
ABSTRACT
CRISPR technologies comprising a Cas nuclease and a guide RNA (gRNA) can utilize multiple gRNAs to enact multi-site editing or regulation in the same cell. Nature devised a highly compact means of encoding gRNAs in the form of CRISPR arrays composed of conserved repeats separated by targeting spacers. However, the capacity to acquire new spacers keeps the arrays longer than necessary for CRISPR technologies. Here, we show that CRISPR arrays utilized by the Cas9 nuclease can be shortened without compromising and sometimes even enhancing targeting activity. Using multiplexed gene repression in E. coli, we found that each region could be systematically shortened to varying degrees before severely compromising targeting activity. Surprisingly, shortening some spacers yielded enhanced targeting activity, which was linked to folding of the transcribed array prior to processing. Overall, shortened CRISPR-Cas9 arrays can facilitate multiplexed editing and gene regulation from a smaller DNA footprint across many bacterial applications of CRISPR technologies.
Introduction
The machinery of CRISPR-Cas systems has been co-opted to form diverse tools used in the fields of synthetic biology, medicine, agriculture and more. In nature, these immune systems comprise a CRISPR array comprising alternating conserved repeats and targeting spacers and a cluster of cas genes. Some of these genes encode the effector complex responsible for immune defence comprising either a large multidomain Cas protein (a single-effector nuclease) or a multi-subunit effector complex. In either case, the effector is bound to guide RNAs (gRNAs) encoded within the CRISPR arrays [Citation1]. The gRNAs direct the ribonucleoprotein complex to a target nucleic acid complementary to the gRNA guide sequence and flanked by a prescribed protospacer-adjacent motif (PAM) in the DNA [Citation2], instructing the effector to cleave the nucleic acid or induce cell dormancy. This modularity of an effector protein or complex and a programmable gRNA has made CRISPR tools invaluable for varying application such as gene regulation and editing. Gene regulation can be achieved by utilizing catalytically-dead effectors that modulate transcription of the adjacent gene [Citation3,Citation4]. For gene editing, several tools have been developed, including base editors [Citation5–8], CRISPR-guided transposons [Citation9–11] or prime editors [Citation12,Citation13].
Both gene regulation and genome editing benefit from multiplexing by expressing multiple gRNAs and directing the effector complex to different DNA sequences at one time. For example, multiple gRNAs can be expressed to differentially regulate or edit multiple genes at once [Citation14]. Separately, the efficiency of gene inhibition (CRISPRi) and activation (CRISPRa) can be improved by directing multiple gRNAs to the target region [Citation15,Citation16]. Several strategies exist to express multiple gRNAs in vivo. Single-guide RNAs (sgRNAs), which normally represents an engineered Cas9 gRNA that does not need to undergo processing [Citation17], can be assembled into an array, with each construct under the control of its own promoter [Citation18,Citation19]. Synthetic sgRNA arrays can also be assembled with intervening RNA cleavage sequences, such as Csy4 sites, self-cleaving ribozymes or tRNAs [Citation20–23].
Another multiplexing strategy involves designing CRISPR arrays derived from natural CRISPR-Cas systems [Citation15,Citation16,Citation24]. Similar to sgRNA arrays, CRISPR arrays enable the expression of multiple gRNAs from a single promoter in a single construct and are compatible with different nucleases. The spacer-repeat subunits in CRISPR arrays are considerably smaller compared to those in sgRNA arrays though, enabling multiple gRNAs to occupy a shorter DNA footprint. Still, CRISPR array designs derived from nature could potentially be further condensed. CRISPR arrays have a common architecture with a leader sequence followed by alternating repeat and spacer sequences. The specific length of the array is determined in part by the acquisition machinery, as the processed gRNAs, also called CRISPR RNAs (crRNAs), are often shorter than a full spacer-repeat subunit [Citation25]. This suggests that sequence parts essential for spacer acquisition can be neglected to design shorter array formats yielding functional gRNAs.
Cas12a, the single-effector nuclease from Type V CRISPR-Cas systems, has been successfully employed for multiplexed genome editing and regulation using shortened CRISPR arrays. Trimming the direct repeat from 36 nucleotides (nts) to 19 nts was shown to even increase the editing efficiency in mammalian cells and yeast [Citation24,Citation26]. By integrating trimmed gRNAs with different spacer lengths in a synthetic array, orthogonal transcriptional gene regulation and editing with Cas12a was achieved [Citation15]. Due to Cas12a’s ability to process its own gRNA, such synthetic arrays have been employed for multi-gene regulation and editing in various organisms like yeast [Citation27], plants [Citation28], bacteria [Citation16,Citation29], Drosophila [Citation30] and mammalian cells [Citation15,Citation28],[Citation31]
Cas9, the single-effector from the Type II CRISPR-Cas systems, has been developed into a broad spectrum of tools for gene editing and regulation and is widely established in many laboratories, partly due to the straightforward guide design and the availability of Cas9 variants with a broad targeting range. In contrast to Type V systems, crRNA biogenesis from CRISPR arrays in Type II systems requires a trans-activating crRNA (tracrRNA), RNase III and host ribonucleases to process the transcribed CRISPR array into mature crRNAs [Citation32,Citation33]. Therefore, Cas9-based gene editing in eukaryotes relies on sgRNAs to circumvent the need to express an additional tracrRNA and process crRNAs. Up to now, Cas9 remains the most widely used nuclease for gene editing and regulation in bacteria, where CRISPR arrays offer the most compact means to encode for several crRNAs. This motivated us to evaluate the extent to which we can condense Cas9 arrays for multiplexed targeting in bacteria. In this work, we show that CRISPR-Cas9 arrays used for gene silencing can be shortened while remaining functional and even resulting in higher targeting activity. Such shortened arrays could benefit various multiplexing applications, ranging from the analysis of CRISPR-based screens with shorter Next-Generation Sequencing (NGS) reads to enhancing CRISPRi/a efficiencies or rapid strain engineering.
Results
CRISPR arrays offer the potential to be shortened for multiplexed targeting with Cas9
To condense CRISPR-Cas9 arrays, we first considered the functional role of each spacer and repeat within an array and their dispensability as part of multiplexed gene targeting. During crRNA biogenesis, the tracrRNA forms a duplex with the precursor CRISPR repeat-spacer transcript through hybridization between the anti-repeat sequence of the tracrRNA and the transcribed CRISPR repeat. In a first processing event, the duplex is cleaved by RNase III to create a 75-nt tracrRNA and 66-nt intermediate crRNA species. In a second processing event thought to be mediated by other host ribonucleases [Citation33], the intermediate crRNA species undergoes further trimming at the 5’ end, resulting in the 39–42 nt long mature crRNA composed of a 20-nt guide and 19–22-nt processed repeat () [Citation33]. At some point in this process, Cas9 binds the RNA duplex to form the ribonucleoprotein effector complex.
Figure 1. crRNA processing suggests the potential of CRISPR-Cas9 arrays to be further shortened.
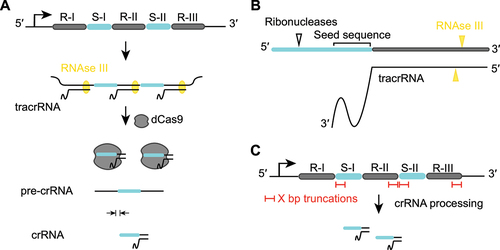
For efficient DNA cleavage, Cas9 must undergo a conformational change that is dependent on the proper binding to the 20–24 nt guide sequence [Citation34]. However, binding to the DNA is possible with as few as 9 nts complementary to the seed sequence [Citation35] and has been shown to lead to strong repression if the coding strand of an open-reading frame (ORF) is targeted with a 12-nt matching guide sequence [Citation4]. Additionally, the DNA binding strength depends on the crRNA guide sequence, where targeting different gene loci can lead to variations in the gene repression efficiency [Citation3]. As mature crRNAs do not consist of the full-length repeats and spacers, we reasoned that the initial array sequence can be shortened from the 5’ end of the spacer and the 3’ end of the repeat – mimicking natural processing – while maintaining efficient DNA targeting with the resulting crRNAs.
We utilized a previously published CRISPR array cloning method [Citation16,Citation36] that takes advantage of the fact that the 5’ end of the spacer does not contribute to DNA targeting. As part of the assembly method, defined 4-bp assembly junctions are used to efficiently clone spacer-repeat subunits into a plasmid backbone, with the position of each subunit specified within the array. For example, this means that in a single-spacer array derived from the native design (i.e. spacer of 30 nts and repeat of 36 nts), the first 4 nts of the spacer from the 5’ end serve as the junction, and the following 26 nts directly come from the target sequence ().
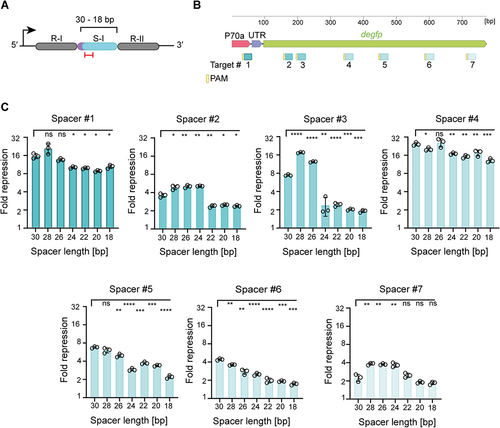
Figure 2. Systematic shortening of single-spacer CRISPR arrays demonstrates that smaller truncations can maintain targeted gene repression by dCas9 in E. coli.
Moderately shortened spacers generally maintain targeted gene repression by dCas9
We began by evaluating the impact of trimming the spacer, the repeat, or both as part of single-spacer CRISPR arrays. Shortened CRISPR arrays were evaluated via CRISPRi by employing a catalytically-dead Cas9 from Streptococcus pyogenes (SpdCas9) targeted to a fluorescence reporter sequence in Escherichia coli (). The introduced mutations D10A and H840A in the RuvC and HNH nuclease domains, respectively, turn it into a programmable DNA-binding protein [Citation17] that will either inhibit transcription initiation when targeted to the promoter region of the gene-of-interest or block transcription elongation when targeted to the coding region [Citation3,Citation4]. We co-transformed E. coli with plasmids expressing SpdCas9, a targeted degfp reporter [Citation37] and the tracrRNA as well as the specific CRISPR array to be analysed.
To determine the extent to which the spacer sequence can be shortened without impeding functionality, we measured the gene repression efficiencies of seven single-spacer arrays with varying spacer truncations (original 30 nts to 18 nts, in 2 nts steps). Spacers were trimmed from the 5’ end following the 4-bp junction, targeting the coding strand at different loci of the degfp gene (), reflecting the presence of the critical PAM-proximal seed region on the 3’ end of the spacer [Citation38]. The repression efficiency of each of the degfp-targeting CRISPR arrays was then assessed in comparison to the no-spacer array plasmid. The repression efficiencies varied between 2.3-fold (single-spacer #7, spacer length = 30 nts) and 24-fold (single-spacer #4, spacer length = 30 nts) ().
We observed a drop in repression efficiencies with larger spacer truncations, starting already at 24-nts spacers (). When considering the 4-bp junction at the 5’ spacer end of each array, only the remaining nucleotides are perfectly complementary to the target DNA (). Two different scenarios can possibly explain the decreasing gene repression efficiency. Firstly, for arrays with less than 20 nts complementary to the target, an increasing number of PAM-distal mismatches has been shown to increase the probability of dCas9 being removed by the RNA polymerase during transcription elongation [Citation39]. Second, for arrays with 20 nts complementary to the target DNA (similar to a full-length sgRNA guide), factors like crRNA stability, secondary structure formation and/or binding to Cas9 could affect the gene repression. Interestingly, in two cases, shortening the spacer significantly boosted gene repression (e.g. single-spacer array #3, 7.4-fold for the 30-nt spacer vs. 18-fold for the 28-nt spacer, P = 1.0E–5, n = 3) (). This boost was limited to either 24-nt or 26-nt spacers and was lost as the spacer was further shortened.
Trimming the downstream repeat in single-spacer arrays has minor effects on targeted gene repression
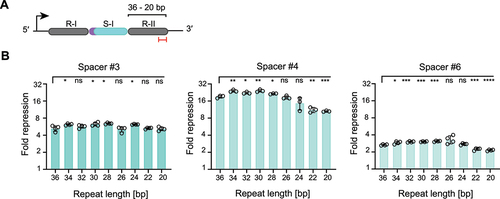
Beyond trimming the spacer, we investigated the effect of systematically trimming the downstream repeat on gene repression. For these experiments, we chose three single-spacer arrays from the previous set targeting different degfp locations. We performed 2-nt stepwise truncations from the 3’ end of the repeat, as the 5’ end is known to be essential for hybridization with the tracrRNA and thus proper crRNA processing (). We observed variations in gene repression depending on the spacer-determined target site. However, the stepwise 2-nt truncations of the downstream repeat sequence, which ranged from the original 36 nts to 20 nts, had only a minor impact on the gene repression efficiency (). This robustness may be attributed to the tracrRNA forming sufficient base pairing interactions with the truncated repeat to drive processing and Cas9 binding. Therefore, trimming the 3’ end of the downstream repeat offers a flexible option for reducing the array size without impacting the targeting activity in the arrays tested.
Figure 3. Truncation of the downstream CRISPR array repeat has only a minor impact on gene repression by dCas9 in E. coli.
Condensing multi-spacer CRISPR array formats can enhance targeted gene repression
When switching from single-spacer arrays to multi-spacer arrays, surrounding repeat-spacer subunits could influence each other, affecting the targeting efficiencies of the individual crRNAs. As examples for this phenomenon, the spatial position of the spacers within a Cas12a array has been shown to lead to varying levels of mature crRNAs [Citation15,Citation16], while modifications that prevented inhibitory base-pairs improved the targeting efficiency for some Cas12a arrays [Citation40,Citation41].
To investigate the effect of array truncations for Cas9 multi-spacer arrays, we began by comparing degfp repression efficiencies of dual- and triple-spacer arrays with either the original 30-nt spacers or more compact 24-nt spacers. We selected six different spacer sequences in six combinations per dual-spacer or triple-spacer array (). Each spacer targets a different locus within the degfp, which is expected to increase the overall repression efficiency compared to single-spacer arrays. Moderately shortening the spacers from 30 nts to 24 nts did not change the repression efficiencies in most arrays. However, we also observed a slight decrease in the array efficiency for some arrays with shorter spacers (i.e. triple-spacer array #4/5/6 and #4/5/7). Interestingly, other arrays yielded a boost in gene repression when using shorter spacer sequences (i.e. dual-spacer array #2/4, #2/7, #4/7, triple-spacer array #2/3/4 and #2/4/5) (). Shortening the spacer sequence to a certain degree thus can still allow for efficient gene repression in the context of multi-spacer arrays.
Figure 4. Condensing multi-spacer CRISPR array formats can enhance gene repression. (A) Fold repression values from GFP-based flow cytometry assays with multi-spacer (dual- or triple-spacer arrays) with truncated spacers (24 nts vs. 30 nts) targeting degfp at different locations. Statistical significance was calculated by comparing the fold repression values between the arrays with the native 30 nts spacer length or the truncated 24 nts spacer length. (B) Fold repression values from GFP-based flow cytometry assays with multi-spacer (dual- or triple-spacer) arrays encoding different spacer lengths (30, 24 and 20 nts) and repeat lengths (36, 28, 20 nts) targeting degfp at different locations. Statistical significance was calculated by comparing the fold repression values of the shortened array formats with the native array encoding for 30-nt spacers and 36-nt repeats. The position of the spacer and repeats within the array is indicated in roman numerals (e.g. R-I, S-I). Error bars in (A) and (B) indicate the mean and standard deviation from independent measurements of three (A) or four (B) individual clones. ****p < 0.0001 ***p < 0.001. **p < 0.01. *p < 0.05. ns: p > 0.05.
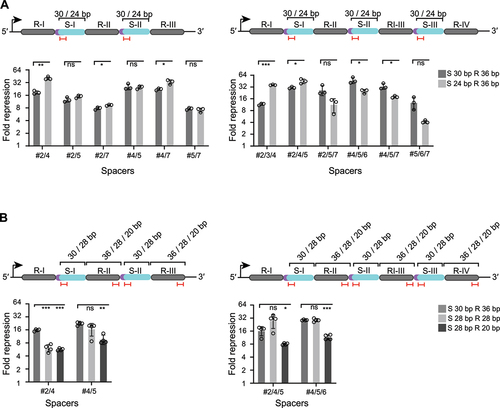
We additionally selected two dual- and triple-spacer arrays and evaluated the effect of trimming the spacer and the repeat simultaneously. A spacer length of 28 nts was chosen, as the extent of gene repression was generally maintained in the previous experiments using single-spacer arrays. For the repeat sequence, which in the single spacer-arrays yielded relatively similar efficiencies for all tested sequence lengths, we now included a 28-nt repeat or a 20-nt repeat in the multi-spacer arrays. When trimming the repeat and spacer sequence, minor deviations from the original format (e.g. 30 nts to 28 nts for the spacer, 36 nts to 28 nts for the repeat) maintained repression, except for the dual-spacer 2/4 array that showed a lower efficiency compared to the native format (P = 2.5E–5, n = 3). Using the shorter repeat sequence of 20 nts, which did not impair the efficiency in the single-spacer array, caused a significant decrease in efficiency for all tested dual- and triple-spacer arrays (e.g. dual-spacer array #2/4, P = 2.2E–5, n = 4; for the, dual-spacer array #7/8, triple-spacer array #4/5/6, P = 1.7E–5, n = 4) (). Despite one case (dual spacer array #2/4) where we observed that the moderately shortened array led to less efficient gene repression, the other arrays with 28-nt spacers and 28-nt repeats performed comparably well to the native arrays. Therefore, shorter multi-spacer arrays can indeed be utilized for efficient multiplexed targeting and in some cases yield enhanced targeting activity.
Enhanced gene repression of shortened spacers may be related to intra-array sequence interactions
We were intrigued why some of the shorter single- and multi-spacer arrays yielded higher targeting activity compared to the native format. We focused on the single-spacer array #3 as one representative example to avoid complex sequence interactions that are likely involved in multi-spacer arrays. One possible explanation is RNA secondary structure, which has a strong influence on crRNA biogenesis, thereby impacting the extent of DNA targeting and gene repression [Citation16,Citation42]. We have therefore employed RNA folding predictions, utilizing the NUPACK software [Citation43], to detect differences in the secondary structure of the single-spacer array #3 that showed the largest difference in gene repression efficiencies when comparing the native array format with the shorter array encoding for a 28-nt spacer ().
Although the predicted secondary structures between the single-spacer array #3 with the native 30-nt spacer and the shortened 28-nt spacer did not differ drastically, we observed smaller differences in the predicted number of base pairing interactions between the upstream repeat and the spacer (). In comparison to the original single-spacer array #3, the array with the shortened 28-nt spacer was predicted to have fewer base pairing interactions between the spacer and the downstream repeat (Table S1). The altered base-pairing interactions – namely, increased interactions between the spacer and upstream repeat – would be expected to prevent spacer interactions with the downstream repeat. Following this reasoning, the improved accessibility of the downstream repeat for tracrRNA binding would consequently enhance crRNA processing and increase the abundance of mature crRNAs ().
Figure 5. Spacer length-dependent regulation depends on the upstream repeat, possibly through changes in RNA folding. (A) The number of base-pairing interactions between the spacer and upstream repeat changes when shortening the single-spacer array #3. Schematic of the RNA secondary structure predictions for the native format (30-nt spacer) and the shortened array (28-nt spacer). Red boxes indicate differences in the base pairing interactions that presumably contribute to the difference in gene repression efficiencies. Predictions of the minimal-free energy structure were made using NUPACK (http://www.nupack.org/). (B) Proposed model on shifting the equilibrium towards a secondary structure where the spacer interacts principally with the upstream repeat, which could potentially make the downstream repeat more accessible for tracrRNA-binding and therefore leading to more efficient crRNA processing. (C) Evaluating the effect of the upstream repeat on the gene repression efficiency with a GFP-based flow cytometry assay with single-spacer arrays #3 encoding different spacer lengths ranging from 30 nts to 18 nts. The boost in gene repression for the shorter array with a 28-nt spacer is abolished when the upstream repeat is absent from the array. Statistical significance was calculated by comparing the fold repression values of arrays with an original upstream repeat with those lacking the upstream repeat. (D) Comparing the effect of a randomly shuffled upstream repeat sequence versus the original upstream repeat on the degfp repression by flow cytometry. The boost in gene repression observed for the single-spacer array #3 with the shorter 28-nt spacer is abolished when the shuffled sequence replaces the original repeat. Statistical significance was calculated by comparing the fold repression values of arrays with an original upstream repeat with those encoding for a randomly shuffled upstream repeat sequence. The predicted secondary structures are depicted in Fig. S3. (E) Assessing the effect on degfp repression when adding 5’ extensions to the upstream repeat of single-spacer arrays #3 (spacer length of 30, 28 and 26 nts). Top graph: 5’ extensions that are predicted to bias the base pairing interactions of the spacer towards the upstream repeat. Bottom graph: 5’ extensions added to the upstream repeat of single-spacer arrays #3 (spacer length of 30, 28 and 26 nts) that are predicted to mostly maintain the base-pairing interactions present in the original format (30-nt spacer and 36-nt repeat). Statistical significance was calculated by comparing the fold repression values of arrays with the original upstream repeat with arrays encoding 5’-modified repeats. The extensions added to the 5’ end of the upstream repeat are coloured in green, mutations within the original repeat sequence are coloured in red. Error bars in (C), (D), (E) indicate the mean and standard deviation from measurements of four individual clones. ****p < 0.0001 ***p < 0.001. **p < 0.01. *p < 0.05. ns: p > 0.05
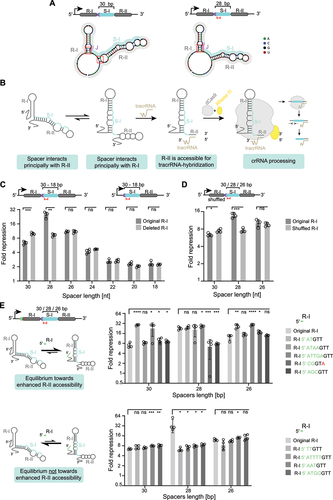
Even though RNA secondary structure predictions yielded no obvious difference in the binding configuration between the tracrRNA and single-spacer arrays #3 with 30- or 28-nt spacers (Fig. S1), we wanted to evaluate if the predicted base pairing interactions between the spacer and the first repeat contributed to the boost in gene repression. For this, we deleted the first repeat, which is not part of the mature crRNA derived from single-spacer arrays, and performed the degfp repression assay. In single-spacer arrays #3 lacking the upstream repeat, gene repression remained at the original level for all tested spacer lengths except for the array format with the shortened 28-nt spacer, which lost the boost in gene repression that we observed when including the upstream repeat (). This finding is in line with higher targeting efficiency stemming from interactions between the shortened 28-nt spacer and the upstream repeat. In addition, deleting the first repeat is a plausible option to shorten the Cas9 array, although doing so could impinge on the targeting efficiency of the array.
To further explore the contribution of the upstream repeat, we compared the abundances of crRNAs derived from single-spacer arrays #3 with 30-, 28- or 26-nt spacers with or without the upstream repeat. Northern blotting analysis revealed higher abundances of the mature crRNAs derived from the array with a shortened spacer length of 28 nts and showed that the higher crRNA levels were dependent on the presence of the upstream repeat (Fig. S2). Deleting the first repeat also resulted in small-sized products of ~ 80 to 90 nts, potentially representing crRNAs without 5’ end trimming, with similar abundances regardless of the spacer length. To further assess whether sequence-specific interactions between the spacer and the upstream repeat were responsible for the boost in gene repression for the array with the shortened 28-nt spacer, we shuffled the upstream repeat sequence (Fig. S3). The gene repression efficiencies remained similar for arrays encoding a 30-nt and 26-nt spacer but decreased for the array with the 28-nt spacer (). These experiments further support base pairing interactions of the upstream repeat with the shortened 28-nt spacer playing a role in enhancing the targeting efficiency.
We next reasoned that biasing the base pair interactions of the spacer towards the upstream repeat could also enhance the targeting efficiency for the single-spacer array #3. To further explore this possibility, we introduced extensions of the 5’ end of the upstream repeat in the single-spacer arrays #3 with 30- or 26-nt spacers yielding lower gene repression (Fig. S4, Table S2). The extensions were intended to increase interactions between the spacer and the upstream repeat based on RNA-folding predictions (Fig. S4, Table S1). Two extensions, 5’ AT and ATTGA, significantly enhanced gene repression with the 30-nt spacer from 7.1-fold to 29-fold and 23-fold, respectively (P = 2.8E–6, n = 4 for the 5’ AT extension; P = 0.0499, n = 4 for the 5’ ATTGA extension). Those extensions also improved the targeting activity of the single-spacer array #3 with a 26-nt spacer from the original 12-fold to 26-fold and 29-fold, respectively (P = 1.1E–3, n = 4 for the 5’ AT extension; P = 7.5E–5, n = 4 for the 5’ ATTGA extension) (, top graph). We then used northern blotting analysis to investigate whether the observed boost in gene repression for the arrays with the 5’ AT and ATTGA extensions was correlated with higher crRNA levels [Citation16], but we could not detect a direct effect on the levels of the mature crRNA (Fig. S2). When we designed 5’ extensions that were not supposed to increase the number of base-pairing interactions between the spacer and upstream repeat (Fig. S4, Table S1), we did not observe a boost in gene repression (, bottom graph). These results suggest that interactions between the upstream repeat and the spacer underlie enhanced repression with the 28-nt spacer, in line with our working model ().
The impact of altering spacer length could be due to other factors as well, such as disrupting RNase sites inadvertently encoded within the synthetic array as well as structures that promote Cas9 binding the crRNA:tracrRNA duplex. In the presence of multiple encoded crRNAs, competitive binding for Cas9 could further exacerbate issues with folding and subsequent targeting efficiency [Citation16]. Overall, the results and the model further support the importance of how the transcribed CRISPR array folds that can later be taken into account when designing standard and condensed CRISPR arrays.
Array length can impact the contribution of each encoded spacer
As gene repression efficiencies varied for multi-spacer arrays targeting the degfp gene, we asked how each spacer contributes to gene repression. Using the triple-spacer array #2/4/5 evaluated in as a case study, we designed a series of triple-spacer arrays in which the original spacer at each position was replaced by a non-targeting spacer generated by scrambling the associated spacer sequence (). We also included dual-spacer arrays #2/4 and #4/5 to evaluate the impact of adding a non-targeting spacer at the front or end of the array. Because the non-targeting sequence itself could cause differences in the targeting efficiency of the array, we created two different versions (sa or sb).
Figure 6. Gene repression efficiencies of crRNAs derived from multi-spacer arrays are influenced by the array context.
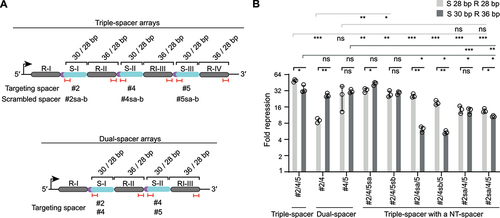
One notable observation was that adding a non-targeting spacer to either end of the dual-spacer arrays often altered gene repression (). Adding the non-targeting spacers at the end of the dual-spacer array #2/4 enhanced repression in almost all instances, particularly for the shortened arrays (shortened dual-spacer array #2/4: fold repression of 9; shortened triple-spacer array #2/4/5sa-b: fold repression of 33 and 28, p = 0.0024 and 0.011, n = 3). However, adding the non-targeting spacers at the beginning of the dual-spacer #4/5 array reduced repression, at least significantly with the native format (native dual-spacer #4/5 array: fold repression of 31 compared to the native triple-spacer array #2sa-b/4/5: fold repression of 14 and 11, p = 0.0042 and 0.0009, n = 3; shortened dual-spacer #4/5 array: fold repression of 26 compared to the shortened triple-spacer array #2sa-b/4/5: fold repression of 14 and 14, p = 0.25 and 0.25, n = 3). The effect was similar between the non-targeting spacers, ruling out any sequence dependencies. These results indicate that the length of the array and associated placement of the spacers impact targeting activity.
Turning to the contribution of each spacer in the triple-spacer array, swapping individual spacers with their non-targeting counterparts revealed a strong dependency on array trimming. For the native array, spacer #4 contributed the most followed by spacer #2, while spacer #5 did not contribute (triple-spacer array #2/4/5: fold repression of 35 versus triple-spacer array #2/4/5sa-b: fold repression of 43 and 29, p = 0.24 and 0.11, n = 3; versus triple-spacer array #2/4sa-b/5: fold repression of 6 and 5, p = 0.013 and 0.013, n = 3; versus triple-spacer array #2sa-b/4/5: fold repression of 14 and 11, p = 0.019 and 0.02, n = 3) (). For the shortened array, spacer #2 contributed the most followed by similar contributions from spacers #4 and #5 (triple-spacer array #2/4/5: fold repression of 49 versus triple-spacer array #2/4/5sa-b: fold repression of 33 and 28, p = 0.0022 and 0.0022, n = 3; triple-spacer array #2/4sa-b/5: fold repression of 26 and 19, p = 0.00053 and 0.0003, n = 3; versus triple-spacer array #2sa-b/4/5: fold repression of 14 and 14, p = 0.00011 and 0.00058, n = 3). The difference between the native and shortened arrays could be traced to spacer #4, which yielded the largest swing in gene repression out of the three spacers when shortening the array. Similar to the dual-spacer array, either non-targeting spacer yielded similar effects. These results indicate that shortening a CRISPR-Cas9 array can change the contributions of the individual spacers in the array.
Shortened arrays enable multiplexed targeting of endogenous and heterologous targets in E. coli
After exploring the properties of shortened CRISPR-Cas9 arrays, we wanted to apply these arrays to one important application of CRISPRi: multi-gene silencing. We chose the plasmid-encoded degfp and the chromosomally-encoded lacZ gene of E. coli, considering future possible applications for shortened Cas9 arrays, as target genes. Three different target sites per gene were tested (). As multiple guides against the same gene have been expressed to enhance CRISPRi-mediated silencing [Citation15], we tested the impact of targeting with arrays encoding one spacer (dual-spacer array) or two spacers (4-spacer array) per gene ().
Figure 7. Shortened CRISPR-Cas9 arrays can be employed for multiplexed targeting of plasmid and chromosomally encoded genes in E. coli.
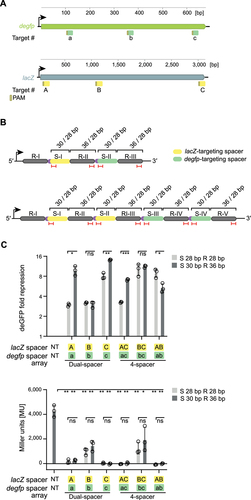
Starting with degfp, we found that either degfp-targeting spacer in a native dual-spacer array already yielded strong repression, with fold repression values of 10 (spacer a) and 14 (spacer c), respectively. In contrast, spacer b showed the lowest efficiency with a fold repression of 3 (, top graph). Interestingly, combining two degfp-targeting spacers did not increase the repression efficiencies achieved with spacer a and c alone. However, including spacer c in the 4-spacer array BC/bc rescued the previous low activity of the dual-spacer array with spacer b, indicating that the addition of a highly-active spacer can at least compensate for a poorly active spacer (, top graph). Trimming the best performing dual-spacer arrays encoding spacers a or c reduced repression activity (dual-spacer array A/a: Fold repression of 10 versus 3 for the native format and shortened format, respectively, p = 0.017, n = 3; dual-spacer array C/c: Fold repression of 14 versus 8 for the native format and shortened format, respectively, p = 0.017, n = 3). Amongst the 4-spacer arrays, only one array yielded lower repression efficiencies when trimmed (4-spacer array AC/ac: Fold repression of 7 versus 3 for the native format and shortened format, respectively, p = 0.0004, n = 3) while the two other 4-spacer arrays led to similar (4-spacer array BC/bc: Fold repression of 11 and 10 for the native format and shortened format, respectively, p = 0.47, n = 3) or even higher repression activities (4-spacer array AB/ab: Fold repression of 5 versus 9 for the native format and shortened format, respectively, p = 0.022, n = 3) when trimmed (, top graph).
lacZ expression was quantified with a β-galactosidase activity assay based on Miller Units (MU) (see methods). All tested arrays achieved a significant reduction in β-galactosidase expression in comparison to a non-targeting control (4,300 MU) ranging from no measurable expression (for the dual-spacer array C/c to 2,000 MU for the 4-spacer array BC/bc) (p = 0.0017–0.0402, n = 3) (, bottom graph). We detected only minor differences in repression efficiencies for dual- or 4-spacer arrays or between the native and shortened formats (, bottom graph). Overall, we demonstrated that shortened arrays can be applied for multi-gene targeting, including chromosomal targets, where the target location and array format both influenced the targeting activities.
Shortened multi-spacer arrays lend to short-read next-generation sequencing
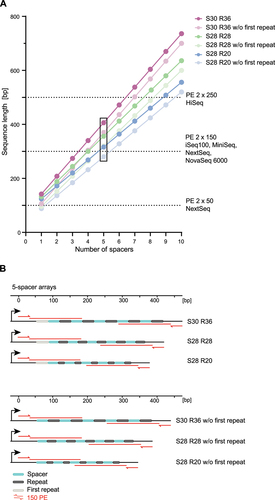
The ability to shorten CRISPR-Cas9 arrays could be important for multiplexing applications, particularly if short-read Next-Generation Sequencing (NGS) is used to read out the array sequence. This is the case, for example, for CRISPR screens in bacteria relying on multi-spacer array libraries [Citation16], such as for combinatorial screens aiming to identify synthetic lethal gene pairs or disentangle genetic interactions of virulence genes from pathogenic bacteria. Even though current long-read sequencing technologies [Citation44,Citation45] can generate reads up to 100 kb, these technologies often come with lower throughput and accuracy [Citation46]. As a result, short-read sequencing technologies are currently better positioned for analysing larger libraries derived from high-throughput CRISPR screens [Citation35,Citation47–50]. Shorter array formats could encode larger numbers of spacers that serve as unique barcodes and can easily be analysed. To prepare the extracted array sequences for NGS analysis, flow cell-binding sites, sequencing primer-binding sites and unique index sequences are added. As CRISPR-Cas9 arrays encode a repeat sequence at the 5’ and 3’ end of the array, the sequencing primer-binding site must be located outside of the array at a non-repetitive unique sequence.
For multi-spacer arrays, even small truncations within the spacer and/or repeat sequence can affect whether all spacers can be fully or even partially covered with standard 50-, 150- or 250-bp NGS reads (). A poignant example is a 5-spacer array in its native format with a length of 366 nts, which cannot be fully read by 150 PE particularly when accounting for the 20-nt primer sites at either end for PCR amplification and addition of NGS indices. Shortening the spacers and repeats to 28 nts condenses the array to 316 nts (356 nts with primer sites), and further shortening the repeats to 20 nts condenses the array to 276 nts (316 nts with primer sites). With 150 PE sequencing of this shortened 5-spacer array, all but 14 of the 28 nts of the middle spacer can be read, allowing all spacers to be reasonably identified (). Deleting the first repeat can further condense the array, allowing the middle spacer of arrays with 28-nt repeats to be partially sequenced and the middle spacer of arrays with 20-nt repeats to be entirely sequenced (). Thus, shortened arrays could allow the sequencing of additional spacers with existing NGS short-read technologies, potentially increasing the targeting activity of the array or the number of target genes as part of CRISPR screens.
Figure 8. The smaller DNA footprint of shortened multi-spacer arrays lends to downstream short-read NGS analyses. (A) Correlation of the Sequence length, including the array and additional 2 × 20bp flanking the array to incorporate unique primer-binding sites for library preparation, with the number of encoded spacers for arrays with native formats and shortened formats (S for spacer length, R for repeat length). Dashed lines indicate the cut-off size for sequences analysed by different Illumina sequencing technologies. The black box shows an example (5-spacer array shown in detail in (B)) where shorter array formats can be covered by 150-PE, but the original format exceeds the read length. For identification, sequencing the unique parts of the array is sufficient, meaning that repeats and even parts of the spacers do not necessarily have to be fully covered. This allows for some flexibility in the cut-off size for an array. (B) A representative example of a 5-spacer array with different spacer and repeat lengths and with (top) or without (bottom) the first repeat. Indicated by red lines are the sequence parts covered by 150 PE sequenci
Discussion
In summary, we found that CRISPR-Cas9 arrays can be shortened to different degrees without compromising functionality. For the spacer region, 26 nts to 28 nts was sufficient to maintain high repression in most single- and multi-spacer arrays, with some shortened arrays performing better than the native format (). For the repeat region, shortening the original 36 nts to 28 nts was well tolerated in the context of multi-spacer arrays, although the repeats could be further shortened based on 20-nt repeats in the context of single-spacer arrays (). We also showed that eliminating the first repeat can shorten CRISPR-Cas9 arrays, although we observed one instance in which removing this repeat negatively affected targeting activity (). As the repeat length, the spacer length, and the presence of the first repeat can be altered independently, there is ample flexibility to devise shortened arrays that maintain targeting activity while meeting the length requirements of different applications.
One surprising observation was the drop in the repression efficiency for arrays with 22-nt and 24-nt spacers (). The spacers in these arrays encode a 5’ assembly junction of 4 nts not designed to be complementary to the target, which shrinks the target-complementary length of each spacer to 18 and 20 nts, respectively. While shorter, these lengths should not affect the repression efficiency, as 20 nts is the standard guide length for SpCas9, and 18 nts should still promote strong DNA binding and transcriptional repression based on prior work [Citation39]. Therefore, we posit that other mechanisms such as secondary structure formation, crRNA stability and/or binding to Cas9 could contribute to the lower gene repression efficiencies. Even though CRISPRi offers some flexibility for spacer truncations at the 5’ end, applications that involve DNA cleavage by Cas9 would be more susceptible to spacer truncations below 20 nts, as this could disrupt the conformational change of the nuclease necessary to catalyse DNA cleavage [Citation34].
The observed impact of shortening CRISPR-Cas9 arrays highlights how the array sequence itself can impact multiplexed gene targeting with CRISPR technologies. Traditionally, guide sequences are selected based solely on target-specific features, such as GC content [Citation51], proximity to the transcriptional start site [Citation4,Citation35], the flanking PAM sequence [Citation52,Citation53] and folding of the guide sequence [Citation53]. While these factors remain crucial for guide design, our work shows that altering features such as spacer lengths and repeat lengths can either enhance or suppress targeting activity. Furthermore, prior work has shown how interactions between a spacer and a repeat as well as spacer placement impact targeting activity [Citation16,Citation41], underscoring the need to account for the array sequence when designing CRISPR arrays. The underlying mechanisms likely involve not only RNA secondary structure impacting crRNA processing and ribonucleoprotein complex formation but also co-transcriptional folding, RNA stability, and how partial processing impacts the activity of the bound Cas nuclease. Given the difficulty in predicting many of these contributions in in vivo settings, future work could focus on performing high-throughput screens combined with machine learning to develop CRISPR array design algorithms, similar to what has been done for CRISPR guide design [Citation51,Citation52]. Overall, we posit that the context of the spacers within an array () should be weighted just as heavily as guide/target selection when designing CRISPR arrays and CRISPR array libraries and should be a focus of future work.
One benefit of shortened CRISPR-Cas9 arrays is the ability to read out more spacers as part of short-read NGS, increasing the number of target sites for each array in a library. However, other considerations may limit this number to below the cut-off posed by NGS read length. One prominent example involves combinatorial screens using libraries of CRISPR arrays, which can quickly give rise to astronomical library sizes even for smaller arrays. For instance, to screen three-way genetic interactions between 100 target genes using one spacer per gene, the resulting library would comprise 161,700 unique 3-spacer arrays. While an actual library would be much larger (e.g. changing spacer positions, multiple spacers per gene, adding a set of non-targeting guides as controls), this minimal library already runs into current bottlenecks posed by plasmid cloning and transformation [Citation53–55]. Therefore, other factors come into play when considering the size limit of CRISPR-Cas9 arrays as well as the importance of condensing the arrays.
While we demonstrated the use of shortened CRISPR-Cas9 arrays for gene silencing in E. coli, these arrays have the potential to be applied to for multiplexed gene editing and regulation in a broad range of bacterial species. One reason is that bacteria commonly encode RNase III necessary for crRNA processing through the tracrRNA, even if the host ribonucleases that complete processing remain unknown [Citation33,Citation56]. Another reason is that Cas9 has already been widely applied in diverse bacteria for different forms of gene editing, gene repression and gene activation [Citation57], laying a foundation for the incorporation of shortened arrays into existing constructs and technologies. In total, our findings demonstrate the potential of shortening CRISPR-Cas9 arrays for efficient CRISPR-based multiplexing with a smaller DNA footprint in bacteria.
Methods
Strains, plasmids and growth conditions
All strains, plasmids, gBlocks and primers are listed in Supplementary Table S3-S6. Primers and gBlocks were synthesized by Integrated DNA Technologies (IDT), plasmids were propagated and maintained in E. coli TOP10, and experiments were carried out using the E. coli MG1655 derived strain CL471. E. coli cells were grown in Luria Bertani (LB) medium (5 g/l NaCl, 5 g/l yeast extract, 10 g/l tryptone) at 37°C with shaking at 250 rpm. To maintain any plasmids, the antibiotics ampicillin, chloramphenicol, and/or kanamycin were added at 100 µg/ml, 34 µg/ml, and 50 µg/ml, respectively.
Cloning of CRISPR arrays
A fragment encoding the tracrRNA was PCR-amplified from a gBlock and joined with the backbone fragment amplified from plasmid pCL239 by Gibson assembly using NEBuilder HiFi DNA Assembly Master Mix (New England Biolabs, cat. #E2621). The resulting plasmid (pSG001) encoded for the tracrRNA and a rfp drop-out region, both under the control of a J23119 promoter. The CRISPR arrays were cloned according to the CRATES method [Citation16,Citation36]. Shortly, forward and reverse oligonucleotides encoding one repeat, one spacer and a 4-nt junction were 5’ phosphorylated with polynucleotide kinase (New England Biolabs) and annealed to form a dsDNA fragment with 4-nt overhangs at the 5’ and 3’ terminal end. In a PCR tube, 2 µl of T4 ligation buffer (New England Biolabs), 1 µl of each inserts, 50 ng of backbone plasmid, 1 µl of T4 ligase (New England Biolabs) and 1 µl of BsmBI-HFv2 (New England Biolabs) were mixed with the appropriate amount of water to a final volume of 20 µl. The approximate ratio of backbone to insert is 1:20. A thermocycler was used to perform 25 cycles of digestion and ligation (42°C for 2 min, 16°C for 5 min) followed by a final digestion step (60°C for 10 min), and a heat inactivation step (80°C for 10 min). The ligation mix (2.5 µl) was directly used to transform chemically competent E. coli TOP10 cells (25 µl). After recovery in SOC medium for 1 h at 37°C with shaking at 250 rpm, cells were plated on LB agar plates with the appropriate antibiotics and incubated ov/n at 37°C. White colonies were verified by Sanger sequencing of the PCR-amplified CRISPR-array region.
Fluorescence assays in E. coli
To demonstrate the gene repression efficiencies of the CRISPR arrays to be tested, the E. coli MG1655 derived strain CL471 was transformed with three compatible plasmids encoding a dCas9, a gfp-targeting CRISPR array and a deGFP reporter gene. For normalization purposes a positive control strain harbouring a non-spacer array and a negative control strain lacking the degfp encoding reporter plasmid was included. Overnight cultures of cells harbouring the above mentioned plasmids were back-diluted to OD600 of ~ 0.01 in LB medium with ampicillin, chloramphenicol and/or kanamycin and were incubated with shaking at 250 rpm, at 37°C until reaching an OD600 of 1. Cultures were then diluted 1:25 in 1× phosphate-buffered saline (PBS) and analysed on an Accuri C6 flow cytometer with C6 sampler plate loader (Becton Dickinson) equipped with CFlow plate sampler, a 488-nm laser, and a 530 ± 15-nm bandpass filter. Briefly, forward scatter (cut-off of 15,500) and side scatter (cut-off of 600) were used to eliminate non-cellular events. The mean value within FL1-H of at least 25,000 events within a gate set for E. coli was used for data analysis. For each experiment, triplicate or quadruplicate cultures were measured, and their standard deviation was indicated as the error bar.
Determining GFP repression
GFP fold-repression was calculated using the mean GFP fluorescence values. The value for the no-spacer array was divided by that for the array to be tested, after subtracting the fluorescence value from cells lacking the GFP reporter plasmid from both.
β-galactosidase activity assay
To quantify the β-galactosidase activity, an overnight culture of the E. coli MG1655-derived strain CL471 co-transformed with a dCas9 and the lacZ targeting CRISPR array constructs was set up in 5 ml of LB medium supplemented with the appropriate antibiotics and incubated at 37°C, while shaking at 250 rpm. As a control, a non-targeting sgRNA was included.
For the β-galactosidase assay, the ov/n cultures were diluted to OD600 = 0.1 in fresh LB medium containing the appropriate antibiotics and 1 mM IPTG and incubated at 37°C, while shaking at 250 rpm until reaching OD600 of ~ 0.5. 2 ml of each culture was collected by centrifugation at 4°C, the supernatant was discarded and the pellet was stored on ice until all samples were collected. Next, the cell pellets were resuspended in cold Z-buffer (60 mM Na2HPO4·7 H2O, 38.7 mM NaH2PO4·H2O, 10 mM KCL, 1 mM MgSO4) with freshly added β-mercaptoethanol (34.5 mM), so that the cell suspension was 1 OD600/ml. 150 µl of chloroform and 100 µl of 0.1% SDS was added to each tube, the tube was vortexed for 15 s and put on ice. Dilutions (1:2, 1:10) were prepared from each sample by mixing the specific volumes of the sample (upper phase) with Z-buffer. 200 µl of each sample and dilution was loaded onto a 96-well plate. The spectrophotometer was set to measure at OD420 and OD600 every 2 min for the duration of 45 min and pre-heated to 28°C. Lastly, 50 µl ortho-nitrophenyl-β-galactoside (ONPG) (4 mg/ml in Z-buffer without β-mercaptoethanol) was added to each well using a multi-channel pipette and the measurement was started immediately. The assay was performed in triplicates, using single colonies to start the ov/n cultures. The β-galactosidase activity was calculated according to the Miller assay protocol [Citation58]. The original equation was modified to incorporate the slope (ΔABS420/Δt[min]) obtained from linear regression analyses performed with the ABS420 values from the first 16 min of the experiment (linear range), resulting in 1000 × ((ΔABS420/Δt[min])/(V [ml] x ABS600)).
Phenol-chloroform RNA extraction
Overnight cultures of cells harbouring the dCas9 and CRISPR array plasmids were back-diluted to OD600 ~ 0.01 in LB medium with appropriate antibiotics and incubated with shaking at 250 rpm, at 37°C until reaching an OD600 of ~ 0.7. The cell culture volume corresponding to an overall OD of 4 was mixed with 20% cold EtOH/phenol stop solution (95% of 100% w/v EtOH with 5% phenol (Roti-Aqua phenol #A980.1)), inverted once, snap frozen in liquid nitrogen and stored at −80°C. For the RNA extraction, the cell suspension was thawed on ice and centrifuged at 8,000 rpm for 2 min at 4°C. The cell pellet was resuspended in 600 µl of 0.5 mg/ml lysozyme TE pH 8.0 solution, transferred to an RNase-free 2-ml Eppendorf tube and mixed with 60 µl of 10% w/v SDS. The tube was placed into a water bath set to 64°C for 2 min until the sample turned clear and mixed by inversion with 66 µl of 3 M NaOAc, pH 5.2. For the hot phenol extraction, ~720 µl phenol (Roti-Aqua phenol, #A980.3) was added and the tube was incubated in the water bath at 64°C for 6 min with shortly vortexing the tube every 30 s. The sample was then chilled on ice for 1 min and centrifuged at 11,000×g for 15 min at 4°C. For the chloroform extraction, the upper aqueous layer was transferred into a 2 ml Phase Lock Gel (PLG) tube (VWR International, #733–2478) and mixed by inversion with ~750 µl of chloroform (Roth, #Y015.2). The tube was centrifuged at maximum speed for 10 min at 4°C and the aqueous layer was transferred into a fresh RNase-free Eppendorf tube to be further purified by ethanol precipitation.
Northern blotting analysis
The E. coli MG1655 derived strain CL471 was transformed with two compatible plasmids: the dCas9 plasmid and the single-spacer array #3 plasmid with the respective modifications on the upstream repeat. Overnight cultures of cells harbouring the two plasmids were back-diluted to an OD600 of ~ 0.01 in LB medium with ampicillin and chloramphenicol and shaken at 250 rpm at 37°C until reaching an OD600 of ~ 0.7. Cells were harvested by centrifugation, and total RNA was extracted using the hot phenol-chloroform RNA extraction method. For Northern blot analysis, 10 μg of each RNA sample was resolved on an 8% polyacrylamide gel containing 7 M urea at 300 V for 2 h and 25 min using a gel transfer system (Doppel-Gelsystem Twin L, PerfectBlue). Using an Electroblotter with an applied voltage of 50 V for 1 h at 4°C (Tank-Elektroblotter Web M, PerfectBlue), the RNA was transferred onto Hybond-XL membranes (GE Healthcare, #RPN203S), crosslinked with UV-light for a total of 0.12 Joules (UV-lamp T8C; 254 nm, 8 W) and hybridized overnight in 15 ml of Roti-Hybri-Quick buffer at 42°C with 5 µL γ-32P-ATP end-labelled oligodeoxyribonucleotides (5 pmol/µl) (Table S5). The labelled RNA was visualized with a Phosphorimager (Typhoon FLA 7000, GE Healthcare). Gel images from a replicate experiment are shown in Supplementary Fig. S2.
RNA-folding predictions
To identify RNA sequence regions or specific nucleobases potentially altering the secondary structure of the CRISPR arrays to be tested, the NUPACK software was utilized (http://www.nupack.org) [Citation43].
Statistical analyses
The statistical analyses were performed using a Welch’s t-test assuming unequal variances for the comparison of two groups. P-value (P) > 0.05 is shown as ns, P < 0.05 is shown as *, P < 0.01 is shown as **, P < 0.001 is shown as ***. P < 0.0001 is shown as ****.
Author contributions
S.G. and C.L.B. conceived this study and designed the experiments; S.G. and C.L. designed and cloned the CRISPR arrays; S.G. performed the flow cytometry experiments; T.A. performed the Northern blotting analysis; and S.G. and C.L.B. analysed the data. S.G. and C.L.B. wrote the manuscript, which was read and approved by all authors.
Supplemental Material
Download PDF (21.2 MB)Acknowledgments
We thank Christophe Toussaint for assistance with cloning. This work was supported by a European Research Council Consolidator Award (865973 to C.L.B.).
Disclosure statement
C.L.B. is a co-founder and member of the Scientific Advisory Board member for Locus Biosciences and is a member of the Scientific Advisory Board for Benson Hill. The other authors declare no competing interests.
Data availability statement
Relevant constructs will be made available through Addgene. All original data are available upon reasonable request.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/15476286.2023.2247247
Additional information
Funding
References
- Makarova KS, Wolf YI, Iranzo J, et al. Evolutionary classification of CRISPR-Cas systems: a burst of class 2 and derived variants. Nat Rev Microbiol. 2020;18:67–83. doi: 10.1038/s41579-019-0299-x
- Leenay, Deciphering RT, Beisel CL. Communicating, and engineering the CRISPR PAM. J Mol Biol. 2017;429:177–191. doi: 10.1016/j.jmb.2016.11.024
- Bikard D, Jiang W, Samai P, et al. Programmable repression and activation of bacterial gene expression using an engineered CRISPR-Cas system. Nucleic Acids Res. 2013;41(15):7429–7437. doi: 10.1093/nar/gkt520
- Qi LS, Larson MH, Gilbert LA, et al. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell. 2021;184(3):844. doi: 10.1016/j.cell.2021.01.019
- Gaudelli NM, Komor AC, Rees HA, et al. Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature. 2017;551(7681):464–471. doi: 10.1038/nature24644
- Grünewald J, Zhou R, Lareau CA, et al. A dual-deaminase CRISPR base editor enables concurrent adenine and cytosine editing. Nat Biotechnol. 2020;38(7):861–864. doi: 10.1038/s41587-020-0535-y
- Komor AC, Kim YB, Packer MS, et al. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature. 2016;533(7603):420–424. doi: 10.1038/nature17946
- Zhang X, Zhu B, Chen L, et al. Dual base editor catalyzes both cytosine and adenine base conversions in human cells. Nat Biotechnol. 2020;38(7):856–860. doi: 10.1038/s41587-020-0527-y
- Klompe SE, Vo PLH, Halpin-Healy TS, et al. Transposon-encoded CRISPR–Cas systems direct RNA-guided DNA integration. Nature. 2019;571(7764):219–225. doi: 10.1038/s41586-019-1323-z
- Strecker J, Ladha A, Gardner Z, et al. RNA-guided DNA insertion with CRISPR-associated transposases. Science. 2019;365(6448):48–53. doi: 10.1126/science.aax9181
- PLH V, Ronda C, Klompe SE, et al. CRISPR RNA-guided integrases for high-efficiency, multiplexed bacterial genome engineering. Nat Biotechnol. 2021;39:480–489. doi: 10.1038/s41587-020-00745-y
- Anzalone AV, Randolph PB, Davis JR, et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature. 2019;576(7785):149–157. doi: 10.1038/s41586-019-1711-4
- Tong Y, Jørgensen TS, Whitford CM, et al. A versatile genetic engineering toolkit for E. coli based on CRISPR-prime editing. Nat Commun. 2021;12(1):5206. doi: 10.1038/s41467-021-25541-3
- Feng X, Zhao D, Zhang X, et al. CRISPR/Cas9 Assisted Multiplex genome editing technique in escherichia coli. Biotechnol J. 2018;13(9):e1700604. doi: 10.1002/biot.201700604
- Campa CC, Weisbach NR, Santinha AJ, et al. Multiplexed genome engineering by Cas12a and CRISPR arrays encoded on single transcripts. Nat Methods. 2019;16(9):887–893. doi: 10.1038/s41592-019-0508-6
- Liao C, Ttofali F, Slotkowski RA, et al. Modular one-pot assembly of CRISPR arrays enables library generation and reveals factors influencing crRNA biogenesis. Nat Commun. 2019;10(1):1–14. doi: 10.1038/s41467-019-10747-3
- Jinek M, Chylinski K, Fonfara I, et al. A Programmable dual-RNA–Guided DNA endonuclease in adaptive bacterial immunity. Science2012. 2012;337(6096):816–821. doi: 10.1126/science.1225829 Available from.
- Kabadi AM, Ousterout DG, Hilton IB, et al. Multiplex CRISPR/Cas9-based genome engineering from a single lentiviral vector. Nucleic Acids Res. 2014;42(19):e147. doi: 10.1093/nar/gku749
- Reis AC, Halper SM, Vezeau GE, et al. Simultaneous repression of multiple bacterial genes using nonrepetitive extra-long sgRNA arrays. Nat Biotechnol. 2019;37(11):1294–1301. doi: 10.1038/s41587-019-0286-9
- Ferreira R, Skrekas C, Nielsen J, et al. Multiplexed CRISPR/Cas9 genome editing and gene regulation using Csy4 in Saccharomyces cerevisiae. ACS Synth Biol. 2018;7:10–15. doi: 10.1021/acssynbio.7b00259
- He Y, Zhang T, Yang N, et al. Self-cleaving ribozymes enable the production of guide RNAs from unlimited choices of promoters for CRISPR/Cas9 mediated genome editing. J Genet Genomics. 2017;44(9):469–472. doi: 10.1016/j.jgg.2017.08.003
- Kurata M, Wolf NK, Lahr WS, et al. Highly multiplexed genome engineering using CRISPR/Cas9 gRNA arrays. PLoS One. 2018;13(9):e0198714. doi: 10.1371/journal.pone.0198714
- Zhang Y, Wang J, Wang Z, et al. A gRNA-tRNA array for CRISPR-Cas9 based rapid multiplexed genome editing in Saccharomyces cerevisiae. Nat Commun. 2019;10(1):1053. doi: 10.1038/s41467-019-09005-3
- Zetsche B, Heidenreich M, Mohanraju P, et al. Multiplex gene editing by CRISPR-Cpf1 using a single crRNA array. Nat Biotechnol. 2017;35:31–34. doi: 10.1038/nbt.3737
- McGinn J, Marraffini LA. Molecular mechanisms of CRISPR–Cas spacer acquisition. Nat Rev Microbiol. 2018;17:7–12. doi: 10.1038/s41579-018-0071-7
- Swiat MA, Dashko S, den Ridder M, et al. FnCpf1: a novel and efficient genome editing tool for Saccharomyces cerevisiae. Nucleic Acids Res. 2017;45(21):12585–12598. doi: 10.1093/nar/gkx1007
- Ciurkot K, Gorochowski TE, Roubos JA, et al. Efficient multiplexed gene regulation in Saccharomyces cerevisiae using dCas12a. Nucleic Acids Res. 2021;49(13):7775–7790. doi: 10.1093/nar/gkab529
- Zhang Y, Ren Q, Tang X, et al. Expanding the scope of plant genome engineering with Cas12a orthologs and highly multiplexable editing systems. Nat Commun. 2021;12(1):1944. doi: 10.1038/s41467-021-22330-w
- Ao X, Yao Y, Li T, et al. A Multiplex genome editing method for Escherichia coli based on CRISPR-Cas12a. Front Microbiol. 2018 [cited 2022 Feb 2]. Internet. 10.3389/fmicb.2018.02307
- Port F, Starostecka M, Boutros M. Multiplexed conditional genome editing with Cas12a in Drosophila. Proc Natl Acad Sci U S A. 2020;117(37):22890–22899. doi: 10.1073/pnas.2004655117
- Bryson JW, Auxillos JY, Rosser SJ. Multiplexed activation in mammalian cells using a split-intein CRISPR/Cas12a based synthetic transcription factor. Nucleic Acids Res. 2021;50:549–560. doi: 10.1093/nar/gkab1191
- Charpentier E, Richter H, van der Oost J, et al. Biogenesis pathways of RNA guides in archaeal and bacterial CRISPR-Cas adaptive immunity. FEMS Microbiol Rev. 2015;39(3):428–441. doi: 10.1093/femsre/fuv023
- Deltcheva E, Chylinski K, Sharma CM, et al. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature. 2011;471(7340):602–607. doi: 10.1038/nature09886
- Sternberg SH, LaFrance B, Kaplan M, et al. Conformational control of DNA target cleavage by CRISPR-Cas9. Nature. 2015;527:110–113. doi: 10.1038/nature15544
- Cui L, Vigouroux A, Rousset F, et al. A CRISPRi screen in E. coli reveals sequence-specific toxicity of dCas9. Nat Commun. 2018;9(1):1–10. doi: 10.1038/s41467-018-04209-5
- Liao C, Slotkowski RA, Beisel CL. CRATES: a one-step assembly method for class 2 CRISPR arrays. Methods Enzymol. 2019;629. Available from: https://pubmed.ncbi.nlm.nih.gov/31727255/
- Shin J, Noireaux V. Efficient cell-free expression with the endogenous E. Coli RNA polymerase and sigma factor 70. J Biol Eng. 2010;4(1):8. doi: 10.1186/1754-1611-4-8
- Jiang W, Bikard D, Cox D, et al. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat Biotechnol. 2013;31:233–239. doi: 10.1038/nbt.2508 Available from.
- Vigouroux A, Oldewurtel E, Cui L, et al. Tuning dCas9’s ability to block transcription enables robust, noiseless knockdown of bacterial genes. Mol Syst Biol. 2018;14(3):e7899. doi: 10.15252/msb.20177899
- Magnusson JP, Rios AR, Wu L, et al. Enhanced Cas12a multi-gene regulation using a CRISPR array separator. Elife 2021;10. Available from 10.7554/eLife.66406.
- Creutzburg SCA, Wu WY, Mohanraju P, et al. Good guide, bad guide: spacer sequence-dependent cleavage efficiency of Cas12a. Nucleic Acids Res. 2020;48(6):3228–3243. doi: 10.1093/nar/gkz1240
- Reimann V, Alkhnbashi OS, Saunders SJ, et al. Structural constraints and enzymatic promiscuity in the Cas6-dependent generation of crRnas. Nucleic Acids Res. 2017;45(2):915–925. doi: 10.1093/nar/gkw786
- Zadeh JN, Steenberg CD, Bois JS, et al. NUPACK: Analysis and design of nucleic acid systems. J Comput Chem. 2011;32:170–173. doi: 10.1002/jcc.21596
- Rhoads A, Au KF. PacBio sequencing and its applications. Int J Genomics Proteomics. 2015;13(5):278–289. doi: 10.1016/j.gpb.2015.08.002
- Lu H, Giordano F, Ning Z. Oxford nanopore MinION sequencing and genome assembly. Int J Genomics Proteomics. 2016;14(5):265–279. doi: 10.1016/j.gpb.2016.05.004
- Pollard MO, Gurdasani D, Mentzer AJ, et al. Long reads: their purpose and place. Hum Mol Genet. 2018;27(R2):R234–41. doi: 10.1093/hmg/ddy177
- Wang T, Guan C, Guo J, et al. Pooled CRISPR interference screening enables genome-scale functional genomics study in bacteria with superior performance. Nat Commun. 2018;9(1):2475. doi: 10.1038/s41467-018-04899-x
- Rousset F, Cui L, Siouve E, et al. Genome-wide CRISPR-dCas9 screens in E. coli identify essential genes and phage host factors. PLoS Genet. 2018;14(11):e1007749. doi: 10.1371/journal.pgen.1007749
- de Bakker V, Liu X, Bravo AM, et al. Crispri-seq for genome-wide fitness quantification in bacteria. Nat Protoc. 2022;17(2):252–281. doi: 10.1038/s41596-021-00639-6
- Liu Y, Wang R, Liu J, et al. Base editor enables rational genome-scale functional screening for enhanced industrial phenotypes in Corynebacterium glutamicum. Sci Adv. 2022;8(35):eabq2157. doi: 10.1126/sciadv.abq2157
- Konstantakos V, Nentidis A, Krithara A, et al. CRISPR-Cas9 gRNA efficiency prediction: an overview of predictive tools and the role of deep learning. Nucleic Acids Res. 2022;50:3616–3637. doi: 10.1093/nar/gkac192
- Chuai G, Ma H, Yan J, et al. DeepCRISPR: optimized CRISPR guide RNA design by deep learning. Genome Biol. 2018;19(1):80. doi: 10.1186/s13059-018-1459-4
- Diehl V, Wegner M, Grumati P, et al. Minimized combinatorial CRISPR screens identify genetic interactions in autophagy. Nucleic Acids Res. 2021;49(10):5684–5704. doi: 10.1093/nar/gkab309
- Joung J, Konermann S, Gootenberg JS, et al. Genome-scale CRISPR-Cas9 knockout and transcriptional activation screening. Nat Protoc. 2017;12(4):828–863. doi: 10.1038/nprot.2017.016
- Bock C, Datlinger P, Chardon F, et al. High-content CRISPR screening. Nat Rev Methods Primers. 2022;2:1–23. doi: 10.1038/s43586-021-00093-4
- Liao C, Beisel CL. The tracrRNA in CRISPR Biology and technologies. Ann Rev Genet. 2021;55(1):161–181. doi: 10.1146/annurev-genet-071719-022559
- Volke DC, Orsi E, Nikel PI. Emergent CRISPR-Cas-based technologies for engineering non-model bacteria. Curr Opin Microbiol. 2023;75:102353. doi: 10.1016/j.mib.2023.102353
- Miller JH, editor. Assay of β-galactosidase. In: Experiments in Molecular Genetics. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1972. p. 352–355.