com). HeLa-FUCCI cells were cultivated in Dulbecco’s modified Eagle’s medium supplemented with 10% FCS and appropriate antibiotics at 37°C in a humidified atmosphere containing 5% CO2.
The HeLa cells were treated with several different inhibitors listed in . This table summarizes the used compounds and their final concentration. It also contains the time of treatment duration and a short description of the main inhibition targets.
Table 1. Chemical compounds used in DNA repair studies.
Local micro‑irradiation of the genome and analysis of DNA lesions with laser scanning confocal microscopy
For local laser microirradiation, cells were seeded on uncoated, γ-irradiated, gridded microscope dishes (#81166, Ibidi, USA). At 50% confluence, cells were either transiently transfected with mCherry-tagged 53BP1 (mCherry-BP1-2 pLPC-Puro; a fragment of human 53BP1, aa 1220–1711; #19835, Addgene, USA [Citation81],) or were treated with the list of inhibitors see and/or pre-sensitized with 10 μM 5-Bromo-2′-deoxyuridine (BrdU; #11296736001, Merck, Germany) for 16–18 h and Hoechst 33,342 solution to final concentration 20 nM (#62249, Thermo Fischer Scientific, USA) see [Citation82–84].
The mCherry-53BP1 plasmid’s DNA was amplified in chemically competent E. coli DH5α bacteria, and DNA was isolated using Qiagen Plasmid Maxi Kit (#121693, QIAGEN, Bio-Consult, the Czech Republic). Transfections were performed using METAFECTENETMPRO reagent (#T040-2.0, Biontex Laboratories GmbH, Germany), see [Citation55].
The cells were irradiated using a TCS SP5-X confocal microscope system (Leica, Germany) equipped with the 355-nm and 405-nm laser and 63x oil objective (HCX PL APO, lambda blue) with a numerical aperture (NA) = 1.4. The irradiation conditions were optimized according to Refs. [Citation28,Citation55,Citation85].
The cells were maintained under optimal cultivation conditions in an incubation chamber (EMBL, Germany) at 37°C, and the cell culture hood was supplemented with 5% CO2. Image acquisition for the induction of DSBs was performed with the following settings: 1024 × 1024 pixels, 400 Hz, bidirectional mode, zoom 2. Micro-irradiated cells were monitored for 30 min or at the exact time points of 1 h and 3 hours. Immunofluorescence staining was performed at the following intervals (in minutes): 0–5, 5–10, 10–20, 20–30, or in the time points 60 min and 180 min. After the staining procedure, identical irradiated cells were found on microscope slides using the registered coordinates at gridded microscopic dishes. We quantified the relative fluorescence intensity of either Alexa Fluor 594 dye in HeLa cells or Cy5 dye in HeLa-FUCCI cells, which were used to visualize the protein accumulations in the regions of interest (ROIs). We used Leica LAS X software to analyse fluorescence intensity or the number of foci per cell. The statistical analysis was performed using GraphPad Prism 9 software (USA) and the nonparametric Mann–Whitney U-test. The asterisks in the figures represent statistical significance with a p-value ≤0.01. The data are presented as the mean ± standard error (SE), representing average values from 40 to 60 evaluated cell nuclei per each time point.
Immunofluorescence staining
Immunofluorescence was modified following [Citation26]. The cells were fixed in 4% formaldehyde (PFA; #AAJ19943K2, Fisher Scientific, USA) for 10 min at room temperature (RT), permeabilized with 0.2% Triton X-100 (#194854, MP Biomedicals, USA) for 8 min, and 0.1% saponin (#S7900, Merck, Germany) for 13 min. In the next step, the microscopic dishes were washed twice in phosphate buffer saline (PBS) for 15 min. We used 10% goat serum (#G9023, Merck, Germany) dissolved in 1x PBS-Triton X-100 (0.1%) as a blocking solution.
The samples were incubated for 1 h at room temperature and then washed in 1x PBS for 15 min. For immunofluorescence analysis, the following antibodies were used: anti-m8A (#ab211498, Abcam, UK), anti-γH2AX (#ab2893, Abcam, UK), anti-XRCC1 (#sc56254, Santa Cruz Biotechnology Inc., USA), anti-XPC (#sc74410, Santa Cruz Biotechnology Inc., USA), anti-BRCA1 (#sc6954, Santa Cruz Biotechnolgy Inc., USA), anti-RAD51 (ab88572, Abcam, UK), anti-MDC1 (#NBP2-12,890, Novus Biological, UK), anti-53BP1 (#MAB3802, Merck, Germany), anti-53BP1pS1778 (#S2675, Cell Signalling Technology, USA), anti-RIF1 (#GTX131889, GeneTex, Taiwan), anti-PARP1 (#A2432, ABclonal, USA), and anti-APE1 (#10519S, Cell Signalling Technology, USA). The indicated primary antibodies were diluted at 1:100 in blocking buffer and incubated at 4°C overnight.
For m6A, 5-methylcytosine (5mC), 5-hydroxymethylcytosine (5hmC), 5-carboxylcytosine (5caC), TET1, TET2, and TET3 proteins, the immunostaining was performed following [Citation28]. Briefly, the cells were fixed in 4% PFA for 10 min at RT, then permeabilized in 0.25% Triton X-100 for 30 min. After washing with PBS. As a blocking solution, we used a PBS containing 1% Bovine Serum Albumin (BSA; #A2153, Meck, Germany), 10% Foetal Bovine Serum (FBS; FB-1090/500, BioTech, the Czech Republic), and 0.25% Triton X-100 for 60 min at RT. The following primary antibodies were used: anti-m [Citation6]A (#202003, Synaptic System, Germany), anti-5mC (#ab10805, Abcam, UK) anti-5hmC (#39999, Active Motif, Germany), anti-5caC (#61225, Active Motif, Germany), anti-TET1 (#1506, ABclonal, USA), anti-TET2 (#16273, ABclonal, USA) and anti-TET3 (#7612, ABclonal, USA). The dilution concentration was 1:100 in blocking buffer, and incubation was at 4°C overnight.
The following secondary antibodies were used: Alexa Fluor 488-conjugated donkey anti-mouse (#A21202, Thermo Fisher Scientific, USA), Alexa Fluor 488-conjugated goat anti-rabbit (#ab150077, Abcam, UK), Alexa Fluor 594-conjugated goat anti-mouse (#A11032, ThermoFisher Scientific, USA), Alexa Fluor 594-conjugated goat anti-rabbit (#A11037, ThermoFisher Scientific, USA), and goat anti-rabbit Cy5 (#ab6564, Abcam, UK). The secondary antibodies were diluted at 1:200, and the incubation time was 60 min at RT.
For Cyclobutane Pyrimidine Dimers (CPDs) staining, the cells were fixed in 4% PFA for 10 min at RT, then permeabilized in 0.5% Triton X-100 for 5 min on ice, followed by denaturation with 2 M HCl for 30 min at RT. After washing with PBS, cells were blocked in a solution containing 20% FBS dissolved in 1x PBS for 60 min at 37°C. After the washing step (five times in 1x PBS), cells were incubated with primary antibody anti-CPDs (#NMDND001, Cosmo Bio Co., Ltd., Japan) for 60 min at 37°C. The cells were rewashed twice with 1x PBS and incubated with a secondary antibody Alexa Fluor 594-conjugated goat anti-mouse (#A11032, ThermoFisher Scientific, USA; dilution 1:100) for 60 minutes at 37°C.
For RNase H treatment, we adapted a protocol according to Kotsantis et al. (2016) [Citation86] and Xiang et al. (2017) [Citation28]. Cells were washed in 1% Triton X-100 and then fixed in 4% formaldehyde for 10 min at room temperature. Followed by permeabilization with 0.3% Triton X-100 in 1x PBS for 30 minutes and washed in 1x PBS for 15 minutes at RT. RNase H (#EN0201, ThermoFisher Scientific, USA) was used at 0.05 U/ml for 2 hours at 37°C. The microscopic dishes were washed 1x PBS for 15 min and blocked with 3% BSA, 10% FCS and 0.1% Triton X-100 in dissolved in 1x PBS for 60 min at RT. The following primary and secondary antibodies were used: anti-m [Citation8]A (#ab211498, Abcam, UK) and Alexa Fluor 594-conjugated goat anti-rabbit (#A11037, ThermoFisher Scientific, USA).
We incubated samples with antibodies dilution buffers without the primary antibodies as negative controls. We did not observe signals due to the non-specific binding of secondary antibodies.
A contour of cell nuclei (condensed chromatin) was visualized by the use of 4′,6-diamidino-2-phenylindole (DAPI; #D9542, Merck, Germany), dissolved in Vectashield (#H-1000, Vector Laboratories, USA).
We acquired images with Leica TCS SP8X SMD confocal microscope (Leica Microsystem, Germany), equipped with HC PL APO 63×/ 1.4 oil CS2 objective. Image acquisition was performed using a white light laser (WLL) with the following parameters: 1024 1024 pixel resolution, 400 Hz, bidirectional mode, and zoom 2. As described above, we used Leica Application Suite (LAS X) software for immunofluorescence analysis.
FLIM-FRET technique
Fluorescence Lifetime Image (FLIM) microscopy combined with Förster Resonance Energy Transfer (FRET) was performed following [Citation87]. Using this method, we studied the interactions between XRCC1 protein (donor) and m8A RNA or m6A RNA (acceptor), γH2AX (donor), and m8A RNA (acceptor), and m8A RNA/m6A RNA (donor) and DNA (acceptor). The protein–protein interactions were studied in fixed, immunostained samples with the use of the following primary antibodies: anti-m [Citation8]A (#ab211498, Abcam, UK), anti-m [Citation6]A (#202003, Synaptic System, Germany), anti-XRCC1 (#sc56254, Santa Cruz Biotechnology Inc., USA), anti-BRCA1 (#sc6954, Santa Cruz Biotechnology Inc., USA) and anti-γH2AX (#05-636, Merck, Germany).
As secondary antibodies, we used goat anti-mouse Cy3 (#ab97035, Abcam, UK), goat anti-rabbit Cy3 (#A10520, Thermo Fisher Scientific, USA), goat anti-rabbit Cy5 (#ab6564, Abcam, UK), and TO-PRO-3 Iodide (#T3506, Thermo Fisher Scientific, USA).
The fluorophores are characterized according to their absorption and fluorescence properties; for example, the higher extinction coefficient (EC) leads to absorbing a more significant amount of light [Citation88]. The specific characteristics of fluorophores used in our FLIM-FRET experiments were adopted from the webpage https://www.fpbase.org/fret/ and are summarized in .
Table 2. Specification of donors and acceptors used for FLIM-FRET experiments.
All samples were mounted in Vectashield (#H-1000, Vector Laboratories, USA). Measurement was performed using Leica TCS SP8 X SMD confocal microscope (Leica Microsystems GmbH, Germany), PicoHarp 300 module (PicoQuant GmbH, Germany), and HyD SMD detectors. For cell visualization, we used a 63× oil immersion objective of numerical aperture 1.4. We used the pulsed white-light laser (WLL) as the excitation source with a repetition rate of 20 MHz. We acquired at least 1500 photons/pixel at the resolution of 512 512 pixels. Results were analysed by SymPhoTime 64 software (PicoQuant GmbH, Germany), and FRET efficiency was calculated following [Citation89,Citation90]. Using the FLIM-FRET technique, we studied up to 40 cell nuclei for each experimental event.
Western blot analysis
The western blot analysis was performed following [Citation91]. Briefly, to achieve identical concentrations of total proteins, we measured protein concentration by µQuant spectrophotometer (BioTek, USA). Then, the proteins were separated using SDS polyacrylamide gel electrophoresis (SDS-PAGE), followed by transfer to polyvinylidene difluoride (PVDF) membranes. In the next step, the membranes were blocked in either 2% non-fat dry milk or 2% BSA for 2 h and then incubated overnight at 4°C with the following primary antibodies: anti-H4K20me2 (#NB21-2089, Novus Biological, UK), anti-H4K20me3 (#A4048, EpigenTek, USA), H3K9ac (#06-942, Merck, Germany) and anti-γH2AX (#05-636, Merck, Germany). Primary antibodies were diluted to 1:1000. Next day, after washing, the membranes were incubated with the following secondary antibodies: goat anti-rabbit IgG (#AP307P, Merck, Germany) and goat anti-mouse IgG1 (#sc-2060, Santa Cruz Biotechnology, USA). Secondary antibodies were diluted to 1:2000 in blocking solutions. The western blot data (density of bands) were measured by ImageJ software (NIH freeware).
Quantitative image-based cytometry
The samples for quantitative image-based cytometry (QIBC) were prepared as describ [Citation92]. Briefly, cells were pre-extracted with ice-cold CSK buffer (10 mM Hepes, pH 7.5, 100 mM, NaCl, 300 mM sucrose, 3 mM MgCl2) for 10 min on ice and then fixed with 4% buffered formaldehyde (#9713.1000, VWR Chemicals, USA) for 30 min at room temperature. Primary antibody against PCNA (human, dilution 1:1000, #2037, Immunoconcepts, USA) and secondary antibody Alexa Fluor 647-conjugated donkey anti-human (dilution 1:1000, #709-605-149, Jackson Immuno Research, USA) were diluted in DMEM containing 10% FBS. The incubation times were 90 and 30 minutes for the primary and secondary antibodies, respectively. The secondary antibody solution was supplemented with 0.5 µg/ml of DAPI. After staining, samples were washed three times in 1x PBS and twice in double distilled water, dried, and mounted into a Mowiol-based mounting medium (12 % Mowiol 4-88 (#81381, Merck, Czech Republic), 30 % glycerol, 0.12 M Tris-HCl pH 8.5).
Images were captured using a ScanR high-content screening station (Olympus, Czech Republic) equipped with IX83 inverted microscope, wide-field optics, UPLSAPO dry objective (20×, 0.75 NA), Lumencor Spectra X LED fluorescence light source, filters for wavelengths of DAPI, FITC, Cy3 and Cy5, and digital Hamamatsu ORCA-R2 CCD camera. Images were acquired and analyzed automatically using ScanR acquisition and analysis software (v3.3.0, Olympus). Data were then exported to TIBCO Spotfire software (v11.8.0, TIBCO, USA) for further analysis. At least 1000 cells were compared for each condition within one experimental data set.
Authors’ contribution
SL is responsible for most experimental results and has written the methodology section. ASK is responsible for experimental results at active DNA demethylation in microirradiated chromatin. JBS is responsible for optimizing and improving the protocol for local micro-irradiation experiments. HPS performed quantitative image-based cytometry. EB suggested all experiments, coordinated experimental progress, finalized all images, and wrote this paper.
Statistical analysis
For statistical analyses, we used GraphPad Prism 9 software (USA). If the data passed the normality test, the Student’s test was applied. If not, the Mann-Whitney U-test was used. The results of the statistical tests were mentioned directly in the figure legend.
Acknowledgments
We thank The Czech Science Foundation, grant No.: 22-20303M and the Laboratory of Cellular Biophysics, a core facility of the Institute of Biophysics, for the help with microscopy techniques. On access demands ([email protected]), see depository files at https://www.ibp.cz/en/research/departments/cellular-biology-and-epigenetics/open-data
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data Availability statement
https://www.ibp.cz/en/research/departments/molecular-cytology-and-cytometry/research-profile
Additional information
Funding
References
- Hirota K, Ooka M, Shimizu N, et al. XRCC1 counteracts poly(ADP ribose)polymerase (PARP) poisons, olaparib and talazoparib, and a clinical alkylating agent, temozolomide, by promoting the removal of trapped PARP1 from broken DNA. Genes Cells. 2022;27:331–344.
- Essers J, Theil AF, Baldeyron C, et al. Nuclear dynamics of PCNA in DNA replication and repair. Mol Cell Biol. 2005;25:9350–9359.
- Cappelli E, Taylor R, Cevasco M, et al. Involvement of XRCC1 and DNA ligase III gene products in DNA base excision repair. J Biol Chem. 1997;272:23970–23975.
- Pascucci B, Stucki M, Jonsson ZO, et al. Long patch base excision repair with purified human proteins. DNA ligase I as patch size mediator for DNA polymerases delta and epsilon. J Biol Chem. 1999;274:33696–33702.
- Shuck SC, Short EA, Turchi JJ. Eukaryotic nucleotide excision repair: from understanding mechanisms to influencing biology. Cell Res. 2008;18:64–72.
- G-M L. Mechanisms and functions of DNA mismatch repair. Cell Res. 2008;18(1):85–98.
- Mao Z, Bozzella M, Seluanov A, et al. Comparison of nonhomologous end joining and homologous recombination in human cells. DNA Repair (Amst). 2008;7:1765–1771.
- Shrivastav M, De Haro LP, Nickoloff JA. Regulation of DNA double-strand break repair pathway choice. Cell Res. 2008;18(1):134–147.
- Isono M, Niimi A, Oike T, et al. BRCA1 directs the repair pathway to homologous recombination by promoting 53BP1 dephosphorylation. Cell Rep. 2017;18:520–532.
- Li S, Chen P-L, Subramanian T, et al. Binding of ctIP to the BRCT repeats of BRCA1 involved in the transcription regulation of p21 is disrupted upon DNA Damage. J Biol Chem. 1999;274(16):11334–11338.
- Shinohara M, Bishop DK, Shinohara A. Distinct functions in regulation of meiotic crossovers for DNA damage response clamp loader rad24(Rad17) and Mec1(ATR) kinase. Genetics. 2019;213(4):1255–1269.
- Hartlerode AJ, Guan Y, Rajendran A, et al. Impact of histone H4 lysine 20 methylation on 53BP1 responses to chromosomal double strand breaks. PLoS One. 2012;7(11):e49211.
- Hsiao KY, Mizzen CA. Histone H4 deacetylation facilitates 53BP1 DNA damage signaling and double-strand break repair. J Mol Cell Biol. 2013;5:157–165.
- Ataian Y, Krebs JE. Five repair pathways in one context: chromatin modification during DNA repair. Biochem Cell Biol. 2006;84:490–504.
- Bao Y. Chromatin response to DNA double-strand break damage. Epigenomics. 2011;3:307–321.
- Downs JA, Allard S, Jobin-Robitaille O, et al. Binding of chromatin-modifying activities to phosphorylated histone H2A at DNA damage sites. Mol Cell. 2004;16:979–990.
- Rogakou EP, Pilch DR, Orr AH, et al. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem. 1998;273:5858–5868.
- Svobodova Kovarikova A, Legartova S, Krejci J, et al. H3K9me3 and H4K20me3 represent the epigenetic landscape for 53BP1 binding to DNA lesions. Aging (Albany NY). 2018;10:2585–2605.
- Game JC, Chernikova SB. The role of RAD6 in recombinational repair, checkpoints and meiosis via histone modification. DNA Repair (Amst). 2009;8:470–482.
- Game JC, Williamson MS, Baccari C. X-ray survival characteristics and genetic analysis for nine Saccharomyces deletion mutants that show altered radiation sensitivity. Genetics. 2005;169:51–63.
- Game JC, Williamson MS, Spicakova T, et al. The RAD6/BRE1 histone modification pathway in Saccharomyces confers radiation resistance through a RAD51-dependent process that is independent of RAD18. Genetics. 2006;173:1951–1968.
- Grenon M, Costelloe T, Jimeno S, et al. Docking onto chromatin via the Saccharomyces cerevisiae Rad9 Tudor domain. Yeast. 2007;24:105–119.
- Ikura T, Ogryzko VV, Grigoriev M, et al. Involvement of the TIP60 histone acetylase complex in DNA repair and apoptosis. Cell. 2000;102:463–473.
- Murr R, Loizou JI, Yang YG, et al. Histone acetylation by Trrap-Tip60 modulates loading of repair proteins and repair of DNA double-strand breaks. Nat Cell Biol. 2006;8:91–99.
- Miller KM, Tjeertes JV, Coates J, et al. Human HDAC1 and HDAC2 function in the DNA-damage response to promote DNA nonhomologous end-joining. Nat Struct Mol Biol. 2010;17:1144–1151.
- Bartova E, Sustackova G, Stixova L, et al. Recruitment of Oct4 protein to UV-damaged chromatin in embryonic stem cells. PLoS One. 2011;6:e27281.
- Svobodova Kovarikova A, Stixova L, Kovarik A, et al. N(6)-adenosine methylation in RNA and a reduced m3G/TMG level in non-coding RNAs appear at microirradiation-induced DNA lesions. Cells. 2020;9:360.
- Xiang Y, Laurent B, Hsu CH, et al. RNA m(6)A methylation regulates the ultraviolet-induced DNA damage response. Nature. 2017;543:573–576.
- Jimeno S, Balestra FR, Huertas P. The emerging role of RNA modifications in DNA double-strand break repair. Front Mol Biosci. 2021;8:664872.
- Zhang M, Wang L, Zhong D. Photolyase: dynamics and mechanisms of repair of sun-induced DNA damage. Photochem Photobiol. 2017;93:78–92.
- Zhang C, Chen L, Peng D, et al. METTL3 and N6-methyladenosine promote homologous recombination-mediated repair of DSBs by modulating DNA-RNA hybrid accumulation. Mol Cell. 2020;79:425–42 e7.
- Costantino L, Koshland D. Genome-wide map of r-loop-induced damage reveals how a subset of r-loops contributes to genomic instability. Mol Cell. 2018;71:487–97 e3.
- Garcia-Muse T, Aguilera A. R loops: from physiological to pathological roles. Cell. 2019;179:604–618.
- Ohle C, Tesorero R, Schermann G, et al. Transient RNA-DNA hybrids are required for efficient double-strand break repair. Cell. 2016;167:1001–13 e7.
- Paull TT. RNA-DNA hybrids and the convergence with DNA repair. Crit Rev Biochem Mol Biol. 2019;54:371–384.
- Puget N, Miller KM, Legube G. Non-canonical DNA/RNA structures during transcription-coupled double-strand break repair: roadblocks or Bona fide repair intermediates? DNA Repair (Amst). 2019;81:102661.
- Zhang A, Peng B, Huang P, et al. The p53-binding protein 1-Tudor-interacting repair regulator complex participates in the DNA damage response. J Biol Chem. 2017;292:6461–6467.
- Qu F, Tsegay PS, Liu Y. N(6)-methyladenosine, DNA repair, and genome stability. Front Mol Biosci. 2021;8:645823.
- Giessing AM, Jensen SS, Rasmussen A, et al. Identification of 8-methyladenosine as the modification catalyzed by the radical SAM methyltransferase Cfr that confers antibiotic resistance in bacteria. RNA. 2009;15:327–336.
- Demin AA, Hirota K, Tsuda M, et al. XRCC1 prevents toxic PARP1 trapping during DNA base excision repair. Mol Cell. 2021;81:3018–30 e5.
- Fisher AE, Hochegger H, Takeda S, et al. Poly(ADP-ribose) polymerase 1 accelerates single-strand break repair in concert with poly(ADP-ribose) glycohydrolase. Mol Cell Biol. 2007;27:5597–5605.
- Kutuzov MM, Belousova EA, Kurgina TA, et al. The contribution of PARP1, PARP2 and poly(ADP-ribosyl)ation to base excision repair in the nucleosomal context. Sci Rep. 2021;11:4849.
- Ström CE, Johansson F, Uhlén M, et al. Poly (ADP-ribose) polymerase (PARP) is not involved in base excision repair but PARP inhibition traps a single-strand intermediate. Nucleic Acids Res. 2010;39:3166–3175.
- Murai J, Huang SY, Das BB, et al. Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res. 2012;72:5588–5599.
- Zhang J. Brothers in arms: emerging roles of RNA epigenetics in DNA damage repair. Cell Biosci. 2017;7:24.
- Bromberg KD, Mitchell TR, Upadhyay AK, et al. The SUV4-20 inhibitor A-196 verifies a role for epigenetics in genomic integrity. Nat Chem Biol. 2017;13:317–324.
- Paquin KL, Howlett NG. Understanding the histone DNA repair code: H4K20me2 makes its mark. Mol Cancer Res. 2018;16:1335–1345.
- Suchankova J, Kozubek S, Legartova S, et al. Distinct kinetics of DNA repair protein accumulation at DNA lesions and cell cycle-dependent formation of gammaH2AX- and NBS1-positive repair foci. Biol Cell. 2015;107:440–454.
- Budhavarapu VN, Chavez M, Tyler JK. How is epigenetic information maintained through DNA replication? Epigenetics Chromatin. 2013;6:32.
- Bartova E, Legartova S, Dundr M, et al. A role of the 53BP1 protein in genome protection: structural and functional characteristics of 53BP1-dependent DNA repair. Aging (Albany NY). 2019;11:2488–2511.
- Yankova E, Blackaby W, Albertella M, et al. Small-molecule inhibition of METTL3 as a strategy against myeloid leukaemia. Nature. 2021;593:597–601.
- Poczta A, Rogalska A, Marczak A. Treatment of multiple myeloma and the role of melphalan in the era of modern therapies-current research and clinical approaches. J Clin Med. 2021;10:1841.
- Sehnalova P, Legartova S, Cmarko D, et al. Recruitment of HP1beta to UVA-induced DNA lesions is independent of radiation-induced changes in A-type lamins. Biol Cell. 2014;106:151–165.
- San Martin Alonso M, Noordermeer SM. Untangling the crosstalk between BRCA1 and R-loops during DNA repair. Nucleic Acids Res. 2021;49:4848–4863.
- Suchankova J, Legartova S, Ruckova E, et al. Mutations in the TP53 gene affected recruitment of 53BP1 protein to DNA lesions, but level of 53BP1 was stable after gamma-irradiation that depleted MDC1 protein in specific TP53 mutants. Histochem Cell Biol. 2017;148:239–255.
- Yu D, Horton JR, Yang J, et al. Human MettL3-MettL14 RNA adenine methyltransferase complex is active on double-stranded DNA containing lesions. Nucleic Acids Res. 2021;49:11629–11642.
- Caron MC, Sharma AK, O’Sullivan J, et al. Poly(ADP-ribose) polymerase-1 antagonizes DNA resection at double-strand breaks. Nat Commun. 2019;10:2954.
- Trott DA, Porter AC. Hypothesis: transcript-templated repair of DNA double-strand breaks. Bioessays. 2006;28:78–83.
- Vågbø CB, Slupphaug G. RNA in DNA repair. DNA Repair (Amst). 2020;95:102927.
- Domingo-Prim J, Bonath F, Visa N. RNA at DNA double-strand breaks: the challenge of dealing with DNA:RNA hybrids. Bioessays. 2020;42:e1900225.
- Lu S, Dong W, Zhao P, et al. lncRNA FAM83H-AS1 is associated with the prognosis of colorectal carcinoma and promotes cell proliferation by targeting the Notch signaling pathway. Oncol Lett. 2018;15:1861–1868.
- Noordermeer SM, Adam S, Setiaputra D, et al. The shieldin complex mediates 53BP1-dependent DNA repair. Nature. 2018;560:117–121.
- Abakir A, Giles TC, Cristini A, et al. N(6)-methyladenosine regulates the stability of RNA:DNA hybrids in human cells. Nat Genet. 2020;52:48–55.
- Adamowicz M, Hailstone R, Demin AA, et al. XRCC1 protects transcription from toxic PARP1 activity during DNA base excision repair. Nat Cell Biol. 2021;23:1287–1298.
- Stixova L, Sehnalova P, Legartova S, et al. HP1beta-dependent recruitment of UBF1 to irradiated chromatin occurs simultaneously with CPDs. Epigenetics Chromatin. 2014;7:39.
- Hegazy YA, Fernando CM, Tran EJ. The balancing act of R-loop biology: the good, the bad, and the ugly. J Biol Chem. 2020;295:905–913.
- Marnef A, Legube G. m(6)A RNA modification as a new player in R-loop regulation. Nat Genet. 2020;52:27–28.
- Bartova E, Legartova S, Krejci J, et al. Depletion of A-type lamins and Lap2alpha reduces 53BP1 accumulation at UV-induced DNA lesions and Lap2alpha protein is responsible for compactness of irradiated chromatin. J Cell Biochem. 2018;119:8146–8162.
- Michelini F, Pitchiaya S, Vitelli V, et al. Damage-induced lncRNAs control the DNA damage response through interaction with DDRNAs at individual double-strand breaks. Nat Cell Biol. 2017;19:1400–1411.
- Zhu JK. Active DNA demethylation mediated by DNA glycosylases. Annu Rev Genet. 2009;43:143–166.
- Ito S, Shen L, Dai Q, et al. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science. 2011;333:1300–1303.
- Zhao H, Zhu M, Limbo O, et al. RNase H eliminates R-loops that disrupt DNA replication but is nonessential for efficient DSB repair. EMBO Rep. 2018;19. DOI:10.15252/embr.201745335
- Horakova AH, Bartova E, Galiova G, et al. SUV39h-independent association of HP1 beta with fibrillarin-positive nucleolar regions. Chromosoma. 2010;119:227–241.
- Sakaue-Sawano A, Ohtawa K, Hama H, et al. Tracing the silhouette of individual cells in S/G2/M phases with fluorescence. Chem Biol. 2008;15:1243–1248.
- Komurkova D, Svobodova Kovarikova A, Bartova E. G-Quadruplex structures colocalize with transcription factories and nuclear speckles surrounded by acetylated and dimethylated histones H3. Int J Mol Sci. 2021;23:22.
- Hickson I, Zhao Y, Richardson CJ, et al. Identification and characterization of a novel and specific inhibitor of the ataxia-telangiectasia mutated kinase ATM. Cancer Res. 2004;64:9152–9159.
- Yao TT, Mo SM, Liu LY, et al. 5-Aza-2’-deoxycytidine may influence the proliferation and apoptosis of cervical cancer cells via demethylation in a dose- and time-dependent manner. Genet Mol Res. 2013;12:312–318.
- Stixova L, Bartova E, Matula P, et al. Heterogeneity in the kinetics of nuclear proteins and trajectories of substructures associated with heterochromatin. Epigenetics Chromatin. 2011;4:5.
- Prasad CB, Prasad SB, Yadav SS, et al. Olaparib modulates DNA repair efficiency, sensitizes cervical cancer cells to cisplatin and exhibits anti-metastatic property. Sci Rep. 2017;7:12876.
- Spies L, Koekemoer TC, Sowemimo AA, et al. Caspase-dependent apoptosis is induced by Artemisia afra Jacq. ex Willd in a mitochondria-dependent manner after G2/M arrest. S Afr J Bot. 2013;84:104–109.
- Dimitrova N, Chen YC, Spector DL, et al. 53BP1 promotes non-homologous end joining of telomeres by increasing chromatin mobility. Nature. 2008;456:524–528.
- Luijsterburg MS, Dinant C, Lans H, et al. Heterochromatin protein 1 is recruited to various types of DNA damage. J Cell Biol. 2009;185:577–586.
- Sustackova G, Kozubek S, Stixova L, et al. Acetylation-dependent nuclear arrangement and recruitment of BMI1 protein to UV-damaged chromatin. J Cell Physiol. 2012;227:1838–1850.
- Dinant C, de Jager M, Essers J, et al. Activation of multiple DNA repair pathways by sub-nuclear damage induction methods. J Cell Sci. 2007;120:2731–2740.
- Jawad MM, Qader STA, Zaidan AA, et al. An overview of laser principle, laser-tissue interaction mechanisms and laser safety precautions for medical laser users. Int J Pharmacol. 2011;7:149–160.
- Kotsantis P, Silva LM, Irmscher S, et al. Increased global transcription activity as a mechanism of replication stress in cancer. Nat Commun. 2016;7:13087.
- Legartova S, Lochmanova G, Zdrahal Z, et al. DNA damage changes distribution pattern and levels of HP1 protein isoforms in the nucleolus and increases phosphorylation of HP1beta-Ser88. Cells. 2019;1097:8.
- Kumar S, Alibhai D, Margineanu A, et al. FLIM FRET technology for drug discovery: automated multiwell-plate high-content analysis, multiplexed readouts and application in situ. Chemphyschem. 2011;12:609–626.
- Lakowicz JR, Gryczynski Z. High throughput screening with multiphoton excitation. J Biomol Screen. 1999;4:355–362.
- Sillen A, Engelborghs Y. The correct use of “average” fluorescence parameters. Photochem Photobiol. 1998;67:475–486.
- Legartova S, Jugova A, Stixova L, et al. Epigenetic aspects of HP1 exchange kinetics in apoptotic chromatin. Biochimie. 2013;95:167–179.
- Sedlackova H, Rask MB, Gupta R, et al. Equilibrium between nascent and parental MCM proteins protects replicating genomes. Nature. 2020;587:297–302.