
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
Precursor mRNA (pre-mRNA) splicing is an essential step in human gene expression and is carried out by a large macromolecular machine called the spliceosome. Given the spliceosome's role in shaping the cellular transcriptome, it is not surprising that mutations in the splicing machinery can result in a range of human diseases and disorders (spliceosomopathies). This review serves as an introduction into the main features of the pre-mRNA splicing machinery in humans and how changes in the function of its components can lead to diseases ranging from blindness to cancers. Recently, several drugs have been developed that interact directly with this machinery to change splicing outcomes at either the single gene or transcriptome-scale. We discuss the mechanism of action of several drugs that perturb splicing in unique ways. Finally, we speculate on what the future may hold in the emerging area of spliceosomopathies and spliceosome-targeted treatments.
Introduction
Nearly every human gene is first transcribed into a precursor RNA molecule (such as pre-mRNAs) that must be extensively modified before it becomes functional. These modifications can include additions at the 5' and 3' ends (capping, poly-adenylation), modification of the RNA nucleotides (adenosine to inosine editing, base methylation) and the removal of introns by splicing. Defects in these processes can have serious consequences and lead to human disease. In this review, we will focus on mutations in the spliceosome machinery that are associated with a variety of medical conditions and recently developed therapeutics designed to restore, block, or alter splicing to correct these diseases. Our goal is not to provide an exhaustive list of these diseases or therapies but instead to provide a foundation for thinking about how splicing-related diseases can arise and the different therapeutic approaches being developed to target them.
The roles of pre-mRNA splicing in human gene expression
While humans have only a modest number of protein coding genes (~25,000) [Citation1], comparable to most other animals [Citation2], human gene architecture and usage are extremely complex. A large part of this complexity comes from pre-mRNA splicing. Most human genes are composed of small exons (~145 bp) separated from one another by large introns (~3400 bp) that must be removed from nascent RNAs by splicing [Citation3]. On average, each human gene contains eight of these large introns [Citation3]—meaning most of a typical pre-mRNA transcript is intronic and the majority of the transcript must be removed to generate a mature mRNA. How and when these introns are removed affords many potential mechanisms to regulate gene expression [Citation4]. While some of these introns are removed in precisely the same way each time, the removal of others is highly regulated and leads to the production of multiple mRNA isoforms by alternative pre-mRNA splicing [Citation5].
Alternative splicing is a pervasive feature of human gene expression with at least 90% of genes being differentially spliced to produce at least two unique transcripts [Citation6]. Recent advances in mass spectrometry have now shown that the majority of alternative mRNA isoforms are translated to produce different protein variants or proteoforms [Citation7]. Consequently, alternative splicing plays a key role in diversifying the human proteome and regulating many downstream biological processes [Citation8].
To understand alternative splicing and how it can be impacted by disease, it is helpful to first define the chemical steps that occur during any splicing reaction. Splicing is catalysed by a large macromolecular machine called the spliceosome (see below). The spliceosome functions to identify the sites of bond cleavage and formation in the substrate RNA, juxtapose those sites, and activate the nucleophiles and leaving groups to carry out two sequential transesterification reactions. The first transesterification in splicing is the cleavage of the 5' splice site (SS), often referred to simply as the first step. The 5'-SS is located at the boundary between the 5' exon and the intron (). During splicing, an intronic nucleotide (almost invariably an adenosine) becomes activated such that its 2'-hydroxy group can act as a nucleophile for attacking the phosphorous atom of the phosphodiester at the 5'-SS via an SN2 mechanism [Citation9]. This frees the 5' exon and creates a lariat structure at the adenosine by the formation of a 2', 5' phosphodiester linkage. The adenosine is now ‘branched’ leading to its identification as the branch point (BP). The adenosine that is chosen as the BP is informed by the surrounding sequence elements (BP sequence, BPS; ). The second transesterification in splicing is exon ligation (the second step). The freed 3'-hydroxy group of the 5' exon attacks the 3'-SS (the boundary between the intron and the 3' exon) to form a new phosphodiester bond and concomitantly free the lariat-intron (). 5'- and 3'-SS have conserved sequence motifs with the 5'-SS nearly always containing a ‘GU’ at the start of the intron. The BPS is usually located near the 3'-SS and is followed by a pyrimidine-rich region (polypyrimidine tract, PPT). The 3'-SS follows the PPT and almost always contains an ‘AG’ as the terminal nucleotides of the intron. How these particular RNA sequences are recognized by the splicing machinery has been the focus of many decades of research and is reviewed elsewhere [Citation10,Citation11].
Figure 1. Mechanisms of pre-mRNA splicing. (A) Splicing occurs in two sequential transesterification steps to remove introns and ligate exons together. In the first step, the 2'-OH group of the branch point adenosine attacks the 5' splice site. This results in the formation of a lariat where the 5' end of the intron is linked to the branch point adenosine. In the second step, the 3'-OH group at the end of the upstream exon attacks the 3' splice site, resulting in the excision of the intron and splicing together of the two exons. (B) Examples of alternative splicing discussed in the text. (C) Alternative splicing that results in inclusion of a poison exon leads to mRNA degradation via NMD.
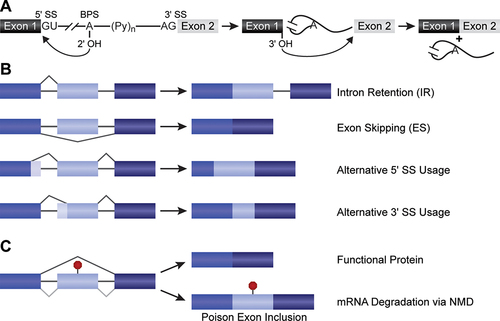
Changes in mRNA isoform production due to alternative splicing generally arise from changes in how the splice sites mentioned above are recognized and used by the splicing machinery. While there are many different types of alternative splicing [Citation5], we focus here on those most relevant to the diseases discussed below. Two of the most common changes in alternative splicing found in human diseases involve the failure to remove an intron (intron retention, IR; ) or failure to recognize an exon and causing it to be 'skipped' (exon skipping, ES). Alternatively, splicing changes can arise from activation of alternative 5' or 3'-SS (), leading to shortening or lengthening of a particular exon. Poison exons present a unique scenario in which exon skipping can result in protein function, whereas canonical splicing can lead to its loss [Citation12]. Poison exons are those that contain an in-frame, premature termination codon (PTC) when included within the mRNA (). In humans, mRNAs that contain PTCs are frequently destined for destruction by a process called nonsense mediated decay (NMD) [Citation13]. As a result, the inclusion or exclusion of a poison exon by alternative splicing can lead to either transcript destruction (inclusion) or stabilization (skipping).
Mechanisms of pre-mRNA splicing and spliceosome assembly
The pre-mRNA splicing machinery is highly complex and dynamic. Spliceosomes are composed of both small nuclear ribonucleoproteins (snRNPs) and a large number of additional proteins (splicing factors). Each snRNP is composed of a U-rich small nuclear RNA (snRNA) and associated proteins. There are two distinct types of spliceosomes that carry out splicing in humans, the major (U2 type) and minor (U12 type) spliceosomes. The major, U2-dependent spliceosome consists of the U1, U2, U4, U5 and U6 snRNPs and is responsible for processing over 99% of human introns that contain typical ‘GU’ and ‘AG’ 5' and 3'-SS (). The minor spliceosome is made up of the U11, U12, U4atac, U5, and the U6atac snRNPs and is responsible for removing less than 1% of introns that contain a different set of splice site consensus sequences [Citation14]. Spliceosomes are single turnover enzymes that assemble in a stepwise manner on nascent pre-mRNAs, meaning that each intron that is removed by its own bespoke spliceosome.
Spliceosome assembly begins when the U1 snRNP and U2 auxiliary factors (U2AF) recognize and bind the 5'-SS, PPT and 3'-SS (). Next, the U2 snRNP is recruited to the BPS to form the spliceosome A complex. This is followed by binding of the U4/U6.U5 tri-snRNP (a pre-organized complex of U4, U5 and U6 snRNPs) to form the spliceosome pre-B and B complexes. The resulting MDa-sized complexes of RNAs and proteins must then undergo extensive rearrangements to form the spliceosome active site (Bact complex) in a process called activation [Citation15]. Activation includes, among many other steps, expulsion of both the U1 and U4 snRNPs. Further changes in conformation and composition result in juxtaposition of the 5'-SS and BPS followed by 5'-SS cleavage (C complex). After completion of the first step of splicing, the spliceosome active site is then remodelled to juxtapose the 5' and 3'-SS for exon ligation and generation of a spliced mRNA and intron lariat. Many other non-snRNP splicing factors are needed for splicing to occur including the large, protein-only NineTeen Complex (NTC) [Citation16], several DExD/H-box ATPases [Citation17,Citation18] and a host of transiently-interacting factors that associate and dissociate from the spliceosome at defined stages. Cryo-EM combined with genetics, biochemistry and mass spectrometry have revealed the large-scale structural rearrangements that take place during splicing as well as the molecular mechanism of the splicing reaction itself [Citation10,Citation11,Citation19]. Single-molecule and biochemical studies of yeast and human spliceosomes have also shown that many, but not all, steps in splicing are reversible including both transesterification steps [Citation20–25]. This reversibility suggests multiple points at which the spliceosome can be regulated to control splicing fates.
Figure 2. Assembly of splicing factors onto pre-mRNA. (A) the spliceosome assembles stepwise by formation of several intermediate complexes with defined compositions and by recognition of RNA sequence elements. (B) Cis-acting regulatory sequences in the pre-mRNA transcript and trans-acting protein factors regulate splice site usage. Splicing regulatory (SR) proteins tend to bind exonic and intronic enhancers (ESE, ISE) while heterogeneous nuclear ribonucleoproteins (hnRNP) tend to bind exonic and intronic silencers.
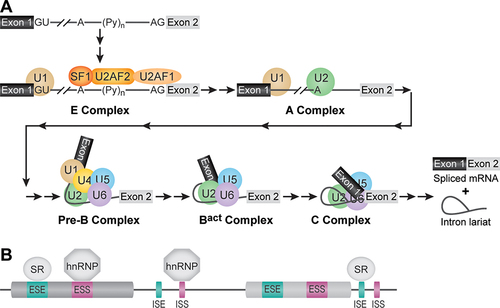
While many of the core factors and features of spliceosome assembly are well conserved among eukaryotes [Citation26,Citation27], others appear to be restricted or much more frequently utilized in animals. For example, the pathway described above holds true for spliceosomes that assemble on small introns (intron definition); however, spliceosomes that assemble on large introns, like those found in human transcripts, tend to first assemble across exons (exon definition) before transitioning to later splicing complexes [Citation28]. The mechanistic details of exon definition and the transition between exon-defined and intron-defined splicing complexes are not well understood but at least some features appear to be conserved between S. cerevisiae and humans [Citation29]. In addition, the human splicing machinery frequently uses auxiliary splicing factors that bind to the nascent transcript to either facilitate or impede spliceosome formation. Two of the most common types of these factors are the splicing regulatory (SR; tend to activate splicing) proteins and heterogenous ribonucleoproteins (hnRNPs; tend to repress splicing) [Citation30]. These factors bind RNA sequences to enhance or silence splicing: exonic splicing enhancers (ESEs), for example, are found in exons and enhance splicing of that exon when bound to an SR protein, while an intronic splicing silencer (ISS) can prevent removal of an intron when bound to an hnRNP protein (). While SR and hnRNP proteins can be found in many eukaryotes, their role in splicing and regulating gene expression programmes is best understood in humans.
Mutations in the splicing machinery and associated diseases
Our knowledge of the mutational landscape of human disease has increased substantially over the past two decades due to advancements in next-generation sequencing. Cis-acting splicing mutations occur in specific genes and impact how that gene is processed by the spliceosome. There are potentially an extremely large number of disease-associated, cis-acting mutations that impact splicing. It is estimated that ~14% of all disease-associated mutations impact splice sites [Citation31] and that as many as 50% of mutations perturb splicing through modification of exonic or intronic silencers or enhancers [Citation32]. Thorough reviews of cis-acting mutations have been published elsewhere [Citation33–37], and a discussion of these is beyond the scope of this text.
In addition, mutations in several different protein and snRNA splicing factors are also associated with a wide range of human conditions. Trans-acting mutations are those that occur in the splicing machinery itself that impact the processing of potentially numerous cellular transcripts. Cells that have trans-acting splicing mutations can display various phenotypes including drug sensitivities that can be exploited for therapy. This has led to a description of these cells as being ‘splicing sick’ [Citation38] or possessing spliceosomopathies [Citation39]. Here, we focus on trans-acting mutations that illustrate the diversity of spliceosomopathies ().
Table 1. Splicing factor mutations and diseases discussed.
U1 snRNA mutations and medulloblastomas
The U1 snRNP binds the 5'-SS during the earliest stages of spliceosome assembly (). U1 recognizes the 5'-SS by base-pairing interactions between the pre-mRNA and the highly conserved 5' end of the U1 snRNA (). Humans have multiple copies of the major spliceosome snRNA genes scattered around their genome. In the case of U1, humans possess 178 copies of the snRNA with an average sequence identity of 77% [Citation40]. Not all of these copies are expressed in every cell, and not all encode functional U1 snRNAs [Citation40,Citation41]. Nonetheless, the high copy number of human snRNA genes has made molecular and bioinformatic analysis of snRNA mutations in humans quite challenging. Despite these challenges, Suzuki and co-workers were able to identify recurrent hotspot mutations in U1 snRNA genes in 10 out of 114 cases of medulloblastoma, a type of brain cancer [Citation42]. The affected U1 snRNA genes were mostly RNU1-1 and RNU1-2, which normally encode identical copies of the snRNA.
Figure 3. Trans-acting mutations in spliceosomal components implicated in human disease. (A) Schematic of U1 snRNA base pairing with a 5'-SS. U1 snRNA mutations in cancer can activate cryptic splice site usage by changing this base pairing. (B) SF3B1 (grey) binds the intronic BPS (blue)/U2snRNA (orange) duplex. Mutations frequently observed in blood cancers are shown in red and interact with the intronic RNA just downstream of the BPS. PDB: 5Z56 (C) Schematic representation of coupling between BPS and 3'-SS usage. (D) Interactions between the PRP8 JAB/MPN1 domain (purple) and Brr2 helicase (grey). RP mutations are shown in red. PDB:4KIT.
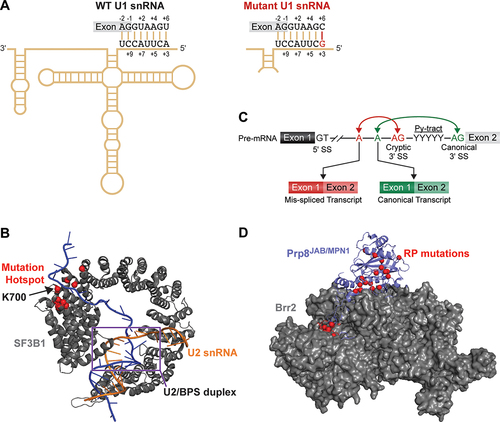
The authors identified a hotspot A>G transition mutation at the third nucleotide of the snRNA, in the region that pairs with the 5'-SS and with the +6 position within the intron () [Citation42]. There are four distinct subgroups of medulloblastoma [Citation43], and the hotspot was only found in the subgroup in which the sonic hedgehog signalling pathway is thought to drive tumorigenesis (the sonic hedgehog subgroup, SHH). Amazingly, 97% of cases of the SHH adult and 25% of the SHHα adolescent subtypes were associated with this snRNA mutation. Moreover, these mutations were correlated with both poor survival and relapse. Given the mutation changes how U1 pairs with the 5'-SS, it is perhaps not surprising that these snRNA variants activated a wide range of cryptic 5'-SS. Two of these sites were in the PTCH1 and GLI2 genes, which cause a frameshift mutation and loss of a repressor protein domain, respectively. The loss of PTCH1 protein in cancer is consistent with its absence causing derepression of Hedgehog signalling [Citation44], while removal of the repressor domain of GLI2 is known to activate Hedgehog signalling [Citation45]. These results provide links between alternative mRNA isoforms generated by U1 snRNA mutants and the high frequencies of these mutants in SHH-type medulloblastomas.
The study described above was published alongside work by Shuai et al. in which U1 snRNA mutations in cancers were analysed more broadly [Citation46]. Here, the authors found that the same hotspot mutation described above for SHH medulloblastomas along with an A>C transversion mutation at the same position were observed in five different types of tumours including pancreatic (pancreatic adenocarcinomas) and blood (chronic lymphocytic leukaemia, CLL) cancers. In the case of CLL, the authors observed that the most significantly misspliced gene was that for musashi RNA binding protein 2 (MSI2). In this case, the snRNA mutations cause activation of a cryptic, poison exon () with a G at the +6 position, suggesting that it is likely activated by the A>C mutation and pairing with the mutant snRNA. This finding is interesting because musashi proteins are key regulators of the stem cell state, and they control several essential signalling pathways implicated in a variety of cancers [Citation47]. In sum, these two studies illustrate how mutations in an snRNA gene can lead to cancer despite their high copy numbers in the genome. Further, they suggest that snRNA sequencing can be an important diagnostic tool for the treatment of cancers and patient prognosis.
U2 SF3B1 mutations and blood cancers
In addition to the snRNA components of the spliceosome, mutations that cause human disease can also be found in a variety of protein splicing factors. Probably, the most frequently mutated splicing factor in cancer and the best studied is the U2 SF3B1 protein. Like U1, the U2 snRNP also binds during the earliest stages of spliceosome assembly (); however, the pairing reaction between the U2 snRNA and the BPS is much more complex than the U1/5'-SS interaction. When U2 identifies and pairs with the BPS, it involves both large conformational rearrangements within the snRNP and the activity of multiple DExD/H-box ATPases [Citation48–50]. Many of these rearrangements are centred around SF3B1—a large, HEAT repeat protein that functions as both a clamp that closes upon the U2 snRNA/BPS duplex and a scaffold for splicing factors [Citation48,Citation51,Citation52] ().
In 2011, a ground-breaking study by Yoshida et al. analysed whole-exome sequencing data from 29 patients with myelodysplasia, a type of myeloid neoplasm that along with myelodysplastic syndrome (MDS) causes age-associated defects in blood cell production and a strong predisposition towards acute myeloid leukaemia (AML) [Citation53]. The authors noted that 16 out of 29 patients had mutations in pre-mRNA splicing factors, most of which are involved in recognition of the BPS and 3'-SS during early stages of spliceosome assembly. By extending their analysis to 582 patient specimens, the authors supported these observations and identified mutational hotspots in several proteins including SF3B1. Remarkably, ~75% of patients with the MDS phenotype of refractory anaemia with ring sideroblasts (RARS) had mutations in SF3B1, and the majority had the same K700E missense mutation (). These results were followed just a few months later by another study that also identified frequent SF3B1 mutations in chronic lymphocytic leukaemia [Citation54]. Much work since has identified splicing factor mutations (SF3B1 and U2AF1 proteins in particular) as being common among many different types of cancers but especially hematopoietic malignancies [Citation55–57].
A splicing phenotype frequently observed due to SF3B1 mutation is altered 3'-SS selection (). Mechanistically, this occurs due to changes in branch site usage where movement of the branch site up- or downstream can result in selection of a nearby cryptic 3'-SS [Citation58,Citation59] (). How the mutations change branch site selection during splicing is not yet clear. Studies using the S. cerevisiae (yeast) homolog, Hsh155, show that MDS mutations change how the splicing machinery utilizes weak branch sites that contain mismatches between the intron and U2 snRNA as well as change interactions between Hsh155 and a conserved DEAD-box ATPase (Prp5/DDX46) that regulates the process [Citation60,Citation61]. In fact, cryo-EM structures of the human U2 snRNP show that the N-terminal region of Prp5/DDX46 nearly encircles SF3B1 and binds near the MDS mutation hotspot [Citation62].
In humans, the process is more complex and involves additional factors, one of which (SUGP1) has an interaction with SF3B1 that is weakened by the K700E mutation [Citation63]. Recent work indicates that SUGP1 is a G-patch protein that likely regulates the activity of the DHX15/Prp43 helicase in disrupting U2/branch site complexes [Citation64–66]. This suggests that MDS mutations could function to promote usage of weak branch sites by preventing disassembly of U2 snRNPs erroneously bound at these locations due to failure to efficiently bind SUGP1/DHX15. Alternatively, it is possible that SUGP1/DHX15 are responsible for clearing proteins away from the branch site (SF1, U2AF1/2; ) to allow U2 to bind. In this case, the MDS mutations lead to less efficient recognition of the canonical branch site and weaken its competitiveness for spliceosome formation relative to other sites.
How altered splicing in cells with mutant SF3B1 proteins (or other splicing factors) leads to MDS or other cancers is also a matter of active debate and research (see Pellagatti and Boultwood for one recent review on this topic) [Citation67]. In brief, SF3B1 mutations have been associated with altered splicing in many genes and pathways associated with controlling cell growth and proliferation [Citation67]. For example, a comparison of altered splicing patterns from cells with either SF3B1-K700E or SRSF2-P95H (another splicing factor) mutations showed that both could result in hyperactivation of NF-B signalling albeit by aberrant splicing of different genes [Citation68]. Some of the splicing changes observed due to these mutations are remarkably switch-like: essentially all spliced or unspliced depending on the presence of the mutation. One such change discovered by Inoue and co-workers involves a poison exon in the BRD9 gene which is efficiently included in the spliced mRNA only in the presence of SF3B1-K700E [Citation69]. BRD9 protein is part of the non-canonical BAF chromatin remodelling complex and a potent tumour suppressor. Loss of BRD9 due to the destruction of its mRNA by NMD may therefore contribute to cancer growth. In an interesting application of these findings, North and co-workers have now developed artificial introns whose splicing is strictly dependent on the expression of SF3B1-K700E [Citation70]. Such splicing switches may be useful for therapeutic targeting of toxic protein-encoding genes to cancer cells containing the splicing factor mutations required for their processing.
U4atac snRNA mutations and MOPD1
Splicing factor mutations do not always result in cancer. Mutations in components of the spliceosome have been associated with many other diseases including those that impact development [Citation71]. In addition, these mutations are not restricted to the components of the major spliceosome. U4atac snRNA mutations that give rise to microcephalic osteodysplastic primordial dwarfism type 1 (MOPD1, also called Taybi-Linder syndrome) are a good example of mutations in the minor splicing machinery that lead to drastic changes in human growth and development.
MOPD1 is an autosomal recessive disease, meaning that patients must inherit a mutation from both parents to be afflicted. Patients with MOPD1 exhibit a wide range of pathologies including small head size (microcephaly), learning disorders, extreme growth defects, organ abnormalities and death in childhood or early adulthood. The cause of MOPD1 was identified by He et al. by focusing on patients among the Ohio Amish community due to their high degree of consanguinity and common patient phenotypes [Citation72]. The authors discovered that these patients shared mutations within the RNU4ATAC gene, which unlike the snRNA components of the major spliceosome is only present in a single copy. U4atac pairs with the U6atac snRNA and functions as an antisense RNA regulator. When U4atac is unwound, U6atac can fold and create the minor spliceosome active site. U4 snRNA functions in an identical manner in the major spliceosome when paired to the U6 snRNA.
He et al. found several U4atac mutations in different populations: 30 G>A, 51 G>A, 55 G>A and 111 G>A [Citation72]. All of these mutations are predicted to disrupt either the 5' or 3' stem loop of the snRNA. These disruptions in turn lead to splicing defects in genes containing minor introns. Subsequent mechanistic studies by Jafarifar et al. indicate that, while at least one mutation (124 G>A) leads to decreased abundance of the U4atac snRNA, others (30 G>A, 50 G>A, 50 G>C and 51 G>A) cause defects in snRNP assembly [Citation73]. This suggests that MOPD1 is caused by a decreased abundance of minor spliceosome components and that the 5' or 3' stem loops of the snRNA are also essential for the assembly of U4atac-containing splicing factors. Even though the minor spliceosome is responsible for processing a small fraction of introns (<1%), it is nonetheless essential, and defects in its function can have serious consequences for human health.
U5 Prp8 mutations and retinitis pigmentosa
One of the first splicing factors associated with a human disease was the U5 snRNP protein Prp8. Prp8 is the largest and most highly conserved protein with the spliceosome [Citation74]. It is responsible for scaffolding the active site as well as orchestrating a large number of conformational and compositional changes that take place during splicing [Citation10,Citation11,Citation15]. Prp8's connection with a subset of cases of autosomal dominant retinitis pigmentosa (RP; a progressive, loss-of-vision disease and the leading genetic cause of blindness [Citation75]) came to light through haplotype analysis of South African, Dutch and British families in which RP had been inherited across several generations [Citation76]. This work, as well as subsequent analyses, has shown that at least 64 different RP-causing mutations in Prp8 tend to cluster in the c-terminal region of the protein in the Jab1/MPN domain [Citation77,Citation78].
The Jab1/MPN domain directly binds the Brr2 DExD/H-box ATPase – the RNA helicase responsible for unwinding U4/U6 and U4atac/U6atac di-snRNAs (). This domain appears to regulate the unwinding and ATPase activities of Brr2. Brr2's regulation by Prp8 is complex, and work in this area has been summarized by Mayerle and Guthrie [Citation77]. Therefore, we will only focus on a few of the major findings. Prp8 is extremely well conserved between yeast and humans, and much of the mechanistic work into the impact of RP mutations on splicing has been carried out in yeast. RP mutations in yeast Prp8 result in viable yeast displaying a range of temperature-sensitive phenotypes [Citation77,Citation79,Citation80]. This indicates that they are not complete loss of function but instead change specific steps in splicing or snRNP assembly. Biochemical reconstitution of Brr2 unwinding activity showed that the RP mutants changed both interactions between the Jab1/MPN domain and Brr2. This also changes the ability for the Jab1/MPN domain or the adjacent very C-terminal tail of Prp8, to regulate Brr2’s unwinding of U4/U6 [Citation80,Citation81]. When splicing was analysed in yeast, the RP mutations also appeared to cause accumulation of unspliced pre-mRNAs (intron retention, ) and a wide range of synthetic genetic interactions with other splicing factors. This suggests that the RP mutants impact multiple steps in splicing beyond U4/U6 unwinding [Citation77,Citation79,Citation80]. Consistent with this hypothesis is the observation that RP mutations impact yeast snRNP assembly and reduce the levels of functional, nuclear U5 snRNP and U4/U6.U5 tri-snRNPs [Citation79].
It is unclear how exactly body-wide, somatic mutations in Prp8 present from conception result in age-related, disease onset in the retina. Blood samples from patients with Prp8 mutations reveal that splicing defects are not limited to the retina and show a remarkable change in splicing of ~20% of exons [Citation82]. In addition, recent work in a mouse model shows that mice homozygous for RP mutant Prp8 proteins exhibit neural atrophy, dysregulation of circular RNAs produced by the spliceosome and a decrease in splicing factor expression in general [Citation83]. Importantly, mutations in many different factors, many of which are not directly involved in pre-mRNA splicing, can cause RP [Citation78]. This includes the Prp31 protein which is also a component of the U4/U6.U5 tri-snRNP and was linked to RP contemporaneously with Prp8 [Citation84]. RP mutants in Prp31 have been more extensively characterized than those in Prp8 and show similar splicing defects [Citation85]. PRP31F knockdown also results in altered splicing of genes involved in phototransduction, suggesting a mechanism for disease development in the eye [Citation85]. Finally, it is possible that a common link between the multitude of factors that cause RP is in the function of cilia within photoreceptor cells. Mutation of many factors can result in ciliopathies due to changes in retinal gene regulation and expression [Citation85,Citation86].
U6 snRNA post-transcriptional processing and genodermatosis
Human disease can also be caused by mutations in factors that interact with snRNPs but do not directly participate in the splicing reaction per se. For example, mutations in snRNP chaperones, assembly factors and enzymes involved in post-transcriptional modification of snRNAs can also be detrimental. One example of this is a relatively recently described genodermatosis (hereditary skin disease) called poikiloderma with neutropenia (PN). Patients with this autosomal-recessive disease typically acquire a rash on their limb and face as infants that then evolves into poikiloderma (reddish-brown spots) along with chronic neutropenia and susceptibility to infections [Citation87]. Next-generation sequencing of 29 members of an Italian family with heredity PN identified mutations in C16orf57 as the gene responsible [Citation88]. Subsequent biochemical studies showed that the protein product of this gene (renamed Mutated in Poikiloderma with Neutropenia 1, MPN1) carries out post-transcriptional processing of the 3' end of the U6 and U6atac snRNAs. MPN1 is responsible for the formation of the terminal 2',3' cyclic phosphate [Citation89–92] found on U6 and U6atac. All of the MPN1 mutations are predicted to produce truncated proteins and result in loss of function [Citation87].
In cells, loss of MPN1 activity results in the formation of incorrectly processed U6 snRNAs which are often extended by the addition of several uridines or even oligoadenylated [Citation89,Citation92,Citation93]. This appears to correlate with slightly reduced stability of the U6 snRNA and, in a S. pombe model, reduced levels of U4/U6 di-snRNAs [Citation89]. Interestingly, PN patients do not appear to possess major splicing defects, making it difficult to connect U6 processing with disease phenotypes [Citation89,Citation93]. It is possible that MPN1 has other unknown, RNA substrates that contribute to PN. Alternatively, perturbations in splicing may only be apparent early in infant development when the rash first develops. Recently, a zebrafish model of PN in which MPN1 was knocked down using morpholinos showed many developmental defects and changes in splicing of genes involved in neutrophil, but not other blood cell, development [Citation94].
Drugs that target splicing factors
Drugs that either target some step in RNA processing or are RNA themselves are poised to revolutionize medicine [Citation95–98]. Here, we focus on the former class of molecules – small molecules that in some way influence splicing outcomes by direct interactions with splicing factors. These small molecules can function in diverse ways – some act as general inhibitors of the spliceosome and alter, or prevent, splicing of hundreds or thousands of transcripts. Conversely, it is now possible to design molecules that change alternative splicing patterns of certain RNAs. There are advantages to both approaches. Global changes in the splicing programme of a cell can be useful for triggering apoptosis while correcting the splicing of just a single gene can restore protein and cell function.
5'-SS correctors target U1/RNA interactions
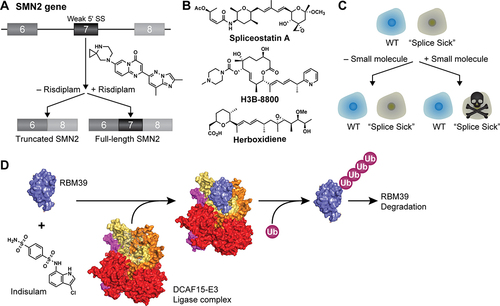
Spinal muscular atrophy (SMA) is a devastating neuromuscular disease and a leading genetic cause of death in infants. Patients with SMA have a mutation in the SMN1 gene, which codes for the Survival of Motor Neuron 1 protein (SMN1). SMN1 is part of a large, essential complex that assembles snRNPs, including the components of the spliceosome. Patients with SMA have a deletion in some parts of the SMN1 gene that results in failure to produce stable, active protein. Humans have a second copy of SMN—the SMN2 gene. However, exon 7 of SMN2 has a weak 5'-SS signal that results in its skipping; consequently, no functional protein is typically made from SMN2 () [Citation99]. One potential therapeutic strategy for SMA is to restore SMN2 splicing so that functional SMN2 protein be produced. The first FDA-approved treatment for SMA was in fact an antisense oligonucleotide (ASO, nusinersen) that accomplishes this feat [Citation100]. Nusinersen binds to an ISS just downstream of SMN2 exon 7 and prevents hnRNP proteins from binding that normally repress splicing of that exon [Citation99]. Bennett, Krainer and Cleveland have published a review that describes both the action of nusinersen and therapeutic ASOs in general [Citation99].
Figure 4. Small molecule therapeutics that target splicing factors. (A) Risdiplam causes SMN2 exon 7 inclusion for treatment of SMA. The target of risdiplam is the U1/5'-SS interaction. (B) Chemical structures of spliceosome inhibitors that target SF3B1. (C) Cells with splicing factor mutations are ‘splice sick’. Splice sick cells can be more sensitive to chemotherapeutics than cells without these mutations. (D) Molecular glues like indisulam recruit the DCAF15-E3 ligase complex to the RBM39 splicing factor. This causes RBM39 to become poly-ubiquitinated and targeted for degradation. PDB:6Q0W. Note that only a small domain of RBM39 is shown.
While nusinersen is a ground-breaking drug, its use in SMA treatment is challenging due to the high cost of its manufacture, need for intrathecal administration and difficulties in correcting splicing defects in both the CNS and peripheral tissues and organs. Consequently, much effort has been spent on identifying small-molecule splicing correctors that promote SMN2 exon 7 inclusion. One of the first hints that this was even feasible came from a discovery in 2009 by the Krainer and Hastings groups and Paratek Pharmaceuticals that tetracyclines can activate exon 7 splicing [Citation101]. In 2014 and 2015, groups from Roche, PTC Therapeutics and Novartis published the discovery of two molecules, SMN-C3 and branaplam, that increased the inclusion of exon 7 in a mini-gene model of SMN2 and in mice [Citation102,Citation103]. In both cases, these molecules were discovered using a high-throughput SMN2 mini-gene assay in which exon 7 inclusion was coupled with the expression of firefly luciferase. While branaplam or other splicing modulators such as PTC518 may eventually prove useful for treatment of Huntington's disease rather than SMA [Citation104,Citation105], studies of an analog of SMN-C3 for SMA were halted due to observations of retinal toxicity in monkeys [Citation106]. However, the Roche and PTC Therapeutics groups were subsequently able to identify risdiplam as a drug candidate (), and this molecule was approved by the FDA for treatment of SMA in 2020 [Citation106,Citation107].
Both branaplam and risdiplam function by changing how the U1 snRNP interacts with the SMN2 exon 7 5'-SS [Citation102,Citation108]. This splice site is unusual in that it contains an adenosine as the terminal nucleotide of the exon rather than the more common guanosine (GGA/GUAAGUCU; where the ‘/’ indicates the exon/intron boundary). As a result, it is possible that when the U1 snRNA pairs with the 5'-SS, the adenosine at the splice junction is bulged out of the resulting duplex, weakening the splice site strength. Campagne and co-workers used solution NMR to study the binding of a risdiplam analog (SMN-C5) to both the human U1 snRNP and an RNA-only model system [Citation109]. They observed that drug binding facilitated a conformational switch within the snRNA/5'-SS duplex, resulting in stacking of the bulged adenosine. The authors hypothesized that this conformation is more compatible with binding of U1 snRNP protein factors. The stronger binding of U1 in the presence of the drug effectively converts the weak 5'-SS into a strong one and promotes exon 7 inclusion. The molecular details of how branaplam and risdiplam differ from one another with respect to this model are not yet clear nor is it clear how the U1 structure accommodates the drug-bound RNA duplex. However, recent transcriptome-wide studies have begun to provide more insight into the splice-site selectivity of branaplam and risdiplam in vivo [Citation110,Citation111].
It is an open question as to how many other 5'-SS are ‘druggable’. However, it is quite likely that flexibility in U1 snRNA/5'-SS interactions extends to other nucleotides within the duplex along with alternate base pairing registers [Citation112]. In addition, it is also unknown if other snRNA/intron duplexes formed during splicing can similarly be targeted by drugs. One obvious candidate is the U2 snRNA/BPS duplex which is often quite weak in humans and likely highly malleable. In any case, there is likely much still to be learned from risdiplam and branaplam themselves. For example, Monteys and co-workers recently co-opted branaplam and the poison exon 7 of SMN2 as a transportable, splicing regulatory switch [Citation113]. When inserted into a gene of interest, these switches allow for branaplam-dependent splicing and protein expression. In even more recent work, Zhang and co-workers synthesized a diazenyl-containing risdiplam analog that is photoswitchable [Citation114]. In this case, cis-trans isomerization of the drug can be light-catalysed and allows the drug to be switched from a form that does not bind RNA (cis) to one that does (trans).
Splicing is strongly inhibited by SF3B1-binding small molecules
Some of the best characterized and most widely used general splicing inhibitors target the U2 snRNP SF3B1 protein – the same protein frequently mutated in MDS and CLL (). Three different classes of these inhibitors are currently in general use: derivatives of the natural product FR901464 (such as spliceostatin, SSA), pladienolides (PB) and herboxidienes (HB) (). These inhibitors and their structure–activity relationship have been recently reviewed by Effenberger, Urabe and Jurica [Citation115]. Here, we describe their discovery, general mechanisms of inhibition and clinical use.
FR901464, PB and HB were all isolated as natural products with potent anti-tumour activities from Pseudomonas (FR901464) [Citation116] or Streptomyces (PB, HB) [Citation117,Citation118]. In 2014, a group at Pfizer re-characterized the origin of FR901464 as Burkholderia [Citation119]. SSA is a synthetic intermediate in the total synthesis of FR901464 and can be prepared from the natural product [Citation120]. Subsequently, the biological target of these drugs (SF3B1) was identified using similar, chemical biology-based approaches [Citation121–123]. In each case, a biotin derivative of the molecule was synthesized (in some cases also containing a photoreactive crosslinker) and interacting proteins identified by mass spectrometry. Amazingly and despite their structural differences, each compound was subsequently shown to inhibit splicing in human cells by binding to the same site on SF3B1 [Citation124,Citation125]. Interactions between SF3B1 and these drugs can lead to potent splicing inhibition with GI50/IC50 values in the nM range [Citation115]. One of the most potent inhibitors yet characterized is meayamycin, an FR901464 analog, that exhibits GI50 values of ~20 pM for MCF-7 cells [Citation126]. Interestingly, not all splicing events are equally susceptible to inhibition by these drugs, and different drugs show different specificities [Citation127]. This has led to their being described as ‘splicing modulators’ [Citation115,Citation124] rather than ‘splicing inhibitors’ due to their ability to change alternative splicing outcomes for transcripts (rather than complete abolishment of all splicing) when used at therapeutically relevant doses.
The mechanism of splicing inhibition for PB and HB is relatively straightforward. These compounds bind to SF3B1 in the same protein cleft that accommodates the bulged, extrahelical branch point adenosine of the U2 snRNA/BPS duplex [Citation128,Citation129]. Thus, they effectively act as competitive inhibitors of RNA binding to that site on SF3B1. The inhibition mechanism of SSA and other FR901464 derivatives is more complex and involves irreversible covalent modification of the splicing machinery [Citation51]. Here, the bulk of the molecule still binds to SF3B1, but the epoxide moiety is juxtaposed with a zinc finger motif of another U2 snRNP protein, PHF5A [Citation51]. In a remarkable set of structures, Cretu et al. were able to show that SSA and sudemycin FR901464 analogs form covalent adducts with Cys26 of PHF5A explaining the extremely strong splicing inhibition observed for FR901464 derivatives. Interestingly, the zinc finger of PHF5A remains intact within their structures despite loss of the cysteine ligand. This suggests that the covalent modification does not result in destabilization and unfolding of PHF5A but rather trapping of the protein and the small molecule within the U2 snRNP.
Outstanding questions related to the inhibition mechanism include identifying the conformations of SF3B1 and U2 snRNP that are most susceptible to the drugs. SF3B1 undergoes a number of conformational changes, and it may be that drug binding occurs preferentially in one state or another [Citation51,Citation52]. Such dynamics could explain how a large number of mutations within SF3B1 and PHF5A can result in resistance to these drugs [Citation124]. It is also interesting to note that the S. cerevisiae SF3B1 homolog, Hsh155, is also resistant to these drugs [Citation130]. Sensitivity can be conferred by mutation of just a single amino acid within the drug-binding pocket [Citation131]. SSA, HB and PB are, in a sense, species-specific splicing inhibitors that fortuitously inhibit the human splicing machinery. Whether or not fungal or other species-specific analogs of these compounds exist naturally or can be synthesized is an interesting question.
Among SF3B1-binding inhibitors, a PB derivative (H3B–8800, ) has advanced the furthest in clinical trials for cancer treatment. One treatment strategy is to specifically target ‘splicing sick’ cancer cells (). Splicing sick cells contain cancer-associated splicing factor mutations and also have much greater sensitivity to splicing inhibitors [Citation132]. A recent study has also shown that the treatment of cancer cells by H3B–8800 leads to the accumulation of cytosolic double-stranded RNAs (due to unspliced introns) [Citation133]. This, in turn, leads to activation of the cellular antiviral immune response and triggering of apoptosis. Activation of the antiviral immune response pathway by unspliced introns may provide a rationale for why some cancer cells show enhanced sensitivity to splicing inhibition. While this is promising, a recent patient trial of H3B–8800 for treatment of myeloid neoplasms failed to produce significant positive outcomes – suggesting much work is needed to connect favourable performance of these drugs in model systems to their use in the clinic [Citation134]. A possible future strategy is to use molecules like H3B–8800 in combination cancer therapy or to sensitize cancer cells to other chemotherapeutics [Citation135–139].
Molecular glues can target splicing factors for destruction
An alternate method to inhibit the function of a protein is to eliminate that protein entirely. ‘Molecular glues’ or protein-targeting chimeric molecules (PROTACs) can achieve this by acting as a bridge between a given protein and ubiquitin ligase complexes, similar to the ways in which auxin functions in plants [Citation140,Citation141]. This results in drug-dependent targeting of the ubiquitin ligase to the protein and the protein's subsequent poly-ubiquitination and degradation. PROTACs have been developed for a variety of cellular proteins [Citation142].
Currently, molecular glues designed against the splicing factor RBM39 are showing the greatest promise for potential use in patients. In addition to roles in transcription, RBM39 (also known as CAPERα) is important for regulating alternative splicing of genes in response to steroid hormones such as prostaglandin [Citation143]. Mechanistically, RBM39's role in splicing regulation is not well understood. It can interact with U2AF2 () and SF3B1, which suggests a function in promoting the earliest stages of spliceosome assembly and recognition of the BPS and 3'-SS [Citation144,Citation145].
Remarkably, while many PROTACs are highly engineered for use, the discovery of an RBM39 molecular glue was serendipitous. Indisulam, an aryl sulphonamide, was discovered in a small-molecule screen for compounds with anticancer activity [Citation146]. However, its biological target was unknown. To identify its target, Han and co-workers screened for indisulam-resistance mutations appearing in HCT-116 human colorectal carcinoma cells, which are DNA mismatch repair defective and particularly adept and acquiring drug-resistance [Citation147]. Three of six indisulam-resistant clones showed mutations in the same codon (Gly268) of RBM39, suggesting that it is a direct target for indisulam. Subsequent work showed that the addition of indisulam resulted in rapid degradation of RBM39 and that this occurs by drug-dependent recruitment of the CUL4-DCAF15 ubiquitin ligase complex (). Similar results were simultaneously reported by Uehara and co-workers using quantitative mass spectrometry to identify proteome changes associated with sulphonamide treatment [Citation148]. Structural work from several groups confirmed that indisulam and related sulphonamides bind at the interface between DCAF15 and RBM39 (satisfyingly, Gly268 is in the binding pocket) and that the drugs effectively knit the proteins together by establishing a large number of hydrogen bonds and other interactions [Citation149–151].
Most relevant for clinical applications, RBM39 has also been shown to be upregulated in most cancers [Citation152]. In one study, Wang and co-workers showed that RBM39 is upregulated in acute myeloid leukaemia patients (AML), and AML cell lines are dependent on RBM39 for growth [Citation153]. RBM39 appeared to regulate the alternative splicing of genes required for AML cell growth, and that degradation of RBM39 with indisulam resulted in changes in expression of these genes. Interestingly, a number of these genes also encode RNA binding proteins, suggesting that RBM39 regulates expression of a network of RNA regulatory factors [Citation153]. It is possible that similar RBM39-regulated networks will be found in other cancers, such as neuroblastomas which are also highly sensitive to expression of RBM39 [Citation154].
It is even possible that RBM39 molecular glues can be used to indirectly treat other cancers that do not have a RBM39-dependency. Lu and co-workers showed that RBM39 degradation with indisulam resulted in alternative splicing changes that in turn produced novel proteins that act as a source of neoantigens on cancer cells [Citation155]. These neoantigens are immunogenic and can enhance immune checkpoint blockade therapy. Thus, patients receiving checkpoint blockade therapies may benefit from splicing modulation as a means to enhance immune function.
Conclusions and perspectives
As we have shown with the examples in this review, dysregulated splicing due to splicing factor mutations contributes to disease development in a variety of ways. A key driver of these discoveries has been the use of next-generation sequencing to identify splicing factor mutations, their effects on the transcriptome and druggable targets. It is quite likely that sequencing will lead to many more breakthroughs [Citation156]. In one possible breakthrough, the splicing machinery itself has been suggested to be an ‘Achilles' heel’ for several diseases [Citation157,Citation158]. While future splicing-targeting therapies hold great promise, several key areas need to be addressed to unlock the full potential of this approach. It is essential that we obtain a better understanding of how mis-splicing and changes in alternative splicing networks give rise to disease. As evidenced by studies in RP and MDS, much work still needs to be done in this area to pinpoint how splicing factor mutations that cause changes in splicing in hundreds of RNAs result in particular disease phenotypes in specific tissues. By investigating these splicing events and their consequences, researchers can identify critical targets for intervention and develop better therapies. As these splicing-targeted therapies become more common, it is quite likely that resistance mutations will also begin to appear. In fact, many such mutations have already been identified in SF3B1 that result in drug insensitivity [Citation124]. It will be interesting to learn how drug resistance arises and can be mitigated to improve treatment outcomes. It is even possible that changes in alternative splicing patterns themselves to include/exclude or alter a drug-targeted protein domain may be involved in the resistance mechanism. Finally, gene-editing may prove therapeutically useful for the treatment of single-gene diseases like MOPD1 and SMA. Recent work by the Liu group has shown that targeted base editing by CRISPR-based methods can rescue SMA in mouse models [Citation159]. This approach could even be used to correct spliceosomopathies even before they start.
Acknowledgments
We thank members of the Hoskins and Koide Labs for feedback on this review.
Disclosure statement
AAH is carrying out sponsored research for and is a member of the scientific advisory board for Remix Therapeutics, Inc. (Watertown, MA).
Additional information
Funding
References
- Venter JC, Adams MD, Myers EW, et al. The sequence of the human genome. Science. 2001;291(5507):1304–1351. doi: 10.1126/science.1058040
- Pertea M, Salzberg SL. Between a chicken and a grape: estimating the number of human genes. Genome Biol. 2010;11(5):206. doi: 10.1186/gb-2010-11-5-206
- Hnilicova J, Stanek D. Where splicing joins chromatin. Nucleus. 2011;2(3):182–188. doi: 10.4161/nucl.2.3.15876
- Gehring NH, Roignant JY. Anything but ordinary – emerging splicing mechanisms in eukaryotic gene regulation. Trends Genet. 2021;37(4):355–372. doi: 10.1016/j.tig.2020.10.008
- Matlin AJ, Clark F, Smith CWJ. Understanding alternative splicing: towards a cellular code. Nat Rev Mol Cell Biol. 2005;6(5):386–398. doi: 10.1038/nrm1645
- Wang ET, Sandberg R, Luo S, et al. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008;456(7221):470–476. doi: 10.1038/nature07509
- Sinitcyn P, Richards AL, Weatheritt RJ, et al. Global detection of human variants and isoforms by deep proteome sequencing. Nat Biotechnol. 2023. doi:10.1038/s41587-023-01714-x.
- Nilsen TW, Graveley BR. Expansion of the eukaryotic proteome by alternative splicing. Nature. 2010;463(7280):457–463. doi: 10.1038/nature08909
- Moore MJ, Sharp PA. Evidence for two active sites in the spliceosome provided by stereochemistry of pre-mRNA splicing. Nature. 1993;365(6444):364–368. doi: 10.1038/365364a0
- Plaschka C, Newman AJ, Nagai K. Structural basis of nuclear pre-mRNA splicing: lessons from yeast. Cold Spring Harb Perspect Biol. 2019;11(5):11. doi: 10.1101/cshperspect.a032391
- Kastner B, Will CL, Stark H, et al. Structural insights into nuclear pre-mRNA splicing in higher eukaryotes. Cold Spring Harb Perspect Biol. 2019;11(11):11. doi: 10.1101/cshperspect.a032417
- Carvill GL, Mefford HC. Poison exons in neurodevelopment and disease. Curr Opin Genet Dev. 2020;65:98–102. doi: 10.1016/j.gde.2020.05.030
- Kurosaki T, Popp MW, Maquat LE. Quality and quantity control of gene expression by nonsense-mediated mRNA decay. Nat Rev Mol Cell Biol. 2019;20(7):406–420. doi: 10.1038/s41580-019-0126-2
- Turunen JJ, Niemela EH, Verma B, et al. The significant other: splicing by the minor spliceosome. Wiley Interdiscip Rev RNA. 2013;4(1):61–76. doi: 10.1002/wrna.1141
- Brow DA. Allosteric cascade of spliceosome activation. Ann Rev Genet. 2002;36(1):333–360. doi: 10.1146/annurev.genet.36.043002.091635
- Hogg R, McGrail JC, O'Keefe RT. The function of the NineTeen Complex (NTC) in regulating spliceosome conformations and fidelity during pre-mRNA splicing. Biochem Soc Trans. 2010;38(4):1110–1115. doi: 10.1042/BST0381110
- Staley JP, Guthrie C. Mechanical devices of the spliceosome: motors, clocks, springs, and things. Cell. 1998;92(3):315–326. doi: 10.1016/S0092-8674(00)80925-3
- De Bortoli F, Espinosa S, Zhao R. DEAH-Box RNA helicases in pre-mRNA splicing. Trends Biochem Sci. 2021;46(3):225–238. doi: 10.1016/j.tibs.2020.10.006
- Wahl MC, Will CL, Luhrmann R. The spliceosome: design principles of a dynamic RNP machine. Cell. 2009;136(4):701–718. doi: 10.1016/j.cell.2009.02.009
- Hoskins AA, Friedman LJ, Gallagher SS, et al. Ordered and dynamic assembly of single spliceosomes. Science. 2011;331(6022):1289–1295. doi: 10.1126/science.1198830
- Hoskins AA, Rodgers ML, Friedman LJ, et al. Single molecule analysis reveals reversible and irreversible steps during spliceosome activation. Elife. 2016;5: doi: 10.7554/eLife.14166
- Fu X, Kaur H, Rodgers ML, et al. Identification of transient intermediates during spliceosome activation by single molecule fluorescence microscopy. Proc Natl Acad Sci U S A. 2022;119(48):e2206815119. doi: 10.1073/pnas.2206815119
- Braun JE, Friedman LJ, Gelles J, et al. Synergistic assembly of human pre-spliceosomes across introns and exons. Elife. 2018;7: doi: 10.7554/eLife.37751
- Abelson J, Blanco M, Ditzler MA, et al. Conformational dynamics of single pre-mRNA molecules during in vitro splicing. Nat Struct Mol Biol. 2010;17(4):504–512. doi: 10.1038/nsmb.1767
- Tseng CK, Cheng SC. Both catalytic steps of nuclear pre-mRNA splicing are reversible. Science. 2008;320(5884):1782–1784. doi: 10.1126/science.1158993
- Sales-Lee J, Perry DS, Bowser BA, et al. Coupling of spliceosome complexity to intron diversity. Curr Biol. 2021;31(22):4898–4910.e4. doi: 10.1016/j.cub.2021.09.004
- Fabrizio P, Dannenberg J, Dube P, et al. The evolutionarily conserved core design of the catalytic activation step of the yeast spliceosome. Mol Cell. 2009;36(4):593–608. doi: 10.1016/j.molcel.2009.09.040
- Robberson BL, Cote GJ, Berget SM. Exon definition may facilitate splice site selection in RNAs with multiple exons. Mol Cell Biol. 1990;10(1):84–94. doi: 10.1128/MCB.10.1.84
- Li X, Liu S, Zhang L, et al. A unified mechanism for intron and exon definition and back-splicing. Nature. 2019;573(7774):375–380. doi: 10.1038/s41586-019-1523-6
- Takeiwa T, Mitobe Y, Ikeda K, et al. Roles of splicing factors in hormone-related cancer progression. Int J Mol Sci. 2020;21(5):21. doi: 10.3390/ijms21051551
- Soemedi R, Cygan KJ, Rhine CL, et al. Pathogenic variants that alter protein code often disrupt splicing. Nat Genet. 2017;49(6):848–855. doi: 10.1038/ng.3837
- Cartegni L, Chew SL, Krainer AR. Listening to silence and understanding nonsense: exonic mutations that affect splicing. Nat Rev Genet. 2002;3(4):285–298. doi: 10.1038/nrg775
- Srebrow A, Kornblihtt AR. The connection between splicing and cancer. J Cell Sci. 2006;119(13):2635–2641. doi: 10.1242/jcs.03053
- Anna A, Monika G. Splicing mutations in human genetic disorders: examples, detection, and confirmation. J Appl Genet. 2018;59(3):253–268. doi: 10.1007/s13353-018-0444-7
- Scotti MM, Swanson MS. RNA mis-splicing in disease. Nat Rev Genet. 2016;17(1):19–32. doi: 10.1038/nrg.2015.3
- Orengo JP, Cooper TA. Alternative splicing in disease. Adv Exp Med Biol. 2007;623:212–223.
- Singh RK, Cooper TA. Pre-mRNA splicing in disease and therapeutics. Trends Mol Med. 2012;18(8):472–482. doi: 10.1016/j.molmed.2012.06.006
- Agrawal AA, Yu L, Smith PG, et al. Targeting splicing abnormalities in cancer. Curr Opin Genet Dev. 2018;48:67–74. doi: 10.1016/j.gde.2017.10.010
- Griffin C, Saint-Jeannet JP. Spliceosomopathies: Diseases and mechanisms. Dev Dyn. 2020;249(9):1038–1046. doi: 10.1002/dvdy.214
- Mabin JW, Lewis PW, Brow DA, et al. Human spliceosomal snRNA sequence variants generate variant spliceosomes. RNA. 2021;27(10):1186–1203. doi: 10.1261/rna.078768.121
- Dvinge H, Guenthoer J, Porter PL, et al. RNA components of the spliceosome regulate tissue- and cancer-specific alternative splicing. Genome Res. 2019;29(10):1591–1604. doi: 10.1101/gr.246678.118
- Suzuki H, Kumar SA, Shuai S, et al. Recurrent noncoding U1 snRNA mutations drive cryptic splicing in SHH medulloblastoma. Nature. 2019;574(7780):707–711. doi: 10.1038/s41586-019-1650-0
- Taylor MD, Northcott PA, Korshunov A, et al. Molecular subgroups of medulloblastoma: the current consensus. Acta Neuropathol. 2012;123(4):465–472. doi: 10.1007/s00401-011-0922-z
- Kogerman P, Krause D, Rahnama F, et al. Alternative first exons of PTCH1 are differentially regulated in vivo and may confer different functions to the PTCH1 protein. Oncogene. 2002;21(39):6007–6016. doi: 10.1038/sj.onc.1205865
- Sasaki H, Nishizaki Y, Hui C, et al. Regulation of Gli2 and Gli3 activities by an amino-terminal repression domain: implication of Gli2 and Gli3 as primary mediators of Shh signaling. Development. 1999;126(17):3915–3924. doi: 10.1242/dev.126.17.3915
- Shuai S, Suzuki H, Diaz-Navarro A, et al. The U1 spliceosomal RNA is recurrently mutated in multiple cancers. Nature. 2019;574(7780):712–716. doi: 10.1038/s41586-019-1651-z
- Kudinov AE, Karanicolas J, Golemis EA, et al. Musashi RNA-Binding proteins as cancer drivers and novel therapeutic targets. Clin Cancer Res. 2017;23(9):2143–2153. doi: 10.1158/1078-0432.CCR-16-2728
- van der Feltz C, Hoskins AA. Structural and functional modularity of the U2 snRNP in pre-mRNA splicing. Crit Rev Biochem Mol Biol. 2019;54(5):443–465. doi: 10.1080/10409238.2019.1691497
- Tholen J, Galej WP. Structural studies of the spliceosome: Bridging the gaps. Curr Opin Struct Biol. 2022;77:102461. doi: 10.1016/j.sbi.2022.102461
- Tholen J, Razew M, Weis F, et al. Structural basis of branch site recognition by the human spliceosome. Science. 2022;375(6576):50–57. doi: 10.1126/science.abm4245
- Cretu C, Gee P, Liu X, et al. Structural basis of intron selection by U2 snRNP in the presence of covalent inhibitors. Nat Commun. 2021;12(1):4491. doi: 10.1038/s41467-021-24741-1
- Schmitzova J, Cretu C, Dienemann C, et al. Structural basis of catalytic activation in human splicing. Nature. 2023;617(7962):842–850. doi: 10.1038/s41586-023-06049-w
- Yoshida K, Sanada M, Shiraishi Y, et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature. 2011;478(7367):64–69. doi: 10.1038/nature10496
- Quesada V, Conde L, Villamor N, et al. Exome sequencing identifies recurrent mutations of the splicing factor SF3B1 gene in chronic lymphocytic leukemia. Nat Genet. 2011;44(1):47–52. doi: 10.1038/ng.1032
- Hahn CN, Scott HS. Spliceosome mutations in hematopoietic malignancies. Nat Genet. 2011;44(1):9–10. doi: 10.1038/ng.1045
- Dvinge H, Kim E, Abdel-Wahab O, et al. RNA splicing factors as oncoproteins and tumour suppressors. Nat Rev Cancer. 2016;16(7):413–430. doi: 10.1038/nrc.2016.51
- Seiler M, Peng S, Agrawal AA, et al. Somatic mutational landscape of splicing factor genes and their functional consequences across 33 cancer types. Cell Rep. 2018;23(1):282–296.e4. doi: 10.1016/j.celrep.2018.01.088
- Darman RB, Seiler M, Agrawal AA, et al. Cancer-associated SF3B1 hotspot mutations induce cryptic 3′ splice site selection through use of a different branch point. Cell Rep. 2015;13(5):1033–1045. doi: 10.1016/j.celrep.2015.09.053
- DeBoever C, Ghia EM, Shepard PJ, et al. Transcriptome sequencing reveals potential mechanism of cryptic 3' splice site selection in SF3B1-mutated cancers. PLoS Comput Biol. 2015;11(3):e1004105. doi: 10.1371/journal.pcbi.1004105
- Tang Q, Rodriguez-Santiago S, Wang J, et al. SF3B1/Hsh155 HEAT motif mutations affect interaction with the spliceosomal ATPase Prp5, resulting in altered branch site selectivity in pre-mRNA splicing. Genes Dev. 2016;30(24):2710–2723. doi: 10.1101/gad.291872.116
- Carrocci TJ, Zoerner DM, Paulson JC, et al. Sf3b1 mutations associated with myelodysplastic syndromes alter the fidelity of branchsite selection in yeast. Nucleic Acids Res. 2017;45:4837–4852. doi: 10.1093/nar/gkw1349
- Zhang Z, Will CL, Bertram K, et al. Molecular architecture of the human 17S U2 snRNP. Nature. 2020;583(7815):310–313. doi: 10.1038/s41586-020-2344-3
- Zhang J, Ali AM, Lieu YK, et al. Disease-causing mutations in SF3B1 alter splicing by disrupting interaction with SUGP1. Mol Cell. 2019;76(1):82–95.e7. doi: 10.1016/j.molcel.2019.07.017
- Zhang J, Huang J, Xu K, et al. DHX15 is involved in SUGP1-mediated RNA missplicing by mutant SF3B1 in cancer. Proc Natl Acad Sci U S A. 2022;119(49):e2216712119. doi: 10.1073/pnas.2216712119
- Beusch I, Rao B, Studer M, et al. Targeted high-throughput mutagenesis of the human spliceosome reveals its in vivo operating principles. Mol Cell. 2023;83(14):2578–2594.e9. Online Ahead of Print. doi: 10.1016/j.molcel.2023.06.003
- Feng Q, Krick K, Chu J, et al. Splicing quality control mediated by DHX15 and its G-patch activator, SUGP1. bioRxiv 2022:2022.11.14.516533.
- Pellagatti A, Boultwood J. SF3B1 mutant myelodysplastic syndrome: Recent advances. Adv Biol Regul. 2021;79:100776. doi: 10.1016/j.jbior.2020.100776
- Lee SC, North K, Kim E, et al. Synthetic lethal and convergent biological effects of cancer-associated spliceosomal gene mutations. Cancer Cell. 2018;34(2):225–241.e8. doi: 10.1016/j.ccell.2018.07.003
- Inoue D, Chew GL, Liu B, et al. Spliceosomal disruption of the non-canonical BAF complex in cancer. Nature. 2019;574(7778):432–436. doi: 10.1038/s41586-019-1646-9
- North K, Benbarche S, Liu B, et al. Synthetic introns enable splicing factor mutation-dependent targeting of cancer cells. Nat Biotechnol. 2022;40(7):1103–1113. doi: 10.1038/s41587-022-01224-2
- Wood KA, Eadsforth MA, Newman WG, et al. The role of the U5 snRNP in genetic disorders and cancer. Front Genet. 2021;12:636620. doi: 10.3389/fgene.2021.636620
- He H, Liyanarachchi S, Akagi K, et al. Mutations in U4atac snRNA, a component of the minor spliceosome, in the developmental disorder MOPD I. Science. 2011;332(6026):238–240. doi: 10.1126/science.1200587
- Jafarifar F, Dietrich RC, Hiznay JM, et al. Biochemical defects in minor spliceosome function in the developmental disorder MOPD I. RNA. 2014;20(7):1078–1089. doi: 10.1261/rna.045187.114
- Grainger RJ, Beggs JD. Prp8 protein: at the heart of the spliceosome. RNA. 2005;11(5):533–557. doi: 10.1261/rna.2220705
- Francis PJ. Genetics of inherited retinal disease. J R Soc Med. 2006;99(4):189–191. doi: 10.1177/014107680609900417
- McKie AB, McHale JC, Keen TJ, et al. Mutations in the pre-mRNA splicing factor gene PRPC8 in autosomal dominant retinitis pigmentosa (RP13). Hum Mol Genet. 2001;10(15):1555–1562. doi: 10.1093/hmg/10.15.1555
- Mayerle M, Guthrie C. Prp8 retinitis pigmentosa mutants cause defects in the transition between the catalytic steps of splicing. RNA. 2016;22(5):793–809. doi: 10.1261/rna.055459.115
- Daiger SP, Sullivan LS, Bowne SJ. Genes and mutations causing retinitis pigmentosa. Clin Genet. 2013;84(2):132–141. doi: 10.1111/cge.12203
- Boon KL, Grainger RJ, Ehsani P, et al. Prp8 mutations that cause human retinitis pigmentosa lead to a U5 snRNP maturation defect in yeast. Nat Struct Mol Biol. 2007;14(11):1077–1083. doi: 10.1038/nsmb1303
- Maeder C, Kutach AK, Guthrie C. ATP-dependent unwinding of U4/U6 snRnas by the Brr2 helicase requires the C terminus of Prp8. Nat Struct Mol Biol. 2009;16(1):42–48. doi: 10.1038/nsmb.1535
- Mozaffari-Jovin S, Wandersleben T, Santos KF, et al. Inhibition of RNA helicase Brr2 by the C-terminal tail of the spliceosomal protein Prp8. Science. 2013;341(6141):80–84. doi: 10.1126/science.1237515
- Korir PK, Roberts L, Ramesar R, et al. A mutation in a splicing factor that causes retinitis pigmentosa has a transcriptome-wide effect on mRNA splicing. BMC Res Notes. 2014;7(1):401. doi: 10.1186/1756-0500-7-401
- Krausova M, Kreplova M, Banik P, et al. Retinitis pigmentosa–associated mutations in mouse Prpf8 cause misexpression of circRnas and degeneration of cerebellar granule cells. Life Sci Alliance. 2023;6(6):6. doi: 10.26508/lsa.202201855
- Vithana EN, Abu-Safieh L, Allen MJ, et al. A human homolog of yeast pre-mRNA splicing gene, PRP31, underlies autosomal dominant retinitis pigmentosa on chromosome 19q13.4 (RP11). Mol Cell. 2001;8(2):375–381. doi: 10.1016/S1097-2765(01)00305-7
- Yang C, Georgiou M, Atkinson R, et al. Pre-mRNA processing factors and retinitis pigmentosa: RNA splicing and beyond. Front Cell Dev Biol. 2021;9:700276. doi: 10.3389/fcell.2021.700276
- Maxwell DW, O'Keefe RT, Roy S, et al. The role of splicing factors in retinitis pigmentosa: links to cilia. Biochem Soc Trans. 2021;49(3):1221–1231. doi: 10.1042/BST20200798
- Shchepachev V, Azzalin CM. The Mpn1 RNA exonuclease: cellular functions and implication in disease. FEBS Lett. 2013;587(13):1858–1862. doi: 10.1016/j.febslet.2013.05.005
- Volpi L, Roversi G, Colombo EA, et al. Targeted next-generation sequencing appoints c16orf57 as clericuzio-type poikiloderma with neutropenia gene. Am J Hum Genet. 2010;86(1):72–76. doi: 10.1016/j.ajhg.2009.11.014
- Shchepachev V, Wischnewski H, Missiaglia E, et al. Mpn1, mutated in poikiloderma with neutropenia protein 1, is a conserved 3′-to-5′ RNA exonuclease processing u6 small nuclear RNA. Cell Rep. 2012;2(4):855–865. doi: 10.1016/j.celrep.2012.08.031
- Didychuk AL, Butcher SE, Brow DA. The life of U6 small nuclear RNA, from cradle to grave. RNA. 2018;24(4):437–460. doi: 10.1261/rna.065136.117
- Shchepachev V, Wischnewski H, Soneson C, et al. Human Mpn1 promotes post-transcriptional processing and stability of U6atac. FEBS Lett. 2015;589(18):2417–2423. doi: 10.1016/j.febslet.2015.06.046
- Mroczek S, Krwawicz J, Kutner J, et al. C16orf57 , a gene mutated in poikiloderma with neutropenia, encodes a putative phosphodiesterase responsible for the U6 snRNA 3′ end modification. Genes Dev. 2012;26(17):1911–1925. doi: 10.1101/gad.193169.112
- Hilcenko C, Simpson PJ, Finch AJ, et al. Aberrant 3′ oligoadenylation of spliceosomal U6 small nuclear RNA in poikiloderma with neutropenia. Blood. 2013;121(6):1028–1038. doi: 10.1182/blood-2012-10-461491
- Patil P, Uechi T, Kenmochi N. Incomplete splicing of neutrophil-specific genes affects neutrophil development in a zebrafish model of poikiloderma with neutropenia. RNA Biol. 2015;12(4):426–434. doi: 10.1080/15476286.2015.1017240
- Childs-Disney JL, Yang X, Gibaut QMR, et al. Targeting RNA structures with small molecules. Nat Rev Drug Discov. 2022;21(10):736–762. doi: 10.1038/s41573-022-00521-4
- Desterro J, Bak-Gordon P, Carmo-Fonseca M. Targeting mRNA processing as an anticancer strategy. Nat Rev Drug Discov. 2020;19(2):112–129. doi: 10.1038/s41573-019-0042-3
- Bhat B, Karve S, Anderson DG. mRNA therapeutics: beyond vaccine applications. Trends Mol Med. 2021;27(9):923–924. doi: 10.1016/j.molmed.2021.05.004
- Rupaimoole R, Slack FJ. MicroRNA therapeutics: towards a new era for the management of cancer and other diseases. Nat Rev Drug Discov. 2017;16(3):203–222. doi: 10.1038/nrd.2016.246
- Bennett CF, Krainer AR, Cleveland DW. Antisense oligonucleotide therapies for neurodegenerative diseases. Annu Rev Neurosci. 2019;42(1):385–406. doi: 10.1146/annurev-neuro-070918-050501
- Finkel RS, Mercuri E, Darras BT, et al. Nusinersen versus sham control in infantile-onset spinal muscular atrophy. N Engl J Med. 2017;377(18):1723–1732. doi: 10.1056/NEJMoa1702752
- Hastings ML, Berniac J, Liu YH, et al. Tetracyclines that promote SMN2 exon 7 splicing as therapeutics for spinal muscular atrophy. Sci Transl Med. 2009;1(5):5ra12. doi: 10.1126/scitranslmed.3000208
- Palacino J, Swalley SE, Song C, et al. SMN2 splice modulators enhance U1–pre-pre-mRNA association and rescue SMA mice. Nat Chem Biol. 2015;11(7):511–517. doi: 10.1038/nchembio.1837
- Naryshkin NA, Weetall M, Dakka A, et al. Motor neuron disease. SMN2 splicing modifiers improve motor function and longevity in mice with spinal muscular atrophy. Science. 2014;345(6197):688–693. doi: 10.1126/science.1250127
- Krach F, Stemick J, Boerstler T, et al. An alternative splicing modulator decreases mutant HTT and improves the molecular fingerprint in Huntington's disease patient neurons. Nat Commun. 2022;13(1):6797. doi: 10.1038/s41467-022-34419-x
- https://classic.clinicaltrials.gov/ct2/show/NCT05358717
- Ratni H, Ebeling M, Baird J, et al. Discovery of Risdiplam, a Selective Survival of Motor Neuron-2 (SMN2) gene splicing modifier for the treatment of Spinal Muscular Atrophy (SMA). J Med Chem. 2018;61(15):6501–6517. doi: 10.1021/acs.jmedchem.8b00741
- Ratni H, Scalco RS, Stephan AH. Risdiplam, the first approved small molecule splicing modifier drug as a blueprint for future transformative medicines. ACS Med Chem Lett. 2021;12(6):874–877. doi: 10.1021/acsmedchemlett.0c00659
- Sivaramakrishnan M, McCarthy KD, Campagne S, et al. Binding to SMN2 pre-mRNA-protein complex elicits specificity for small molecule splicing modifiers. Nat Commun. 2017;8(1):1476. doi: 10.1038/s41467-017-01559-4
- Campagne S, Boigner S, Rudisser S, et al. Structural basis of a small molecule targeting RNA for a specific splicing correction. Nat Chem Biol. 2019;15(12):1191–1198. doi: 10.1038/s41589-019-0384-5
- Bhattacharyya A, Trotta CR, Narasimhan J, et al. Small molecule splicing modifiers with systemic HTT-lowering activity. Nat Commun. 2021;12(1):7299. doi: 10.1038/s41467-021-27157-z
- Ishigami Y, Wong MS, Martí-Gómez C, et al. Specificity, synergy, and mechanisms of splice-modifying drugs. 2023. bioRxiv 2022:2022.12.30.522303.
- Roca X, Akerman M, Gaus H, et al. Widespread recognition of 5′ splice sites by noncanonical base-pairing to U1 snRNA involving bulged nucleotides. Genes Dev. 2012;26(10):1098–1109. doi: 10.1101/gad.190173.112
- Monteys AM, Hundley AA, Ranum PT, et al. Regulated control of gene therapies by drug-induced splicing. Nature. 2021;596(7871):291–295. doi: 10.1038/s41586-021-03770-2
- Zhang L, Xie X, Djokovic N, et al. Reversible control of RNA splicing by photoswitchable small molecules. J Am Chem Soc. 2023;145(23):12783–12792. doi: 10.1021/jacs.3c03275
- Effenberger KA, Urabe VK, Jurica MS. Modulating splicing with small molecular inhibitors of the spliceosome. Wiley Interdiscip Rev RNA. 2017;8(2):e1381. doi: 10.1002/wrna.1381
- Nakajima H, Hori Y, Terano H, et al. New antitumor substances, FR901463, FR901464 and FR901465. II. Activities against experimental tumors in mice and mechanism of action. J Antibiot (Tokyo). 1996;49(12):1204–1211. doi: 10.7164/antibiotics.49.1204
- Mizui Y, Sakai T, Iwata M, et al. Pladienolides, new substances from culture of Streptomyces platensis Mer-11107 III. In vitro and in vivo antitumor activities. J Antibiot. 2004;57(3):188–196. doi: 10.7164/antibiotics.57.188
- Sakai Y, Tsujita T, Akiyama T, et al. GEX1 compounds, novel antitumor antibiotics related to herboxidiene, produced by Streptomyces sp. II. The effects on cell cycle progression and gene expression. J Antibiot (Tokyo). 2002;55(10):863–872. doi: 10.7164/antibiotics.55.863
- Eustaquio AS, Janso JE, Ratnayake AS, et al. Spliceostatin hemiketal biosynthesis in Burkholderia spp. is catalyzed by an iron/α-ketoglutarate–dependent dioxygenase. Proc Natl Acad Sci U S A. 2014;111(33):E3376–85. doi: 10.1073/pnas.1408300111
- Masato H, Hajime M, Hidenori W, et al. A synthesis of FR901464. Tetrahedron Lett. 2001;42(46):8207–8210. doi: 10.1016/S0040-4039(01)01763-4
- Kotake Y, Sagane K, Owa T, et al. Splicing factor SF3b as a target of the antitumor natural product pladienolide. Nat Chem Biol. 2007;3(9):570–575. doi: 10.1038/nchembio.2007.16
- Hasegawa M, Miura T, Kuzuya K, et al. Identification of SAP155 as the target of GEX1A (Herboxidiene), an antitumor natural product. ACS Chem Biol. 2011;6(3):229–233. doi: 10.1021/cb100248e
- Kaida D, Motoyoshi H, Tashiro E, et al. Spliceostatin a targets SF3b and inhibits both splicing and nuclear retention of pre-mRNA. Nat Chem Biol. 2007;3(9):576–583. doi: 10.1038/nchembio.2007.18
- Teng T, Tsai JH, Puyang X, et al. Splicing modulators act at the branch point adenosine binding pocket defined by the PHF5A–SF3b complex. Nat Commun. 2017;8(1):15522. doi: 10.1038/ncomms15522
- Effenberger KA, Urabe VK, Prichard BE, et al. Interchangeable SF3B1 inhibitors interfere with pre-mRNA splicing at multiple stages. RNA. 2016;22(3):350–359. doi: 10.1261/rna.053108.115
- Osman S, Albert BJ, Wang Y, et al. Structural requirements for the antiproliferative activity of pre-mRNA splicing inhibitor FR901464. Chemistry. 2011;17(3):895–904. doi: 10.1002/chem.201002402
- Vigevani L, Gohr A, Webb T, et al. Molecular basis of differential 3′ splice site sensitivity to anti-tumor drugs targeting U2 snRNP. Nat Commun. 2017;8(1):2100. doi: 10.1038/s41467-017-02007-z
- Finci LI, Zhang X, Huang X, et al. The cryo-EM structure of the SF3b spliceosome complex bound to a splicing modulator reveals a pre-mRNA substrate competitive mechanism of action. Genes Dev. 2018;32(3–4):309–320. doi: 10.1101/gad.311043.117
- Cretu C, Agrawal AA, Cook A, et al. Structural basis of splicing modulation by antitumor macrolide compounds. Mol Cell. 2018;70(2):265–273.e8. doi: 10.1016/j.molcel.2018.03.011
- Carrocci TJ, Paulson JC, Hoskins AA. Functional analysis of Hsh155/SF3b1 interactions with the U2 snRNA/branch site duplex. RNA. 2018;24(8):1028–1040. doi: 10.1261/rna.065664.118
- Hansen SR, Nikolai BJ, Spreacker PJ, et al. Chemical inhibition of pre-mRNA splicing in living saccharomyces cerevisiae. Cell Chem Biol. 2019;26(3):443–8 e3. doi: 10.1016/j.chembiol.2018.11.008
- Seiler M, Yoshimi A, Darman R, et al. H3B-8800, an orally available small-molecule splicing modulator, induces lethality in spliceosome-mutant cancers. Nat Med. 2018;24(4):497–504. doi: 10.1038/nm.4493
- Bowling EA, Wang JH, Gong F, et al. Spliceosome-targeted therapies trigger an antiviral immune response in triple-negative breast cancer. Cell. 2021;184(2):384–403.e21. doi: 10.1016/j.cell.2020.12.031
- Steensma DP, Wermke M, Klimek VM, et al. Phase I first-in-human dose escalation study of the oral SF3B1 modulator H3B-8800 in myeloid neoplasms. Leukemia. 2021;35(12):3542–3550. doi: 10.1038/s41375-021-01328-9
- Ten Hacken E, Valentin R, Regis FFD, et al. Splicing modulation sensitizes chronic lymphocytic leukemia cells to venetoclax by remodeling mitochondrial apoptotic dependencies. JCI Insight. 2018;3(19):3. doi: 10.1172/jci.insight.121438
- Wheeler EC, Martin BJE, Doyle WC, et al. Splicing modulators impair DNA damage response and induce killing of cohesin-mutant MDS/AML. Blood. 2022;140(Supplement 1):6888–6889. bioRxiv 2022:2022.09.26.509430. doi: 10.1182/blood-2022-170996
- Gao Y, Koide K. Chemical perturbation of Mcl-1 pre-mRNA splicing to induce apoptosis in cancer cells. ACS Chem Biol. 2013;8(5):895–900. doi: 10.1021/cb300602j
- Salton M, Kasprzak WK, Voss T, et al. Inhibition of vemurafenib-resistant melanoma by interference with pre-mRNA splicing. Nat Commun. 2015;6(1):7103. doi: 10.1038/ncomms8103
- Gao Y, Trivedi S, Ferris RL, et al. Regulation of HPV16 E6 and MCL1 by SF3B1 inhibitor in head and neck cancer cells. Sci Rep. 2014;4(1):6098. doi: 10.1038/srep06098
- Sakamoto KM, Kim KB, Kumagai A, et al. Protacs: Chimeric molecules that target proteins to the Skp1–Cullin–F box complex for ubiquitination and degradation. Proc Natl Acad Sci U S A. 2001;98(15):8554–8559. doi: 10.1073/pnas.141230798
- Calderon-Villalobos LI, Tan X, Zheng N, et al. Auxin perception–structural insights. Cold Spring Harb Perspect Biol. 2010;2(7):a005546. doi: 10.1101/cshperspect.a005546
- Liu Z, Hu M, Yang Y, et al. An overview of PROTACs: a promising drug discovery paradigm. Mol Biomed. 2022;3(1):46. doi: 10.1186/s43556-022-00112-0
- Dowhan DH, Hong EP, Auboeuf D, et al. Steroid hormone receptor coactivation and alternative RNA Splicing by U2AF65-related proteins CAPERα and CAPERβ. Mol Cell. 2005;17(3):429–439. doi: 10.1016/j.molcel.2004.12.025
- Stepanyuk GA, Serrano P, Peralta E, et al. UHM–ULM interactions in the RBM39–U2AF65 splicing-factor complex. Acta Crystallogr D Struct Biol. 2016;72(4):497–511. doi: 10.1107/S2059798316001248
- Loerch S, Maucuer A, Manceau V, et al. Cancer-relevant Splicing Factor CAPERα Engages the essential splicing factor SF3b155 in a specific ternary complex. J Biol Chem. 2014;289(25):17325–17337. doi: 10.1074/jbc.M114.558825
- Owa T, Yoshino H, Okauchi T, et al. Discovery of novel antitumor sulfonamides targeting G1 phase of the cell cycle. J Med Chem. 1999;42(19):3789–3799. doi: 10.1021/jm9902638
- Han T, Goralski M, Gaskill N, et al. Anticancer sulfonamides target splicing by inducing RBM39 degradation via recruitment to DCAF15. Science. 2017;356(6336). doi: 10.1126/science.aal3755
- Uehara T, Minoshima Y, Sagane K, et al. Selective degradation of splicing factor CAPERα by anticancer sulfonamides. Nat Chem Biol. 2017;13(6):675–680. doi: 10.1038/nchembio.2363
- Faust TB, Yoon H, Nowak RP, et al. Structural complementarity facilitates E7820-mediated degradation of RBM39 by DCAF15. Nat Chem Biol. 2020;16(1):7–14. doi: 10.1038/s41589-019-0378-3
- Du X, Volkov OA, Czerwinski RM, et al. Structural basis and kinetic pathway of RBM39 Recruitment to DCAF15 by a sulfonamide molecular glue E7820. Structure. 2019;27(11):1625–1633.e3. doi: 10.1016/j.str.2019.10.005
- Bussiere DE, Xie L, Srinivas H, et al. Structural basis of indisulam-mediated RBM39 recruitment to DCAF15 E3 ligase complex. Nat Chem Biol. 2020;16(1):15–23. doi: 10.1038/s41589-019-0411-6
- Xu C, Chen X, Zhang X, et al. RNA-binding protein 39: a promising therapeutic target for cancer. Cell Death Discov. 2021;7(1):214. doi: 10.1038/s41420-021-00598-7
- Wang E, Lu SX, Pastore A, et al. Targeting an RNA-Binding protein network in acute myeloid leukemia. Cancer Cell. 2019;35(3):369–384.e7. doi: 10.1016/j.ccell.2019.01.010
- Singh S, Quarni W, Goralski M, et al. Targeting the spliceosome through RBM39 degradation results in exceptional responses in high-risk neuroblastoma models. Sci Adv. 2021;7(47):eabj5405. doi: 10.1126/sciadv.abj5405
- Lu SX, De Neef E, Thomas JD, et al. Pharmacologic modulation of RNA splicing enhances anti-tumor immunity. Cell. 2021;184(15):4032–4047.e31. doi: 10.1016/j.cell.2021.05.038
- Dahui Q. Next-generation sequencing and its clinical application. Cancer Biol Med. 2019;16(1):4–10. doi: 10.20892/j.issn.2095-3941.2018.0055
- Anczukow O, Krainer AR. The spliceosome, a potential Achilles heel of MYC-driven tumors. Genome Med. 2015;7(1):107. doi: 10.1186/s13073-015-0234-3
- Maji D, Grossfield A, Kielkopf CL. Structures of SF3b1 reveal a dynamic Achilles heel of spliceosome assembly: Implications for cancer-associated abnormalities and drug discovery. Biochim Biophys Acta, Gene Regul Mech. 2019;1862(11–12):194440. doi: 10.1016/j.bbagrm.2019.194440
- Arbab M, Matuszek Z, Kray KM, et al. Base editing rescue of spinal muscular atrophy in cells and in mice. Science. 2023;380(6642):eadg6518. doi: 10.1126/science.adg6518