
ABSTRACT
Traffic of molecules across the bacterial membrane mainly relies on porins and transporters, whose expression must adapt to environmental conditions. To ensure bacterial fitness, synthesis and assembly of functional porins and transporters are regulated through a plethora of mechanisms. Among them, small regulatory RNAs (sRNAs) are known to be powerful post-transcriptional regulators. In Escherichia coli, the MicF sRNA is known to regulate only four targets, a very narrow targetome for a sRNA responding to various stresses, such as membrane stress, osmotic shock, or thermal shock. Using an in vivo pull-down assay combined with high-throughput RNA sequencing, we sought to identify new targets of MicF to better understand its role in the maintenance of cellular homoeostasis. Here, we report the first positively regulated target of MicF, the oppA mRNA. The OppA protein is the periplasmic component of the Opp ATP-binding cassette (ABC) oligopeptide transporter and regulates the import of short peptides, some of them bactericides. Mechanistic studies suggest that oppA translation is activated by MicF through a mechanism of action involving facilitated access to a translation-enhancing region in oppA 5ʹUTR. Intriguingly, MicF activation of oppA translation depends on cross-regulation by negative trans-acting effectors, the GcvB sRNA and the RNA chaperone protein Hfq.
Introduction
Bacterial cells are in direct contact with their environment, which can be harsh and unpredictable. To protect themselves against the extracellular medium, bacteria have developed a complex and layered structure, the cell envelope. In Gram-negative bacteria, this envelope is composed of three main elements: the inner membrane (IM), the peptidoglycan-containing periplasm and the outer membrane (OM) [Citation1].
The periplasm is a dense, viscous and oxidizing cellular compartment containing more than 60 known proteins [Citation2]. Previously, several groups have focused on the quality-control role of the periplasm, identifying folding chaperones such as SurA [Citation3] and Skp [Citation4], and unravelling the chaperone properties of the protease DegP [Citation5]. Since then, many other proteins, such as the substrate-binding protein OppA, have been shown to also possess chaperone activity [Citation6,Citation7].
Due to their crucial importance for cellular membrane integrity, viability, signal transduction and virulence, the expression of membrane proteins and transporters is tightly regulated [Citation8]. Membrane proteins are often under the control of transcriptional regulators responding to specific stresses [Citation9]. Post-translational control of membrane proteins occurs xmainly in the periplasm, where misfolded proteins can be degraded by proteases such as DegP [Citation10–12]. Both membrane proteins and transporters can also be regulated at the post-transcriptional level by small regulatory RNAs (sRNAs) [Citation8,Citation13,Citation14].
sRNAs are usually non-coding RNAs, which can either originate from their own promoter in intergenic regions or derive from 5’ [Citation15,Citation16] and 3ʹUTRs [Citation17,Citation18], the coding sequence (CDS) of mRNA transcripts, or other non-coding RNAs such as tRNAs [Citation19]. sRNAs can negatively or positively regulate their target mRNA stability and/or translation [Citation20–22]. Negative regulation mostly occurs by hindering translation initiation and favouring mRNA degradation. In contrast, positive regulation generally results from the disruption of inhibitory secondary structures in the mRNA 5′UTR, facilitating translation initiation. sRNAs often act in concert with a chaperone protein, such as Hfq [Citation23–27]. A plethora of new and unconventional regulatory mechanisms have been emerging over the last few years. For example, sRNAs are capable of regulation through base-pairing in the coding region of their targets, or they can even target translation enhancer elements to modulate protein synthesis.
Many sRNAs are well-known membrane-stress response effectors. In E. coli, RybB, MicF, MicA, MicC and RseX are responsible for the coordinated repression of porin-encoding mRNAs ompF, ompC, ompW and ompA during membrane stress [Citation28]. Additional sRNAs, such as GcvB, also repress membrane proteins and transporters [Citation29]. When glycine is abundant, GcvB expression is induced through the GcvA regulator and represses more than 30 target mRNAs, including dppA, tcyJ and oppA, all encoding subunits of ABC transporters [Citation30,Citation31].
The MicF sRNA was identified decades ago by the group of Inouye as a repressor of ompF expression [Citation32]. Since then, its targetome in E. coli has been expanded by only three repressed mRNAs: two transcriptional regulators, lrp and cpxR and phoE, which encodes a porin importing phosphorus-containing compounds [Citation33].
Here, we combined results from multiple-screening assays to identify new target mRNAs of MicF to better understand its role during membrane stress [Citation34]. Data from both high-throughput techniques MAPS (MS2 affinity purification coupled with RNA sequencing) and RIL-Seq (RNA interaction by ligation and sequencing) [Citation35] were combined together with in silico predictions from CopraRNA [Citation36–38] to identify two new targets of MicF in E. coli, namely tcyJ (formerly fliY) and oppA mRNAs. Notably, both newly identified targets of MicF are also targeted by the GcvB sRNA. Our results indicate that MicF increases oppA mRNA translational activity only in the presence of both translational repressors GcvB and Hfq. Additional in vivo and in vitro data indicate that MicF promotes translation initiation of oppA mRNA probably by rendering accessible a translational enhancer site in the 5ʹUTR. Translation activation of oppA mRNA depends on the presence of active negative regulators and suggests an unusual interplay between opposite effectors.
Material and methods
Reagents
Enzymes EcoRI (R3101), BamHI (R3136), SphI (R3182), MscI (R0534), T4 Polynucleotide kinase (M0201), Protoscript II (M0368), calf intestinal phosphatase (M0290), Q5 High fidelity DNA polymerase (M0491), XRN-1 (M0338) and Vent (exo–) DNA Polymerase (M0257) were purchased from New England Biolabs (Ipswich, MA, USA). Ampicillin (AMP201.5), Chloramphenicol (CLR201.5), kanamycin (KAN201.5), Ortho-Nitrophenyl-β-galactoside (ONP301.5), Polymyxin B (POL435.5), tetracyclin (TET701) and Phenol (PHE509.1) were purchased from BioShop (Burlington, ON, Canada). Acrylamide:Bisacrylamide 19:1 and 29:1 (A0006 and A0007) and PCR product purification kit (BS-365) were purchased from BioBasic (Markham, ON, Canada). Turbo DNase (AM2238) was purchased from ThermoFisher Scientific (Waltham, MA, USA). QIAprep Spin Miniprep Kit was purchased from QIAGEN (Hilden, Germany). Pyrophosphatase (10108987001) was purchased from Millipore Sigma (Burlington, MA, USA). T4 DNA ligase and RNase Inhibitor were produced by the Sherbrooke University Protein Purification Platform. T7 RNA polymerase was purified in house (Massé Laboratory).
Biological resources
All strains are described in Supplementary Table S1. All plasmids and oligonucleotides (oligos) are described in Supplementary Table S2 and Supplementary Table S3, respectively. For construction of lacZ fusions, PCR fragments were obtained with oligos EM4513-EM1321 (oppA), EM1320-EM1321 (P2_oppA), EM4513-EM5064 (P1_oppAprom), EM3895-EM3896 (ompF) or EM4860-EM4861 (yjbJ). The PCR fragments were digested with EcoRI and BamHI and ligated into an EcoRI/BamHI-digested pRS1551 (translational fusion) or an EcoRI/BamHI-digested pFRΔ (transcriptional fusion). To obtain oppAUGC, a three-step PCR was performed with oligos EM4513-4467 (PCR1), EM4466-EM1321 (PCR2) and EM4513-EM1321 (PCR3) using PCR1+ PCR2 as a template. PCR3 was digested with EcoRI and BamHI and ligated into an equally digested pRS1551. To obtain oppAΔ399, a three-step PCR was performed with oligos EM4513-5096 (PCR4), EM5095-EM1321 (PCR5) and EM4513-EM1321 (PCR6) using PCR4+ PCR5 as a template. PCR6 was digested with EcoRI and BamHI and ligated into an equally digested pRS1551. All fusions were then inserted into the chromosome of relevant strains at the λ attI site [Citation39]. Lysogens were screened by PCR for selection of single insertion recombinant using oligos EM111‐EM112‐EM113 and then sequenced using oligos EM194-195 [Citation40].
Plasmid pBAD-micFCGA was generated by digestion of a PCR fragment (oligos EM4426-EM2681) with MscI and SphI followed by ligation into an equally digested pNM12 vector. Plasmids pFRΔ-oppA+term and pFRΔ-oppA-MS2+ term were obtained by first performing sequential PCR reactions with oligos EM4513-EM3996 (PCR7, oppA+term), EM4513 + 1577 (PCR8 on PCR7, oppA+term), EM4513-EM3995 (PCR9, oppA-MS2+ term), EM4513-EM1576 (PCR10 on PCR9, oppA-MS2+ term) and EM4513-EM1577 (PCR11 on PCR10, oppA-MS2+ term). PCR8 and PCR11 were then digested with EcoRI and BamHI and ligated into an equally digested pFRΔ vector.
Chromosomal mutations or knockouts were obtained as previously described [Citation41]. For gcvBΔR1::cat, PCR12 was first performed on pBAD-gcvBΔR1 with oligos EM168-EM4587. Then, PCR13 was performed on PCR12 with oligos EM4592-EM4587. Finally, PCR13 was used as a primer, along with oligo EM4588 on pKD3 to generate PCR14. For generation of Δlrp::cat, PCR15 was performed on pKD3 with oligos EM3647-EM3648. For generation of oppA-1xFLAG::kan, PCR16 was performed with oligos EM4393-EM4091 on pKD4. Then, PCR17 was performed with oligos EM4394-EM4091 using PCR16 as a template. PCR14, PCR15 and PCR17 were transformed into a wild-type (WT) strain expressing the λ Red system from the pKD46 plasmid [Citation41].
Chromosomal deletion of gcvB (gcvB::tet) was obtained as follows. First, PCR18 was performed on strain GD641 with oligos EM215-EM216 to amplify the tetracycline resistance cassette. Then, PCR19 was performed with oligos EM1247-EM1248 using PCR18 as a template. PCR19 was transformed into DY330 after induction of λ‐Red [Citation42].
The Hfq-link-kan constructs were obtained as follows. PCR20 was performed on pKD4 with oligos EM4835-4836 and transformed into a WT strain, expressing the λ Red system from the pKD46 plasmid [Citation41] to give rise to strain KP2242. The Hfq-link-kan construct was transferred to a WT strain (EM1055) by P1 transduction (KP2258). For the Hfq Y25D-link-kan construct, PCR21 was performed on strain KP2242 with oligos EM1810-EM1690. PCR22 was then carried out on PCR21 with oligos EM1810-4473. PCR22 was transformed into a Hfq Y25D strain expressing the λ Red system from the pKD46 plasmid [Citation41] to give rise to strain KP2257. The Hfq Y25D-link-kan construct was transferred to a WT strain (EM1055) by P1 transduction (KP2261).
The addition of the Flag tag to Hfq was done by first performing PCR23 on pKD4 using oligos EM1689-EM1690. PCR24 was then performed on PCR23 using oligos EM1689-EM1691. PCR24 was transformed into DY330 after induction of λ‐Red [Citation42].
For Hfq Y25D-3x-Flag construct, PCR25 was carried on pKD4 with oligos EM1689-EM1690. PCR26 was performed on PCR23 with oligos EM1691-EM1690. PCR26 was transformed into the Hfq Y25D strain expressing the λ Red system from the pKD46 plasmid [Citation41]. The Hfq Y25D-3x-Flag construct was transferred to a WT strain (EM1055) by P1 transduction (KP1758).
Strains were selected with chloramphenicol, kanamycin, or tetracycline and verified by PCR. Transfer of all chromosomal mutations to different strains was achieved by P1 transduction, and recombinants were selected with appropriate antibiotics and verified by PCR. When necessary, FRT-flanked antibiotic resistance cassettes were eliminated after transformation with pCP20 (FLIP), as described before [Citation41].
Growth conditions
All strains derive from Escherichia coli strain K-12 MG1655. Unless stated otherwise, the cells were grown in Luria-Bertani (LB) medium at 37°C. When required, the medium was supplemented with 50 μg/ml ampicillin, 30 μg/ml chloramphenicol, or 10 μg/ml tetracycline.
MS2 affinity purification couples with RNA sequencing (MAPS)
The MAPS was performed as described before [Citation19,Citation43,Citation44]. The rne131 background was used to maximize target recovery. Cells grown in the LB medium were harvested at OD600nm = 0.5 and 1.0 following induction of either pBAD-micF or pBAD-MS2-micF expression with 0.1% arabinose for 15 min. Raw data was analysed as described previously, with the only modification being that due to the low quality of reads in the R2 sequencing dataset, analysis was done single-ended using R1 [Citation43,Citation44]. Selection of the putative targets was done according to the following cut-offs: RNAs with >25 reads (before normalization) and a ratio ≥3 were selected. Among these, only the RNAs with >50 reads (before normalization) and a ratio of <6.5 in the pBAD-MS2 control were selected (Supplementary Data Table S4). GalaxyProject [Citation45] and UCSC Microbial Genome Browser [Citation46] were used to analyse and visualize the data. MS2‐micF GEO accession number is GSE113584. MS2-control was previously published with GEO accession number GSE67606 [Citation47]. For MS2 affinity purification, followed by Northern blot analysis in different backgrounds (WT, ΔgcvB, hfq Y25D), cells were harvested at OD600nm = 2.0. Input samples were taken prior to any manipulation of the culture. The output samples result from MS2 affinity purification. RNA was analysed by Northern blot as described below.
β-Galactosidase assays
β-Galactosidase assays were performed as described previously [Citation48]. Cells from overnight cultures were diluted 1/1000 in 50 ml fresh medium and grown at 37°C with agitation. Antibiotics were used at appropriate concentrations when required. MicF was expressed either endogenously or from a BAD promoter induced by addition of 0.25–0.75% arabinose at OD600nm = 0.5, as specified. Results are presented as mean ± Standard Deviation (mean ± SD) of the Vmax/OD600nm calculated values.
Total RNA extraction and Northern blot analysis
Cells were grown in the LB medium at 37°C, and sample were collected at different OD600nm. For rne3071 experiments, cultures were grown at 30°C until an OD600nm = 2.0 before half was transferred at 44°C for 15 minutes before sampling. For half-life assays, cells were treated with 250 µg/ml Rifampicine before total RNA extraction was performed at specific time points post-induction. Total RNA was extracted following the hot phenol procedure described previously [Citation49]. For polyacrylamide gels, 5 μg RNA was loaded on 5–10% acrylamide 29:1, 8 M urea gels and migrated at 100 V. RNAs were electro-transferred to Hybond‐XL membranes. Membranes were prehybridized in Church buffer (0.36 M Na2HPO4, 0.14 M NaH2PO4, 1 mM EDTA, 7% SDS) [Citation50] at 42°C and radiolabeled DNA probes were then added. After incubation, membranes were washed, exposed to phosphor screens and visualized using Typhoon Trio (GE Healthcare) instrument. Image Studio Lite software (LICOR) was used for densitometry analysis when applicable.
In vitro transcription
Templates for in vitro transcriptions were generated by PCR using oligos EM4528-EM3438 (T7-oppA), EM2982-EM2983 (T7-micF), EM1215-EM1216 (T7-gcvB), EM4436-EM2983 (T7-micFCGA) and ΕΜ4528-ΕΜ4665 (T7-oppADRBS) on genomic DNA. T7-oppAUCG was obtained by performing a three-step mutagenesis. PCR1 (EM4528-EM4467) and PCR2 (EM4466-EM3438) were performed on genomic DNA and served as a template for PCR3 (EM4528-EM3438). T7-oppAΔ399 was obtained by performing a three-step mutagenesis. PCR4 (EM4528-EM5096) and PCR5 (EM5095-EM3438) were performed on genomic DNA and served as a template for PCR6 (EM4528-EM3438). Transcription reactions were carried out as described previously [Citation51]. When necessary, RNA fragments were dephosphorylated with calf intestinal phosphatase and 5’-radiolabeled with γ-32P using T4 polynucleotide kinase (NEB).
Lead acetate probing assays
Lead acetate probing assay was performed as described previously, with modifications [Citation51]. About 0.2 pmol of 5’-radiolabeled oppA RNA was incubated with or without 1 μM MicF sRNA for 10 min at 37°C before treatment with 10 mM PbAc for 2 min. H2O was used for controls. Reactions were stopped by the addition of 10 μl Loading Buffer II (LBII: 95% formamide, 18 mM EDTA, 0.025% SDS, 0.025% xylene cyanol-bromophenol blue). For in-line probing, 0.2 pmol 5’-radiolabeled oppA RNA was incubated with or without 1 μM MicF sRNA for 46 h at 22°C in in-line probing buffer (50 mM Tris-Cl pH 8.0, 100 mM KCl, 25 mM MgCl2). Reactions were stopped by the addition of 10 μl LBII. Samples were migrated on polyacrylamide gels (8% acrylamide:bisacrylamide 19:1, 8 M urea) in TBE 1X at 38 W. Gels were dried, exposed to phosphor screens and visualized using the Typhoon Trio (GE Healthcare) instrument.
Footprint assays
Footprinting assays were performed as described previously, with modifications [Citation51]. About 0.2 pmol of 5’-radiolabeled oppA RNA was incubated with or without 1 μM purified Hfq for 10 min at 37°C before treatment with 10 mM PbAc for 2 min. H2O was used for controls. Reactions were stopped by addition of STOP solution (50 mM Tris-Cl pH 8.0, 0.1% SDS). RNA was extracted using phenol-chloroform. Samples were migrated on polyacrylamide gels (8% acrylamide:bisacrylamide 19:1, 8 M urea) in TBE 1X at 38 W. Gels were dried, exposed to phosphor screens and visualized using the Typhoon Trio (GE Healthcare) instrument.
Electrophoretic mobility shift assays
Electrophoretic mobility shift assays (EMSAs) were performed as described by Morita and colleagues, with slight modifications [Citation52]. Radiolabeled oppA or oppAUCG RNA was heated for 1 min at 90°C and put on ice for 1 min. The RNA was diluted at 5 nM or 20 nM, as indicated, in binding buffer (10 nM Tris-HCl pH 8.0, 1 mM DTT, 1 mM MgCl2, 20 mM KCl, 10 mM Na2HPO4-NaH2PO4 pH 8.0, 12.5 μg/mL yeast tRNA) and mixed with specific concentrations of MicF (0–500 nM). Samples were incubated for 15 min at 37°C, and reactions were stopped by addition of 1 μL of non-denaturing loading buffer (1X TBE, 50% glycerol, 0.1% bromophenol blue, 0.1% xylene cyanol). Samples were resolved on native polyacrylamide gels (5% acrylamide:bisacrylamide 29:1) in cold TBE 1X and migrated at 50 V, at 4°C. Gels were dried, exposed to phosphor screens and visualized using the Typhoon Trio (GE Healthcare) instrument. Image Studio Lite software (LICOR) was used for densitometry analysis when required.
Primer extension assays
Reverse transcription (RT) was carried out with radiolabeled primer EM4516 or EM4518 on 20 μg of total RNA extracted from the rne3071 mutant strain at 30°C and at 44°C or from the WT and ΔmicF strains at 37°C following a previously described protocol [Citation48]. Briefly, RNA was incubated 5 min at 65°C in the presence of the primer and dNTPs. Reaction was cooled down, and an RNase inhibitor (in house) and ProtoScript II (NEB) were added to the reactions. RT was carried out for 1 h at 42°C before inactivation of the enzyme 10 min at 90°C. The resulting radioactive complementary DNA was migrated on polyacrylamide gels (8% acrylamide:bisacrylamide 19:1, 8 M urea) in TBE 1X at 38 W. Gels were dried, exposed to phosphor screens and visualized using the Typhoon Trio (GE Healthcare) instrument.
Toeprinting assays
Toeprinting assays were performed as described previously [Citation53]. RNAs were transcribed from PCR products described above. Radiolabeled primer EM3551 and ProtoScript II (NEB) were used for reverse transcription. The resulting radioactive complementary DNA was migrated on polyacrylamide gels (8% acrylamide:bisacrylamide 19:1, 8 M urea) in TBE 1X at 38 W. Gels were dried, exposed to phosphor screens and visualized using the Typhoon Trio (GE Healthcare) instrument.
Computational resources
The following online resources have been used in this study:
CopraRNA (http://rna.informatik.uni-freiburg.de/CopraRNA/Input.jsp) [Citation36–38], UCSC Microbial Genome Browser (http://microbes.ucsc.edu/) [Citation46], GalaxyProject (https://usegalaxy.org/) [Citation45], GWIPS-viz (http://gwips.ucc.ie/) [Citation54].
Statistical analyses
The number of biological replicates (N) is indicated for each experiment in figure caption. When applicable, statistical analyses were performed using an unpaired two-tailed Student’s t test or a two-way ANOVA multiple-comparison test (adjusted P value) on the GraphPad Prism 9 software, as detailed in each figure caption. For all experiments, P < 0.05 was considered statistically significant. Specific P values are indicated in the figure caption of each experiment, when applicable.
Results
In vivo analysis of MicF targetome reveals new mRNA targets
We previously used the MAPS technique to identify new target mRNAs of various sRNAs [Citation19,Citation30,Citation47,Citation55]. Here, we tagged the 5’ end of the MicF sRNA with an MS2 RNA aptamer, expressed the construct in a ΔmicF strain and proceeded to affinity purify the MS2-tagged MicF sRNA. Experiments were performed in a rne131 background, a mutation truncating the C-terminal of RNase E that serves as the scaffold for assembly of the RNA degradosome complex [Citation56]. The rne131 background was used to limit the degradation of sRNA-mRNA complexes. Samples taken during the exponential growth phase and the early stationary growth phase were combined for the purification steps. After affinity purification, RNA sequencing was performed. Enriched RNAs were identified as described previously [Citation43,Citation44]. Data analysis revealed that three of the four previously characterized targets of MicF were enriched (cpxR, ompF and lrp), as well as new putative targets (Supplementary Table S4). The fourth previously characterized target of MicF, phoE, might not be expressed in our experimental conditions. Considering the high number of hits obtained, we compared the MAPS data to in silico interaction predictions obtained with the CopraRNA algorithm [Citation34,Citation36–38]. Also, we compared both data sets (MAPS and CopraRNA) to the RIL-Seq data obtained from the stationary phase condition [Citation34,Citation35]. RIL-Seq exploits the role of Hfq in sRNA-dependent regulation to identify sRNA-mRNA complexes. We observed a slight overlap of putative targets between the different methods, with only five mRNAs identified by all three: ompF, lrp, oppA, tcyJ and yjbJ (, Supplementary Table S4). Two of those RNAs (ompF and lrp) are previously characterized targets of MicF, suggesting that combining these methods can efficiently identify sRNA targets [Citation34]. We therefore focused on oppA, tcyJ and yjbJ mRNAs as new candidate targets of MicF.
Figure 1. Identification of new targets of MicF. (A) Overlap of putative MicF targets obtained from RIL-Seq, CopraRNA and MAPS (enrichment ≥ 3, reads ≥ 25) experiments represented in a Venn Diagram. Previously characterized targets of MicF are indicated in red. (B) Visualization of MicF and MS2-MicF MAPS reads of oppA (top) and tcyJ (bottom) using the UCSC microbial genome browser (http://archaea.ucsc.edu) [Citation46]. CopraRNA-predicted base-pairing sites are represented below each graphical representation of RNA-seq reads. Blue lines represent predicted base-pairing sites. Red lines represent fragments recovered by RIL-Seq [Citation35]. For oppA, the validated pairing site (see ) is underlined in black. (C) Translational β-galactosidase assay of OppA-LacZ in WT and ΔmicF, of TcyJ-LacZ in WT, ΔmicF, ΔgcvB and ΔmicFΔgcvB and of YjbJ-LacZ in WT and ΔmicF. Samples (N = 3, mean ± SD) were taken at OD600nm = 2.5. *p = 0.01, **p = 0.0019, ***p = 0.0007, ns: p > 0.05, unpaired two-tailed Student’s t test.
![Figure 1. Identification of new targets of MicF. (A) Overlap of putative MicF targets obtained from RIL-Seq, CopraRNA and MAPS (enrichment ≥ 3, reads ≥ 25) experiments represented in a Venn Diagram. Previously characterized targets of MicF are indicated in red. (B) Visualization of MicF and MS2-MicF MAPS reads of oppA (top) and tcyJ (bottom) using the UCSC microbial genome browser (http://archaea.ucsc.edu) [Citation46]. CopraRNA-predicted base-pairing sites are represented below each graphical representation of RNA-seq reads. Blue lines represent predicted base-pairing sites. Red lines represent fragments recovered by RIL-Seq [Citation35]. For oppA, the validated pairing site (see Fig. 2A) is underlined in black. (C) Translational β-galactosidase assay of OppA-LacZ in WT and ΔmicF, of TcyJ-LacZ in WT, ΔmicF, ΔgcvB and ΔmicFΔgcvB and of YjbJ-LacZ in WT and ΔmicF. Samples (N = 3, mean ± SD) were taken at OD600nm = 2.5. *p = 0.01, **p = 0.0019, ***p = 0.0007, ns: p > 0.05, unpaired two-tailed Student’s t test.](/cms/asset/104c7704-a378-4347-b8b8-18adaa6492c0/krnb_a_2179582_f0001_oc.jpg)
OppA is the main periplasmic substrate-binding protein (SBP) of the Opp ABC transporter, responsible for the import of oligopeptides. Similarly, tcyJ encodes the periplasmic SBP of the cystine ABC transporter [Citation57]. The location of MAPS reads enrichment on both oppA and tcyJ mRNAs correlate with CopraRNA base-pairing predictions, suggesting that MicF pairs with oppA mRNA at more than 100 nucleotides upstream of the initiator codon, and tcyJ at the RBS sequence (). Notably, both oppA and tcyJ mRNAs are targeted by the sRNA GcvB (Fig. S1A, B) [Citation29,Citation30]. The reads profile for the yjbJ mRNA did not allow base-pairing prediction, but CopraRNA locates a putative base-pairing site of MicF in yjbJ CDS, outside of the five-codon window [Citation58]. The role of YjbJ has not been characterized yet.
We constructed translational fusions of oppA, tcyJ and yjbJ genes to the lacZ reporter and performed β-galactosidase assays in both wild-type (WT) and ΔmicF backgrounds. The specific activity of OppA-LacZ is decreased by 40% in the absence of MicF. For TcyJ-LacZ, a small 18% increase is observed ().
Previous reports demonstrated that GcvB pairs near MicF predicted pairing site on tcyJ mRNA to achieve downregulation (Fig. S1C) [Citation30]. We therefore hypothesized that MicF base pairing could be hindered by the presence of GcvB. We performed TcyJ-LacZ β-galactosidase assay in ΔgcvB and ΔgcvBΔmicF mutant backgrounds and observed a 72% increase of TcyJ-LacZ activity when micF is not expressed, suggesting that the tcyJ mRNA is a negative target of MicF (). The activity of the YjbJ-LacZ fusion was not affected by the ΔmicF mutation ().
We decided to focus on the oppA mRNA, the first positively regulated mRNA identified in MicF targetome. To confirm the specific co-purification of the oppA mRNA with MicF, we performed MS2 affinity purification (MAP) using either MicF or a truncated oppA RNA as baits and analysed the purified RNAs in Northern blot assays (Fig. S1D, E). In both cases, MicF and oppA, co-purified. The ompF mRNA and sRNAs GcvB and SgrS served as positive or negative controls in these assays. The longer transcripts observed in the oppA panel are most probably the result of termination read-through from both oppA and oppA-MS2 constructs.
Even though direct interaction between MicF and the oppA mRNA is suggested by in vivo and in silico results, we considered the possibility that the regulatory effect could be indirect, via the leucine-responsive regulatory protein (Lrp). Transcription of oppA is well characterized as regulated by Lrp [Citation59] and expression of lrp is negatively regulated at the post-transcriptional level by MicF [Citation33,Citation37]. Using a lacZ reporter gene fused to oppA promoter (refer to Fig. S3), we demonstrate that oppA promoter activity is not affected by the knock-out of micF gene (Fig. S1F). Moreover, we show that MicF retains its regulatory effect on OppA-LacZ in a Δlrp background (Fig. S1G). These results confirm that MicF does not achieve regulation of oppA mRNA through regulation of lrp.
Recently, a study published by Vogel’s team and performed in Salmonella enterica describes a sRNA, OppX, deriving from oppABCDF 5ʹUTR [Citation60]. The OppX sRNA is present in three different forms, depending on processing: OppX-L (410 nts), OppX-M (190 nts) and OppX-S (109 nts). The OppX sRNA was characterized as a MicF sRNA sponge, sequestering MicF to derepress ompF mRNA. Even though the binding site of MicF in the 5ʹUTR of oppA mRNA is well conserved in Enterobacteriaceae, our results obtained in E. coli clearly suggest a different story as we were unable to detect any stable RNA fragments deriving from oppA 5ʹUTR (Fig. S1H). Furthermore, our results are supported by an RIL-Seq study performed in E. coli [Citation35] in which MicF is interacting with fragments of oppA that are not contained to the OppX boundaries reported in S. enterica (, red lines). Therefore, in E. coli, MicF does not interact with a fragment deriving from the oppA mRNA but rather interacts with the oppA mRNA itself.
Moreover, the various results presented here, obtained in E. coli, do not correlate with the findings in S. enterica, suggesting different roles and mechanisms involving the MicF sRNA and the oppA 5ʹUTR in these closely related bacteria.
MicF sRNA pairs far upstream within oppA 5’ UTR
Next, to synthesize the oppA transcript in vitro, we first sought to determine the transcription start site (TSS) of oppA. In 1997, Igarashi and colleagues identified three putative TSS for oppA in the polyamine-requiring mutant strain MA261 [Citation61]. To confirm the presence of these TSSs in E. coli K-12 MG1655, we performed primer extension (PE) assays and observed two TSS identical to those identified by Igarashi and colleagues, P1 (+1) and P2 (+246) (Fig. S3A). P1 and P2 have also been identified in genome-wide mapping of TSS in E. coli [Citation62]. The results of our assays, however, indicate that the third TSS identified by Igarashi and colleagues, P3 (+341) is actually an RNase E-dependent processing site (Fig. S3A, B). The data is also supported by the analysis of XRN-1-treated RNA (Fig. S3C). Finally, our in vivo data indicate that P1 leads to the strongest OppA-LacZ β-galactosidase activity (Fig. S3D, E) and that the transcript is then processed by RNase E at the identified cleavage site (Fig. S3AB).
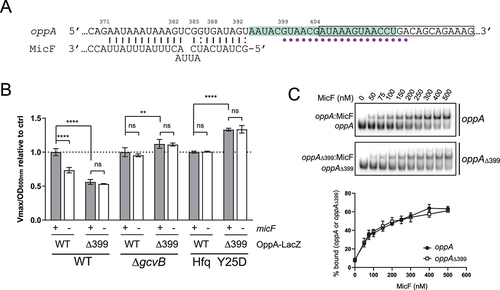
Using in vitro transcribed oppA RNA starting at the cleavage site (+341), we performed an in vitro probing assay with lead acetate (PbAc) in the absence or presence of MicF. We observed a clear protection of nucleotides +372 to +392 in the presence of MicF, suggesting a potential pairing site more than 100 nts upstream of oppA mRNA initiator codon (, S4A). To validate that the protection is due to MicF base pairing to oppA mRNA, we introduced point mutations in MicF to obtain MicFCGA (). We performed electrophoretic mobility shift assays (EMSAs) of radiolabeled oppA RNA (γ-oppA) and results showed that MicF interacts with γ-oppA, but the shift observed with MicFCGA required a higher concentration of sRNA (Fig. S5). We then introduced compensatory mutations in oppA to obtain oppAUCG () and EMSAs were performed on γ-oppAUCG in the presence of either MicF or MicFCGA. While MicF's capability to induce a shift of γ-oppAUCG was greatly hindered, the complex formation was restored with MicFCGA (Fig. S5). This suggests that the interaction between oppA mRNA and MicF is base pairing-dependent in vitro.
Figure 2. MicF regulates oppA through direct base pairing. (A) Lead acetate (PbAc) probing of γ-oppA with MicF. γ-oppA was incubated for 10 min with or without MicF (0.1 μM) prior to the addition of PbAc. Ctrl; non-reacted controls, OH; alkaline ladder, T1; and RNase T1 ladder. Numbers on the left indicate nucleotide position relative to P1 transcriptional start site (see ). The putative MicF pairing site is indicated with a bracket. Competing yeast tRNA was used at a concentration of 0.15 mg/ml. (B) Schematization of MicF pairing site on the oppA mRNA. Mutations introduced in MicF to obtain MicFCGA are indicated in red. Compensatory mutations introduced in oppA to obtain oppAUCG are indicated in green. (C) β-galactosidase assay of OppA-LacZ (left) or OppAUCG-LacZ (right). Expression of micF and micFCGA was induced by the addition of 0.025% and 0.75% arabinose, respectively, at OD600nm = 0.5. Samples (N = 3, mean ± SD) were taken at OD600nm = 2.0. ***p = 0.0010, ns: p > 0.05, unpaired two-tailed Student’s t test. Representative Northern blots of MicF are shown. 5S rRNA was used as a loading control.
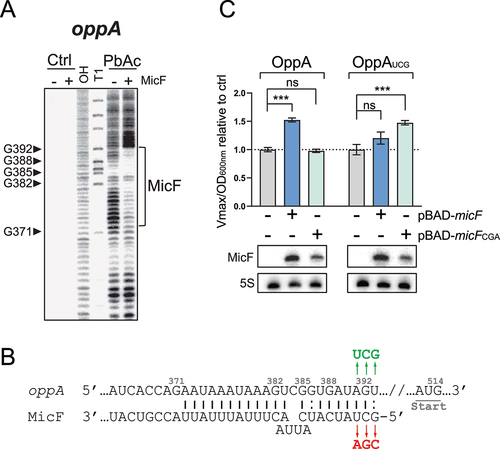
To assess if the MicF-dependent regulation of oppA mRNA observed previously () directly results from the base pairing between MicF and oppA, we overproduced either MicF or MicFCGA in a strain carrying an OppA-LacZ fusion and performed β-galactosidase assays. Whereas the overexpression of MicF led to a 50% increase in OppA-LacZ activity, MicFCGA had no effect (). We then constructed the mutant OppAUCG-LacZ fusion, which restores base pairing to MicFCGA (). Although a slight regulation of OppAUCG-LacZ was observed when MicF was overproduced (20%), only the overproduction of MicFCGA fully restored the upregulation of OppAUCG-LacZ (48%) (). This demonstrates that the in vivo regulation of oppA mRNA by MicF depends on direct RNA–RNA interaction. Note that the induction of micF expression was performed with 0.025% and 0.75% arabinose for WT MicF and MicFCGA, respectively, to obtain more comparable levels of MicF and MicFCGA sRNAs. One could argue that inducing WT MicF expression with higher arabinose concentration than 0.025% may activate translation of the OppAUCG-LacZ mutant construct due to massive MicF concentration. To assess if higher MicF concentration would lead to OppAUCG-LacZ regulation, we induced expression of micF with 0.1% arabinose (normal concentration) and observed regulation of WT OppA-LacZ but not of OppAUCG-LacZ, confirming the result obtained previously (Fig. S6).
MicF activates oppA mRNA translation through a non-canonical mechanism of action
To characterize the mechanism by which MicF activates oppA mRNA translation, we monitored the effect of MicF on oppA mRNA at OD600nm of 0.5 (exponential phase) and at 2.5 (stationary phase) by Northern blot (). Analysis of four replicates confirmed that the knockout of micF does not affect oppA mRNA levels. Stability of oppA mRNA is also unaffected by micF knockout, as observed in half-life assays performed in WT and ΔmicF backgrounds (). We then constructed the oppA-lacZ transcriptional fusion and performed β-galactosidase assays in both WT and ΔmicF backgrounds, using the translational fusion OppA-LacZ as a control. As expected, the activity of the transcriptional fusion remained stable (, left), suggesting that MicF specifically activates oppA mRNA translation without affecting mRNA levels. By contrast, β-galactosidase activity of the translational fusion significantly decreased in the absence of MicF, indicative of positive regulation (, right).
Figure 3. MicF regulates oppA at the translational level through a non-canonical mechanism. (A) Northern blot analysis of oppA mRNA in WT and ΔmicF backgrounds. Samples were taken at 0.5 and 2.5 of OD600nm. 16S rRNA was used as a loading control. The data are representative of four independent experiments. (B) Half-life assays of oppA. 250 µg/ml Rifampicine was added at 2.0 OD600nm, after which samples were collected at specified time points. 16S rRNA was used as a loading control. The data are representative of four independent experiments. (C) β-galactosidase assay of OppA-LacZ translational and oppA-lacZ transcriptional, fusions in WT and ΔmicF. Sample (N = 3, mean ± SD) were taken at OD600nm = 3.7 and relativized to their respective WT conditions. **p = 0.0011, ns: p > 0.05, unpaired two-tailed Student’s t test. (D) Primer extension analysis of oppA mRNA extracted from WT and ΔmicF strains. Lane 1–4: sequencing ladder. P1 (+1) and the cleavage site (+341) are indicated with arrows. (E) Toeprinting assay of γ-oppA in the presence of increasing concentration of MicF. Lane 1–4: sequencing ladder, lane 5: negative control. Annotation +15 represents the ribosome toeprint. The weak MicF pairing site is indicated in grey (see Figure S7B).
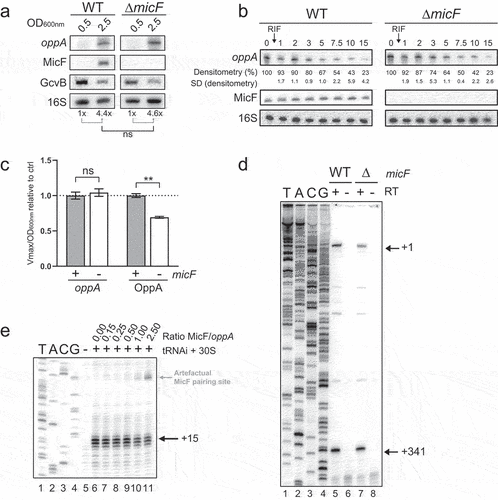
Our data identified an RNase E-dependent cleavage site in the 5ʹUTR of the oppA mRNA, at position +341 (Fig. S3). This cleavage site is located approximately 30 nucleotides upstream of the MicF pairing site (see ). We hypothesized that the pairing of MicF could modulate accessibility to the cleavage site, which could potentially lead to facilitated translation initiation of the unprocessed oppA mRNA. To determine if this was the case, we used total RNA extracted from the WT and ΔmicF strains and performed primer extension assays. We observed no difference in the primer extension signals between WT and ΔmicF RNAs, indicating that MicF does not modify the processing of oppA 5ʹUTR ().
To assess whether MicF pairing to oppA mRNA increases binding of the 30S ribosomal subunit to the RBS of oppA RNA, we performed toeprinting assays. Using an in vitro transcribed oppA RNA starting at the cleavage site (+341) (Fig. S3), toeprinting was performed with increasing concentrations of MicF (). Surprisingly, MicF did not facilitate binding of the 30S subunit and even acted as a weak inhibitor at higher ratios (lanes 10–11). Further investigations have led to the identification of a weak MicF base-pairing site, near the Shine–Dalgarno sequence of oppA mRNA, responsible for this competition at the RBS. However, this binding is only detectable at high concentration of MicF and in the absence of competing Yeast tRNA (Fig. S7). Considering the absence of MicF-dependent effect on the 30S subunit binding in toeprinting assays, our results suggest that MicF increases oppA mRNA translation through a mechanism independent of SD-sequestering structures.
MicF activation of oppA translation depends on the negative regulation by GcvB sRNA
A previous report described the GcvB-dependent repression of the oppA mRNA [Citation31]. GcvB has an extensive targetome composed of mRNAs encoding proteins involved in amino acid transport or biosynthesis [Citation37]. It is strongly expressed in rich medium during the exponential growth phase as well as in the stationary phase [Citation30]. GcvB is expressed in our experimental conditions and represses oppA mRNA (Fig. S1B).
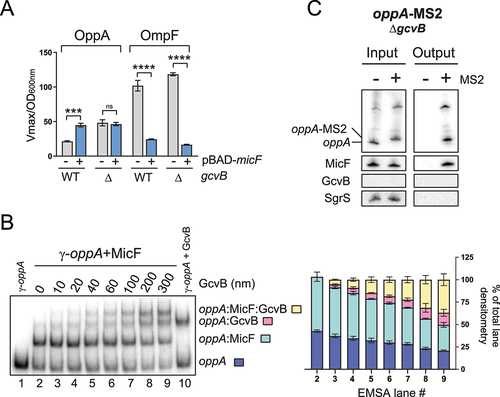
We hypothesized that an indirect interplay between MicF and GcvB could potentially dictate the regulation of the oppA mRNA. Both sRNAs interact at very distinct regions on the oppA mRNA: MicF pairs far upstream in the 5ʹUTR and GcvB interacts at the translation initiation region (, S2). If both sRNAs antagonize each other following their pairing on the oppA mRNA, we would expect a stronger effect of MicF in a ΔgcvB background compared to a WT background. We therefore verified the effect of MicF on oppA mRNA in the absence of GcvB. We overexpressed MicF in ΔmicF and ΔmicFΔgcvB strains harbouring the OppA-LacZ fusion or the OmpF-LacZ fusion, the latter being a positive control of the MicF activity (). Surprisingly, the positive regulation of OppA-LacZ by MicF is completely lost in the ΔgcvB background. Regulation of the control fusion OmpF-LacZ by MicF was detected in the absence of GcvB, dismissing the possibility of the ΔgcvB mutation generally disrupting the ability of MicF to regulate mRNAs. Very similar results were obtained using the gcvBΔR1 mutant (Fig. S8A), which lacks the region base pairing to the oppA mRNA [Citation63]. This suggests that loss of the direct regulation of OppA-LacZ by GcvB causes the loss of MicF regulation.
Figure 4. Regulation by GcvB is required to observe the MicF-dependent regulation of oppA. (A) β-galactosidase assay of OppA-LacZ (left) or OmpF-LacZ (right) in WT and ΔgcvB backgrounds. Expression of micF was induced by addition of 0.1% arabinose at OD600nm = 0.5. Samples (N = 3, mean ± SD) were taken at OD600nm = 2.0. ***p = 0.0001, ****p < 0.0001, ns: p > 0.05, unpaired two-tailed Student’s t test. (B) Left – Electrophoretic mobility shift assay of the pre-bound γ-oppA:MicF (5 nM: 300 nM, incubation 15 min at 37°C) complex incubated in the presence of increasing concentration of GcvB for 15 min at 37°C. γ-oppA (5 nM, lane 1), γ-oppA:MicF (5 nM: 300 nM, lane 2) and γ-oppA:GcvB (5 nM: 300 nM, lane 10) were used as controls to identify the different populations. Right – Densitometry analysis (N = 3, mean ± SD) of the different RNA complexes. (C) MS2 affinity purification of oppA and oppA-MS2 in a ΔgcvB background. Cells were harvested at OD600nm = 2.0. SgrS sRNA served as a negative control. The results are representative of two independent experiments.
These results can be explained if (i) MicF acts by sequestering GcvB, (ii) MicF prevents GcvB from interacting with oppA mRNA, or (iii) MicF is unable to interact with oppA mRNA in the ΔgcvB background. To address the first possibility, we used CopraRNA in silico predictor [Citation36–38] for potential base pairing between MicF and GcvB. No putative interaction was detected between both sRNAs. Then, we sought a putative in vivo interaction between MicF and GcvB using the RIL-Seq data [Citation35]. No interaction between GcvB and MicF was detected in all tested conditions (log phase, stationary phase and iron limitation). Finally, we looked at the enrichment of MicF in the MS2-GcvB MAPS data [Citation30] and that of GcvB in the MS2-MicF MAPS data. Even though a few reads can be visualized on the BedGraph representation of the data (Fig. S8B), the number of reads, or the enrichment ratio of the MS2-sRNA/sRNA, was below our cut-offs. Moreover, GcvB seemed to bind non-specifically to either the chromatography column or to the MS2 aptamer, as suggested by the MS2 control experiment (Fig. S8C). However, this non-specific binding was not detected in Northern blots performed on affinity-purified samples (Fig. S1F). Overall, in silico predictions and in vivo experiments do not support a direct interaction between MicF and GcvB. To confirm that MicF does not act by titrating GcvB, we performed an EMSA of radiolabeled GcvB (γ-GcvB) with increasing concentration of MicF. No interaction between GcvB and MicF was detected. As a positive control, γ-GcvB was incubated with the oppA mRNA, and a clear shift was observed (Fig. S8D). Finally, the toeprinting assays confirmed that MicF does not interfere with GcvB-dependant regulation of oppA mRNA (Fig. S8E).
We next used EMSAs to verify if GcvB can interact with a MicF-bound oppA RNA molecule (). γ-oppA, γ-oppA+MicF and γ-oppA+GcvB were used as controls to distinguish between the different shifted populations. Densitometry analysis of the EMSA shows that GcvB interacts with γ-oppA and with the γ-oppA-MicF complex (, right panel). Furthermore, the knockout of gcvB gene results in the upregulation OppA-LacZ in a WT or a ΔmicF background (Fig. S8F). Finally, the oppA-MS2 construct was used to validate whether MicF could still be co-purified with oppA in a ΔgcvB background. The results show that even in the absence of GcvB, MicF still co-purifies with the oppA mRNA, indicating that the ΔgcvB mutation does not hinder base pairing of MicF to the oppA mRNA (). These results suggest that MicF does not act on the oppA mRNA translation by competing with the GcvB-dependent regulation.
Figure 5. The Hfq chaperone is a negative regulator of oppA translation. (A) Footprinting analysis of γ-oppA in presence or absence of 1 μM purified Hfq protein. Ctrl; non-reacted controls, OH; alkaline ladder, T1; RNase T1 ladder. Numbers on the left indicate nucleotide position relative to the P1 transcriptional start site. The Hfq binding site and the Shine–Dalgarno (SD) are indicated with brackets in blue and green, respectively. Competing yeast tRNA was used at a concentration of 0.15 mg/ml. (B) Toeprinting assay of γ-oppA in presence or absence of purified Hfq protein (1 μM). Lane 1–4: sequencing ladder; lane 5–8: negative controls. Position +15 from the start codon is indicated by an arrow. Annotation +15 represents the ribosome toeprint. (C) β-galactosidase assay of OppA-LacZ translational fusion in three hfq backgrounds (WT, Δhfq, hfq Y25D) and in two sRNA backgrounds (WT or ΔmicFΔgcvB). Samples (N = 2, mean ± SD) were taken at OD600nm = 2.0. Fold changes are indicated. (D) β-galactosidase assay of OppA-LacZ (left) or OmpF-LacZ (right) in ΔmicF and ΔmicF/hfq Y25D backgrounds. Expression of micF was induced by addition of 0.1% arabinose at OD600nm = 0.5. Samples (N = 3, mean ± SD) were taken at OD600nm = 2.0. The data were relativized to the pNM12 empty vector control for each condition. *p = 0.02, ***p = 0.0001, ****p < 0.0001, ns: p > 0.05, unpaired two-tailed Student’s t test. (E) MS2 affinity purification of oppA and oppA-MS2 in a hfq Y25D background. Cells were harvested at OD600nm = 2.0. GcvB and SgrS sRNAs served as positive and negative controls, respectively. The results are representative of two independent experiments.
Hfq directly regulates oppA mRNA and is indirectly required for MicF-dependent positive regulation of oppA mRNA
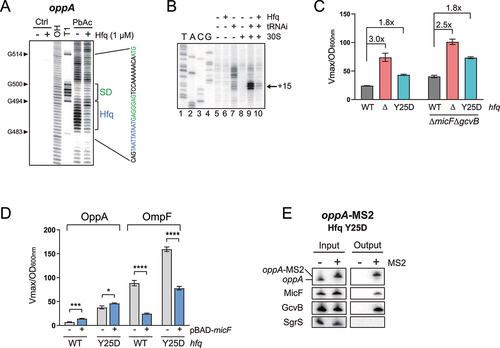
Hfq is a major RNA chaperone that also acts as a direct regulator of translation [Citation64–67]. This hexamer protein harbours multiple RNA binding sites, mainly the proximal and distal faces, preferring a poly(U) region or an (A-R-N)n motif, respectively [Citation68,Citation69]. The rim of Hfq is also able to interact with RNA, mostly through AU-rich regions. Since the distal face is generally involved in interaction with mRNAs [Citation69–71], we sought (A-R-N)n-like motifs in the 5ʹUTR of oppA that could serve as binding sites for the distal face of Hfq. We identified a putative Hfq binding region at position +484 to +492, near the SD. Using an in vitro footprinting assay, we confirmed that this region is protected in the presence of Hfq (, S4B). We then asked if Hfq binding could directly regulate oppA mRNA in the absence of sRNA partners. In a toeprinting assay, we observed a clear decrease in 30S ribosomal subunit binding to oppA mRNA in the presence of Hfq, suggesting that Hfq alone blocks oppA mRNA translation initiation in vitro ().
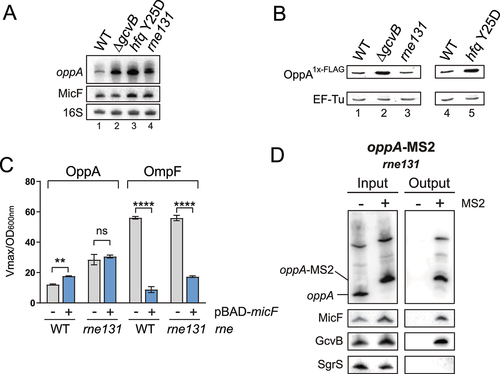
Figure 6. MicF-dependent regulation of oppA depends on oppA cellular mRNA concentration. (A) Northern blot analysis of oppA and MicF in WT, ΔgcvB, hfq Y25D and rne131 backgrounds. Samples were taken at OD600nm = 2.0. 16S rRNA was used as a loading control. The data are representative of two independent experiments. (B) Western blot analysis of the OppA1x-FLAG protein in WT, ΔgcvB, rne131, or hfq Y25D backgrounds. EF-Tu was used as a loading control. The data are representative of two independent experiments. (C) β-galactosidase assay of OppA-LacZ (left) and OmpF-LacZ (right) in ΔmicF and ΔmicF/rne131 backgrounds. Expression of micF was induced by addition of 0.1% arabinose at OD600nm = 0.5. Samples (N = 3, mean ± SD) were taken at OD600nm = 2.0. **p = 0.0029, ****p < 0.0001, ns: p > 0.05, unpaired two-tailed Student’s t test. (D) MS2 affinity purification of oppA and oppA-MS2 in a rne131 background. Cells were harvested at OD600nm = 2.0. GcvB and SgrS sRNAs served as positive and negative controls, respectively. The results are representative of two independent experiments.
To assess the role of Hfq in oppA mRNA translational regulation in vivo, we performed β-galactosidase assays of OppA-LacZ in backgrounds addressing the role of Hfq (WT, Δhfq and hfq Y25D), in two sRNA backgrounds (WT and ΔmicFΔgcvB). The Y25D mutation in the distal face of Hfq alters its binding to AU-rich sequences, such as the Hfq binding site in the oppA mRNA but does not affect binding to class 1 sRNAs, such as MicF and GcvB [Citation72]. We observed a significant increase in β-galactosidase activity in the Δhfq and in the hfq Y25D backgrounds when compared to the WT (, left). This indicates that the absence of Hfq, or its inability to interact with the oppA mRNA, alleviates translational repression of the mRNA. Similar regulation ratios were observed in the ΔmicFΔgcvB background, suggesting that Hfq regulates the oppA mRNA independently of the known sRNAs interacting with the oppA mRNA (, right).
We then asked if hindering Hfq binding to oppA mRNA would affect the MicF-dependent regulation in vivo. We opted to test the effect of MicF overproduction on both the OppA-LacZ and OmpF-LacZ fusions in ΔmicF and ΔmicF/hfq Y25D backgrounds. We favoured the use of Hfq Y25D over a Δhfq strain because the Y25D mutation does not decrease MicF () or GcvB [Citation73] steady-state levels. Whereas MicF-dependent regulation of OppA-LacZ was partially lost in the hfq Y25D strains, regulation of OmpF-LacZ remained (, S9). We then used oppA-MS2 MAP followed by Northern blot analysis to show that MicF still pairs with oppA mRNA in the hfq Y25D background (). Thus, MicF-dependent regulation of the oppA mRNA seems to depend on the repression of translation by Hfq.
Figure 7. MicF potentially regulates oppA through a translational enhancer. (A) Schematic representation of a portion of oppA 5ʹUTR. Pairing site of MicF is shown. In green: region more accessible to PbAc cleavage in the presence of MicF (Fig. S4A). Boxed: region comprised in the Ribo-seq peak (Fig. S10). Nucleotides deleted in oppAΔ399 are indicated by a dot. Nucleotides are numbered relative to P1. (B) β-galactosidase assay of OppA-LacZ and OppAΔ399-LacZ in WT and ΔmicF, in ΔgcvB and ΔgcvBΔmicF, or in hfq Y25D and hfq Y25D/ΔmicF backgrounds. Samples (N = 3, mean ± SD) were taken at OD600nm = 2.0. Data of each ΔmicF condition (white bars) were relativized to their corresponding WT micF condition (grey bars). **p = 0.0089, ****p < 0.0001, ns: p > 0.05, two-way ANOVA, multiple-comparison test. (C) Electrophoretic mobility shift assay of γ-oppA or γ-oppAΔ399 incubated in the presence of increasing concentration of MicF for 15 min. Below, densitometry analysis is plotted as the fraction of γ-oppA or γ-oppAΔ399 bound to MicF. The data are representative of two independent experiments.
MicF-dependent regulation of oppA depends on the mRNA cellular concentration
Previously, we showed that two negative regulators of oppA mRNA, GcvB and Hfq, are individually essential for the MicF-dependent activation of oppA mRNA. We therefore sought to determine a common characteristic shared by the ΔgcvB and hfq Y25D mutants. Northern blot analyses show increased levels of oppA mRNA in both backgrounds compared to WT (, lanes 1 to 3). Moreover, both ΔgcvB and hfq Y25D strains present higher levels of the OppA protein, as shown by Western blot (, lanes 1, 2 and 4, 5).
We then determined if the increase in oppA mRNA or OppA protein levels caused the loss of MicF-dependent regulation. We observed that the rne131 mutation causes an accumulation of the oppA mRNA transcript (, lanes 1, 4) without affecting OppA protein levels (, lanes 1, 3). Therefore, using the rne131 mutant, we assessed the impact of increased oppA mRNA levels on the MicF-dependent regulation of oppA mRNA. We overproduced MicF and monitored the activity of the OppA-LacZ fusion in ΔmicF and ΔmicF/rne131 backgrounds. The overproduction of MicF did not affect the activity of the OppA-LacZ fusion in the rne131 background (). To confirm that MicF could still pair to the oppA mRNA in rne131 strains, we performed Northern blot on MAP samples. The results show specific enrichment of MicF and GcvB with oppA-MS2, demonstrating that the rne131 allele does not affect the pairing of these sRNAs to oppA mRNA (). Moreover, we confirmed previously that the rne131 mutation did not affect GcvB-dependent regulation of the oppA mRNA (Fig. S1B). These results suggest that MicF can regulate the oppA mRNA only when the oppA mRNA is expressed below a certain level.
MicF potentially upregulates the oppA mRNA by modulating accessibility to a translation-enhancing region
Our data suggest that MicF positively regulates oppA mRNA translation by an unknown mechanism. Among the cis-encoded mRNA elements involved in modulation of translation initiation are the translational enhancers (TEs). TEs are usually AU- or CA-rich sequences located upstream or downstream of the SD sequence that facilitate translation initiation [Citation74–76]. Based on our probing assay of oppA mRNA, we noticed a region seemingly more sensitive to cleavage in the presence of MicF. This AU-rich stretch of 23 nts is located immediately downstream of the MicF pairing site (, Fig. S4A). We asked if this region of the oppA mRNA could act as a TE whose accessibility depends on the pairing of MicF. Ribosome profiling data obtained from our lab (Geffroy et al., in preparation) indicated a modest but clearly defined peak corresponding to the AU-rich stretch located immediately downstream of the MicF base-pairing site (, S10).
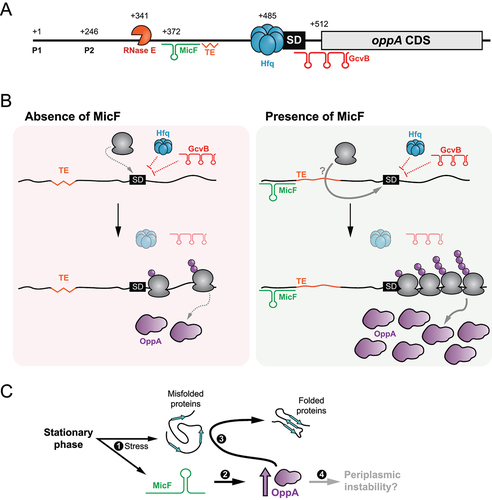
Figure 8. Schematic representation of the post-transcriptional regulation of oppA expression. (A) Scheme of oppA mRNA. Transcriptions can originate from both transcription start sites (P1 and P2). The mRNA is cleaved in its 5ʹUTR by the RNase E endonuclease at position +341. The Hfq protein binds just upstream of the SD to impede translation. The GcvB sRNA binds the RBS to hinder translation. MicF positively regulates oppA translation by base pairing at positions +372 to +393. A putative translational enhancer is located immediately downstream of the MicF base-pairing site. The numbers indicate the nucleotide position relative to P1. (B) Proposed mechanism of oppA regulation. Left – in the absence of MicF, the TE is hardly accessible (represented by a jagged line). Hfq and GcvB repress translation of oppA. Translation occurs at a minimal rate. Right – in the presence of MicF, the sRNA renders the TE more accessible (regular line) and favours translation through an unknown mechanism directly involving the ribosome or not. Even in the presence of Hfq and GcvB, which hinder translation, the now accessible TE allows efficient translation of oppA. (C) Proposed physiological role of the MicF-dependent regulation of oppA. 1 – The stationary phase of growth causes membrane stress. MicF is expressed and misfolded proteins start accumulating in the periplasm. 2 – MicF increases the translation of oppA to a specific threshold. 3 – OppA can act as a chaperone in the periplasm, facilitating protein folding. 4 – An excessive increase in OppA protein levels could lead to periplasmic instability.
To address the potential role of this mRNA region as a TE, we designed a deletion mutant where nucleotides deleted to generate oppAΔ399 are indicated in . In silico structure predictions are presented in Supplementary Figure S11. β-Galactosidase assays were performed on OppAΔ399-LacZ in WT and ΔmicF strains. First, we observed a decrease of 46% for OppAΔ399-LacZ β-galactosidase activity compared to OppA-LacZ (, left, grey bars). This suggests that deleting the AU-rich region between nucleotides 399 and 413 of the oppA mRNA negatively impacts translation, supporting the idea of this 5ʹUTR region acting as a translational enhancer. Furthermore, a complete loss of the MicF-dependent regulation was observed for OppAΔ399-LacZ (, left). To exclude the possibility of MicF being unable to interact with oppAΔ399, we performed EMSAs in the presence of an increasing concentration of MicF (, top). The results show that the oppAΔ399 mutant interacts similarly with MicF than the WT oppA (, bottom).
Since the MicF-dependent regulation of oppA mRNA is inhibited by the ΔgcvB mutation or by the hfq Y25D mutation, and that the function of the TE seems to be intimately linked to that of MicF, we asked whether the deletion of the TE would affect β-galactosidase activity in those mutants. Thus, we performed β-galactosidase assays of the OppA-LacZ and OppAΔ399-LacZ fusions in the ΔgcvB and hfq Y25D backgrounds. After deleting the oppA mRNA TE, no decrease of translational activity occurs in the absence of GcvB (, middle, grey bars) or in the hfq Y25D mutant (, right, grey bars). Moreover, as expected, the ΔmicF mutation has no effect in both mutant backgrounds ΔgcvB and hfq Y25D (, middle and right). These results support a model in which the MicF-dependent regulation of oppA mRNA can occur through the modulation of a TE element, at low oppA mRNA cellular concentrations.
Discussion
In the present work, a combination of in vivo and in silico techniques led to the identification of two new targets of MicF, the tcyJ and oppA mRNAs. While the tcyJ mRNA is the fifth negative target of MicF, the oppA mRNA is its first positively regulated target. We demonstrate that the pairing of MicF in the 5ʹUTR of the oppA mRNA promotes mRNA translation without affecting transcript stability.
The regulatory effect of MicF on oppA mRNA translation does not appear to be conserved between closely related species, even though the interaction between the two entities is conserved. Indeed, Vogel’s team recently identified an sRNA deriving from Salmonella enterica oppA 5ʹUTR that acts as a sponge RNA to sequester MicF. In E. coli, however, two major differences must be noted. First, no stable RNA fragment originating from oppA 5ʹUTR could be detected. Second, we clearly demonstrate that MicF is a true regulator of oppA mRNA translation. Here, we propose that MicF can positively regulate oppA mRNA translation by modulating the accessibility of a translational enhancer located approximately 100 nts upstream of oppA RBS. To date, this major difference between the roles of MicF and of the oppA mRNA 5ʹUTR in S. enterica and E. coli cannot be explained. However, this would not be the first instance of differential regulation of gene expression between these two closely related bacteria. This is not surprising considering the different environmental pressures dictating the evolution of S. enterica and E. coli.
Many mechanisms of positive regulation have been previously described, with two of them standing out. First, sRNAs are known to base-pair in the 5ʹUTR of their target to disrupt inhibitory structures, resulting in higher translation rates. This mechanism was first uncovered in the Gram-positive bacterium Staphylococcus aureus, in which RNAIII upregulates hla [Citation77]. Since then, many sRNAs have been shown to employ a similar mechanism, as it is the case for the regulation of shiA by the sRNA RyhB [Citation48]. In the pathogen Listeria monocytogenes, the pairing of sRNA Rli27 far upstream of the RBS (−151 to −129 relative to the start codon) is hypothesized to modify the structure of the entire 5ʹUTR, liberating the RBS and increasing translation initiation efficiency [Citation78]. This last example is reminiscent of MicF pairing 120 nts upstream of oppA start codon. However, in vitro structural probing of oppA RNA showed no disruption of mRNA secondary structures near the RBS. Additionally, we demonstrated that MicF is unable to promote binding of the 30S ribosomal subunit to oppA RNA in vitro.
In the second mechanism of positive regulation, sRNAs have previously been shown to interfere with mRNA decay, mainly by masking endonuclease cleavage sites. In E. coli, the SgrS sRNA positively affects the yigL mRNA by base pairing in the coding sequence masking an RNase E cleavage site and stabilizing the mRNA [Citation79]. Contrasting with this example, MicF knockout or overproduction has no influence on the oppA mRNA levels or stability, ruling out the possibility of MicF interfering with RNases cleavage sites. Furthermore, sRNAs are known to be able to promote or obstruct the processing of their targets [Citation80], but this explanation was rapidly dismissed in the present case, based on the analysis of oppA 5ʹUTR processing.
The roles of other cis-encoded elements of the oppA mRNA were considered throughout our investigation of the MicF-dependent regulation of oppA mRNA (Fig. S2). Polyamines, such as spermidine, have been shown to promote oppA mRNA translation through a riboswitch located in the mRNA 5ʹUTR (Fig. S2) [Citation61,Citation81,Citation82]. While MicF could participate in riboswitch regulation, several data point otherwise. Indeed, the MicF pairs approximately 60 nts upstream of the riboswitch, limiting the possibility of direct interference. Moreover, no structural modification, apart from the TE region located immediately downstream of the MicF pairing site, was observed in oppA 5ʹUTR following MicF pairing.
In E. coli, an oppA deletion mutant is more sensitive to heat shock compared to a wild-type strain, suggesting a role for OppA in the heat shock response [Citation83]. A variation in temperature could therefore increase, or impair, MicF capability to regulate oppA mRNA through structural modifications. However, the regulation observed here is at constant temperature, suggesting that the MicF-dependent regulation of oppA does not depend on temperature-modulated modifications of oppA 5ʹUTR structure.
We therefore explored the trans elements involved in oppA mRNA expression. The oppA mRNA has previously been shown as a direct negative target of the GcvB trans-acting sRNA [Citation29]. In our experimental conditions, the loss of GcvB expression (ΔgcvB) causes the loss of MicF-dependent regulation even though no interaction or indirect competition between both sRNAs was detected. The dual regulation of the oppA mRNA by both MicF and GcvB is especially intriguing. There are only a few examples of mRNAs that are both positively and negatively regulated by sRNAs. The first case of antagonistic regulation concerns flhD, which is negatively regulated by ArcZ, OmrA, OmrB and OxyS [Citation84] and positively regulated by McaS [Citation85]. Another well-characterized case is rpoS mRNA, which is upregulated by DsrA, ArcZ and RprA [Citation86–88] and downregulated by CyaR [Citation89,Citation90]. In both situations, though, three regulators or more act in concert versus a single antagonistic regulator, suggesting that only a highly specific set of environmental conditions would lead to a switch in regulation of the target. Moreover, both sRNAs McaS and ArcZ share a base-pairing site on flhD target, creating competition between both sRNAs [Citation84,Citation85]. As for the regulation of rpoS, the sRNA ArcZ specifically pairs and promotes degradation of the CyaR, creating an exclusive regulation: CyaR can only regulate rpoS in the absence of ArcZ [Citation90]. Finally, the cirA mRNA is positively regulated by RyhB and negatively by OmrA and OmrB sRNAs [Citation91–93]. While RyhB is mainly expressed during iron-starvation conditions [Citation94], OmrA/B are mostly expressed in high osmolarity conditions [Citation92]. Again, only a specific set of conditions would cause expression of RyhB and OmrA/B at the same time.
The dual regulation of flhD, rpoS and cirA target mRNAs contrasts with that of the oppA mRNA by MicF and GcvB sRNAs. Indeed, MicF and GcvB are concurrently expressed during the early stationary phase of growth (experimental condition of this study), and they pair at distinct sites on the oppA mRNA. Moreover, both MicF and GcvB can accumulate without promoting each other’s degradation. Our initial results suggest that GcvB repression is a prerequisite to observe the positive regulation of oppA mRNA by MicF. Further investigation revealed that a combination of GcvB, Hfq and the RNA degradosome complex reduces oppA mRNA expression to enable translational activation by MicF.
Previously, Hfq was shown to directly regulate specific mRNA translation. For example, it directly binds the RBS of the IS10 transposase mRNA to block translation initiation [Citation65], interacts with cis-encoded elements of the cirA mRNA to modulate translation [Citation91]. More recently, a study found that the binding of Hfq in the 5ʹUTR of the mutS mRNA causes remodelling of the secondary structure, hindering mutS translation [Citation66]. Here, we demonstrate that Hfq interacts with oppA mRNA in the translation initiation region, just upstream of the RBS to prevent 30S ribosomal subunit binding to the mRNA, probably through steric inhibition. Like GcvB, the presence of Hfq is required to observe the MicF-dependent regulation of oppA mRNA. Taken together, these results indicate that MicF can only regulate oppA mRNA in a narrow range of cellular mRNA levels, which is made possible by the concerted action of GcvB, Hfq and RNase E. This is highly reminiscent of the regulation of RyhB by the 3’ external transcribed spacer of the glyW-cysT-leuZ pre-tRNA transcript (3ʹETSleuZ), which is only capable of exerting its effect on RyhB at low cellular RyhB levels [Citation19].
Investigation of the oppA mRNA sequence revealed a putative translational enhancer rendered more accessible by the presence of MicF. We were able to show that this region of oppA mRNA was important to achieve optimal translation rates. More importantly, we presented evidence that MicF might act by modulating the access of this putative TE site to promote translation initiation. This positive mechanism of regulation is surprising as it contrasts with other examples of sRNAs interacting with TEs. Indeed, the GcvB sRNA represses translation of both gltI and dppA mRNAs by masking CA-rich TEs, and SgrS sRNA obstructs the U-rich TE of manX mRNA to hinder translation [Citation95]. However, the mechanisms involved in TE-dependent translation modulation are not yet fully understood. In some cases, the S1 ribosomal protein contacts the TE and stabilizes the ribosome at the translation initiation site. The S1 protein can also contact TEs to help promote initiating ribosomes to the elongation step [Citation76].
The S1 protein, the largest of the ribosomal proteins, is composed of six domains. While domains 1 to 4 are essential for cell viability [Citation96], truncation of domains 5 and 6 (rpsAΔ56) results in a viable strain. Although their role is not yet fully understood, deletion of domains 5 and 6 in E. coli presents a strong growth defect. Domain 5 is highly similar to domain 4 and might be involved in RNA binding [Citation96–98]. We asked whether the putative oppA TE we identified was S1-dependent. If this is the case, truncation of both domains 5 and 6 could affect the binding of S1 to oppA mRNA, and we would expect a reduced effect of MicF. Thus, we performed β-galactosidase assays of OppA-LacZ in WT or rpsAΔ56 backgrounds following the overexpression of MicF. Whereas the translational activity of oppA strongly decreases in rpsAΔ56 background, we observed no difference in the MicF regulation of oppA mRNA compared to WT (Fig. S10B). While the data presented here do not reveal the mechanism by which TE increases oppA mRNA translation, evidence demonstrate that oppA TE is fully functional when assisted by MicF ().
MicF-dependent activation of oppA mRNA in the stationary phase of growth, while GcvB is active, seems to be counter-intuitive. OppA is the SBP of the oligopeptide (Opp) ABC transporter, responsible for the import of charged tripeptides and tetrapeptides [Citation99–101]. The Opp ABC transporter is especially useful in conditions of amino acid starvation, a stress present in the late stationary phase of growth or in minimal medium. GcvB, however, is highly expressed in rich medium, and repressed when amino acids are scarce, alleviating the repression of many transporters, such as the Opp ABC transporter, the cytidine ABC transporter [Citation30], the dipeptide (Dpp) ABC transporter [Citation31], or the D-serine/alanine/glycine/-H+ symporter [Citation102]. Therefore, why would MicF activate OppA when GcvB is still highly synthesized in the cells? We hypothesize that the answer could reside in the role of OppA as a periplasmic chaperone [Citation6,Citation7]. During the stationary phase of growth, an abundance of stresses, including the accumulation of misfolded proteins in the periplasm, induces the expression of the envelope stress response (ESR) effectors, especially the transcriptional factors σS and σE [Citation103,Citation104]. In such stressful conditions, σS triggers MicF synthesis, which helps upregulate OppA levels. OppA could then act as a periplasmic chaperone, preventing protein denaturation in the periplasm ().
Previous work performed on the uropathogenic E. coli (UPEC) strain indicates that OppA is essential during infection [Citation105]. This has been attributed to the central role of OppA in peptide uptake, possibly the main source of carbon for bacterial cells during urinary tract infections. Later, OppA was shown to be involved in the sensitivity to antibiotics in UPEC, particularly Polymyxin B [Citation106]. Other toxic compounds, such as the synthetic tri-peptide tri-L-ornithine [Citation107] or the recently developed antibiotic GE81112 [Citation108], also rely on the presence of OppA to enter the cell. The second target of MicF we identified in this study, the tcyJ mRNA, also encodes a periplasmic substrate-binding protein, the cystine ABC transporter. Like the Opp ABC transporter, the cystine ABC transporter is involved in the uptake of the toxic analogues of two amino acids, L-selenaproline (SCA) and L-selenocystine (SeCys) [Citation109]. Further experiments are needed to assess the role of MicF in antibiotic sensitivity in relation to the regulation of oppA and tcyJ mRNAs.
Data Availability
Galaxy is an open source, community-driven and web-based platform accessible at https://usegalaxy.org/ and can be found in a GitHub repository (https://github.com/galaxyproject).
The UCSC Microbial Genome Browser is a web-based genome browser hosted by the University of California, Santa Cruz (http://microbes.ucsc.edu/).
MicF MAPS data has been deposited to the NCBI Gene Expression Omnibus (GEO) under the accession number GSE113584 (token for reviewers: mpifekaarjsrpsx).
MS2-control MAPS data is available on the NCBI Gene Expression Omnibus (GEO) under the accession number GSE67606.
Supplemental Material
Download Zip (9.1 MB)Acknowledgments
We thank Jörg Vogel, Maude Guillier, Cari Vanderpool, Jingyi Fei and Karine Prévost for useful comments and discussions. M-CC is grateful for the financial support received from the Natural Sciences and Engineering Research Council of Canada (NSERC) and from the Fonds de Recherche du Québec - Nature et technologies (FRQ-NT) during her PhD. This work was funded by an operating grant BMB 389354 from the Canadian Institutes of Health Research (CIHR) to EM.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/15476286.2023.2179582
Additional information
Funding
References
- Silhavy TJ, Kahne D, Walker S. The bacterial cell envelope. Cold Spring Harb. Perspect. Biol. 2010;2:a000414–a000414.
- Schmidt TM. Encyclopedia of Microbiology. Cambridge MA, United States: Academic Press; 2019.
- Rouvière PE, Gross CA. SurA, a periplasmic protein with peptidyl-prolyl isomerase activity, participates in the assembly of outer membrane porins. Genes Dev. 1996;10:3170–3182.
- Walton TA, Sousa MC. Crystal structure of Skp, a prefoldin-like chaperone that protects soluble and membrane proteins from aggregation. Mol Cell. 2004;15:367–374.
- Spiess C, Beil A, Ehrmann M. A temperature-dependent switch from chaperone to protease in a widely conserved heat shock protein. Cell. 1999;97:339–347.
- Richarme G, Caldas TD. Chaperone Properties of the Bacterial Periplasmic Substrate-binding Proteins. J Biol Chem. 1997;272:15607–15612.
- Lennon CW, Thamsen M, Friman ET, et al. Folding optimization in vivo uncovers new chaperones. J Mol Biol. 2015;427:2983–2994.
- De la Cruz MA, Calva E. The complexities of porin genetic regulation. J Mol Microbiol Biotechnol. 2010;18:24–36.
- Hews CL, Cho T, Rowley G, et al. Maintaining integrity under stress: envelope stress response regulation of pathogenesis in gram-negative bacteria. Front Cell Infect Microbiol. 2019;9. DOI:10.3389/fcimb.2019.00313
- Lyu ZX, Zhao XS. Periplasmic quality control in biogenesis of outer membrane proteins. Biochem Soc Trans. 2015;43:133–138.
- Miot M, Betton J-M. Protein quality control in the bacterial periplasm. Microb Cell Factories. 2004;3:4.
- Narita S, Tokuda H. Bacterial lipoproteins; biogenesis, sorting and quality control. Biochim. Biophys. Acta BBA - Mol. Cell Biol. Lipids. 2017;1862:1414–1423.
- Fröhlich KS, Gottesman S. Small regulatory RNAs in the enterobacterial response to envelope damage and oxidative stress. Microbiol Spectr. 2018;6. DOI:10.1128/microbiolspec.RWR-0022-2018
- Vogel J, Papenfort K. Small non-coding RNAs and the bacterial outer membrane. Curr Opin Microbiol. 2006;9:605–611.
- Loh E, Dussurget O, Gripenland J, et al. A trans-acting riboswitch controls expression of the virulence regulator PrfA in Listeria monocytogenes. Cell. 2009;139:770–779.
- Thomason MK, Voichek M, Dar D, et al. A rhlI 5’ UTR-derived sRNA regulates RhlR-dependent quorum sensing in Pseudomonas aeruginosa. mBio. 2019;10. DOI:10.1128/mBio.02253-19
- Chao Y, Papenfort K, Reinhardt R, et al. An atlas of Hfq-bound transcripts reveals 3′ UTRs as a genomic reservoir of regulatory small RNAs. Embo J. 2012;31:4005–4019.
- Chao Y, Vogel J. A 3’ UTR-derived small RNA provides the regulatory noncoding arm of the inner membrane stress response. Mol Cell. 2016;61:352–363.
- Lalaouna D, Carrier M-C, Semsey S, et al. A 3’ external transcribed spacer in a tRNA transcript acts as a sponge for small RNAs to prevent transcriptional noise. Mol Cell. 2015;58:393–405.
- Carrier M-C, Lalaouna D, Massé E. Broadening the definition of bacterial small RNAs: characteristics and mechanisms of action. Annu Rev Microbiol. 2018;72:141–161.
- Jørgensen MG, Pettersen JS, Kallipolitis BH. sRNA-mediated control in bacteria: an increasing diversity of regulatory mechanisms. Biochim Biophys Acta Gene Regul Mech. 2020;1863:194504.
- Papenfort K, Vanderpool CK. Target activation by regulatory RNAs in bacteria. FEMS Microbiol Rev. 2015;39:362–378.
- Sledjeski DD, Whitman C, Zhang A. Hfq is necessary for regulation by the untranslated RNA DsrA. J Bacteriol. 2001;183:1997–2005.
- Møller T, Franch T, Højrup P, et al. Hfq: a bacterial sm-like protein that mediates RNA-RNA interaction. Mol Cell. 2002;9:23–30.
- Massé E, Escorcia FE, Gottesman S. Coupled degradation of a small regulatory RNA and its mRNA targets in Escherichia coli. Genes Dev. 2003;17:2374–2383.
- Updegrove TB, Zhang A, Storz G. Hfq: the flexible RNA matchmaker. Curr Opin Microbiol. 2016;30:133–138.
- Santiago‐Frangos A, Woodson SA. Hfq chaperone brings speed dating to bacterial sRNA. WIREs RNA. 2018;9:e1475.
- Guillier M, Gottesman S, Storz G. Modulating the outer membrane with small RNAs. Genes Dev. 2006;20:2338–2348.
- Urbanowski ML, Stauffer LT, Stauffer GV. The gcvB gene encodes a small untranslated RNA involved in expression of the dipeptide and oligopeptide transport systems in Escherichia coli. Mol Microbiol. 2000;37:856–868.
- Lalaouna D, Eyraud A, Devinck A, et al. GcvB small RNA uses two distinct seed regions to regulate an extensive targetome. Mol Microbiol. 2019;111:473–486.
- Pulvermacher SC, Stauffer LT, Stauffer GV. The role of the small regulatory RNA GcvB in GcvB/mRNA posttranscriptional regulation of oppA and dppA in Escherichia coli. FEMS Microbiol Lett. 2008;281:42–50.
- Mizuno T, Chou MY, Inouye M. A unique mechanism regulating gene expression: translational inhibition by a complementary RNA transcript (micRNA). Proc Natl Acad Sci U S A. 1984;81:1966–1970.
- Holmqvist E, Unoson C, Reimegård J, et al. A mixed double negative feedback loop between the sRNA MicF and the global regulator Lrp. Mol Microbiol. 2012;84:414–427.
- Georg J, Lalaouna D, Hou S, et al. The power of cooperation: experimental and computational approaches in the functional characterization of bacterial sRNAs. Mol Microbiol. 2020;113:603–612.
- Melamed S, Peer A, Faigenbaum-Romm R, et al. Global mapping of small RNA-target interactions in bacteria. Mol Cell. 2016;63:884–897.
- Raden M, Ali SM, Alkhnbashi OS, et al. Freiburg RNA tools: a central online resource for RNA-focused research and teaching. Nucleic Acids Res. 2018;46:W25–W29.
- Wright PR, Richter AS, Papenfort K, et al. Comparative genomics boosts target prediction for bacterial small RNAs. Proc Natl Acad Sci U S A. 2013;110:E3487–3496.
- Wright PR, Georg J, Mann M, et al. CopraRNA and IntaRNA: predicting small RNA targets, networks and interaction domains. Nucleic Acids Res. 2014;42:W119–W123.
- Simons RW, Houman F, Kleckner N. Improved single and multicopy lac-based cloning vectors for protein and operon fusions. Gene. 1987;53:85–96.
- Powell BS, Rivas MP, Court DL, et al. Rapid confirmation of single copy lambda prophage integration by PCR. Nucleic Acids Res. 1994;22:5765–5766.
- Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci. 2000;97:6640–6645.
- Yu D, Ellis HM, Lee EC, et al. An efficient recombination system for chromosome engineering in Escherichia coli. Proc Natl Acad Sci U S A. 2000;97:5978–5983.
- Carrier M-C, Laliberté G, Massé E. Identification of new bacterial small RNA targets using MS2 affinity purification coupled to RNA sequencing. Methods Mol Biol Clifton NJ. 2018;1737:77–88.
- Lalaouna D, Prévost K, Eyraud A, et al. Identification of unknown RNA partners using MAPS. Methods San Diego Calif. 2017;117:28–34.
- Afgan E, Baker D, Batut B, et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 2018;46:W537–W544.
- Chan PP, Holmes AD, Smith AM, et al. The UCSC archaeal genome browser: 2012 update. Nucleic Acids Res. 2012;40:D646–D652.
- Lalaouna D, Morissette A, Carrier M-C, et al. DsrA regulatory RNA represses both hns and rbsD mRNAs through distinct mechanisms in Escherichia coli. Mol Microbiol. 2015;98:357–369.
- Prévost K, Salvail H, Desnoyers G, et al. The small RNA RyhB activates the translation of shiA mRNA encoding a permease of shikimate, a compound involved in siderophore synthesis. Mol Microbiol. 2007;64:1260–1273.
- Aiba H, Adhya S, de Crombrugghe B. Evidence for two functional gal promoters in intact Escherichia coli cells. J Biol Chem. 1981;256:11905–11910.
- Church GM, Gilbert W. Genomic sequencing. Proc Natl Acad Sci. 1984;81:1991–1995.
- Desnoyers G, Morissette A, Prévost K, et al. Small RNA-induced differential degradation of the polycistronic mRNA iscRSUA. Embo J. 2009;28:1551–1561.
- Morita T, Maki K, Aiba H. Detection of sRNA-mRNA interactions by electrophoretic mobility shift assay. Methods Mol Biol Clifton NJ. 2012;905:235–244.
- Salvail H, Lanthier-Bourbonnais P, Sobota JM, et al. A small RNA promotes siderophore production through transcriptional and metabolic remodeling. Proc Natl Acad Sci U S A. 2010;107:15223–15228.
- Michel AM, Kiniry SJ, O’Connor PBF, et al. GWIPS-viz: 2018 update. Nucleic Acids Res. 2018;46:D823–D830.
- Lalaouna D, Prévost K, Laliberté G, et al. Contrasting silencing mechanisms of the same target mRNA by two regulatory RNAs in Escherichia coli. Nucleic Acids Res. 2018;46:2600–2612.
- Lopez PJ, Marchand I, Joyce SA, et al. The C-terminal half of RNase E, which organizes the Escherichia coli degradosome, participates in mRNA degradation but not rRNA processing in vivo. Mol Microbiol. 1999;33:188–199.
- Moussatova A, Kandt C, O’Mara ML, et al. ATP-binding cassette transporters in Escherichia coli. Biochim. Biophys. Acta BBA - Biomembr 2008;1778:1757–1771.
- Bouvier M, Sharma CM, Mika F, et al. Small RNA binding to 5′ mRNA coding region inhibits translational initiation. Mol Cell. 2008;32:827–837.
- Cho B-K, Barrett CL, Knight EM, et al. Genome-scale reconstruction of the Lrp regulatory network in Escherichia coli. Proc Natl Acad Sci. 2008;105:19462–19467.
- Matera G, Altuvia Y, Gerovac M, et al. Global RNA interactome of Salmonella discovers a 5′ UTR sponge for the MicF small RNA that connects membrane permeability to transport capacity. Mol Cell. 2022;82:629–644.e4.
- Igarashi K, Saisho T, Yuguchi M, et al. Molecular mechanism of polyamine stimulation of the synthesis of oligopeptide-binding protein. J Biol Chem. 1997;272:4058–4064.
- Thomason MK, Bischler T, Eisenbart SK, et al. Global transcriptional start site mapping using differential RNA sequencing reveals novel antisense RNAs in Escherichia coli. J Bacteriol. 2015;197:18–28.
- Sharma CM, Papenfort K, Pernitzsch SR, et al. Pervasive post-transcriptional control of genes involved in amino acid metabolism by the Hfq-dependent GcvB small RNA. Mol Microbiol. 2011;81:1144–1165.
- Moll I, Afonyushkin T, Vytvytska O, et al. Coincident Hfq binding and RNase E cleavage sites on mRNA and small regulatory RNAs. RNA. 2003;9:1308–1314.
- Ellis MJ, Trussler RS, Haniford DB. Hfq binds directly to the ribosome‐binding site of IS10 transposase mRNA to inhibit translation. Mol Microbiol. 2015;96:633–650.
- Chen J, Gottesman S. Hfq links translation repression to stress-induced mutagenesis in E. coli. Genes Dev. 2017;31:1382–1395.
- Andrade JM, Dos Santos RF, Chelysheva I, et al. The RNA-binding protein Hfq is important for ribosome biogenesis and affects translation fidelity. Embo J. 2018;37:e97631.
- Schumacher MA, Pearson RF, Møller T, et al. Structures of the pleiotropic translational regulator Hfq and an Hfq-RNA complex: a bacterial Sm-like protein. Embo J. 2002;21:3546–3556.
- Link TM, Valentin-Hansen P, Brennan RG. Structure of Escherichia coli Hfq bound to polyriboadenylate RNA. Proc Natl Acad Sci. 2009;106:19292–19297.
- Soper TJ, Woodson SA. The rpoS mRNA leader recruits Hfq to facilitate annealing with DsrA sRNA. RNA. 2008;14:1907–1917.
- Robinson KE, Orans J, Kovach AR, et al. Mapping Hfq-RNA interaction surfaces using tryptophan fluorescence quenching. Nucleic Acids Res. 2014;42:2736–2749.
- Zhang A, Schu DJ, Tjaden BC, et al. Mutations in interaction surfaces differentially impact E. coli Hfq association with small RNAs and their mRNA targets. J Mol Biol. 2013;425:3678–3697.
- Schu DJ, Zhang A, Gottesman S, et al. Alternative Hfq-sRNA interaction modes dictate alternative mRNA recognition. Embo J. 2015;34:2557–2573.
- Hook-Barnard IG, Brickman TJ, McIntosh MA. Identification of an AU-rich translational enhancer within the Escherichia coli fepB leader RNA. J Bacteriol. 2007;189:4028–4037.
- Sharma CM, Darfeuille F, Plantinga TH, et al. A small RNA regulates multiple ABC transporter mRNAs by targeting C/A-rich elements inside and upstream of ribosome-binding sites. Genes Dev. 2007;21:2804–2817.
- Takahashi S, Furusawa H, Ueda T, et al. Translation enhancer improves the ribosome liberation from translation initiation. J Am Chem Soc. 2013;135:13096–13106.
- Morfeldt E, Taylor D, von Gabain A, et al. Activation of alpha-toxin translation in Staphylococcus aureus by the trans-encoded antisense RNA, RNAIII. Embo J. 1995;14:4569–4577.
- Quereda JJ, Ortega ÁD, Pucciarelli MG, et al. The listeria small RNA Rli27 regulates a cell wall protein inside eukaryotic cells by targeting a long 5′-UTR variant. PLoS Genetics. 2014;10:e1004765.
- Papenfort K, Sun Y, Miyakoshi M, et al. Small RNA-mediated activation of sugar phosphatase mRNA regulates glucose homeostasis. Cell. 2013;153:426–437.
- Obana N, Nomura N, Nakamura K. Structural requirement in Clostridium perfringens collagenase mRNA 5′ leader sequence for translational induction through small RNA-mRNA base pairing. J Bacteriol. 2013;195:2937–2946.
- Yoshida M, Meksuriyen D, Kashiwagi K, et al. Polyamine stimulation of the synthesis of oligopeptide-binding protein (OppA) involvement of a structural change of the Shine-Dalgarno sequence and the initiation codon AUG in OppA mRNA. J Biol Chem. 1999;274:22723–22728.
- Igarashi K, Kashiwagi K. Effects of polyamines on protein synthesis and growth of Escherichia coli. J Biol Chem. 2018;293:18702–18709.
- Krisko A, Copic T, Gabaldón T, et al. Inferring gene function from evolutionary change in signatures of translation efficiency. Genome Biol. 2014;15:R44.
- De Lay N, Gottesman S. A complex network of small non-coding RNAs regulate motility in Escherichia coli. Mol Microbiol. 2012;86:524–538.
- Thomason MK, Fontaine F, Lay ND, et al. A small RNA that regulates motility and biofilm formation in response to changes in nutrient availability in Escherichia coli. Mol Microbiol. 2012;84:17–35.
- Majdalani N, Cunning C, Sledjeski D, et al. DsrA RNA regulates translation of RpoS message by an anti-antisense mechanism, independent of its action as an antisilencer of transcription. Proc Natl Acad Sci U S A. 1998;95:12462–12467.
- Majdalani N, Hernandez D, Gottesman S. Regulation and mode of action of the second small RNA activator of RpoS translation, RprA. Mol Microbiol. 2002;46:813–826.
- Mandin P, Gottesman S. Integrating anaerobic/aerobic sensing and the general stress response through the ArcZ small RNA. Embo J. 2010;29:3094–3107.
- Zhang A, Altuvia S, Tiwari A, et al. The OxyS regulatory RNA represses rpoS translation and binds the Hfq (HF-I) protein. Embo J. 1998;17:6061–6068.
- Kim W, Lee Y. Mechanism for coordinate regulation of rpoS by sRNA-sRNA interaction in Escherichia coli. RNA Biol. 2020;17:176–187.
- Salvail H, Caron M-P, Bélanger J, et al. Antagonistic functions between the RNA chaperone Hfq and an sRNA regulate sensitivity to the antibiotic colicin. Embo J. 2013;32:2764–2778.
- Guillier M, Gottesman S. Remodelling of the Escherichia coli outer membrane by two small regulatory RNAs. Mol Microbiol. 2006;59:231–247.
- Guillier M, Gottesman S. The 5’ end of two redundant sRNAs is involved in the regulation of multiple targets, including their own regulator. Nucleic Acids Res. 2008;36:6781–6794.
- Massé E, Gottesman S. A small RNA regulates the expression of genes involved in iron metabolism in Escherichia coli. Proc Natl Acad Sci U S A. 2002;99:4620–4625.
- Azam MS, Vanderpool CK. Translation inhibition from a distance: the small RNA SgrS silences a ribosomal protein S1-dependent enhancer. Mol. Microbiol. 2020;114:391–408.
- Duval M, Korepanov A, Fuchsbauer O, et al. Escherichia coli ribosomal protein S1 unfolds structured mRNAs onto the ribosome for active translation initiation. PLoS Biol. 2013;11:e1001731.
- Takeshita D, Yamashita S, Tomita K. Molecular insights into replication initiation by Qβ replicase using ribosomal protein S1. Nucleic Acids Res. 2014;42:10809–10822.
- Byrgazov K, Grishkovskaya I, Arenz S, et al. Structural basis for the interaction of protein S1 with the Escherichia coli ribosome. Nucleic Acids Res. 2015;43:661–673.
- Higgins CF, Hardie MM. Periplasmic protein associated with the oligopeptide permeases of Salmonella typhimurium and Escherichia coli. J Bacteriol. 1983;155:1434–1438.
- Abouhamad WN, Manson M, Gibson MM, et al. Peptide transport and chemotaxis in Escherichia coli and Salmonella typhimurium: characterization of the dipeptide permease (Dpp) and the dipeptide-binding protein. Mol Microbiol. 1991;5:1035–1047.
- Klepsch MM, Kovermann M, Löw C, et al. Escherichia coli peptide binding protein OppA has a preference for positively charged peptides. J Mol Biol. 2011;414:75–85.
- Pulvermacher SC, Stauffer LT, Stauffer GV. Role of the sRNA GcvB in regulation of cycA in Escherichia coli. Microbiol. Read. Engl 2009;155:106–114.
- Jaishankar J, Srivastava P. Molecular basis of stationary phase survival and applications. Front Microbiol. 2017;8. DOI:10.3389/fmicb.2017.02000
- Mitchell AM, Silhavy TJ. Envelope stress responses: balancing damage repair and toxicity. Nat Rev Microbiol. 2019;17:417–428.
- Alteri CJ, Smith SN, Mobley HLT. Fitness of Escherichia coli during urinary tract infection requires gluconeogenesis and the TCA Cycle. PLoS Pathog. 2009;5:e1000448.
- Subashchandrabose S, Smith SN, Spurbeck RR, et al. Genome-wide detection of fitness genes in uropathogenic Escherichia coli during systemic infection. PLoS Pathogens. 2013;9:e1003788.
- Lin B, Short SA, Eskildsen M, et al. Functional testing of putative oligopeptide permease (Opp) proteins of Borrelia burgdorferi: a complementation model in opp− Escherichia coli. Biochim. Biophys. Acta BBA - Mol. Cell Res 2001;1499:222–231.
- Maio A, Brandi L, Donadio S, et al. The oligopeptide permease opp mediates illicit transport of the bacterial P-site decoding inhibitor GE81112. Antibiotics. 2016;5. DOI:10.3390/antibiotics5020017
- Deutch CE, Spahija I, Wagner CE. Susceptibility of Escherichia coli to the toxic L-proline analogue L-selenaproline is dependent on two L-cystine transport systems. J Appl Microbiol. 2014;117:1487–1499.