
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
In Streptomyces species, the cell cycle involves a switch from an early and vegetative state to a later phase where secondary products including antibiotics are synthesized, aerial hyphae form and sporulation occurs. AdpA, which has two domains, activates the expression of numerous genes involved in the switch from the vegetative growth phase. The adpA mRNA of many Streptomyces species has a UUA codon in a linker region between 5’ sequence encoding one domain and 3’ sequence encoding its other and C-terminal domain. UUA codons are exceptionally rare in Streptomyces, and its functional cognate tRNA is not present in a fully modified and acylated form, in the early and vegetative phase of the cell cycle though it is aminoacylated later. Here, we report candidate recoding signals that may influence decoding of the linker region UUA. Additionally, a short ORF 5’ of the main ORF has been identified with a GUG at, or near, its 5’ end and an in-frame UUA near its 3’ end. The latter is commonly 5 nucleotides 5’ of the main ORF start. Ribosome profiling data show translation of that 5’ region. Ten years ago, UUA-mediated translational bypassing was proposed as a sensor by a Streptomyces phage of its host’s cell cycle stage and an effector of its lytic/lysogeny switch. We provide the first experimental evidence supportive of this proposal.
1 Introduction
The filamentous gram-positive bacteria Streptomyces are part of the phylum Actinobacteria, one of the most deeply branching clades on the phylogenetic tree [Citation1]. Streptomyces are important in part because of the array of secondary metabolites including antibiotics, they naturally produce. Its study has also opened a new appreciation of complex multifaceted life cycles in bacteria. Streptomyces possess a distinct genetic codon usage bias in part due to their high genomic GC content of [Citation2,Citation3]. Even accounting for this high GC content, as initially highlighted by Chater, utilization of the UUA codon is unusually low, with only
of coding sequences containing a UUA codon [Citation4]. The few UUA codons present show extreme bias for being close to the start of coding sequences [Citation5], where the energy wastage of ribosomes encountering them in the absence of cognate aminoacyl-tRNA would be minimal. Further, most of those present derive from species-specific horizontal gene transfer [Citation6]. Many UUA codons are conserved and the products from coding sequences in which they occur are involved in processes such as sporulation and antibiotic production, both features of late growth-phase development in S. coelicolor [Citation7]. Intriguingly however, work by Ostash and others has highlighted an in-frame UUA at an internal position of sequence encoding AdpA, which regulates the expression of an enormous number of genes required for the late growth phase. The adpA mRNA of many Streptomyces species has a UUA between 5’ sequence encoding a GATase-1 domain and 3’ sequence encoding an AraC-like family transcriptional regulator (AraC) adpA genes [Citation8,Citation9].
To our knowledge, the function that has driven selection for UUA being positioned between sequence encoding the two domains has not been discussed. In the absence of functional charged cognate tRNA, translation of the upstream sequence would likely lead to proteolysis of a substantial proportion of the upstream-encoded product unless ‘special’ contextual features are involved. When a bacterial ribosome encounters a codon whose cognate aminoacyl-tRNA is absent or sparse, it stalls allowing a following ribosome to collide with it. Such collided ribosomes are recognized by the endonuclease SmrB, which cleaves upstream of stalled ribosomes to generate a truncated mRNA. tmRNA then acts to rescue upstream-stalled ribosomes and often mRNA degradation and proteolysis of the incomplete protein [Citation10].
However, absence or greatly reduced amounts of a functional form of the tRNA or release factor needed to decode the codon in the ribosomal A-site can lead to a non-standard decoding event. One type involves peptidyl-tRNA anticodon dissociating from its codon and re-pairing to mRNA at an overlapping codon mediating a shift in reading frame. Thousands of such frameshift events occur with very high efficiency in gene expression in Euplotes ciliates, and the number is predicted to increase over evolutionary time to between 17 and 71 thousand depending on the species [Citation11]. Aside from that exceptional situation, except where programmed events occur (see below), frameshifting efficiency is commonly dramatically lower and in some cases occurs by the alternative of an incoming aminoacyl-tRNA pairing with an out-of-frame overlapping codon in the A-site [Citation12,Citation13]. For pragmatic reasons, the word ‘frameshifting’ is generally reserved for shifts of frame to an overlapping codon. When the re-pairing occurs to a non-overlapping codon, the descriptor ‘bypassing’ is commonly used. When a block of non-coding nucleotides is avoided by this process, a reading frame shift may or may not occur depending on the frame of the codon to which the tRNA re-pairs to mRNA. Contextual signals often known as recoding signals can greatly stimulate such events and can be selected to generate functionally useful products or for regulatory purposes. Such ‘translational recoding’ is in competition with standard decoding [Citation14–18]. In the absence of functional UUA-decoding tRNA, might there be some frameshifting at the UUA between sequence encoding the two domains of AdpA which quickly leads to termination in a new frame and so release of functional upstream encoded product? Though the efficiency of frameshifting may be optimal at low levels and apparently not involve recoding signals, for instance the +1 frameshifting important for Influenza virus expression [Citation19], the great majority of productive occurrences do involve recoding signals, i.e. they are programmed. In the absence, or highly limiting amounts, of functional UUA-decoding tRNA, the presence of stimulatory signals for hypothetical frameshifting at the adpA UAA could influence the proportion of GATase 1 that is immediately degraded or released in a functional state following frameshifting mediated termination (or out-of-frame bypassing). An alternative possibility for liberating functional upstream-encoded product, GATase 1, is that when a UUA codon is in the ribosomal A-site and functional cognate tRNA is unavailable, peptidyl-tRNA drops off the ribosome and the ester linkage of the peptide to the tRNA is cleaved by a peptidyl-tRNA hydrolase (pth) [Citation20,Citation21]. One of the recoding signals for the 30% to 50% efficient translational bypassing of 50 nucleotides required for synthesis of a phage T4-encoded topoisomerase subunit is relevant to the extent of drop-off in that system [Citation22]. Without preconceptions about the potential recoding mechanism involved, we sought to find putative contextual features linked to the UUA that occurs in the short ‘linker’ between the parts of the ORF encoding the two separate domains of the product AdpA protein. Before embarking on that, we reassessed chromosomal UUAs using a bioinformatics software we previously used to discover frameshifting in the decoding of bacterial magnesium chelatase mRNAs [Citation23]. The aim is to enable future work directed to discerning the function of such UUAs and the mechanism(s) involved, to use cassettes including any candidate recoding signals identified.
Support for the merits of the task comes from the properties of mutants of the gene specifying the UUA-decoding tRNA and of the genes encoding some of its modifying enzymes. Instead of fuzzy, deeply pigmented WT Streptomyces colonies, a relevant mutant, S48, described in pioneering work with S. coelicolor in 1967 by Hopwood was unable to erect aerial, spore-bearing, hyphae and was smooth [Citation24]. The ‘bald’ phenotype later prompted the corresponding gene being termed bldA, with several additional bld genes now known to be also important for the transition from vegetative to reproductive growth [Citation25]. AdpA binds to the promoter of its own gene and that of bldA [Citation26], providing reciprocity of functional interactions since the bldA product influences adpA expression. The bldA product is relevant to the general restriction of standard decoding of UUA during early growth phase [Citation7,Citation27,Citation28]. While the structural gene specifying the tRNA cognate for UUA is transcribed even early in the vegetative state, expression at a later growth stage of the modification enzymes MiaA and MiaB is important for classically functional tRNA being formed [Citation29–32]. MiaA and MiaB catalyse reactions in the conversion of the residue at anticodon adjacent position 37 of UUA-decoding tRNA, and certain other tRNAs, to hypermodified 2-methylthio-N6-(cis-hydroxyisopentenyl) adenosine (ms2io6A). Variant forms of the modification also occur, and much is unknown about the timing of their synthesis and function as well as that of potentially relevant modifications at other tRNA positions [Citation33].
The affinity of EF-Tu for deacylated tRNAs tested is ca. 1000-fold less than for acylated tRNA [Citation34] and where the measured, ribosomal A-site dwell time for uncharged tRNA is less than for charged tRNA [Citation35]. Nevertheless, ribosomal A-sites for which charged cognate tRNA is sparse or absent can be occupied by uncharged cognate tRNA resulting in relA-mediated synthesis of the alarmone (p)ppGpp and the stringent response [Citation36,Citation37]. While certain other physiological conditions can induce ppGpp synthesis [Citation37], it is a paradox that the known broad-ranging effects of (p)ppGpp on Streptomyces are at the later growth stages [Citation38,Citation39] (and also in Caulobacter [Citation40]). The presence during the vegetative state of preformed UUA-cognate tRNA only needing the final base modification to be a substrate for aminoacylation is expected to facilitate a quick transition to the formation of functional acylated tRNA. However, it may be pertinent for the discussion below to keep in mind the curious presence of incompletely modified tRNA throughout the vegetative phase. Further, guanosine tetraphosphate ppGpp influences the lytic/lysogeny decision of phage Mu [Citation41] and relA mutants affect ribosomal frameshifting [Citation42] and probably some other forms of recoding.
Streptomyces minimal use of UUA, which may have facilitated potential regulatory use of some remaining occurrences, could have been aided by the selection for enhanced survival against phage with UUA in essential coding sequences. To counter this unusual codon usage bias, Streptomyces phage genomes usually entirely lack UUA codons [Citation43,Citation44]. However, phage Hau3 mRNA has two UUA codons. It has been hypothesized that decoding of these UUA codons, which occur in its terminase mRNA, serves as a sensor of its host’s life cycle state [Citation45]. In the early and vegetative cell growth state, the phage enters its lytic cycle for which terminase is required [Citation46]. In the late cell growth phase, the phage instead enters the lysogenic state for which terminase is not required. Sequestration into spores as a prophage during an unfavourable time for phage reproduction would enable it to be ready to take advantage of a future return of its host cell to favourable growth conditions. There are two separate blocks of frame-disrupting 10 nucleotide inserts with UUA at their 5’ ends in Hau3ʹs terminase coding sequence. Sequences flanking these 10 nt blocks match sequences adjacent to each other in counterpart phage sequences that do not have the inserts. In the late phase of cell growth functional cognate UUA-decoding tRNA results in standard translation, a stop codon being encountered shortly after the UUA and so no terminase is synthesized. Nevertheless, it has been hypothesized that in the vegetative phase of cell growth when UUA enters the ribosomal A-site in the absence of functional cognate UUA-decoding tRNA, bypassing of the blocks of 10 nt inserts occurs allowing the synthesis of functional terminase [Citation45]. Low-level bypassing can occur when slow-to-decode or unassigned codons are in the ribosomal A-site [Citation47–52]. In contrast, candidate recoding signals for enabling efficient Hau3 bypassing were identified in 2013, when that bypassing was hypothesized [Citation45]. However, the Hau3 bypassing proposal was not in the title or abstract of the publication that contained it, and apart from being considered in one review [Citation12], has largely escaped attention for the past 10 years despite the extensive departure from standard decoding it would involve.
To complement our chromosomal UUA analysis, we have experimentally investigated the proposed Hau3 bypassing. Bypassing is known to be required in the decoding of E. coli phage T4 [Citation22,Citation53,Citation54]. Fifty nucleotides are bypassed with 40% efficiency with bypassing of the 3’ part of the coding gap involving the scanning of each overlapping triplet in a process that uses
2 GTP per nucleotide bypassed [Citation55,Citation56]. Bypassing also occurs in related phages, one of which features an unusually rare codon in the ribosomal A-site at the initiation of bypassing [Citation57] as well as extensively in the mitochondria of the yeast Magnusiomyces capitatus, where one coding sequence has as many as 12 disruptive inserts bypassed [Citation58–60].
2 Materials and methods
2.1 Development of a dual-tagged reporter for the detection of transframe events in-vivo in Streptomyces
Dual-tag reporter systems are widely used to analyse translational-recoding events [Citation61–64]. The dual-tagged reporter system used for expression in Streptomyces has no UUA codons in its tag-encoding sequences and has a GC content above 65 mol%. The tag, eGFP (27 kDa), is encoded 5’ of the multiple cloning site into which the recoding test cassette sequence is inserted so the 3’ encoded tag SNAP/ hemagglutinin (HA) (19 kDa) is in the predicted new frame translated by ribosomes that performed recoding. After induction of plasmid expression, eGFP fluorescence levels, as measured either by a blue LED light transilluminator (470 nm) or fluorescence microscope [Citation65], reflect ribosome loading prior to the recoding site. HA tag Immunoreactive sensitivity was used to monitor translation in the post-recode-site new frame. A Human Rhino Virus 3C protease site (LEVLFQ↓GP) encoding sequence just 5’ of the site into which the recoding cassettes were cloned permitted removal of the GFP tag from the rest of the protein product. As a control, an empty dual-tagged vector (pGS1) with no test sequence inserted was first analysed, to ensure full-length expression of both tags. Synthesis of GFP was verified in both vegetative mycelium on solid agar under a blue LED light transilluminator and in liquid media under a fluorescent microscope. Full-length expression of the GFP (top) and the SNAP/HA tag (middle) was confirmed by western blot analysis. A composite signal was detected from the overlay of both the GFP (green 800 nm) and HA (red 700 nm) signals. Finally, protease cleavage at the HRV 3C was confirmed by the separation of the GFP tag from the rest of the coding sequence.
2.2 Production of custom antibodies
Custom antibodies were generated against a zero-frame peptide (number 12 – binds a 0 frame peptide in g6) in the g6 sequence and against two other peptides in the g7 sequence (2, 5 – both bind different g7 + 1 frame peptides), in the +1 frame to g6. The specificity of each of the antibodies was verified against an in-frame terminase control.
2.3 Mass spectrometry analysis
To ensure the detection of peptides covering the predicted bypass site following trypsin cleavage, the lysine codon (AAG) 3’ of the landing site was mutated to an asparagine codon (AAC) (pJg6-7_4). To separate the recode and non-recode products from one another, a His tag purification was performed and elutions were analysed via SDS-PAGE, with a full-length recode product of the correct size being detected. Imidazole fractions with a high quantity of recode protein were then utilized for a second round of purification with glutathione sepharose for clean-up purposes and to concentrate the protein that was then used for MS/MS analysis and peptide mapping.
2.4 Data resources
Three core resources were used: GenBank NCBI [Citation66], the Actinobacteriophage Database at PhagesDB [Citation67] and Pfam DB (release 35.0) [Citation68]. Complete genomes, proteomes, coding sequences and annotations were obtained from GenBank. Usually, gene names were absent from the GenBank annotations that contained general description for the corresponding proteins only. For example, products of the adpA genes were annotated as ‘helix-turn-helix domain-containing protein’. However, in our phylogenetic analysis, high similarity between protein sequences, their lengths and domain structures, as well as conservation of the UUA codons, strongly indicated that these were actually adpA genes encoding ‘AraC-like family transcriptional regulators’ (AFTRs). Thus, we will use the adpA notation to refer to these genes throughout the text. The PhagesDB was used to clarify the phages that infect Streptomyces hosts (bacteria). We collected data for 213 Streptomyces bacteria and 258 Streptomyces phages. Annotations for Pfam domains (DJ-1_PfpI, HTH_18, and HTH_AraC) of the target proteins were retrieved from Pfam DB.
2.5 Identification of terminase genes using tBLASTn
The analysis of the ter genes from Hau3 demonstrated that UUA codon may trigger translational bypassing and generation of a full-length protein from a frameshifted gene. Thus, we considered the possibility that orthologous ter genes may also contain frameshifts. On the other hand, genes with frameshifts are frequently mis-annotated. Taking this into account, we utilized the tBLASTn-based approach [Citation23] to identify genes with optional frameshifts near the UUA codons.
The stand-alone tBLASTn [Citation69] tool was used to identify the genomes containing the three parts g6, g7 and g8 of the terminase gene from the Hau3 phage. The following amino acid sequences g6, g7 and g8 of the terminase protein from the
Hau3 phage were used as queries:
>g6
VSPISTLTTEEIEALAPTFLGPTWAKDSLGQWKLPQRTLGWQIAGWCAEYLRAEDGGPWK
FTREQLRFVLWWYAVDENGRFVYRKGVLQRLKGWG
>g7
KDPLLAVVCMVEFVGPSRFSHFDEAGDPVGIPHPQAWVQVAAVSRDQTRNTMTLFPSLMS
DRLIETYGIKAGAELIRANGGRQRLEAVTSSYRALEGARS
>g8
TFVVLNETHHWVTGNNGDKMYSTIDGNATKKDSRYLAITNAYLPGEDSVAERMREAYDKI
REGKAVDIGFMYDSIEAHPSTPLTVEAIRIVLPKIRGDASWLNVETIIQSIMDATIAPSR
SRRMWFNQIVAEEDALYGPEQWADILHEGATLQPGDEIVMGFDGGKSDDATALVAIRVRD
MCAFVLGLWEKPDGPKGDGWSVPRAEVDSAVHDAFRVYDVKAFYADVALWESYISEWDEL
YGEGLSVKSPVGKDRIGWDMRSSLKLSTMAHERLMRSIFDKKLRYDGDLTLRRHALNARR
RSNNYGISFGKESRESPRKVDAYAALMLAHEALYDLRARGKKVRARSGRGYFM
To allow the identification of the other terminase genes in the genomes, three separate tBASTn searches (using launch options: E-value threshold , number of aligned sequences to keep
, number of threads (CPUs) to use in blast search
, genetic code to translate subject sequences
, the length of the found sequence at least 60% (g6), 90% (g7), and 85% (g8)), were performed for each g6, g7, and g8 part of the requesting protein against Streptomyces bacteria and phages nucleotide database. The genome regions with closest neighbour hits (in the correct order) were classified as terminase genes. At the same time, the first best genome region
was selected (by E-value and identity) and duplicates were discarded.
2.6 Cluster classification
Using in-house Perl-script, Streptomyces bacteria and phages GenBank NCBI files were analysed and genes that have UUA codons (UUA-genes) were extracted. The UUA-genes were grouped into clusters (COFs) of orthologous UUA-genes based on sequence similarity. Orthologous UUA-genes and clusters search were executed using stand-alone BLASTp [Citation70] (NCBI BLAST package). Each protein sequence of UUA-gene is used as a query in BLASTp for similarity search against Streptomyces bacteria and phages protein database.
Among the hits, we selected those in which scores have an E-value better than . Such hits were used to assign the query UUA-gene to a cluster database with orthologous UUA-genes. If, for a given query, the E-value of several BLASTp alignments is better than the
threshold, the orthologous UUA-gene (target cluster) that has an alignment with the highest BLASTp score is selected. A query UUA-gene that has no high enough scores and, thus, has not assigned to any cluster is not used in the cluster classification. The final cluster consolidation was done to scores of those having an E-value better than
.
2.7 Identification of adpA genes in 213 Streptomyces genomes
To identify a comprehensive set of adpA genes, complete genomes of 213 Streptomyces bacteria [Citation71] were downloaded from GenBank database. Proteome of each bacteria was obtained by extracting translations of all the coding sequences (CDSs) as they were annotated in the corresponding GenBank files. Sequences of AdpA proteins from S. coelicolor and S. griseus were used as queries to run BLASTp with stringent thresholds (E-value threshold , max_target_seqs = 99,999).
>S_coelicolor:NC_003888:3,047,134–3,048,331(+)
MSHDSTAAPEAAARKLSGRRRKEIVAVLLFSGGPIFESSIPLSVFGIDRQDAGVPRYRLL
VCAGEDGPLRTTGGLELTAPQGLEAISRAGTVVVPAWRSITSPPPEEALDALRRAHEEGA
RIVGLCTGAFVLAAAGLLDGRPATTHWMYAPTLAKRYPSVHVDPRELFVDDGDVLTSAGT
AAGIDLCLHIVRTDHGNEAAGALARRLVVPPRRSGGQERYLDRSLPEEIGADPLAEVVAW
ALEHLHEQFDVETLAARAYMSRRTFDRRFRSLTGSAPLQWLITQRVLQAQRLLETSDYSV
DEVAGRCGFRSPVALRGHFRRQLGSSPAAYRAAYRARRPQGDRQPDPDTAAAGATRPLPP
SDPPASLAPENAVPFQTRRTATPMPAGAASVPGQRSAP
>S_griseus:NC_010572:5,572,624-5,573,842(-)
MSQDSAAATEAARKLTGRRRREVVAVLLFSGGPIFESSIPLSVFGIDRQDAGVPRYRLLV
CGGEEGPLRTTGGLELTAPYGLEAISRAGTVVVPAWRSITSPPPAEALDALRRAHEEGAR
IVGLCTGAFVLAAAGLLDGRPATTHWMYAPTLAKRYPSVHVDPRELFVDDGDVLTSAGTA
AGIDLCLHIVRTDHGTEAAGALARRLVVPPRRSGGQERYLDRSLPEEIGSDPLAEVVAWA
LEHLHEQFDVETLAARAYMSRRTFDRRFRSLTGSAPLQWLITQRVLQAQRLLETSDYSVD
EVAGRCGFRSPVALRGHFRRQLGSSPAAYRAAYRARRPQGVAESAATVVETMVPSQGPPS
GRRGSTLSSAAVAVAASVGSGELSLPGPDAYVPGRPALPGQRSAP
The results of the two searches were merged, and the 222 full-length CDSs (according to GenBank annotation) corresponding to the identified protein hits were collected and used for subsequent analysis.
2.8 Multiple protein sequence alignment, codon alignment, protein domain structure prediction, SynPlot2 and phylogenetic analysis
Multiple protein sequence alignment (MPSA) was performed using stand-alone MAFFT version 7 [Citation72] (linsi tool) with default parameters (linsi – treeout input.fasta > output.algnment). To align the codons of the nucleotide sequences, an indirect MAFFT procedure was used, in which the corresponding amino acid translation is first aligned before reporting the inferred gap positions at the codon level. We used identity as a proxy for accuracy, since all amino acids produced by a codon are expected to be aligned (identical) in the same column.
Sequences for MPSA and codon alignment were prepared, analysed and back-translated from amino acid to nucleotide sequence alignment using EMBOSS tools v.6.6.0.0 (seqret, tranalign) [Citation73] and an in-house Perl-script.
Sequence logos were generated using stand-alone WebLogo-3 tool (version 3.7.11) [Citation74].
Protein domains of the terminase proteins, delineated by coordinates, were identified and extracted by HMMER (hmmsearch version-3.3 with cut_ga option) [Citation75] and in-house Perl scripts along with the Pfam-DB provided profile HMMs. The found protein domains with their coordinates were scaled to codon alignments and used to visualize the SynPlot2 results.
For finding regions of enhanced synonymous site conservation in coding-sequence alignments, we used stand-alone SynPlot2 tool [Citation76]. For each cluster or random sample, SynPlot2 was run with its respective inputs:
phylogenetic tree was obtained from MPSA for a given cluster (sample). The unweighted pair-group method with arithmetic averaging (UPGMA) algorithm and neighbour-joining (NJ) algorithm [Citation77] were used to retrieve the results;
a file containing a list of sequence pairs tracing around a phylogenetic tree was prepared by an in-house Perl-script. These pairwise comparisons covered each branch of the phylogenetic tree exactly twice without putting extra sequence pairs (otherwise p-values would be overestimated);
FASTA-files with individual nucleotide sequences were generated from the MPSA results codon alignment using EMBOSS seqretsplit tool.
To visualize the SynPlot2 results, we used a statistical package such as R and adapted to the location of UUA-codons and protein domains the plot.R script for a 25-codon sliding window (win = 25).
Finally, phylogenetic analyses of the codon alignment were conducted and visualized using the iTOLtool (https://itol.embl.de) [Citation78].
These software tools are freely available to the scientific community and have been benchmarked on diverse types of datasets and utilized verified algorithms. They were selected based on availability and support at the time of analysis.
2.9 Analysis of the ribosome profiling data (Ribo-seq)
The ribo-seq files were obtained from NCBI Gene Expression Omnibus portal (accession GSE138278 and GSE128216). The individual SRA accession ids used were SRR10212831, SRR10212833, SRR10212835, SRR10212837 for S. griseus and SRR8718525, SRR8718527, SRR8718529, SRR8718531 for S. clavuligerus. Processing of these files included clipping of the adapter sequence (AGATCGGAAGAGCACACGTCTGAACTCCAG) with Cutadapt [Citation79] and removal of reads that mapped to rRNA sequence reads with Bowtie [Citation80]. The remaining reads were then aligned to their respective genomes of S. griseus (accession AP009493.1) or S. clavuligerus (accession NZ_CP027858) with Bowtie using the parameters (-m 1 -l 25 -n 2). Thus, only reads that aligned uniquely to genome were retained. The images with profiles were produced using custom python scripts.
2.10 Data availability
All the data files and scripts used to generate figures and tables from the article are available at https://github.com/vanya-antonov/article-tta-codon.
3 Results
3.1 Large-scale bioinformatic search identified a number of gene families with conserved UUA codons
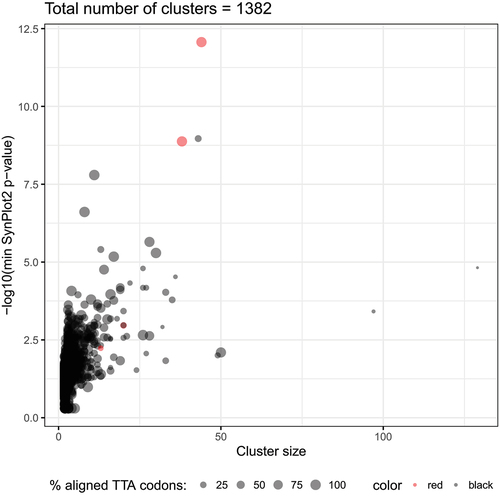
Our first goal was to collect all the UUA-containing genes in the genomes of Streptomyces and their phages. To our knowledge, such a unified analysis of both cellular and viral genes has not been attempted before (though after completion of our work for the revised version of this ms, an in-depth analysis of multiple new bacterial genomes was published [Citation71]). With our focus on UUA codons, we analysed all the annotated genes from the 259 genomes that correponded to 98 Streptomyces bacteria and 161 Streptomyces actinophages (see Methods and Supplementary Data 1). This search revealed 21,487 genes (2.8% of all the annotated genes) with at least one UUA codon. Interestingly, the fraction of the UUA-containing genes was higher in the bacterial rather than in the viral genomes (2.86% vs 1.91%, respectively) – a result that has been observed previously [Citation4,Citation43,Citation44]. It has been well established that sequence features linked to biologically important functions are preserved in evolution [Citation62,Citation81]. Therefore, we expected UUA codon conservation in the families where it may be involved in regulation of gene expression. To identify such cases, all the UUA-containing genes were grouped into clusters based on the similarities between corresponding protein sequences (see Methods). In total, we obtained 1,382 clusters containing 5,596 genes (Supplementary Data 2 and 3). Thus, we identified many putative gene families where rare UUA codons were preserved in evolution and therefore may perform some relevant biological function(s), such as translational recoding [Citation14].
In order to reveal the most promising candidates where the UUA codon may perform a functional role, we analysed the positional conservation of this codon in the identified clusters. Namely, for each of the 1,382 clusters we generated a codon alignment and analysed the locations of the UUA codons (see Methods). This analysis revealed a number of alignments where the position of the UUA codons was highly conserved, i.e. they were located in the same alignment column (Supplementary Figure 1). High positional conservation of the rare UUA codons in some clusters may indicate their possible biological function, such as the site of recoding. It should be noted that functional recoding cases frequently possess cis-stimulatory signals near the recoding site [Citation23,Citation82,Citation83]. On the other hand, it has been shown that regions with additional functions located inside coding sequences (CDS) evolve under purifying selection and can be detected using computational approaches [Citation84]. To identify the possible stimulatory sequences in the UUA-containing genes, the SynPlot2 tool developed in our laboratory [Citation76,Citation85] was applied to the codon alignments of all the clusters (see Methods). Interestingly, several clusters had both positional conservation of the UUA-codons in the alignments and strong SynPlot2 p-values (). Particularly, the two large clusters (with 44 and 38 UUA+ genes) listed as ‘AraC-like family transcriptional regulators’ (AFTRs) in GenBank were among them and contained the adpA genes [Citation9]. The conservation of the rare UUA codon in the Streptomyces adpA genes has been well-documented and thoroughly described elsewhere [Citation8]. The fact that our independent analysis revealed such case supported the validity of our bioinformatics approach.
Figure 1. Statistics on all the identified clusters of UUA-genes. The total number of UUA-containing genes in a cluster (Cluster size) and -log10 of the minimal SynPlot2 p-value observed for the cluster alignment are plotted on the X and Y axis, respectively. The size of each dot indicates the percentage of the UUA codons strictly aligned to each other at the same position in the corresponding codon alignment. The clusters annotated as ‘arac family transcriptional regulator’ are highlighted in red.
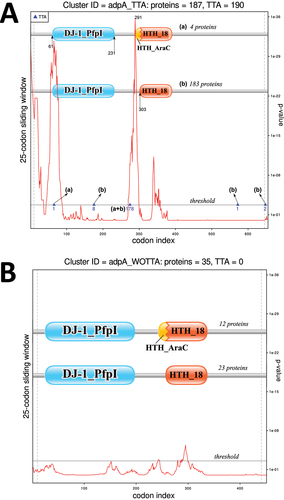
Despite decade-long knowledge of a UUA codon in adpA-coding sequence [Citation86], the functional role of this rare codon remains obscure. AdpA (AFTR) proteins usually consist of two domains – a dimerization domain (such as ‘DJ-1’) and a DNA-binding (helix-turn-helix or HTH) domain [Citation87]. Interestingly, in both clusters, the rare UUA codons were always located in a ‘linker sequence’ between the regions encoding these two domains. Earlier work considered this position of the UUA codon in the adpA gene to be ‘classical’ [Citation8]. Strikingly, SynPlot2 regions with the highest evolutionary constraints (i.e. with the smallest p-values) were located less than 50 nt downstream of the conserved UUA codons (Supplementary Figure 2). This suggested that these regions may be functionally associated with the UUA codons in adpA genes. Thus, we investigated them in depth.
3.2 Putative signal sequences were present in the UUA-containing adpA genes
The protein sequences from the two adpA (AFTR) clusters had similar domain structures as well as SynPlot2 results. To explore the differences between these two gene groups, a phylogenetic tree of a combined codon alignment was generated (Supplementary Figure 3). According to the obtained results, the genes from the two clusters were highly similar to each other and could be considered as a single group. In order to identify a complete list of adpA genes, we analysed a comprehensive collection of 213 Streptomyces genomes that include those just published [Citation71]. In total, 222 adpA genes were identified (see Methods and Supplementary Data 4). The majority (187 out of 222) of the identified genes contained the rare UUA codon inside their coding sequences (Supplementary Figure 4 and Supplementary Data 5). Importantly, the majority (205 out of 213) of the analysed Streptomyces genomes contained at least one adpA gene confirming an essential function of these genes in Streptomyces cells.
To further explore the role of the UUA codon in the adpA genes, we separately analysed sequences with and without an in-frame UUA. We hypothesize that the SynPlot2 signals observed in the two original clusters correspond to cis-stimulators of the putative recoding induced by the rare UUA codon (Supplementary Figure 2). To check the association between the UUA codon and this putative stimulator, two codon alignments were generated – one from the 187 CDSs containing a UUA codon and another from the remaining 35 UUA-free CDSs (Supplementary Data 6 and 7). Importantly, SynPlot2 identified two adjacent highly significant peaks for the UUA-containing alignment only (). This result was recapitulated for a smaller subsets of the UUA-containing genes that matched the number of the UUA-free adpAs (Supplementary Figure 5). These peaks were located immediately downstream of the conserved UUA codon position. Interestingly, the 300 nt sequence corresponding to the SynPlot2 peaks can be folded into a strong RNA secondary structure (Supplementary Figure 6). Thus, our analysis revealed a long region inside the CDS of the UUA-containing adpA genes that were specifically associated with this rare codon and could function as a putative recoding stimulator. This observation supported the possible functional regulatory role of the internal UUA codons in the Streptomyces adpA genes.
Figure 2. SynPlot2 graphs generated from the codon alignments of the adpA genes (A) with UUA or (B) without UUA codons in the main ORF . The locations of these codons in the alignments are marked by the dots, and the number of the UUA codons at corresponding alignment positions is also indicated.
3.3 Ribo-seq data revealed ribosome pausing at the conserved UUA codon upstream of the adpA start-codon
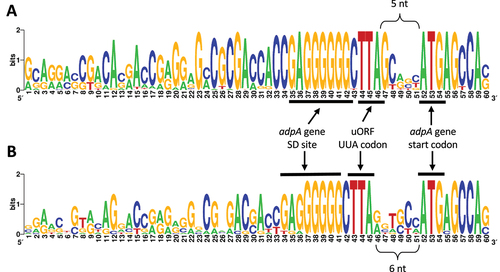
We examined published ribosome profiling data to determine if it contains unidentified pertinent information about UUA decoding. We started by analysing the S. coelicolor ribosome profiling data [Citation88] available from the GWIPS database [Citation89,Citation90]. A relatively high peak at the UUA codon inside adpA confirmed ribosome pausing at this rare codon during mRNA translation (Supplementary Figure 7A). Interestingly, a high ribosomal density was also observed immediately upstream of the adpA start codon. Closer examination of that region revealed another UUA codon located 5 nt upstream of the start codon (Supplementary Figure 7B). Ribo-seq signal outside the adpA CDS suggested the presence of a putative upstream ORF (uORF) that can also be translated. It has been shown that uORFs perform an important regulatory role in some gene families [Citation91–93]. The product of the uORF translation is typically called the ‘leader peptide’. According to our analysis, the putative uORF likely uses GUG as its initiation codon (). Independent ribo-seq experiments from other Streptomyces species [Citation94] confirmed uORF translation as well (Supplementary Figure 8). Thus, regulation of the adpA gene may involve two UUA codons – the internal UUA and the uORF UUA.
Figure 3. The location of the putative ORF upstream of the S. coelicolor adpA CDS.

Next, we analysed the conservation of the UUA codon in the putative uORF in all previously identified adpA genes (with or without an internal UUA). For this analysis, we excluded four adpA genes with atypical (i.e. non-ATG) start codons according to the GenBank annotation. Interestingly, 207 (95%) out of the 218 remaining genes contained a TTA triplet within 51 nt upstream of the annotated ATG start codon. Moreover, in most of the cases, the UUA codon in the putative uORF was located just 5 or 6 nt (172 or 24 genes, respectively) upstream of the main ORF start codon. In almost all cases (171 out of 172) where the gap was 5nt, the uORF UUA was followed by conserved guanine (). Consequently, the overlapping +1 frame codon (relative to uORF frame) is UAG, a stop codon. Additionally, the adpA start codon, AUG is followed by a conserved A where the underlined overlapping UGA is the stop-codon for the uORF (). On the other hand, in the 24 genes where the uORF UUA was 6 nt upstream of the initiation codon (i.e. it is in the adpA reading frame), the guanine downstream the UUA did not have such a high conservation (). These results suggested additional conservation of the in-frame and out-of-frame stop-codons in the putative uORF.
Figure 4. Sequence logos of the regions upstream of the adpA start codon generated from (A) 172 sequences where the distance between uORF UUA codon is 5 nt or (B) the 24 sequences where the distance is 6 nt.
Finally, we checked the co-occurrence between the UUA codons in the main ORF and in the uORF. Out of the 218 adpA genes with annotated ATG start codon, 177 genes had UUA codons in both the main ORF and the uORF (Supplementary Data 8). This observation was highly statistically significant (), suggesting that the functioning of the two rare codons may also be linked to each other. Taken together, conservation of the upstream UUA codon plus the identity of adjacent or nearby nucleotides suggests the existence of an additional mechanism that regulates adpA expression at the translational level.
Table 1. Relationship of occurrence of a UUA at the internal linker position to presence of UUA in the uORF. The observed association is highly significant according to the Fisher’s exact test. Only (main) adpA genes with ATG start codons are included.
3.4 Bioinformatics analysis of the phage ter genes suggested UUA-induced translational bypassing
Our next task was to explore one of the molecular mechanism(s) that may be triggered by the rare UUA codons during translation in Streptomyces cells. One specific case of potential regulatory occurrence has been highlighted, and a hypothesis presented for how UUA may participate in that occurrence is in the decoding of Streptomyces phage Hau3 terminase gene. Thus, we selected that ter as a model for our study. An earlier independent analysis of the 19 genomes (14 phage and 5 prophage sequences) has revealed two interrupted ter genes (from the Hau3 phage and Streptomyces C.1 prophage) with UUA codons [Citation45]. Interestingly, these two atypical ter genes contained UUA-containing noncoding fragments that separated their CDSs into several parts. While the ter gene from Streptomyces C.1 prophage was split into two parts (g6 and g78), the
Hau3 phage gene contained two 10-nt long noncoding fragments that separated it into three parts – g6, g7 and g8 (Supplementary Figure 9 and Supplementary Table 1). To determine whether the utilization of a rare UUA codon in the ter gene was a common phenomenon in various actinophages and prophages, 258 phage and 99 Streptomyces genomes were analysed (see Methods and Supplementary Data 1). Similarly to the adpA analysis (see above), the tBLASTn search was performed independently for the g6, g7 and g8 parts of the
Hau3 large terminase protein that allowed identification of potentially disrupted ter genes (Methods). In total, 100 hits were identified – 85 and 15 terminase genes from phages and bacteria (i.e. prophages), respectively (Supplementary Data 9). It should be noted that both known UUA-containing disrupted ter genes (from
Hau3 and StrepC.1) were recovered by our search indicating the validity of our bioinformatics approach. Further analysis of the remaining 98 sequences indicated that there was only one other UUA-containing (and frameshifted) ter gene. Similarly to the StrepC.1 prophage, the large terminase gene from Streptomyces catenulae prophage included a 7-nt long noncoding region between the g6 and g78 parts (Supplementary Table 2). Thus, only 3 out of the 100 analysed ter genes contained UUA codon(s) and in all cases these rare codons were followed by short non-coding regions that shifted the original reading frame. This indicated that the UUA codon may trigger a recoding event, such as translational bypassing or programmed frameshifting, to restore the correct reading frame and produce the full-length large terminase protein.
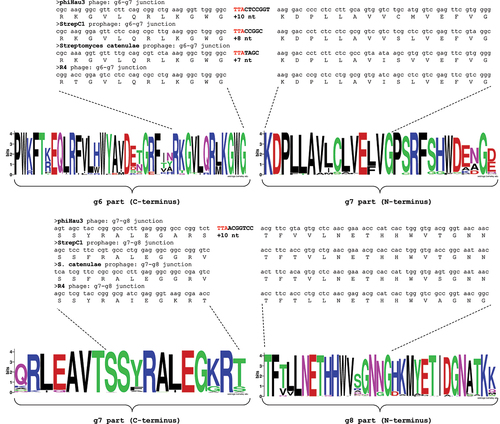
We further analysed the collected ter genes to distinguish between two possible events that can change the reading frame during translation – (i) programmed frameshifting where ribosome moved +1 or −1 nt (or 2 nt) at the frameshifting site or (ii) translational bypassing where a longer block of mRNA sequence was skipped. Analysis of the multiple alignment of the 85 phage terminase proteins demonstrated that amino acid sequences corresponding to the g6/g7 and g7/g8 junctions were highly conserved (). In case of +1 or −1 frameshifting at the UUA codon, translation of the downstream nucleotides would add additional amino acids inside these junctions. Extremely low variability at the junction sites observed in the protein alignment suggested that integrity of these regions was important for terminase function. Thus, we deduce that a frameshifting mechanism which would lead to the insertion of additional amino acids is unlikely. Instead, we favour the proposal of Smith et al. [Citation45] that there are short non-coding sequence blocks following the UUA codons which are translationally bypassed in the absence of acylated cognate tRNA.
Figure 5. The location of the g6-g7 (top) and g7-g8 (bottom) UUA-containing regions relative to the amino acid sequence of the terminase protein. The sequence logos were generated from the alignment of the 85 terminase proteins from various Streptomyces phages. The terminase gene from Hau3 phage contains two UUA and 3’ flanking sequence inserts, while the genes from two prophages included one UUA-chunk only (between the “g6” and “g7” parts). The R4 phage terminase did not contain UUA codons and was shown for comparison purposes only.
The earlier bioinformatics analysis [Citation45] pointed to putative recoding signals that may stimulate the bypassing event. Namely, the internal Shine Dalgarno sequence, local RNA secondary structures around and the ‘landing’ codon have been suggested to play a role in this process. Indeed, our analysis of the four putative bypassing sites (from three genes) revealed strong secondary structures surrounding the rare UUA codons (Supplementary Figure 10). In all cases, the predicted intra-molecular interactions within these mRNAs localized the ‘landing’ codon closer to the rare UUA codon. Significance for the potential pairing involving the coding gap is not implied. These computational analyses prompted experimental investigation.
3.5 Expression of the ter gene from  Hau3 phage in Streptomyces cells produced full-length protein
Hau3 phage in Streptomyces cells produced full-length protein
For experimental investigation of Hau3 g6/7/8 protein in Streptomyces cells from the wild-type gene, a novel dual-tagged reporter system was constructed (see Methods and Supplementary Figure 11). As mentioned above, both the g6/7 and g7/8 junctions of the relevant mRNA have a UUA codon adjacent to the 5’ end of the junction of the junction that presumably performs a key role in the peptidyl-tRNA anticodon: codon dissociation at the initiation of each of the proposed recoding events. A full-length protein (
113.9 kDa) fused to both tags could only be produced if two consecutive +1 frame recoding events occurred (one +1 event at g6/7 and the other at g7/8). Bioinformatic analysis of the sequence spanning both g6/7 and g7/8 sites predicted the recode event to be a +10 bypassing event; however, the potential for a +1 frameshifting event to occur at either site could not be precluded [Citation45], also see above. If a + 1 recode event occurred at the first recode site but not at the second, a g6/7 termination product (
53.8 kDa) would be produced, and if the first +1 recode event was unsuccessful, a zero frame g6 termination product would be produced. Each potential protein output for
Hau3 g6/7/8 can be distinguished based on size.
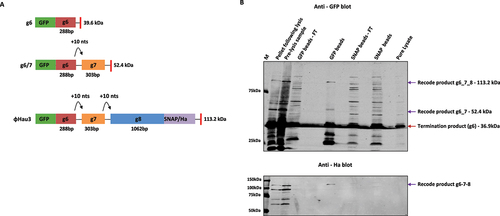
The full-length Hau3 terminase gene sequence (g6/7/8) was inserted into the dual-tagged reporter system for expression in vivo in Streptomyces (). After expression, pure lysate was analysed via SDS-PAGE, and only the g6 termination protein (
42.4 kDa) was detected. When cell pellets before and after lysis were analysed, three distinct bands were detected on the GFP immunoblot, corresponding in size to a g6 termination product, a g6/7 termination product and the full-length g6/7/8, which was also detected in the HA immunoblot (). Further confirmation for the expression of a full-length
Hau3 g6/7/8 product was indicated by the direct overlay of both the GFP and HA signals as seen in the composite blot (Supplementary Figure 12). Due to multiple bands being detected in the composite blot for the pellet samples, we next wanted to verify by purification of the lysate whether these specific bands could be isolated. Both GFP and SNAP tag purified preparations were analysed, and while the SNAP tag purified product was inconclusive (detection of multiple bands), identical bands to the three derived from products in the pellet samples were detected following GFP purification (). Thus, this construct yielded the first experimental evidence that a contiguous g6/7/8 protein is synthesized in Streptomyces .
Figure 6. Streptomyces Hau3 g6/7/8 bypass candidate: (A) A schematic representation of the Streptomyces
Hau3 g6/7/8 terminase test cassette (pGS2), including the protein outcomes for the zero frame and potential transframe events. The N-terminal fluorescent GFP tag is in the 0 frame upstream of
Hau3 g6 and the C-terminal SNAP/HA tag is in the 0 frame to g8. The recode product g6/7/8 is distinguished from both the termination product (g6) and the recode product g6/7 by the production of a dual-tagged product, produced by two consecutive +1 recoding events. Conjugated J1929 S. coelicolor spores (pGS2 vector containing g6/7/8) were grown in YT media for 2 days from a starter culture on the third day cells were induced with 50
thiostrepton. (B) Immunoblot analysis of protein profiles following GFP and SNAP agarose beads purification (top – GFP and middle – Ha). An overlay blot (bottom) proves direct overlay of the HA tag signal with the GFP tag signal and the three products (zero frame g6 – red arrow, and transframe products g6/7 and g6/7/8 – purple arrow) are indicated by arrows.
Despite sustained effort, with the available vectors and techniques, it proved impractical to perform a mutational analysis of the possible recoding signals, which include an mRNA stem loop and a nearby internal Shine Dalgarno sequence [Citation12,Citation45]. We had concerns about embarking on such an analysis in heterologous E. coli, only in part as cognate aminoacyl-tRNA for UUA is not limiting there and more generally codon usage is very different. Nevertheless, we explored whether a cassette of Hau3 6/7 recoding signals could cause bypassing in E. coli (Supplementary Figure 13). When a cassette with
Hau3 g6/7 or
Hau3 g6/7/8 was expressed in E. coli, unsurprisingly only the product expected from g6 coding sequence was detected. In E. coli, stop codons are slow-to-decode codons. On switching the
Hau3 g6/7 leucine UUA codon to a stop-codon UAA, a g6/7 dual-tagged recode product was detected. It can be inferred from these results that recoding at g6/7 requires the presence of a slow-to-decode codon with at least some level of interchangeability between the slow-to-decode codon present.
To ascertain whether the recoding event in E. coli from a g6/g7 cassette with UAA substituted for UUA, custom antibodies were generated (see Methods and Supplementary Figure 14). Notably, detection by either antibodies 2 or 5 would indicate a +1 recoding event (Supplementary Figure 15A). A faint band corresponding to the predicted size of the recode protein was observed when Hau3 g6/7 (UAA) was expressed in the wild-type E. coli K-12 MG1655 strain for all the antibodies generated. This experimental result confirmed recoding at the g6/g7 site.
3.6 Mass spectrometry and mutational analysis supported translational bypassing at the g6/g7 site
To establish the nature of the recoding events (i.e. frameshifting vs bypassing) in the ter gene from Hau3, two complementary approaches were employed. For the first, product mass spectrometry, large-scale protein purification was performed for g6/7 (UAA) (see Methods). The predicted outcome for a bypass event at
Hau3 g6/7 (UAA) was created as a standard against which to map the peptide reads generated from the MS/MS (Supplementary Figure 16). The output of the MS/MS analysis revealed reads that mapped to peptides encoded up to and including the take-off site, as well as peptides encoded further downstream. The output of the MS/MS analysis revealed the sequence of a ‘junction’ encoded peptide whose amino acids were encoded 5’ of the take-off site, the take-off site, the resume codon (3’ adjacent to the ‘landing’ codon) and sequence 3’ of it (GWG
NDPLLAVVCMVEFVGPSR – see Supplementary Figure 16). Thus, the obtained mass spectrometry results provided definitive evidence that in E. coli recoding at g6/7 (UAA) is functioned via bypassing.
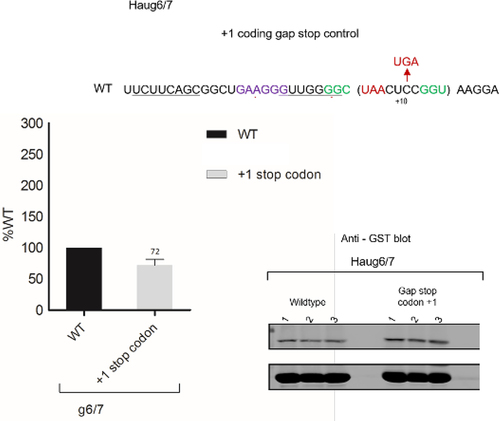
The second approach involved positioning a UGA stop codon in the +1 frame just 3’ of the g6/g7 UUA (within the putative coding gap), so that any ribosomes that underwent +1 frameshifting during the stall at UUA whose cognate tRNA was lacking would terminate (). Although a decrease in recoding was not expected, surprisingly the presence of a frame UGA stop codon led to a twofold increase in recoding efficiency (225% of WT) (). While bypassing was confirmed, the reason for the elevated level was not investigated.
Figure 7. Analysis of the bypassing hypothesis for Hau3 g6/7. To test for +1 frameshifting, a +1 stop codon was placed within the coding gap (+1 Stop codon – top sequence). Constructs were assayed in triplicate, and samples were analysed by immunoblotting. Experiments were performed in duplicate. Densitometry was performed on the termination product and the transframe product for each sample, recoding efficiency was calculated for each test sequence and values were normalized against the wild-type control.
4 Discussion
The discovery of leader-peptide encoding sequences for the Salmonella and E. coli histidine and tryptophan biosynthetic operons with runs of histidine or tryptophan codons respectively [Citation91–93] was an earlier pointer to the more general use in bacteria of the sensing of the translatability of specific codons for related regulatory purposes. This is now known to be also important in eukaryotes. A recently discovered eukaryotic example involves the polyamine pathway-related mRNA for antizyme inhibitor. Under high polyamine conditions, a queue of ribosomes forms behind a lead ribosome stalled at PPW codons near the 3’ end of an upstream ORF and enhances initiation at the otherwise poor initiator of that same ORF. This has regulatory significance for the expression of antizyme inhibitor encoded by the close 3’ main ORF [Citation95,Citation96]. In the present work, we analysed adpA ribosome profiling data from the Cho group [Citation88], initially on our GWIPS-Viz browser [Citation89,Citation90]. The finding of a prominent ribosome peak just 5’ of the annotated adpA start codon and the presence there of UUA illustrates the utility of this type of data resource and prompted bioinformatics follow-up. This led to the finding that many Streptomyces species have an upstream ORF with a UUA codon close, commonly 5 nt, 5’ of the annotated start codon for synthesis of AdpA. Both UUA decoding and adpA expression are key Streptomyces features for exit from the vegetative state of growth. We have not performed the needed follow-up experiments to show regulatory significance, but for clarity and to provoke such follow-up by others, we will discuss the bioinformatics findings in the context of their being a regulatory significant adpA upstream ORF in the great majority of Streptomyces species.
In 177 (96%) of the 184 Streptomyces adpA genes that have a UUA in the linker region of the main ORF, an upstream ORF has a UUA 5 or 6 nt upstream the main ORF start codon (). Interestingly, in the other 34 adpA sequences analysed, 15% of the total adpA genes with an ATG start codon, where there is no UUA in the main ORF ‘linker’, the proportion with a corresponding UUA-containing upstream ORF, is much lower, 56% (i.e. 19 of the 34). For the 177 sequences with a main ORF UUA, the upstream ORF UUA is located 5nt (153 cases) or 6nt (24 cases) 5’ of the main ORF start (). Given the extreme rarity of UUA codons in Streptomyces, this pattern of occurrences 5’ of the main ORF start is highly likely to imply regulatory significance. The most obvious thought is that its regulatory effect is mediated by ribosomes translating the upstream ORF encountering UUA in its ribosomal A-site. While the ribosome profiling data analysed show clear evidence of translation 5’ of the main ORF start, we are cautious about making more specific inferences from it. Despite one of the figures (Supplementary Figure 7 and 8), possibly giving a hint of the 5’ ribosome of a pair of collided ribosomes, disome and polysome profiling at different growth phases is required to assess the situation.
In Streptomyces, the most efficient and the most common initiator is AUG, with the frequency of GUG being approximately half as many [Citation97]. However, for the upstream ORFs, the candidate start codon in all cases is GUG. For GC-rich Streptomyces, the canonical SD sequence, GGAGG, is the same as E. coli [Citation97,Citation98]. While for the candidate upstream ORF candidate GUG initiator, the sequence GAGG(G) is similar to that of the canonical and its spacing is relatively short though within the known range. The combination of SD and GUG initiator is expected to result in only moderately efficient initiation.
In addition to delaying main ORF initiation by initiator occlusion, a prolonged pause at UUA is expected to lead to the formation of a ribosome queue 5’ of it. Would such a queue ‘just’ delay upstream ORF initiation and of itself be of little consequence? If one assumes that bacterial initiation exclusively involves 30S subunits first binding to mRNA at the initiation site to be joined there by the 50S subunit, the answer would probably be yes. However, a number of experiments point to scanning, at least by 30S subunits [Citation99–101], and others directly to 70S ribosome scanning [Citation102,Citation103]. Scanning by preloaded 70S ribosomes was evident even without special signals from bypassing studies and was originally proposed by Sarabhai and Brenner in 1967 [Citation104]. For 70S scanning to be potentially relevant for the adpA upstream ORF, the distance from the 5’ end of the mRNA should be short and free of secondary structure [Citation103]. If this alternative mode of initiation occurs near the 5’ end of adpA mRNA, should the possibility be considered that a ribosome queue serves paradoxically to enhance adpA upstream ORF initiation with subsequent further sensing of the level of functional cognate tRNA at its downstream UUA? Irrespective of whether ribosome pausing at the UUA has any effect on initiation, a major effect of a ribosome paused with UUA in its A-site would be delay of main ORF initiation.
The great majority of bacterial-release factor 2 mRNAs has a UGA early in their coding sequence. When the UGA enters the ribosomal A-site, a +1 ribosomal frameshift is required to synthesize functional-release factor [Citation82,Citation105]. The efficiency of the regulatory frameshift event is greatly stimulated by pairing of the anti-SD sequence of translating ribosomes with an SD sequence whose 3’ end is exactly 6nts 5’ of the UGA [Citation47,Citation82,Citation106,Citation107]. After such an rRNA, mRNA hybrid forms translation can continue for a short distance before the hybrid breaks, and as it approaches that point it acts as a stimulator of −1 frameshifting [Citation108,Citation109].
The sequence, GAGGGGGG, is 9nt 5’ of the adpA main ORF start codon. The two underlined Gs form part of the sequence GGC UUA in the upstream ORFs, where the UUA is 5nt 5’ of the adpA main ORF start. In these cases, the extent to which the UUA is 3’ of the GA6G rather than 5’ of it and the conserved spacing between them UUA is consistent with positive selection. Remarkably in the minority of species that lack UUA at that position, the number of adjacent Gs is less, though the sequence still likely acts as a start for main ORF initiation. Whether future work reveals some novel relationship of the GA6G sequence to the conserved G (or A) 3’ adjacent to UUA remains to be seen. If the GA6G sequence is related to the UUA 3’ adjacent base entering the A-site to create a stop codon, the mechanism would likely be at least substantially different from that involved in release factor 2 regulatory programmed +1 frameshifting. Indicative of potential significance of G (or A) being 3’ adjacent to that upstream ORF UUA is exceptional occurrence of G (or A) at that position. Genome-wide analysis of TTAs in Streptomyces protein coding genes showed a major under-representation of A or G as the 3’ adjacent base for TTA and for other NNA codons [Citation110]. However, A or G is present 3’ adjacent to the UUA in the adpA upstream ORF in many Streptomyces species whose sequence we have examined. While it is formally possible that the overwhelming preponderance of G (or A) 3’ adjacent to the UUA is due to selection at the amino acid level, we consider this to be unlikely. Instead might selection for the identity of 3’ adjacent base have been for an rRNA: mRNA hybrid 1 nt ‘push’ to result in a ribosomal A-site stop codon that results in translation termination? If so, the situation would still be very different from eukaryotic termination, which involves 4 mRNA bases in the ribosomal A-site [Citation111], and from E. coli termination where the identity of the 3’ adjacent base is also important [Citation112].
In the later phases of the cell cycle, abundant functional cognate UUA-decoding tRNA is present. For those adpA mRNAs with the upstream ORF UUA at its most common position, termination will then occur 2 codons 3’ of the UUA at a stop codon that overlaps the main ORF start. A terminating ribosome is likely to have its 30S subunit tethered to the mRNA by SD: anti-SD pairing it formed just before termination. This is expected to affect the speed of main ORF initiation. Any ribosome that terminates at the stop overlapping the UUA would be expected to also be tethered but perhaps not so extensively. However, the timing of derived main ORF initiation is likely to be substantially slowed. If, as expected, experimental data supports a function for the upstream ORF, there will be no shortage of questions arising.
Further to potential ‘epigenetic’ aspects involving the effects of growth conditions on tRNA modifications that influence either or both aminoacylation levels and codon binding, arguably, the varied pattern expression of mRNAs containing UUA merits the consideration of greater decoding versatility than previously considered. This may have relevance for antibiotic production. The UUA in hyg, aad and ccaR mRNAs in at least some species is somehow decoded to different extents under conditions where the UUA in most UUA-containing mRNAs is not [Citation7,Citation113]. In Streptomyces ghanaensis, unlike the situation in several other species, decoding of the UUA in the linker region between sequence encoding the two different domains of AdpA is not fully dependent on the tRNA specified by bldA and there is a variable effect of bldA deletion under different environmental conditions [Citation86]. Irrespective of whether near-cognate decoding is involved, because of the selectivity involved in some cases, context features are implicated. The conserved contextual elements computationally identified 3’ of the adpA linker UUA have features suggestive of their being stimulatory elements for distinctive decoding of that rare UUA codon. Absence of these features from the counterpart gene in a small minority of species that lack the TTA is supportive of the proposition that those are functionally associated with decoding of UUA. Interestingly, the spacer length between the 3’ end of the UUA and the 5’ end of the closest apparent 3’ stem loop structure is 12 nt. This is much more than the 5–6 nt spacing commonly present from bacterial programmed frameshift sites to the start of the intra-mRNA structures known to act as stimulatory signals for those cases of recoding, or 5–9 nt for eukaryotic programmed frameshifting or readthrough [Citation114–116]. These structures start to penetrate the mRNA entrance tunnel before being unwound [Citation117–119]. However, the spacing, 13 nt, for Cardiovirus programmed frameshifting is exceptional; it involves a protein binding to a 3’ intra-molecular mRNA structure increasing the frameshifting efficiency from negligible to 70%–80% [Citation120,Citation121]. This spacing is very similar to the adpA internal candidate counterpart where it is 12 nt. It is tempting to suggest that a ligand binding to the adpA mRNA main ORF internal UUA increases the spacing from the ribosomal A-site to the ‘barrier’ 3’ of the ribosome and that ligand serves a regulatory function for Streptomyces transition from the vegetative state. Determining the relationship of the components of the features identified to each other and their structure transitions will be a worthwhile task, even without expectations of a tandem riboswitch [Citation122]. While reporter systems and approaches for the initial part of this work have long been used in recoding studies in more genetically tractable organisms, advances in genetic manipulation of Streptomyces that are recent compared to the origin of the present work open the door for such studies.
Phage UUA: exploiting an arms race for its lytic/lysogeny decision?
The model proposed for the role of the two UUAs at separate internal position of the sole phage Hau3 terminase encoding sequence requires that in the absence of functional cognate tRNA, both UUAs trigger translational bypassing. And that this bypassing leads to synthesis of the terminase protein required for the phage’s lytic cycle [Citation43,Citation45]. Influenced by the technical difficulties involved in analysing very low-level protein products in Streptomyces, the experimental data generated previously have only been presented in thesis form and involved heterologous E. coli [Citation123–126]. In addition, lack of awareness of the dilemma of blocks of apparently non-coding sequence in an essential gene has also been relevant for the original hypothesis mostly languishing unknown. [Our analysis of phage terminase sequences unknown at the time of the original hypothesis strengthens evidence that the extra blocks of 10 nt sequence would likely be disruptive for terminase function even if both the UUAs resulted in single nucleotide frameshift events.] Though the experimental evidence we have presented does not prove the occurrence of UUA-mediated regulatory translational bypassing, it nevertheless does provide substantial support. The proposed bypassing 5’ stimulatory signals involving a stem loop and flanking Shine Dalgarno interaction are positionally distinct with the latter perhaps being related to the much shorter distance bypassed than for instance in phage T4 gene 60. Their investigation is now merited. Hopefully the visibility will be timely in view of recent advance in genetic tools for certain Streptomyces species and the imminent prospect of single-molecule protein sequencing [Citation127].
Overall, the results presented here point to a likely hidden treasure trove of novel and now investigable, recoding with potential for considerable scientific interest and in some cases of practical significance.
Author contribution
I.A. and A.G. performed the bioinformatics analyses, S.O’L generated the experimental data, P.O’C. analyzed the ribosome profiling data not in GWIPS-Viz, P.B. suggested the use of Synplot and searched GWIPS-viz, J.A. initiated and coordinated the project, identified the candidate GUG initiator and wrote much of the Introduction and Discussion with I.A. dealing with most of the rest of the ms with input from all authors.
Supplemental Material
Download Zip (9.7 MB)Acknowledgments
J.F.A. appreciates the discussions with Drs M. Smith and the late R.W. Hendrix about bypassing and thanks the John Innes Centre scientists, Drs K. Chater, and indirectly D.A. Hopwood, for facilitating the help generously provided by Drs M.J. Bibb and J.P Gomez-Escribano to S. O’L. for Streptomyces work including vectors and hosting her visit. We thank the reviewers for their constructive criticisms.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/15476286.2023.2270812
Additional information
Funding
References
- Moody ERR, Mahendrarajah TA, Dombrowski N, et al. An estimate of the deepest branches of the tree of life from ancient vertically evolving genes. Elife. 2022;11:e66695.
- Gładek A, Zakrzewska J. Genome size of Streptomyces. FEMS Microbiol Lett. 1984;24(1):73–76.
- Wright F, Bibb MJ. Codon usage in the G+C-rich Streptomyces genome. Gene. 1992;113(1):55–65.
- Chater KF, Chandra G. The use of the rare UUA codon to define “expression space” for genes involved in secondary metabolism, development and environmental adaptation in Streptomyces. J Microbiol. 2008;46(1)1–11.
- Zaburannyy N, Ostash B, Fedorenko V. TTA Lynx: a web-based service for analysis of actinomycete genes containing rare tta codon. Bioinformatics. 2009;25(18):2432–2433.
- Chater KF, Chandra G. The evolution of development in Streptomyces analysed by genome comparisons. FEMS Microbiol Rev. 2006;30(5):651–672.
- Leskiw BK, Lawlor EJ, Fernandez-Abalos JM, et al. TTA codons in some genes prevent their expression in a class of developmental, antibiotic-negative, Streptomyces mutants. Proc Nat Acad Sci. 1991;88(6):2461–2465.
- Rabyk M, Yushchuk O, Rokytskyy I, et al. Genomic insights into evolution of AdpA family master regulators of morphological differentiation and secondary metabolism in Streptomyces. J Mol Evol. 2018;86(3–4):204–215.
- Ting L, Xiaohua W, Cao Q, et al. Sulfane sulfur posttranslationally modifies the global regulator AdpA to influence actinorhodin production and morphological differentiation of Streptomyces coelicolor. Mbio. 2022;13(3):e03862–21.
- Saito K, Kratzat H, Campbell A, et al. Ribosome collisions induce mRNA cleavage and ribosome rescue in bacteria. Nature. 2022;603(7901):503–508.
- Gaydukova S, Moldovan M, Vallesi A, et al. Nontriplet feature of genetic code in Euplotes ciliates is a result of neutral evolution. Proc Nat Acad Sci. 2023;120(22). DOI:10.1073/pnas.2221683120
- Atkins JF, Loughran G, Bhatt PR, et al. Ribosomal frameshifting and transcriptional slippage: from genetic steganography and cryptography to adventitious use. Nucleic Acids Res. 2016;44(15):7007–7078.
- Lindsley D, Gallant J. On the directional specificity of ribosome frameshifting at a” hungry” codon. Proc Nat Acad Sci. 1993;90(12):5469–5473.
- Gesteland RF, Weiss RB, Atkins JF. Recoding: reprogrammed genetic decoding. Science. 1992;257(5077):1640–1641.
- Baranov PV, Atkins JF, Yordanova MM. Augmented genetic decoding: global, local and temporal alterations of decoding processes and codon meaning. Nat Rev Genet. 2015;16(9):517–529.
- Rodnina MV. Decoding and recoding of mRNA sequences by the ribosome. Annu Rev Biophys. 2023;52(1):161–182.
- Poulis P, Patel A, Rodnina MV, et al. Altered tRNA dynamics during translocation on slippery mRNA as determinant of spontaneous ribosome frameshifting. Nat Commun. 2022;13(1):4231.
- Atkins JF, Gesteland RF. editors. Recoding: expansion of decoding rules enriches gene expression. New York, NY: Springer; 2010. P. 466.
- Firth AE, Jagger BW, Wise HM, et al. Ribosomal frameshifting used in influenza a virus expression occurs within the sequence UCC_UUU_CGU and is in the +1 direction. Open Biol. 2012;2(10):120109.
- Caplan AB, Menninger JR. Dissociation of peptidyl-tRNA from ribosomes is perturbed by streptomycin and by strA mutations. Mol Gen Genet. 1984;194(3):534–538.
- Tomasi FG, Schweber JTP, Kimura S, et al. Peptidyl tRNA hydrolase is required for robust prolyl-tRNA turnover in Mycobacterium tuberculosis. Mbio. 2023;14(1):e03469–22.
- O’Loughlin S, Capece MC, Klimova M, et al. Polysomes bypass a 50-nucleotide coding gap less efficiently than monosomes due to attenuation of a 5’ mRNA stem–loop and enhanced drop-off. J Mol Biol. 2020;432(16):4369–4387. DOI:10.1016/j.jmb.2020.05.010.
- Antonov IV. Two cobalt chelatase subunits can be generated from a single chlD gene via programed frameshifting. Mol Biol Evol. 2020;37(8):2268–2278.
- Hopwood DA. Genetic analysis and genome structure in Streptomyces coelicolor. Bacteriol Rev. 1967;31(4):373–403.
- Bush MJ, Chandra G, Al-Bassam MM, et al. BldC delays entry into development to produce a sustained period of vegetative growth in Streptomyces venezuelae. MBio. 2019;10(1):e02812–18.
- Higo A, Horinouchi S, Ohnishi Y. Strict regulation of morphological differentiation and secondary metabolism by a positive feedback loop between two global regulators AdpA and BldA in Streptomyces griseus. Mol Microbiol. 2011;81(6):1607–1622.
- Lawlor EJ, Baylis HA, Chater KF. Pleiotropic morphological and antibiotic deficiencies result from mutations in a gene encoding a tRNA-like product in Streptomyces coelicolor A3 (2). Genes Dev. 1987;1(10):1305–1310.
- Trepanier NK, Nguyen GD, Leedell PJ, et al. Use of polymerase chain reaction to identify a leucyl tRNA in Streptomyces coelicolor. Gene. 1997;193(1):59–63.
- Koshla O, Lopatniuk M, Rokytskyy I, et al. Properties of Streptomyces albus J1074 mutant deficient in tRNALeu UAA gene bldA. Arch Microbiol. 2017;199(8):1175–1183.
- Fredrik Pettersson BM, Kirsebom LA. tRNA accumulation and suppression of the bldA phenotype during development in Streptomyces coelicolor. Mol Microbiol. 2011;79(6):1602–1614.
- Koshla O, Yushchuk O, Ostash I, et al. Gene miaA for post-transcriptional modification of tRNAXXA is important for morphological and metabolic differentiation in Streptomyces. Mol Microbiol. 2019;112(1):249–265. DOI:10.1111/mmi.14266.
- Sehin Y, Koshla O, Dacyuk Y, et al. Gene ssfg_01967 (miaB) for tRNA modification influences morphogenesis and moenomycin biosynthesis in Streptomyces ghanaensis ATCC14672. Microbiology. 2019;165(2):233. DOI:10.1099/mic.0.000747.
- Koshla O, Vogt L-M, Rydkin O, et al. Landscape of post-transcriptional tRNA modifications in Streptomyces albidoflavus J1074 as portrayed by mass spectrometry and genomic data mining. J Bacteriol. 2023;205(1):e00294–22.
- Fabiola Janiak VAD, Abrahamson JK, Watson BS, et al. Fluorescence characterization of the interaction of various transfer RNA species with elongation factor Tu.GTP: evidence for a new functional role for elongation factor Tu in protein biosynthesis. Biochemistry. 1990;29(18):4268–4277.
- Gobet C, Dieter Weger B, Marquis J, et al. Robust landscapes of ribosome dwell times and aminoacyl-tRNAs in response to nutrient stress in liver. Proc Nat Acad Sci. 2020;117(17):9630–9641.
- Brown A, Fernández IS, Gordiyenko Y, et al. Ribosome-dependent activation of stringent control. Nature. 2016;534(7606):277–280.
- Dworkin J. Understanding the stringent response: experimental context matters. Mbio. 2023;14(1):e03404–22.
- Hesketh A, Joan Chen W, Ryding J, et al. The global role of ppGpp synthesis in morphological differentiation and antibiotic production in Streptomyces coelicolor A3 (2). Genome Biol. 2007;8(8):1–18.
- Song Y, Zhang X, Zhang Z, et al. Physiology and transcriptional analysis of ppGpp-related regulatory effects in Streptomyces diastatochromogenes 1628. Microbiol Spectr. 2023;11(1):e01200–22.
- Boutte CC, Henry JT, Crosson S. ppGpp and polyphosphate modulate cell cycle progression in Caulobacter crescentus. J Bacteriol. 2012;194(1):28–35.
- Slominska M, Neubauer P, Wegrzyn G. Regulation of bacteriophage lambda development by guanosine 5’-diphosphate-3’-diphosphate. Virology. 1999;262(2):431–441.
- Masucci J, Gallant J, Lindsley D, et al. Influence of the relA gene on ribosome frameshifting. Mol Genet Genomics. 2002;268(1):81–86.
- Smith MCM, Neil Burns R, Wilson SE, et al. The complete genome sequence of the Streptomyces temperate phage φc31: evolutionary relationships to other viruses. Nucleic Acids Res. 1999;27(10):2145–2155.
- Hesketh A, Bucca G, Laing E, et al. New pleiotropic effects of eliminating a rare tRNA from Streptomyces coelicolor, revealed by combined proteomic and transcriptomic analysis of liquid cultures. BMC Genomics. 2007;8(1):1–22.
- Smith MCM, Hendrix RW, Dedrick R, et al. Evolutionary relationships among actinophages and a putative adaptation for growth in Streptomyces spp. J Bacteriol. 2013;195(21):4924–4935. DOI:10.1128/JB.00618-13.
- Hilbert BJ, Hayes JA, Stone NP, et al. The large terminase DNA packaging motor grips DNA with its ATPase domain for cleavage by the flexible nuclease domain. Nucleic Acids Res. 2017;45(6):3591–3605.
- Weiss RB, Dunn DM, Atkins JF, et al. Slippery runs, shifty stops, backward steps, and forward hops: -2, -1, +1, +2, +5, and +6 ribosomal frameshifting. In: Cold Spring Harbor Symposia on Quantitative Biology. Vol. 52. Cold Spring Harbor Laboratory Press; 1987. p. 687–693.
- Gallant J, Bonthuis P, Lindsley D. Evidence that the bypassing ribosome travels through the coding gap. Proc Nat Acad Sci. 2003;100(23):13430–13435.
- Wills NM Translational bypassing – peptidyl-tRNA re-pairing at non-overlapping sites. In: Recoding: expansion of decoding rules enriches gene expression. New York, NY: Springer; 2010. p. 365–381.
- Kane JF, Violand BN, Curran DF, et al. Novel in-frame two codon translational hop during synthesis of bovine placental lactogen in a recombinant strain of Escherichia coli. Nucleic Acids Res. 1992;20(24):6707–6712.
- Natalie Jing M, Hemez CF, Barber KW, et al. Organisms with alternative genetic codes resolve unassigned codons via mistranslation and ribosomal rescue. Elife. 2018;7:e34878.
- Lindsley D, Gallant J, Doneanu C, et al. Spontaneous ribosome bypassing in growing cells. J Mol Biol. 2005;349(2):261–272.
- Huang WM, Shi-Zhou A, Casjens S, et al. A persistent untranslated sequence within bacteriophage T4 DNA topoisomerase gene 60. Science. 1988;239(4843):1005–1012.
- Poulis P, Peske F, Rodnina MV. The many faces of ribosome translocation along the mRNA: reading frame maintenance, ribosome frameshifting and translational bypassing. Biol Chem. 2023;404(8–9):755–767.
- Chen J, Coakley A, O’Connor M, et al. Coupling of mRNA structure rearrangement to ribosome movement during bypassing of non-coding regions. Cell. 2015;163(5):1267–1280.
- Klimova M, Senyushkina T, Samatova E, et al. EF-G–induced ribosome sliding along the noncoding mRNA. Sci Adv. 2019;5(6):eaaw9049.
- Manning G, Atkins JF. An RNA sequence that reprograms ribosomes to bypass a 50 nucleotide coding gap is encoded by a mobile element whose sequence conservation illuminates its bypass mechanisms. bioRxiv 2022.08.31.505936. 2022.
- Lang BF, Jakubkova M, Hegedusova E, et al. Massive programmed translational jumping in mitochondria. Proc Nat Academ Sci. 2014;111(16):5926–5931. doi: 10.1073/pnas.1322190111
- Nosek J, Tomaska L, Burger G, et al. Programmed translational bypassing elements in mitochondria: structure, mobility, and evolutionary origin. Trends Genet. 2015;31(4):187–194.
- Brejová B, Lichancová H, Brázdovič F, et al. Genome sequence of the opportunistic human pathogen Magnusiomyces capitatus. Curr Genet. 2019;65(2):539–560. DOI:10.1007/s00294-018-0904-y.
- Grentzmann G, Ingram JA, Kelly PJ, et al. A dual-luciferase reporter system for studying recoding signals. RNA. 1998;4(4):479–486.
- Antonov I, Baranov P, Borodovsky M. GeneTack database: genes with frameshifts in prokaryotic genomes and eukaryotic mRNA sequences. Nucleic Acids Res. 2012;41(D1):D152–D156.
- Loughran G, Howard MT, Firth AE, et al. Avoidance of reporter assay distortions from fused dual reporters. RNA. 2017;23(8):1285–1289.
- Smith AM, Costello MS, Kettring AH, et al. Ribosome collisions alter frameshifting at translational reprogramming motifs in bacterial mRNAs. Proc Nat Acad Sci. 2019;116(43):21769–21779.
- Sun J, Kelemen GH, Fernández–Abalos JM et al. Green fluorescent protein as a reporter for spatial and temporal gene expression in Streptomyces coelicolor A3 (2). Microbiology. 1999;145(9):2221–2227.
- Benson DA, Cavanaugh M, Clark K, et al. GenBank. Nucleic Acids Res. 2018;46(D1):D41–D47.
- Russell DA, Hatfull GF. PhagesDB: the actinobacteriophage database. Bioinformatics. 2017;33(5):784–786.
- Mistry J, Chuguransky S, Williams L, et al. Pfam: the protein families database in 2021. Nucleic Acids Res. 2021;49(D1):D412–D419. DOI:10.1093/nar/gkaa913.
- Camacho C, Coulouris G, Avagyan V, et al. Blast+: architecture and applications. BMC Bioinformatics. 2009;10(1):421.
- Shiryev SA, Papadopoulos JS, Schäffer AA, et al. Improved BLAST searches using longer words for protein seeding. Bioinformatics. 2007;23(21):2949–2951, 10.
- Nikolaidis M, Hesketh A, Frangou N, et al. A panoramic view of the genomic landscape of the genus Streptomyces. Microb Genom. 2023;9(6):001028.
- Katoh K, Daron MS. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30(4):772–780, 01.
- Rice P, Longden I, Bleasby A. EMBOSS: the European molecular biology open software suite. Trends Genet. 2000;16(6):276–277.
- Crooks GE, Hon G, Chandonia J-M, et al. WebLogo: a sequence logo generator. Genome Res. 2004;14(6):1188–1190.
- Potter SC, Luciani A, Eddy SR, et al. HMMER web server: 2018 update. Nucleic Acids Res. 2018;46(W1):W200–W204.
- Firth AE. Mapping overlapping functional elements embedded within the protein-coding regions of RNA viruses. Nucleic Acids Res. 2014;42(20):12425–12439.
- Sokal RR, Michener CD, Of kansas U. A statistical method for evaluating systematic relationships. Lawrence, KS: University of Kansas science bulletin. University of Kansas; 1958.
- Letunic I, Bork P. Interactive tree of life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021;49(W1):W293–W296.
- Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011;17(1):10–12.
- Langmead B, Trapnell C, Pop M, et al. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10(3):1–10.
- Sharma V, Firth AE, Antonov I, et al. A pilot study of bacterial genes with disrupted ORFs reveals a surprising profusion of protein sequence recoding mediated by ribosomal frameshifting and transcriptional realignment. Mol Biol Evol. 2011;28(11):3195–3211.
- Baranov PV, Gesteland RF, Atkins JF. Release factor 2 frameshifting sites in different bacteria. EMBO Rep. 2002;3(4):373–377.
- Blinkowa AL, Walker JR. Programmed ribosomal frameshifting generates the Escherichia coli DNA polymerase iii γ subunit from within the τ subunit reading frame. Nucleic Acids Res. 1990;18(7):1725–1729.
- Lin MF, Kheradpour P, Washietl S, et al. Locating protein-coding sequences under selection for additional, overlapping functions in 29 mammalian genomes. Genome Res. 2011;21(11):1916–1928.
- Firth AE, Atkins JF. A conserved predicted pseudoknot in the NS2A-encoding sequence of west Nile and Japanese encephalitis flaviviruses suggests NS1ʹmay derive from ribosomal frameshifting. Virol J. 2009;6(1):1–6.
- Makitrynskyy R, Ostash B, Tsypik O, et al. Pleiotropic regulatory genes bldA, adpA and absB are implicated in production of phosphoglycolipid antibiotic moenomycin. Open Biol. 2013;3(10):130121.
- Yang J, Tauschek M, Robins-Browne RM. Control of bacterial virulence by AraC-like regulators that respond to chemical signals. Trends Microbiol. 2011;19(3):128–135.
- Jeong Y, Kim J-N, Woo Kim M, et al. The dynamic transcriptional and translational landscape of the model antibiotic producer Streptomyces coelicolor A3 (2). Nat Commun. 2016;7(1):11605. DOI:10.1038/ncomms11605.
- Michel AM, Kiniry SJ, Patrick BFO, et al. GWIPS-viz: 2018 update. Nucleic Acids Res. 2018;46(D1):D823–D830.
- Zaheed O, Kiniry SJ, Baranov PV, et al. Exploring evidence of non-coding RNA translation with Trips-Viz and GWIPS-viz browsers. Front Cell Dev Biol. 2021;9:703374.
- Kasai T. Regulation of the expression of the histidine operon in Salmonella typhimurium. Nature. 1974;249(5457):523–527.
- Johnston HM, Barnes WM, Chumley FG, et al. Model for regulation of the histidine operon of Salmonella. Proceedings of the National Academy of Sciences. 1980;77(1):508–512. doi: 10.1073/pnas.77.1.508
- Yanofsky C. Attenuation in the control of expression of bacterial operons. Nature. 1981;289(5800):751–758.
- Kim W, Hwang S, Lee N, et al. Transcriptome and translatome profiles of Streptomyces species in different growth phases. Sci Data. 2020;7(1):138.
- Ivanov IP, Loughran G, Atkins JF. uORFs with unusual translational start codons autoregulate expression of eukaryotic ornithine decarboxylase homologs. Proc Nat Acad Sci. 2008;105(29):10079–10084.
- Ivanov IP, Shin B-S, Loughran G, et al. Polyamine control of translation elongation regulates start site selection on antizyme inhibitor mRNA via ribosome queuing. Mol Cell. 2018;70(2):254–264.
- Hwang S, Lee N, Jeong Y, et al. Primary transcriptome and translatome analysis determines transcriptional and translational regulatory elements encoded in the Streptomyces clavuligerus genome. Nucleic Acids Res. 2019;47(12):6114–6129.
- Rudolph MM, Vockenhuber M-P, Suess B. Synthetic riboswitches for the conditional control of gene expression in Streptomyces coelicolor. Microbiology. 2013;159(Pt_7):1416–1422.
- Berkhout B, Schmidt BF, van Strien A, et al. Lysis gene of bacteriophage MS2 is activated by translation termination at the overlapping coat gene. J Mol Biol. 1987;195(3):517–524.
- Adhin MR, van Duin J. Scanning model for translational reinitiation in eubacteria. J Mol Biol. 1990;213(4):811–818.
- Saito K, Mattheakis LC, Nomura M. Post-transcriptional regulation of the str operon in Escherichia coli: ribosomal protein S7 inhibits coupled translation of S7 but not its independent translation. J Mol Biol. 1994;235(1):111–124.
- Yamamoto H, Wittek D, Gupta R, et al. 70S scanning initiation is a novel and frequent initiation mode of ribosomal translation in bacteria. Proc Nat Acad Sci. 2016;113(9):E1180–E1189.
- Qin B, Yamamoto H, Ueda T, et al. The termination phase in protein synthesis is not obligatorily followed by the RRF/EF-G-dependent recycling phase. J Mol Biol. 2016;428(18):3577–3587.
- Sarabhai A, Brenner S. A mutant which reinitiates the polypeptide chain after chain termination. J Mol Biol. 1967;27(1):145–162.
- Craigen WJ, Thomas Caskey C. Expression of peptide chain release factor 2 requires high-efficiency frameshift. Nature. 1986;322(6076):273–275.
- Weiss RB, Dunn DM, Dahlberg AE, et al. Reading frame switch caused by base-pair formation between the 3’ end of 16S rRNA and the mRNA during elongation of protein synthesis in Escherichia coli. EMBO J. 1988;7(5):1503–1507.
- Atkins JF, Weiss RB, Gesteland RF. Ribosome gymnastics — degree of difficulty 9.5, style 10.0. Cell. 1990;62(3):413–423.
- Larsen B, Wills NM, Gesteland RF, et al. rRNA-mRNA base pairing stimulates a programmed-1 ribosomal frameshift. J Bacteriol. 1994;176(22):6842–6851.
- Sharma V, Prère M-F, Canal I, et al. Analysis of tetra-and hepta-nucleotides motifs promoting -1 ribosomal frameshifting in Escherichia coli. Nucleic Acids Res. 2014;42(11):7210–7225.
- Silov S, Zaburannyi N, Anisimova M, et al. The use of the rare TTA codon in Streptomyces genes: significance of the codon context? Indian J Microbiol. 2021;61(1):24–30.
- Brown A, Shao S, Murray J, et al. Structural basis for stop codon recognition in eukaryotes. Nature. 2015;524(7566):493–496.
- Poole ES, Major LL, Mannering SA, et al. Translational termination in Escherichia coli: three bases following the stop codon crosslink to release factor 2 and affect the decoding efficiency of UGA-containing signals. Nucleic Acids Res. 1998;26(4):954–960.
- Trepanier NK, Jensen SE, Alexander DC, et al. The positive activator of cephamycin C and clavulanic acid production in Streptomyces clavuligerus is mistranslated in a bldA mutant. Microbiology. 2002;148(3):643–656.
- Jacks T, Madhani HD, Masiarz FR, et al. Signals for ribosomal frameshifting in the Rous sarcoma virus gag-pol region. Cell. 1988;55(3):447–458.
- Brierley I, Digard P, Inglis SC. Characterization of an efficient coronavirus ribosomal frameshifting signal: requirement for an RNA pseudoknot. Cell. 1989;57(4):537–547.
- Wills NM, Gesteland RF, Atkins JF. Evidence that a downstream pseudoknot is required for translational read-through of the Moloney murine leukemia virus gag stop codon. Proc Nat Acad Sci. 1991;88(16):6991–6995.
- Caliskan N, Katunin VI, Belardinelli R, et al. Programmed -1 frameshifting by kinetic partitioning during impeded translocation. Cell. 2014;157(7):1619–1631.
- Choi J, O’Loughlin S, Atkins JF, et al. The energy landscape of -1 ribosomal frameshifting. Sci Adv. 2020;6(1):eaax6969.
- Bhatt PR, Scaiola A, Loughran G, et al. Structural basis of ribosomal frameshifting during translation of the SARS-CoV-2 RNA genome. Science. 2021;372(6548):1306–1313. DOI:10.1126/science.abf3546.
- Loughran G, Firth AE, Atkins JF. Ribosomal frameshifting into an overlapping gene in the 2B-encoding region of the cardiovirus genome. Proc Nat Acad Sci. 2011;108(46):E1111–E1119. doi: 10.1073/pnas.1102932108
- Napthine S, Bell S, Hill CH, et al. Characterization of the stimulators of protein-directed ribosomal frameshifting in Theiler’s murine encephalomyelitis virus. Nucleic Acids Res. 2019;47(15):8207–8223.
- Sherlock ME, Higgs G, Diane Y, et al. Architectures and complex functions of tandem riboswitches. RNA Biol. 2022;19(1):1059–1076. DOI:10.1080/15476286.2022.2119017.
- O’Loughlin S. Programmed genetic decoding with an emphasis on translational bypassing. PhD thesis, University College Cork, 2020.
- Boyle KN. Investigating phage adaption to growth in Streptomyces. Master’s thesis, University of Oldenburg, 2012.
- Keenan T. Investigating a putative recoding event in the Streptomyces coelicolor phage, φhau3. Master’s thesis, The University of York, 2013.
- Coakley A. Genetic decoding dynamics: from its exploitation to enhance or rescue gene expression to a search for relevance for early evolution. PhD thesis, University College Cork, 2016.
- Eisenstein M. Seven technologies to watch in 2023. Nature. 2023;613(7945):794–797.