
ABSTRACT
Post-transcriptional regulation by RNA binding proteins can determine gene expression levels and drive changes in cancer cell proteomes. Identifying mechanisms of protein-RNA binding, including preferred sequence motifs bound in vivo, provides insights into protein-RNA networks and how they impact mRNA structure, function, and stability. In this review, we will focus on proteins that bind to AU-rich elements (AREs) in nascent or mature mRNA where they play roles in response to stresses encountered by cancer cells. ARE-binding proteins (ARE-BPs) specifically impact alternative splicing, stability, decay and translation, and formation of RNA-rich biomolecular condensates like cytoplasmic stress granules (SGs). For example, recent findings highlight the role of ARE-BPs – like TIAR and HUR – in chemotherapy resistance and in translational regulation of mRNAs encoding pro-inflammatory cytokines. We will discuss emerging evidence that different modes of ARE-BP activity impact leukaemia and lymphoma development, progression, adaptation to microenvironment and chemotherapy resistance.
Introduction
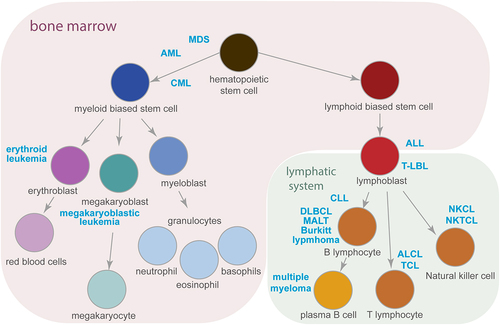
Haematopoiesis, the formation of blood cells of the haematopoietic lineage, occurs mostly in the bone marrow (). Erythrocytes, platelets, and immune cells originate from haematopoietic stem cells (HSCs). Asymmetric divisions of HSCs generate two daughter cells: one is a self-renewed HSC, and the other differentiates into a cell of the myeloid or lymphoid lineage. Cell fate is determined by counteracting processes such as cell cycle progression versus quiescence, cell death versus survival, and self-renewal versus differentiation. In turn, the microenvironment, influenced by cytokines, vesicles, and direct intercellular contact, controls these features. Dysregulation of the microenvironment impairs haematopoiesis, by causing selected cell types over- or under-representation observed in clonal haematopoiesis or bone marrow failure, respectively. The occurrence of genetic alterations disturbs cell physiology and initiates the development of malignancy [Citation1]. The uncontrolled proliferation of cells with mutations causes leukaemia and lymphoma. Different cancer types are classified by the cell type of origin within the haematopoietic lineage, as shown in . Leukaemia develops in the bone marrow, and lymphoma arises from (pre-)lymphocytes in various locations of the lymphatic system, reflecting the importance of the microenvironment in cell fate decisions.
Figure 1. Haematopoiesis and the development of haematological malignancies. A simplified schematic representation of haematopoiesis demonstrating the origin of myeloid and lymphoid lineages. Cancer types described in this review (marked in blue), develop from the cells at a particular step of haematopoiesis that is in the bone marrow (highlighted in purple) or in the lymphatic system (in green). Arrows indicate the direction of cell differentiation. See text for abbreviations.
Malignant transformation affects many aspects of cell physiology. Genomic mutations initiating malignancy result in the expression of oncogenes and/or dysfunction of cancer suppressors [Citation1]. This enhances proliferation and accelerates metabolism to meet the demands of the activated cells. The deregulated activity of signalling pathways results in aneuploidy, DNA damage, and the accumulation of reactive oxygen species. Changes in the microenvironment, such as hypoxia, inflammation, limited nutrient availability, acidification, or metalloproteinase release, contribute to cancer progression. Malignant transformation induces stress by perturbing cellular functions and microenvironment. The ability of cancer cells to adapt to these comprehensive changes plays an essential role in their power to survive and propagate.
The cellular stress responses are triggered by changes in internal and extracellular cell homeostasis. These cellular signalling pathways depend on the type of stress, resolve blocks to cell survival, and prevent activities that would enhance stress (). These mechanisms, used by non-malignant cells and hijacked by cancer cells, generally operate by regulating translation through the integrated stress response (ISR) to adapt to changes in metabolic balance [Citation2,Citation3]. The specific kinases that are sensitive to specific stresses and activate ISR are: amino acid starvation activates the general control nonderepressible 2 kinase (GCN2), heme-regulated inhibitor kinase (HRI) responds to iron deficiency, protein kinase R (PKR) is activated by viral infection, and protein kinase RNA-like endoplasmic reticulum (ER) kinase (PERK) activity is triggered by accumulation of unfolded proteins in the endoplasmic reticulum [Citation4]. These kinases converge on the eukaryotic initiation factor 2 alpha subunit (eIF2α), which binds and sequesters the limiting guanine nucleotide exchange factor eIF2B when phosphorylated [Citation2,Citation3]. Cellular stress also induces activation of mTORC, the mammalian target of rapamycin complex [Citation5]. This results in increased phosphorylation of eIF4E, another translation initiation factor. Consequently, cap-dependent translation of mRNA is inhibited, reducing the synthesis of most proteins.
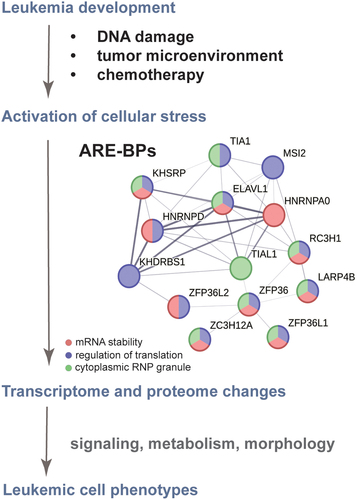
Figure 2. The ARE-BP axis is a functionally related network crucial for leukaemia development and chemotherapy resistance. Evidence-based interaction network retrieved from the string-db.org database. A full STRING network is presented; edges set to confidence (line thickness indicates the strength of data support); sources of interaction: experiments, co-occurrence, and co-expression; minimum required interaction score confidence set to low (0.15). GeneOntology enrichment analysis shows proteins involved in the regulation of mRNA stability (GOBP:0043488 in red), regulation of translation (GOBP:0006417 in blue) and localized in cytoplasmic ribonucleoprotein (RNP) granules (GOCC:0036464 in green).
Activation of ISR and mTORC induces autophagy that supports survival through protein turnover, cell remodelling and/or nutrient recycling [Citation6]. These changes enable cells to limit energy consumption and prevent the accumulation of unfolded nascent peptides. Moreover, phosphorylation of translation initiation factors induces the formation of RNA-rich subcellular compartments. In the cytoplasm, these biomolecular condensates are mainly processing bodies (p-bodies) and stress granules (SG) that stabilize and protect mRNA from degradation. Changes in translation initiation rewire the translation process to facilitate the synthesis of proteins necessary to cope with cellular stress, such as protein chaperones and transcription factors. Reduction of metabolic activity and protein synthesis enforces reduction of proliferation rate [Citation7] and can induce differentiation [Citation8,Citation9]. Thus, activation of stress responses could contribute to cancer cell survival by driving the establishment of a quiescent state and enabling changes in the proteome.
Severe cellular stress can lead to apoptosis, a cellular safety loop involving programmed cell death and the activation of the ATF4 transcription factor, which induces expression of the C/EBP homologous protein (CHOP) transcription factor. In turn, CHOP leads to the activation of a pro-apoptotic program, by modulating the transcription level of relevant genes. Consequently, this causes the inhibition of anti-apoptotic proteins BCL-2, BCL-xl, MCL-1 and stimulation of pro-apoptotic activity of Bax. The occurrence of mutations in some genes that elicit the development of AML (e.g., RUNX1) as well as post-translational modifications may, however, decrease ATF4-dependent pro-apoptotic activity of ISR [Citation10,Citation11]. When the apoptotic process is impaired the balance of cell fate can tip towards cancer development. Therefore, the hijacking of stress responses by cancer cells becomes a useful tool that enables adaptation, while the safety loop-free pathway provides a survival advantage.
RNA binding proteins (RBPs) provide post-transcriptional plasticity, inducing global changes in the proteome of leukaemic cells, including intracellular, transmembrane, or extracellular proteins that drive leukaemic cell phenotypes [Citation12,Citation13]. While transcriptional regulation supports some of these changes, post-transcriptional RNA processing events including precursor mRNA (pre-mRNA) capping, splicing, polyadenylation, RNA editing, and modification, as well as nuclear and cytoplasmic RNA decay are important determinants of gene expression levels [Citation14]. Human cells express approximately one thousand RBPs [Citation15]. Each RBP harbours one or more types of RNA binding domain that specifies sequence-specific binding to 5–7 nucleotides-long sequence motifs. RBPs regulate the processing of all cellular RNAs. The concentration of any given RBP is often kept in homeostasis through autoregulatory mechanisms [Citation16]. For example, many RBPs bind their own (pre-)mRNAs, creating positive and negative feedback loops that enhance or repress mRNA expression. In addition, RBPs bind to (pre-)mRNAs encoding other RBPs, enabling a network to form with multiple layered effects on the cell’s transcriptome and resulting proteome [Citation17–19]. Importantly, the cellular effect of silencing a particular RBP may be due to the perturbation of the entire network. Thus, impaired formation of a particular network might have a broad impact reflecting both direct and indirect targets of the RBP. Direct and indirect targets of RBPs are frequently functionally related, enabling RBP networks to regulate multiple steps within significantly complex pathways. Remarkably, individual RBPs (rather than RBPs at each step) are frequently key master regulators.
A broad range of technologies that crosslink RNA to protein (e.g. CLIP) have been employed to determine the precise sites of RBP interactions with (pre-)mRNA targets in cells [Citation20]. These approaches have helped to reveal that individual RBPs could be involved in specific processing steps for a particular set of RNAs harbouring a target motif. In this review, we focus on the role of RBPs that bind to AU-rich elements (AREs) within mRNAs. AREs are typically located in the 3’ untranslated regions (3’UTRs) and introns, and RBP binding can affect transcript stability and translation [Citation21,Citation22]. The development of ‘omics’ approaches has allowed the field to gain insight into global changes that accompany cancer development at both transcriptional and proteomic levels. Recent studies have demonstrated the significance of AREs and ARE-binding proteins (ARE-BPs) in leukaemia and lymphoma development, progression, and resistance to treatment. This constellation of ARE-BPs forms a distinct network, which we call the ARE-BP axis, with related activities shaping the leukaemic cell transcriptome and proteome (). We will highlight the newest outcomes – from high throughput RNA-sequencing, proteomics, and protein-RNA cross-linking-based approaches – that cause us to reconsider the functions of ARE-binding proteins and their networks in cancer biology.
Interactions of ARE-binding proteins with RNA targets and downstream effects
In this section, we will introduce proteins belonging to the ARE-BP family and present their activity towards the targets, pointing to their general role in the regulation of gene expression. In the subsequent sections, we will show how they trigger downstream effects that are important in either leukaemia or lymphoma, and chemotherapy resistance.
Early studies on mRNA stability identified the ARE consensus sequence motif as ‘WWWU(AUUUA)UUUW’ (where W stands for A or U). Later, studies of ARE-BP interactions with RNA contributed to our understanding of specific binding motifs (). Target sequence motifs were initially divided into two classes: the first encompasses several AUUUA motifs dispersed over the 3′UTR; the second contains multiple, overlapping copies of AUUUA [Citation23]. Subsequent analyses came up with a putative consensus sequence WUAUUUAUW [Citation24] and also added U-rich stretches to these elements [Citation25]. Another class is the constitutive decay element (CDE) in the 3’UTR of some transcripts, which is an AU-rich sequence forming a stem-loop structure [Citation26,Citation27]. Nearly 3300 mRNAs contain at least one consensus ARE-site in their 3’UTRs, constituting approximately 16% of all protein-coding transcripts [Citation24] and making this motif a key cis-regulatory element in 3’UTRs.
Figure 3. ARE-BPs recognize sequences in RNA called AU-rich elements (AREs). A) Sequence motif logos representing the most frequent nucleotide in each position recognized by the indicated ARE-BP from the ENCORE/ENCODE project database for cell-free pulldown experiments (RNA bind-n-seq). B) Results from crystal structure analysis of RNA binding domains (in green/blue) of selected proteins in complex with ARE (in brown). From the left are the two N-terminal RRM domains of HuR bound to poly(U) RNA from crystal structure PDB ID: 4ED5; the ROQ domain from murine roquin-1 bound to a 23-mer tnf-CDE RNA from X-ray structure PDB ID:4QI2; the KH domains of T-STAR dimer bound to AAAUAA RNA from structure PDB ID: 5ELS.
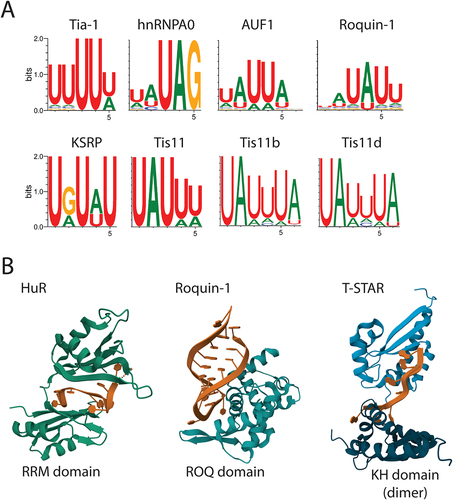
RNA binding proteins usually bind to sequence elements in RNA via various protein domains, including RNA Recognition Motifs (RRMs), La Motifs (LAMs), zinc fingers (zfs), K Homology domains (KH), and the roquin domain (ROQ). The combination of the structural components provides specificity to sequence recognition and activity upon binding. A summary of structural features described in this review is presented in . Examples of selected RNA binding domains observed in interaction with AU-rich elements are shown in . Characteristics of each RNA-binding domain have been reviewed in detail elsewhere [Citation61].
Table 1. Summary of information about the ARE-binding proteins discussed in this review. Provided references cite studies of the ARE-BP in the context of leukaemia or lymphoma. See text for abbreviations. *direct link to cancer development has not been shown.
Genome-wide RNA binding studies provided major clues to the mechanism of action of different ARE-BPs. Initial research was mostly focused on studying small numbers of mRNA targets and the cellular consequences of their dysregulation. Technological developments, allowing the determination of sites of RNA-protein interaction, yielded a broader view of specific ARE-BP RNA targets and the motifs bound in cells (, ). Such approaches helped establish the nuclear activities of ARE-BPs in alternative pre-mRNA splicing and nucleo-cytoplasmic transport, which is regulated by post-transcriptional modifications of ARE-BPs. Methods based on the enrichment of proteins with cross-linked RNA, like CLIP or its derivates, elucidated the role of overlap among trans-acting factors. In the following part of this section, we evaluate specificity and redundancy relative to different ARE-BP functions.
Tia and Tis proteins
In the case of the family of Tia (T-cell induced antigen) proteins, the difference between Tia-1 and Tia-like-1 (TIAL1 or TIAR) paralogs is elusive. Through RRM domain the proteins bind to thousands of transcripts in cells and interact with the same binding sites in the target [Citation62]. Double knock-out resulted in the accumulation of aberrantly spliced transcripts that otherwise were subject to nonsense-mediated decay [Citation62]. In vitro studies showed that Tia proteins can bind U-rich intronic sequences immediately downstream of 5’splice sites [Citation63] and promote the recruitment of U1 small nuclear ribonucleoprotein (U1 snRNP) at 5’ splice sites [Citation64]. This impacts the inclusion of exons in transcripts and changes alternative splicing patterns and was later confirmed in the cellular context [Citation65,Citation66]. Members of the Tis protein family, Tis11 (known as tristetraprolin, TTP), Tis 11d and Tis11b, also share similar motifs and a significant portion of targets that they bind by the zinc finger CCCH (zf-CCCH) type domain. This redundancy may play a crucial role in securing their activity.
HuR and AUF1
HuR is a widely expressed member of a family of ARE-BPs generally referred to as Hu proteins. The HuR binding motif was identified in the 3’UTRs and/or introns in the RNA target; in almost half of the cases of transcripts, the motif is present in both. Upon HuR knock-down, reduced protein synthesis correlated with reduced levels of corresponding transcripts, including pre-mRNA transcripts containing intronic HuR binding motifs [Citation25]. This showed that HuR may be required at early steps of mRNA maturation, coupling alternative splicing of pre-mRNA with stabilization of mRNA [Citation25,Citation67], enabling mRNA export to cytoplasm and translation. High throughput cross-linking-based studies showed that HuR binding sites overlap with Ago2/miRNA binding [Citation68]. Regulation of particular mRNAs by HuR and Ago2 was dependent on the distance between their binding sites in the transcript, suggesting that HuR inhibits Ago2 binding [Citation67,Citation68], and indicating possible dependencies between the stability and decay of transcripts. Competition for binding sites among ARE-BPs might play a role in the regulation of alternative splicing and transcript stability. Such crosstalk was revealed in the case of HuR and the AU-rich element-binding factor 1 (AUF1) binding to transcripts. AUF1 recognizes U-rich and GU-rich motifs to either upregulate mRNA translation or target them for degradation. HuR and AUF1 proteins share many targets, that they bind by the RRM domain (), and their cooperative binding increases the translation rate of selected transcripts [Citation69].
hnRNPA0
The heterogeneous nuclear ribonucleoprotein A0 (hnRNPA0) binds to heterogeneous nuclear RNA (hnRNA) by RRM domain [Citation70]. The protein bound to pre-mRNA is involved in the formation of complexes that regulate RNA processing, stability, transport from the nucleus, and translation; however, its cellular activity has not been extensively studied [Citation71].
KSRP, Sam68, T-STAR
KH-type splicing regulatory protein (KSRP) is a single-stranded nucleic acid (DNA or RNA)-binding protein [Citation72]. KSRP binds to active promotors in DNA and intronic GU-rich sites in RNA. The activity of this protein contributes to the consistency of transcription and alternative splicing regulation [Citation58]. In the cytoplasm, KSRP directs transcripts to degradation upon binding to ARE in the 3’UTR [Citation73]. Other examples of KH-type splicing proteins are Src-associated substrate during mitosis of 68 kDa (Sam68), also known as KH domain containing RNA binding signal transduction associated protein 1 (KHDRBS1), and T-STAR/SLM2 (testis-signal transduction and activation of RNA/Sam68-like mammalian protein 2). The proteins are ARE-BP that recognize polyA or bipartite (A/U)AA-(N > 15nt)-(A/U)AA motif [Citation74]. They possess a single RNA binding domain that upon binding to RNA dimerize, due to the QUA1 domain, which positions the two RNA motifs very close in an anti-parallel orientation (). The dimerization is essential for Sam68 and T-STAR mediated splicing [Citation74].
MSI2, LARPs
Musashi 2 (MSI2) protein by RRM domain binds mRNA targets with the consensus motif ‘UUAG’ in 3’UTRs and intronic regions and thereby regulates the expression of a broad range of genes [Citation75]. La-related protein 4B (LARP4B) is an ARE-BP containing a La motif that stabilizes transcripts by targeted interaction with the 3’UTRs of almost 8000 mRNAs [Citation76]. In the nucleus, some LARPs (La and LARP7) function as tRNA chaperones by binding 3’ end U-tracts and preventing misfolding. In the cytoplasm LARP1, 4, 4B, and 6 bind mRNA polyA tails directly or through interaction with cytosolic poly(A)-binding protein (PABPC1), contributing to stability and translation regulation [Citation77].
Roquin-1, Regnase-1
Roquin-1 and Regnase-1 proteins belong to ARE-BP that recognize the ARE stem-loop motif and share similar targets but act based on different mechanisms and at distinct locations [Citation78]. They bind targeted RNA by different domains, which are the zf-CCCH domain in Regnase-1 and the roquin (ROQ) domain in Roquin-1 (). Regnase-1 is a ribonuclease that acts on endoplasmic reticulum-bound ribosomes inducing degradation of translated mRNAs in cooperation with up-frameshift protein-1 (UPF1) helicase. Roquin-1 is recruited to processing-body/stress granules and induces translationally inactive transcript decay in a UPF1-independent manner [Citation79].
ARE-BPs in response to cellular stress form nucleoprotein complexes. Tia-1, TIAR, and HuR were shown to participate in the formation of stress granules [Citation80]. Cytoplasmic stress granules are sites of mRNA triage, where transcripts are stabilized; excluded mRNAs can be targeted for translation or degradation. Upon activation of cellular stress Tis11, Tis11d, and Roquin-1 join to stress granules and p-bodies to facilitate de-capping and decay of transcripts by recruiting the Ccr4-Caf1-Not deadenylase complex, the decapping enzyme Dcp2 and the exosome [Citation81]. Interestingly, Tis11b in a soluble form, like other Tis proteins, facilitates the decay of transcripts. Upon assembly, Tis11b forms gel-like TIS granules that contact endoplasmic reticulum membranes and facilitate translation [Citation82,Citation83]. The role of TIS granules in the context of stress response is not clear yet.
The network of ARE-BPs regulates various aspects of RNA biology from RNA stability to targeted degradation and translation to the formation of cellular sub-compartments. Recent studies of ARE-BP-mRNA interactome provided recognition of cellular contexts, in which activity of the ARE-BP could play a role. Targets of ARE-BP encode proteins that orchestrate processes critical in the view of haematopoiesis. Some of these are essential in myeloid while others in lymphoid skewed cells, originating from myeloblasts and lymphoblasts, respectively. In the following two sections of this review, we will discuss how the activity of ARE-BP network contributes to haematological malignancy development, considering separately the two main cell lineages. Some of the described examples are schematized in .
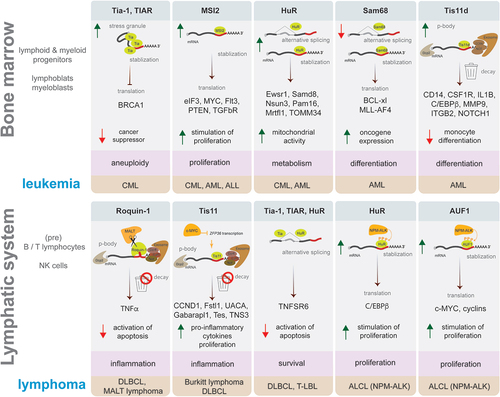
Figure 4. Examples of ARE-binding proteins role in leukaemia and lymphoma development. Schematic representation in each box presents an example of ARE-BP activity discussed in this review that affects a process supporting malignancy (highlighted in pink), which plays a role in the development of leukaemia (upper panel) or lymphoma (lower panel) subtype (highlighted in brown). ARE-BPs change the level of proteins causing stimulation (green arrow up) or inhibition (red arrow down) of cellular processes, by modulating RNA processing like alternative splicing, stabilization, decay, translation, and formation of stress granules or p-bodies. Key to symbols: mRNA decay (trash can), ARE-BP (green), oncogene (dark yellow), CCR4-NOT deadenylase complex with exosome (brown), Dcp2 decapping enzyme (grey), AU-rich element (red line), mRNA 5’ cap (grey dot).
Stress and ARE-BP-axis in the development of myeloid malignancies
Haematopoiesis in bone marrow (see ) is controlled by the balance of extracellular signals. Activation of surface receptors by a repertoire of cytokines stimulates the expression of genes that direct cell proliferation and differentiation in a specific path. During the differentiation process, cells acquire the ability to perform effector functions, such as the secretion of antibodies, and may gain or lose the potential to divide. In the event of differentiation process disturbance, activation of apoptosis protects against the multiplication of improperly maturating cells. Disruption of this mechanism is a key cause of cancer development. If accompanied by impaired control of dormancy and proliferation, this increases the number of disordered cells. Regulation of basic processes controlling cell fate decisions is therefore essential for proper haematopoiesis. This could be affected by inherited genetic alterations, like mutations in ribosomal proteins that cause Diamond – Blackfan anaemia. Hematological malignancies are driven by genetic changes in haematopoietic cells that lead to dysregulated expression of proteins controlling haematopoiesis.
Apart from mutations, disturbed cytokine signalling, and inflammation are the main driving forces behind the development of haematologic malignancies arising in the bone marrow. There are several main inflammatory cytokines produced by various cells such as TNF-α, interleukins IL-1α, IL-1β, IL-6, IL-8, IL-12, IL-17, interferon-gamma (IFNγ) [Citation84]. Cytokines act in a coordinated manner and are involved in both the initiation and resolution of inflammation. Dysregulation of cytokine production or signalling affects the entire immune network, leading to chronic inflammatory conditions and autoimmune diseases. Chronic de-regulated level of inflammatory cytokines is postulated to be one of the causes of bone marrow failure, as summarized in a recent review [Citation85]. Infection-triggered innate immunity can impact not only the maturation of lymphocytes but also haematopoiesis. Lymphocytes and HSPCs have a similar repertoire of surface receptors responding to infection. An appropriate balance of immune stimulation by inflammatory signals orchestrates differentiation and self-renewal of stem cells in the bone marrow. Immune signalling plays a so-called hormetic role in the regulation of the HSPCs proliferation, in low doses it stimulates cell proliferation but disrupts the cellular microenvironment in high dose [Citation86]. Stimulation of immune receptors induces exit from a quiescent state and proliferation. This enhances cell metabolism causing increased levels of reactive oxygen species (ROS). Accumulation of ROS causes DNA damage that can lead to genetic alterations. This is one of the reasons why mitochondria dysfunction that induces the generation of ROS is postulated to be the main driver of myelodysplastic syndrome (MDS) [Citation87], which is a prerequisite of acute myeloid leukaemia (AML) (see ).
Immune stimulation of surface receptors leads to activation of integrated stress response and stimulation of PKR in HSCs. Activation of stress response plays a pro-survival role in HSCs [Citation88]. Many examples demonstrate that this signalling is hijacked by cancer cells. Increased activity of PKR was observed in the MDS [Citation89] and contributed to AML and acute lymphoblastic leukaemia (ALL) development [Citation90]. In line with these findings, elevated expression of PKR in AML cells correlates with poor prognosis [Citation91]. Altogether, this implicates immune stimulation stress-triggered response in the haematologic malignancy development.
Activation of stress responses could be evoked by the expression of an oncogene and accompanies the development of leukaemia. The activity of oncokinase Bcr-Abl1 in a murine model of chronic myeloid leukaemia (CML) perturbs calcium homoeostasis and leads to mild activation of UPR, which is enhanced upon CML progression from chronic to a more aggressive phase, called blast crisis [Citation92,Citation93]. Activation of stress response supported the survival of haematopoietic stem cells and myeloid progenitor cells and correlated with better engraftment of primary AML cells in a mouse model [Citation88]. Blocking stress response activation by small molecule integrated stress response inhibitor (ISRIB) in stressed CML cells sensitized them to imatinib treatment mediated by decreased activation of STAT5 as well as Ras/Raf/MAPK and IFNγ signalling [Citation94]. Another study demonstrated the impact of stress response and stress granule formation on the activity of splicing factors in the context of AML [Citation95]. The authors found that U2AF1 bearing mutations, which are known drivers of AML, regulate splicing of a pool of mRNAs which is enriched in transcripts encoding proteins regulating RNA biology and stress granules formation. Moreover, cells expressing mutated splicing factor U2AF1 were more prone to form SGs in response to stress inducers such as arsenite. The ability to form stress granules in the cells was blocked by ISRIB, which reduced the survival of AML cells upon arsenite-induced stress. Taken together, these studies demonstrate the connection between stress, SG formation, regulation of alternative splicing, and potential for survival of AML cells.
The formation of stress granules by ARE-binding proteins impacts the translation rate of some key cancer suppressors. For instance, UPR induced by Bcr-Abl1 expression and increased formation of stress granules reduced the level of BRCA1 protein accompanied by an unaffected level of corresponding mRNA. In this context, HuR regulated the stability of BRCA1 mRNA, and TIAR had an impact on translation initiation; both effects depended on the recognition of the AU-rich element in the 3’UTR () [Citation28,Citation41]. This activity of ARE-binding proteins was also demonstrated in activated B lymphocytes. Induced formation of stress granules triggered the binding of p53 transcript by Tia-1 protein at the 3’UTR, inhibiting translation of the mRNA but not affecting its level in the cells. Dissociation of the mRNA from Tia-1 complexes reinitiated the translation of p53 mRNA [Citation96]. Thus, ARE-BPs impact the fate of mRNAs encoding cancer suppressors by regulating their stability and translation, thereby dictating specific proteomes that contribute to cancer development.
ARE-BPs impact cancer development and progression by modulating the synthesis of proteins that play key roles in processes detrimental to cell survival. Below we summarize recent findings that implicate SG-independent activities of the ARE-BPs in the development of myeloid malignancies. Selected examples are presented in (upper panel).
HuR
HuR regulates a broad spectrum of targets implicated in various cellular functions. HuR activity affected alternative splicing and upregulated expression of proteins involved in processes related to metabolism, translation, and mitochondrial respiration (such as Ewsr1, Samd8, Nsun3, Pam16, and Mrtfl1, TOMM34), while downregulated – markers of myeloid differentiation () [Citation29]. The range of targets and their role in the regulation of cellular processes important for haematopoiesis implicates the significance of HuR in the view of leukaemia development and progression. Silencing of HuR or blocking its interaction with RNA by MS-444 caused reduced clonogenic potential of AML cells [Citation29], and reduced activation of macrophages, decreasing chemotaxis and pro-inflammatory cytokine release [Citation97]. Apart from that, HuR-mediated stabilization of target transcripts stimulated proliferation and inhibited apoptosis in model AML cell lines [Citation30]. Post-transcriptional regulation by HuR is implicated in NF-κB and JAK/STAT signalling, which is an essential component in haematological malignance development and resistance to chemotherapy [Citation98,Citation99]. The spectrum of HuR targets encompasses transcripts for factors regulating RNA biology and transcription, suggesting a master-regulatory role of HuR [Citation67]. HuR targets a network of 32 RNA-binding proteins critical for leukaemia development, indicating the significance of HuR in cancer development. Increased HuR mRNA level was detected in human primary CML cells progressing to the blast crisis phase, in AML cells with different genetic driving backgrounds and upon cancer relapse [Citation29]. Altogether, alteration in HuR expression might be driven by changes in cellular state evoked by oncogene and seem to support the aggressive phenotype of leukaemia.
MSI2
Transcript stability and translation play a role in various types of leukaemia and involve several other members of the ARE-BP axis. MSI1 and MSI2 control the post-transcriptional expression of genes playing detrimental roles in the development of various cancers, such as MYC, Flt3, PTEN, TGFβR, and Smad3 (), which have been reviewed elsewhere [Citation100]. Moreover, MSI2 regulates post-transcriptional signalling of EGF and HGF pathways that lead to activation of JAK/STAT, PI3K/AKT as well as ERK/MAPK kinases that orchestrate cell death, DNA repair, proliferation, and survival [Citation75]. Thus, all the processes are key for haematopoiesis and malignancy development. More recently it was shown that acute MSI2 downregulation impacts translation of eIF3 [Citation42]. Thus, the overexpression of MSI2 in leukaemic cells could drive changes in the translation of just several factors that in turn enhance the effect of RBP activity [Citation42]. Overexpression of MSI proteins has been observed in various cancers including leukaemia CML, AML, and ALL [Citation43–48]. Silencing of MSI2 blocked the proliferation of primary AML cells and led to increased apoptosis accompanied by a reduced level of anti-apoptotic Bcl2 and increased pro-apoptotic Bax, demonstrating the significance of the protein in AML cell survival [Citation46].
LARP4B
Given the broad range of transcripts stabilized by LARP4B, its main function could be related to the stabilization of transcripts relevant to ‘house-keeping’ functions; an interesting example is the target RALY, an RNA binding protein regulating splicing, EXOSC3 and Glyoxalase 1 [Citation76]. The relevance of LARP4B in leukaemia is not well understood. Depletion of LARP4B inhibited the development of leukaemia and prolonged the survival of the AML murine model (MLL-AF9 mice) by reducing the number of leukaemia stem cells [Citation60]. Yet the large number of LARP4B targets leaves many open questions.
Sam68
The landscape of cellular interactors could determine the activity of RBP, what can also play a role in the view of leukaemia development. This has been observed in the case of Sam68 protein, which regulates the stability and translation of mRNA. For instance, it binds to the MLL-AF4 oncogene transcript and thereby inhibits its translation (). This activity of Sam68 abolished the development of AML/MLL-AF4 leukaemia in a mouse model [Citation55], implicating the anti-cancer role of Sam68. The level of Sam68 was significantly increased in AML patient samples, but this difference did not correlate with clinical outcomes [Citation56]. This discrepancy could result from the fact that the protein activity is modified in a phosphorylation-dependent by various kinases, which might not be present in murine cells.
hnRNPA0
Coupling of transcripts alternative splicing, stability, and translation regulation is also attributed to hnRNPA0. It is expressed at high levels in haematopoietic stem cells and gradually decreases during cell differentiation. Knockdown of hnRNPA0 in a murine model cell line changes the differentiation pattern of monocytes towards granulocytes. The gene encoding hnRNPA0 is located on chromosome 5 in the region that is frequently deleted in MDS and AML. Del(5q) causes broad changes in ARE-regulated levels of transcripts that encode proteins involved in cell cycle regulation, MAPK, and chemokine signalling [Citation33].
Tis proteins
Timely decay of transcripts also plays a significant role in the regulation of cellular processes. Tis11d is upregulated in AML patient samples versus healthy haematopoietic progenitors [Citation18]. A CRISPR-Cas9 screen revealed that Tis11d silencing induced the differentiation of AML cells. Interaction of Tis11d with transcripts encoding proteins promoting myeloid differentiation (like CD14, CSF1R, C/EBPβ, Il-1β, MMP9, ITGB2, NOTCH1) resulted in suppressed differentiation of AML cells () [Citation18]. Interestingly, an inverted correlation was found between the level of Tis11d and its paralogs, Tis11, and Tis11b, that directed the degradation of transcripts encoding the same protein products (i.e. in an auto-regulatory loop). Silencing of Tis11d reduced the clonogenic potential of AML cells in vitro and diminished leukaemia development in a mouse model [Citation18].
The formation of TIS granules by Tis11b could play a role in the acquisition of immune evasion potency by cancer cells. The gel-like granules create a unique environment facilitating the translation of integrin-associated protein (CD47) and its interaction with SET protein for plasma membrane inclusion [Citation82,Citation83]. CD47 plays a role in the ‘don’t eat me’ signal, recognized by the signal-regulatory protein alpha (SIRPα) receptor on macrophages. This myeloid cell immune checkpoint determines survival and CD47 is upregulated in haematopoietic progenitor, MDS, and AML cells that are extravasated to the circulation [Citation101]. The surface level of CD47 was dependent on Tis11b expression [Citation83], implicating the protein activity in support of leukaemia development.
Regnase-1
In haematopoietic stem and progenitor cells, Regnase-1 plays a crucial role in regulating the decay of Gata2 and Tal1 mRNA, key players in cell self-renewal. A heightened level of Regnase-1 is essential for maintaining the quiescent state of progenitor cells in the bone marrow, while the absence of Regnase-1 results in the excessive accumulation of immature HSPCs skewed towards granulocytes. As a result, mice with a Regnase-1 knock-out exhibit decreased levels of lymphocytes. Furthermore, similar changes in gene expression patterns were observed in Regnase1 null CD34- cells and AML patient samples. Regnase-1 oversees the decay of transcription factors Gata2 and Tal1, effectively orchestrating haematopoiesis. Both overexpression and knock-down of Regnase-1 have profound effects on haematopoiesis, leading to the development of MDS and AML [Citation54].
Stress and ARE-BPs in the development of lymphoid malignancies
Targets of ARE-BP are implicated in the development of lymphoid malignancies (). The AU-rich element is in transcripts for cytokines and other proteins that regulate lymphocyte immune response. In the 3’UTR of some key regulatory factors such as tumour necrosis factor α (TNFα), there is an additional AU-rich sequence folded in a stem-loop [Citation26,Citation102]. A study of childhood leukaemia revealed that apart from genetically inherited predisposition, it is the exposition to immune stimulation and dysregulated secretion of cytokines that is a major cause of B cell-derived leukaemia [Citation103]. Chronic inflammation in the lymphatic system contributes to the development of lymphoma. The inflammatory microenvironment increases leukocyte mobility, and propagation and directs differentiation [Citation104]. Activation of lymphocytes (T and B cells) involves a complex cascade of signalling events that ultimately leads to changes in gene expression. Increased transcription in activated cells is attributed to the stimulation of transcription factors such as nuclear factor-kappa B (NF-κB), activator protein-1 (AP-1), nuclear factor of activated T cells (NFAT) and CCAAT/enhancer-binding protein β (C/EBPβ). Translation of pro-inflammatory cytokines in leukocytes is regulated by ARE binding proteins [Citation105,Citation106]. The activity of ARE-BPs also determines the stability and decay of inflammation-related transcripts in leukocytes [Citation78,Citation107]. During inflammation, leukocytes may experience an increased load of mRNAs, and the cell responds by sequestering these mRNAs into stress granules.
The formation of stress granules in leukocytes is part of the broader cellular response to inflammatory signals, and it contributes to the regulation of gene expression and the control of immune responses [Citation108]. For instance, Tia-1 and TIAR-mediated recruitment of IL-1β to SGs suppressed the cytokine expression [Citation109]. SGs prevent excessive innate immune activation protecting lymphocytes from TNFα/NFκB induced cell death [Citation110]. In line with this, impaired stress granule formation leads to hyperactivity of RIG-I-like receptors stimulated upon viral infection [Citation110]. The protective activity of SGs could be hijacked in NK/T cell lymphoma (NKTCL), which is characterized by a high level of Tia-1 expression [Citation40].
Since inflammation is one of the critical factors contributing to the development of lymphoma, the activity of ARE-BPs controlling the response of lymphoid cells to inflammatory stimulation is a significant factor. Below we provide examples of the role of selected ARE-BPs in the context of lymphoma development and highlight activities that are independent of stress granules and p-bodies (, lower panel).
Roquin-1 and Regnase-1
Reducing the production of pro-inflammatory cytokines is attributed to the activity of Roquin-1 and Regnase-1. The cooperative activity of Roquin-1 and Regnase-1 keeps immune-signalling homoeostasis by targeting transcripts encoding pro-inflammatory factors ICOS, CREL, IL-6, NFκB, and cytokines. Central in the activation of immune cells is mucosa-associated lymphoid tissue lymphoma translocation 1 (MALT1) paracaspase, which by proteolytic cleavage at Arg residue suppresses the activity of Roquin-1 and Regnase-1 () [Citation52]. The activity of Regnase-1 is critical in T cell development. In a transgenic mouse model having Regnase-1 mutated at the site of MALT1 cleavage, maturation of T cells was affected resulting in lymphopenia. This effect was caused by impaired T cell receptor (TCR) signalling that resulted in a lack of survival signalling for positive selection of cells in the thymus [Citation111]. Regnase-1 is also involved in the regulation of B cell maturation. Specific knock-out in B cells resulted in the accumulation of immature B cells and increased level of antibodies detected in the sera, which could result from perturbed peripheral differentiation of B cells upon lack of Regnase-1-dependent decay of Tis11b and transcription factor BATF [Citation112]. In DLBCL and MALT lymphoma, the paracaspase is constitutively active [Citation53] leading to a chronic inflammatory microenvironment that contributes to the lymphoma development.
Tis11
Tis11 protein is also involved in the modulation of immune signalling. In T lymphocytes, Tis11 tunes the expression of effector cytokines involved in TCR complex signalling, and Tis silencing leads to a dysregulated cytokine release () [Citation113]. Expression of many ARE-containing genes, as well as ARE-BP, is controlled by the c-Myc transcription factor. The c-Myc oncogene directly suppresses the expression of Tis11, positioning the protein as a potent cancer suppressor. In line with this, restoration of Tis11 overturns malignant phenotype in a mouse model [Citation49]. Overactivation of c-Myc accompanies various types of B lymphoma, including Burkitt lymphoma and DLBCL [Citation114].
Tia-1, TIAR, and HuR
Tia-1, TIAR, and HuR proteins play a key role in regulating the splicing of Fas, the TNF receptor (TNFSR6), a transmembrane protein () [Citation115,Citation116]. The cytoplasmic cell death domain of Fas induces apoptosis upon TNFα signalling. Alternative splicing produces an isoform of Fas that is solely cytoplasmic, effectively blocking the pro-apoptotic activity of Fas. This occurs when one of the exons is skipped during splicing. HuR promotes exon skipping, thereby antagonizing the activity of the Tia protein. Supported by TIAR or Tia-1, the inclusion of this exon in the transcript increases the level of transmembrane Fas expression. This suggests that the regulation of alternative splicing by Tia proteins plays a crucial role in determining lymphocyte survival. A reduced level of Fas contributes to the progression of T cell lymphoblastic lymphomas (TCL) and reinforces resistance to chemotherapy [Citation117]. Fas plays a role in the maturation of B cells, and the loss of its activity contributes to the development of aggressive DLBCL [Citation118].
The development of lymphoblastic leukaemia or lymphoma is driven by abnormal differentiation of lymphoid precursors along with increased proliferation. Knock-down of either HuR or Tia-1 impacts not only immune signalling but also the maturation of B and T lymphocytes [Citation96,Citation119–121]. This results from post-transcriptional control that the proteins are involved in the cytoplasm as well as the nucleus. Studies using HuR knock-out mouse models revealed that HuR’s activity is critical for B lymphocyte functioning in the immunological system, but not for their development or differentiation [Citation119]. Activation of primary B cells increases HuR level, which impacts cell cycle and energy metabolism through alternative splicing. HuR-mediated regulation of splicing and stimulation of translation of selected mRNA supported the proliferation of activated B cells [Citation119]. A recent study demonstrated that Tia-1 represses pluripotency induction during B cell reprogramming by altering the expression of splice isoforms encoding proteins relevant for cell fate decisions [Citation122]. Similarly, the activity of KSRP is implicated in cell differentiation. A recent study showed that KSRP protein localized in the nucleus combines regulation of transcription and alternative splicing of targets that play a pivotal role in the coordination of cell differentiation [Citation58]. The findings demonstrating the role of ARE-binding proteins in the regulation of leukocyte cell fate decisions indicate the significance of the ARE-BP-axis in the early steps of lymphoma development.
The ARE-BP axis contributes to lymphoma development by modulating the level of proteins regulating proliferation. HuR protein was implicated in the expression of a discrete number of transcripts of signalling proteins relevant to the development of various cancers. In the context of lymphoid malignancies, it seems important that HuR regulates the stability and translation of C/EBPβ () [Citation31], a transcription factor controlling the proliferation of lymphoblasts. B cell receptor (BCR) signalling that activates C/EBPβ contributes to the development of B cell acute lymphoblastic leukaemia (B-ALL) [Citation123]. An RNAi-based screen of cells positive for the anaplastic large cell lymphoma (ALCL) main driver, nucleophosmin-anaplastic lymphoma kinase (NPM-ALK), demonstrated that transcription factors C/EBPβ and Bcl2A1 were necessary for cell transformation and to sustain the growth and survival of ALCL cells [Citation124]. The NPM-ALK mediated phosphorylation of HuR enhanced its association with polyribosomes increasing translation of C/EBPβ post-transcriptionally [Citation31].
AUF1
In cooperation with HuR, AUF1 increases the translation of transcripts that preserve chromosome function and maintain DNA integrity [Citation69]. Independently of HuR, AUF1 inhibits cellular senescence by reducing p16 levels and delays the ageing phenotype, by regulating TOP2 and TERT mRNA translation. Intriguingly, AUF1 protein in a phosphorylation-dependent manner can exhibit contradictory effects on transcript stability and direct mRNA to either decay or translation [Citation35,Citation36]. AUF1 is hyperphosphorylated by NPM-ALK onco-kinase in anaplastic large-cell lymphoma. The proteins AUF1 and NMPK-ALK were detected in the same cytoplasmic nucleoprotein granules, and hyperphosphorylation of AUF1 led to increased stability of the ARE-regulated transcripts, including those encoding c-Myc and several cyclins (). This implicated AUF1-mediated stabilization of transcripts in lymphoma development and progression [Citation35] and increased survival of ALCL cells upon transcription inhibition by actinomycin treatment [Citation37]. Apart from transcript stabilization, AUF1 was considered to contribute to the changes in alternative splicing in DLBCL [Citation38]. Interestingly, a mutation in the SF3B1 splicing factor in MDS cells results in the expression of alternative isoforms of AUF1 [Citation39]. The role of the AUF1 isoforms in splicing regulation remains unclear but these observations point to a potential involvement of AUF1 in the interplay of splicing and transcript stability regulation in the context of cancer development.
ARE-BP-axis in therapy resistance of haematological malignancies
The treatment of haematologic malignancies revolves around the administration of drugs designed to target rapidly dividing cells, eliciting cytotoxic effects. Traditional chemotherapeutics, initially developed for other cancer types, form a cornerstone of this approach. In addition, targeted strategies are employed to suppress the activity of enzymes that are overly active in cancer cells. Another avenue involves the emerging use of antibodies to block immune checkpoints, thereby enhancing the immune system’s recognition of cancer cells. However, significant challenges, such as therapy intolerance, adverse effects, and the development of resistance, impede the path to achieving effective cancer treatment.
The crux of cancer survival during therapy lies in the cellular plasticity afforded by the regulation of gene expression. Post-transcriptional regulation, particularly, plays a pivotal role in the integrated response to adverse conditions created by the treatment. Chemotherapy can trigger an integrated stress response, as elaborated elsewhere [Citation80]. To enhance chemotherapy’s efficacy, a proposed strategy involves combining ISR inhibition with targeted therapy. Below we will discuss how various steps of RNA processing, pointing to the role of ARE-BP, could support cell adaptation to chemotherapy and thereby survival.
Modulation of proteome in hypoxic microenvironment
The therapeutic challenge of successful leukaemia treatment comes from the fact that leukaemic cells function in various environments. Leukaemia develops in the bone marrow. When the disease progresses, malignant cells become more aggressive and intensively extravasate. In the case of CML, this advanced phase is called blast crisis. Cells that actively proliferate and metabolize while circulating in the bloodstream are more vulnerable to treatment with drugs targeting metabolic pathways, blocking DNA replication, or translation. Cells that reside in hypoxic niches of the bone marrow or lymph nodes remain insensitive to many therapeutics because the environment enforces quiescence and reduced metabolic activity [Citation125].
Microenvironment determines the proteome of CML cells [Citation12]. In an experimental setup mimicking hypoxic bone marrow conditions cell population growth was suppressed. This was accompanied by a mild induction of eIF2α phosphorylation, substantial reduction in polyribosomes, and decreased translation rates. Hypoxia also changed the selectivity of translation modifying nascent proteome (newly synthesized peptides/proteins). Proteomic data revealed profound changes in the activity of metabolic and signalling pathways that can support the therapy resistance of CML cells. The activity of RNA binding proteins modulated the preference of transcript translation in these conditions. TIAR and FRMP regulated translation of a non-overlapping set of mRNAs, in line with the finding that FMRP/p-bodies and stress granules target transcripts relevant for different cellular processes [Citation126]. This supports a model whereby RBPs determine proteomic outcomes in response to stimuli such as stress. TIAR-mediated regulation had an impact on the expression of factors regulating genome stability and cell cycle progression [Citation12]. Silencing of TIAR supported fitness of CML cells upon inhibition of translation by homoharringtonine treatment in regular cell culture conditions [Citation12], in line with the previous suggestions that Tia proteins play a cancer suppressor role [Citation127]. On the other hand, in hypoxic bone marrow conditions, CML cells with TIAR knock-down were more sensitive to treatment with a ribotoxin homoharringtonine. These data indicated that changes in translation regulation and initiation could play a role in chemotherapy resistance in the bone marrow by enforcing the quiescence of leukaemic cells [Citation12]. Recent data show that TIAR plays a pivotal role in response to ribotoxins-induced p38-MAPK signalling, which arrests cells in the G1 phase [Citation128]. Thus, in hypoxic conditions that stimulate quiescence, the activity of TIAR in leukaemic cells could be critical for maintaining the cell cycle arrest and resistance to ribotoxins. Disabling this resistance mechanism in CML cells by TIAR knock-down led to the observed increased sensitivity under hypoxia to ribotoxins [Citation12].
Establishing of quiescence by modulation of mRNA decay
Transitioning into a quiescent state or differentiating into another cell type can drive therapy resistance by altering the activity of transcription factors, signalling pathways, and metabolic circuits. These modifications can make cells less responsive to the applied treatment. The direction of mRNA degradation by Tis proteins plays a crucial role in cell quiescence and differentiation, processes that contribute to therapy resistance. The following recent examples serve as illustrations of this principle.
Proteins Tis11b and Tis11d promote quiescence in lymphocytes, by decay of transcripts encoding proteins stimulating cell cycling [Citation129]. Other authors confirmed the significance of Tis proteins in acquiring quiescence as a resistance mechanism enabling the survival of CML cells upon imatinib treatment. In CML patient cells Tis11b and Tis11d protein levels were significantly increased compared to healthy donor cells. Cells resistant to imatinib treatment were characterized by overexpression of Tis11b and Tis11d. This effect was accompanied by reduced expansion capability of the resistant cells. Silencing of either of the proteins sensitized CML cells to the treatment [Citation51]. These results implicated Tis11b and Tis11d as key players in the quiescence-based resistance mechanism.
Tis proteins emerge also as essential factors in the resistance to chemotherapy of AML [Citation13]. In this research, AML cells were arrested in the G0 phase of the cell cycle either through starvation or by treatment with chemotherapeutic cytarabine (AraC). Omics analysis of transcriptomes and proteomes showed great similarity in the proteome but not the transcriptome of the two groups of stressed cells. This indicated that a common post-transcriptional mechanism likely orchestrates the proteomic output in these stressed cells. The difference between the short-term effect of stress and the long-term relates to the acquisition of chemoresistance, which is accompanied by increased phosphorylation of eIF2α and reduction of polyribosomes. Additionally, translation initiation was shifted towards mRNAs bearing internal ribosome entry site (IRES), and the profile of translated mRNAs was modified. Upregulated transcripts included a significant portion of mRNAs containing AU-rich motifs in the 3’UTR. Many of these transcripts encoded inflammation-related cytokines like TNFα and CCL2 and their upregulation was caused by the downregulation of Tis11 in the stressed cells. Tis11 directly interacted with TNFα mRNA and directed it for degradation. Moreover, stress evoked activation of the p38/MAPK/MK2 pathway, leading to phosphorylation-mediated inhibition of Tis11. This resulted in increased stability of ARE-BP-regulated transcripts and enhanced expression of TNFα and DUSP1, which are known to stimulate survival through TNF/NFκB axis [Citation50]. This pathway suppresses JNK-dependent apoptosis which stimulates chemotherapy resistance of CML cells [Citation130]. Therefore, blocking of phosphorylation of Tis11 reduced the therapy resistance potential of leukaemic cells, decreasing their survival upon treatment.
Cell cycle arrest by stabilization of mRNA
Induction of DNA damage by cytotoxic drugs is one leukaemia treatment strategy. The ability to increase the expression of cell cycle checkpoint proteins represents a mechanism enabling cancer cells’ resistance to cytotoxic drugs targeting proliferating cells. Activation of checkpoint proteins arrests cell cycle entry, allowing cell survival. Kinases like p38 and MK2 play a key regulatory function in this respect. Apart from TIAR also hnRNPA0 is subject to phosphorylation by p38 and MK2 [Citation34,Citation128]. Activation of p38/MK2 in p53 deficient cancers upon therapy with doxorubicin induces MK2-dependent phosphorylation of hnRNPA0 [Citation34,Citation131]. This increases the stability and translation of p27Kip1 and Gadd45a through an hnRNPA0-dependent post-transcriptional regulation, thereby inducing a cell cycle arrest [Citation34].
Modulation of anti-apoptotic protein level
Silencing of intrinsic mechanisms orchestrating apoptosis represents one of the driving forces of malignancy development. Execution of cell death is modulated by the balance of pro- and anti-apoptotic proteins. The activity of anti-apoptotic proteins belonging to the BCL family is recognized as one of the mechanisms contributing to cancer development and therapy resistance [Citation132]. Targeting anti-apoptotic proteins is one of the leukaemia treatment strategies. Blocking survivin [Citation133] or BCL-2 [Citation134] with inhibitors like Venetoclax sensitizes CML cells to imatinib treatment. Unfortunately, it induces upregulation of other members of the anti-apoptotic Bcl protein family (e.g. MCL1), which is a major route of resistance to this kind of chemotherapy [Citation135]. In line with its activity, the downregulation of MCL-1 sensitized CML cells to imatinib [Citation136]. The activity of HuR contributes to this mechanism. HuR enhances the stability of MCL-1 and survivin mRNAs in cancer cells by binding the 3’UTR [Citation137,Citation138]. Overall, the post-transcriptional modulation of expression of anti-apoptotic proteins by HuR should be seen as a strong obstacle to effective chemotherapy.
Modulation of ROS
The altered metabolism of cancer cells provides a therapeutic opportunity in leukaemia and lymphoma. Elevated oxidative phosphorylation levels contribute to increased production of reactive oxygen species (ROS), making the activity of proteins that neutralize ROS crucial for cell survival. A strategy for treating AML involves the use of hydroquinone, inducing cytotoxicity by disrupting the balance between ROS generation and scavenging. In this process, fine-tuning the level of sirtuins, ROS scavengers play a critical role. Sirtuin 1 and 3 are post-transcriptionally controlled by HuR [Citation32,Citation139]. Hydroquinone treatment induces HuR mRNA decay, leading to protein downregulation. Consequently, the lack of HuR-mediated transcript stabilization results in decreased sirtuin levels. This suggests that combining hydroquinone treatment with the inhibition of HuR activity could enhance the efficacy of AML therapy [Citation32].
The link between HuR activity and ROS may also impact cytokine production. The HuR-dependent stability of mRNAs encoding CCR2 and CCL2 is enhanced by the accelerated formation of ROS [Citation140]. Post-transcriptional upregulation of CCR2/CCL2 stimulates the migration of AML cells [Citation141], a process crucial in the context of AML treatment with 5-aza-2’-deoxycytidine (5-Aza). CCR2/CCL2 signalling in response to 5-Aza is upregulated, promoting the transmigration of AML cells – a recognized main mechanism supporting therapy resistance to 5-Aza [Citation142].
Modulation of immune response
Chimeric Antigen Receptor (CAR) T cell therapy stands as a promising option for the targeted treatment of B-ALL, LBCL, and MM. However, its effectiveness is hindered in some patients due to the development of resistance [Citation143]. One significant challenge is the loss of inflammatory effector functions in CAR-T cells, referred to as T cell exhaustion. Recent studies suggest that the activity of Regnase-1 May play a pivotal role in this mechanism. Regnase-1 was found to be upregulated in T cells that infiltrated melanoma tumours, proving to be essential in suppressing the effector functions of T lymphocytes through the decay of BATF transcripts. The diminished BATF activity resulted in an inhibition of metabolic switch related to enhanced oxidative phosphorylation, which is necessary for T cell activity. The targeted inhibition of Regnase-1 restored the anti-tumour activity of T lymphocytes by reinstating mitochondrial metabolism [Citation144]. Similarly, the dual inhibition of Regnase-1 and Roquin-1 heightened the activity of CAR-T cells [Citation145]. This effect could be attributed to the Regnase-1-dependent decay of T cell factor 1 (TCF1), a transcription factor essential for the formation of persisting memory T cells that sustain the activity of CAR-T cells [Citation146].
Antibody-based targeting of immune checkpoint proteins represents another option for leukaemia and lymphoma treatment. The activity of cytotoxic T lymphocyte antigen 4 (CTLA-4), and programmed cell death 1 (PD-1) receptor and ligand (PD-L1) network represent critical components of immune surveillance. The level of the PD-L1 is upregulated by malignant cells. HuR protein directly upregulates the stability and translation of CMTM6 mRNA encoding the protein that recruits PD-L1 to the cell surface. Therefore, its post-transcriptional activity contributes to the immune resistance of cancer cells. Inhibition of HuR activity with a specific inhibitor reduces the level of PD-L1, increasing the effectiveness of treatment [Citation147].
Another strategy of leukaemia and lymphoma treatment is an antibody-based targeting of the myeloid immune checkpoint protein CD47, whose surface level on the malignant cells is increased [Citation101]. This approach is in the development phase. The activity of HuR and Tis11b proteins that orchestrate translation and therefore surface exposition of CD47 could play a significant role. Noteworthy, the development of cancer is associated with the emergence of so-called tumour-associated macrophages (TAM), characterized by limited phagocytic activity. It has recently been reported, that combining anti-CD47 treatment with drugs activating TAMs supported better clearing of non-Hodgkin lymphoma cells [Citation148], demonstrating new strategic options.
Conclusions
The examples we have detailed here from cancer development and chemotherapy resistance of leukaemic cells illustrate how RNA binding proteins orchestrate the fate of cellular transcripts regulating mRNA and protein levels. Stress response activation, mediated in part by RBPs that stimulate stress granule formation, plays an important role in the development, progression, and therapy resistance of myeloid leukaemia cells. Therefore, the regulation of processes determining mRNA splicing, stability, and translation by ARE-BPs places them in a post-transcriptional network – the ARE-BP axis – that can directly support or prevent cancer development. Their activity can influence the outcome of pharmaceutical interventions in cancer cells, particularly in the case of treatments that induce stress responses. RNA binding protein activity provides cells with the ability to adapt to changed microenvironments, which may impact therapy success.
The provided examples suggest that the cellular transcriptome does not exclusively mirror the cellular proteome. Post-transcriptional control significantly influences the actual repertoire of proteins and their isoforms within cells. This is a very important aspect of gene expression regulation. The protein level or dominance of a specific protein isoform plays a pivotal role in shaping cellular biology and fostering cancer development.
Because ARE-BPs operate within intricate networks, each possessing a protein-interactome, caution is warranted when perturbing single ARE-BPs and drawing specific conclusions. Consequently, experimental results may reflect the activities of network rather than individual protein activities. Moreover, these proteins often substitute for, regulate, and compete for binding sites, introducing complexities that can obscure interpretations of specific ARE-BP activities. The presence of the ARE in a transcript determines its recognition, with the specificity of ARE-binding proteins ARE-BPs rooted in structural characteristics. Other structural components of the protein further delineate its activity towards recognized transcripts. The capacity of multiple proteins recognizing the same motif in the target RNA to regulate the same processes, such as decay, underscores the crucial role of AU-rich element-mediated control. Additionally, the sensitivity to cellular signalling underscores the importance of ARE-BP action in the broader cellular context. The development of future approaches should aim to address these challenges.
Acknowledgments
We are grateful to Drs Manoj Pillai and Stephanie Halene for helpful discussions. We thank Tejita Agarwal and Pernille Bech for critical reading of this manuscript and Reviewer 1 for helpful suggestions.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Hanahan D. Hallmarks of cancer: new dimensions. Cancer Discov. 2022;12(1):31–46. doi: 10.1158/2159-8290.CD-21-1059
- Pakos-Zebrucka K, Koryga I, Mnich K, et al. The integrated stress response. EMBO Rep. 2016;17(10):1374–1395. doi: 10.15252/embr.201642195
- Costa-Mattioli M, Walter P. The integrated stress response: from mechanism to disease. Science. 2020;368(6489). doi: 10.1126/science.aat5314
- Hetz C, Zhang K, Kaufman RJ. Mechanisms, regulation and functions of the unfolded protein response. Nat Rev Mol Cell Biol. 2020;21(8):421–438. doi: 10.1038/s41580-020-0250-z
- Heberle AM, Prentzell MT, van Eunen K, et al. Molecular mechanisms of mTOR regulation by stress. Mol Cell Oncol. 2015;2(2):e970489. doi: 10.4161/23723548.2014.970489
- Li X, He S, Ma B. Autophagy and autophagy-related proteins in cancer. Mol Cancer. 2020;19(1):12. doi: 10.1186/s12943-020-1138-4
- Zhu J, Thompson CB. Metabolic regulation of cell growth and proliferation. Nat Rev Mol Cell Biol. 2019;20(7):436–450. doi: 10.1038/s41580-019-0123-5
- Folmes CD, Dzeja PP, Nelson TJ, et al. Metabolic plasticity in stem cell homeostasis and differentiation. Cell Stem Cell. 2012;11(5):596–606. doi: 10.1016/j.stem.2012.10.002
- Tarazona OA, Pourquie O. Exploring the influence of cell metabolism on cell fate through protein post-translational modifications. Dev Cell. 2020;54(2):282–292. doi: 10.1016/j.devcel.2020.06.035
- Matsumura T, Nakamura-Ishizu A, Muddineni S, et al. Hematopoietic stem cells acquire survival advantage by loss of RUNX1 methylation identified in familial leukemia. Blood. 2020;136(17):1919–1932. doi: 10.1182/blood.2019004292
- Wortel IMN, van der Meer LT, Kilberg MS, et al. Surviving stress: modulation of ATF4-mediated stress responses in normal and malignant cells. Trends Endocrinol Metab. 2017;28(11):794–806. doi: 10.1016/j.tem.2017.07.003
- Wolczyk M, Serwa R, Kominek A, et al. TIAR and FMRP shape pro-survival nascent proteome of leukemia cells in the bone marrow microenvironment. iScience. 2023;26(4):106543. doi: 10.1016/j.isci.2023.106543
- Lee S, Micalizzi D, Truesdell SS, et al. A post-transcriptional program of chemoresistance by AU-rich elements and TTP in quiescent leukemic cells. Genome Biol. 2020;21(1):33. doi: 10.1186/s13059-020-1936-4
- Kilchert C, Strasser K, Kunetsky V, et al. From parts lists to functional significance-RNA-protein interactions in gene regulation. Wiley Interdiscip Rev RNA. 2020;11(3):e1582. doi: 10.1002/wrna.1582
- Hentze MW, Castello A, Schwarzl T, et al. A brave new world of RNA-binding proteins. Nat Rev Mol Cell Biol. 2018;19(5):327–341. doi: 10.1038/nrm.2017.130
- Muller-McNicoll M, Rossbach O, Hui J, et al. Auto-regulatory feedback by RNA-binding proteins. J Mol Cell Biol. 2019;11(10):930–939. doi: 10.1093/jmcb/mjz043
- Quattrone A, Dassi E. The architecture of the human RNA-Binding protein regulatory network. iScience. 2019;21:706–719. doi: 10.1016/j.isci.2019.10.058
- Wang E, Zhou H, Nadorp B, et al. Surface antigen-guided CRISPR screens identify regulators of myeloid leukemia differentiation. Cell Stem Cell. 2021;28(4):718–31 e6. doi: 10.1016/j.stem.2020.12.005
- Izquierdo JM, Valcarcel J. Two isoforms of the T-cell intracellular antigen 1 (TIA-1) splicing factor display distinct splicing regulation activities. Control of TIA-1 isoform ratio by TIA-1-related protein. J Biol Chem. 2007;282(27):19410–19417. doi: 10.1074/jbc.M700688200
- Lee FCY, Ule J. Advances in CLIP technologies for studies of protein-RNA interactions. Mol Cell. 2018;69(3):354–369. doi: 10.1016/j.molcel.2018.01.005
- Garcia-Maurino SM, Rivero-Rodriguez F, Velazquez-Cruz A, et al. RNA binding protein regulation and cross-talk in the control of AU-rich mRNA fate. Front Mol Biosci. 2017;4:71. doi: 10.3389/fmolb.2017.00071
- Otsuka H, Fukao A, Funakami Y, et al. Emerging evidence of translational control by AU-Rich element-binding proteins. Front Genet. 2019;10:332. doi: 10.3389/fgene.2019.00332
- Bakheet T, Williams BR, Khabar KS. ARED 3.0: the large and diverse AU-rich transcriptome. Nucleic Acids Res. 2006;34(Database issue):D111–4. doi: 10.1093/nar/gkj052
- Gruber AR, Fallmann J, Kratochvill F, et al. Aresite: a database for the comprehensive investigation of AU-rich elements. Nucleic Acids Res. 2011;39(Database issue):D66–9. doi: 10.1093/nar/gkq990
- Lebedeva S, Jens M, Theil K, et al. Transcriptome-wide analysis of regulatory interactions of the RNA-binding protein HuR. Mol Cell. 2011;43(3):340–352. doi: 10.1016/j.molcel.2011.06.008
- Binas O, Tants JN, Peter SA, et al. Structural basis for the recognition of transiently structured AU-rich elements by Roquin. Nucleic Acids Res. 2020;48(13):7385–7403. doi: 10.1093/nar/gkaa465
- Tan D, Zhou M, Kiledjian M, et al. The ROQ domain of Roquin recognizes mRNA constitutive-decay element and double-stranded RNA. Nat Struct Mol Biol. 2014;21(8):679–685. doi: 10.1038/nsmb.2857
- Podszywalow-Bartnicka P, Wolczyk M, Kusio-Kobialka M, et al. Downregulation of BRCA1 protein in BCR-ABL1 leukemia cells depends on stress-triggered TIAR-mediated suppression of translation. Cell Cycle. 2014;13(23):3727–3741. doi: 10.4161/15384101.2014.965013
- Vujovic A, de Rooij L, Chahi AK, et al. In vivo screening unveils pervasive RNA-Binding protein dependencies in leukemic stem cells and identifies ELAVL1 as a therapeutic target. Blood Cancer Discov. 2023;4(3):180–207. doi: 10.1158/2643-3230.BCD-22-0086
- Tang YJ, Wu W, Chen QQ, et al. miR-29b-3p suppresses the malignant biological behaviors of AML cells via inhibiting NF-kappaB and JAK/STAT signaling pathways by targeting HuR. BMC Cancer. 2022;22(1):909. doi: 10.1186/s12885-022-09996-1
- Bergalet J, Fawal M, Lopez C, et al. HuR-mediated control of C/EBPbeta mRNA stability and translation in ALK-positive anaplastic large cell lymphomas. Mol Cancer Res. 2011;9(4):485–496. doi: 10.1158/1541-7786.MCR-10-0351
- Wang LJ, Lee YC, Chiou JT, et al. Effects of SIDT2 on the miR-25/NOX4/HuR axis and SIRT3 mRNA stability lead to ROS-mediated TNF-α expression in hydroquinone-treated leukemia cells. Cell Biol Toxicol. 2022;39(5):2207–2225. doi: 10.1007/s10565-022-09705-5
- Young DJ, Stoddart A, Nakitandwe J, et al. Knockdown of Hnrnpa0, a del(5q) gene, alters myeloid cell fate in murine cells through regulation of AU-rich transcripts. Haematologica. 2014;99(6):1032–1040. doi: 10.3324/haematol.2013.098657
- Cannell IG, Merrick KA, Morandell S, et al. A pleiotropic RNA-Binding protein controls distinct cell cycle checkpoints to drive resistance of p53-defective tumors to chemotherapy. Cancer Cell. 2015;28(5):623–637. doi: 10.1016/j.ccell.2015.09.009
- Wilson GM, Lu J, Sutphen K, et al. Phosphorylation of p40AUF1 regulates binding to a + U-rich mRNA-destabilizing elements and protein-induced changes in ribonucleoprotein structure. J Biol Chem. 2003;278(35):33039–33048. doi: 10.1074/jbc.M305775200
- Wilson GM, Lu J, Sutphen K, et al. Regulation of a + U-rich element-directed mRNA turnover involving reversible phosphorylation of AUF1. J Biol Chem. 2003;278(35):33029–33038. doi: 10.1074/jbc.M305772200
- Fawal M, Armstrong F, Ollier S, et al. A “liaison dangereuse” between AUF1/hnRNPD and the oncogenic tyrosine kinase NPM-ALK. Blood. 2006;108(8):2780–2788. doi: 10.1182/blood-2006-04-014902
- Zhang R, Lin P, Yang X, et al. Survival associated alternative splicing events in diffuse large B-cell lymphoma. Am J Transl Res. 2018;10(8):2636–2647.
- Dolatshad H, Pellagatti A, Fernandez-Mercado M, et al. Disruption of SF3B1 results in deregulated expression and splicing of key genes and pathways in myelodysplastic syndrome hematopoietic stem and progenitor cells. Leukemia. 2015;29(5):1092–1103. doi: 10.1038/leu.2014.331
- Lima M. Aggressive mature natural killer cell neoplasms: from epidemiology to diagnosis. Orphanet J Rare Dis. 2013;8(1):95. doi: 10.1186/1750-1172-8-95
- Wolczyk M, Podszywalow-Bartnicka P, Bugajski L, et al. Stress granules assembly affects detection of mRNA in living cells by the NanoFlares; an important aspect of the technology. Biochim Biophys Acta Gen Subj. 2017;1861(5 Pt A):1024–1035. doi: 10.1016/j.bbagen.2017.02.010
- Karmakar S, Ramirez O, Paul KV, et al. Integrative genome-wide analysis reveals EIF3A as a key downstream regulator of translational repressor protein Musashi 2 (MSI2). NAR Cancer. 2022;4(2):zcac015. doi: 10.1093/narcan/zcac015
- Ito T, Kwon HY, Zimdahl B, et al. Regulation of myeloid leukaemia by the cell-fate determinant Musashi. Nature. 2010;466(7307):765–768. doi: 10.1038/nature09171
- Kharas MG, Lengner CJ, Al-Shahrour F, et al. Musashi-2 regulates normal hematopoiesis and promotes aggressive myeloid leukemia. Nat Med. 2010;16(8):903–908. doi: 10.1038/nm.2187
- Griner LN, Reuther GW. Aggressive myeloid leukemia formation is directed by the Musashi 2/Numb pathway. Cancer Biol Ther. 2010;10(10):979–982. doi: 10.4161/cbt.10.10.14010
- Han Y, Ye A, Zhang Y, et al. Musashi-2 silencing exerts potent activity against acute myeloid leukemia and enhances chemosensitivity to Daunorubicin. PLOS ONE. 2015;10(8):e0136484. doi: 10.1371/journal.pone.0136484
- Kwon HY, Bajaj J, Ito T, et al. Tetraspanin 3 is required for the development and propagation of acute myelogenous leukemia. Cell Stem Cell. 2015;17(2):152–164. doi: 10.1016/j.stem.2015.06.006
- Vu LP, Prieto C, Amin EM, et al. Functional screen of MSI2 interactors identifies an essential role for SYNCRIP in myeloid leukemia stem cells. Nat Genet. 2017;49(6):866–875. doi: 10.1038/ng.3854
- Rounbehler RJ, Fallahi M, Yang C, et al. Tristetraprolin impairs myc-induced lymphoma and abolishes the malignant state. Cell. 2012;150(3):563–574. doi: 10.1016/j.cell.2012.06.033
- Kagoya Y, Nakatsugawa M, Ochi T, et al. Transient stimulation expands superior antitumor T cells for adoptive therapy. JCI Insight. 2017;2(2):e89580. doi: 10.1172/jci.insight.89580
- Kaehler M, Dworschak M, Rodin JP, et al. ZFP36L1 plays an ambiguous role in the regulation of cell expansion and negatively regulates CDKN1A in chronic myeloid leukemia cells. Exp Hematol. 2021;99:54–64 e7. doi: 10.1016/j.exphem.2021.05.006
- Jeltsch KM, Hu D, Brenner S, et al. Cleavage of roquin and regnase-1 by the paracaspase MALT1 releases their cooperatively repressed targets to promote T(H)17 differentiation. Nat Immunol. 2014;15(11):1079–1089. doi: 10.1038/ni.3008
- Wimberger N, Ober F, Avar G, et al. Oncogene-induced MALT1 protease activity drives post-transcriptional gene expression in malignant lymphomas. Blood. 2023;142(23):1985–2001. doi: 10.1182/blood.2023021299
- Kidoya H, Muramatsu F, Shimamura T, et al. Regnase-1-mediated post-transcriptional regulation is essential for hematopoietic stem and progenitor cell homeostasis. Nat Commun. 2019;10(1):1072. doi: 10.1038/s41467-019-09028-w
- Okuda H, Miyamoto R, Takahashi S, et al. RNA-binding proteins of KHDRBS and IGF2BP families control the oncogenic activity of MLL-AF4. Nat Commun. 2022;13(1):6688. doi: 10.1038/s41467-022-34558-1
- Sumithra B, Saxena U, Das AB. A comprehensive study on genome-wide coexpression network of KHDRBS1/Sam68 reveals its cancer and patient-specific association. Sci Rep. 2019;9(1):11083. doi: 10.1038/s41598-019-47558-x
- Wang Q, Li Y, Cheng J, et al. Sam68 affects cell proliferation and apoptosis of human adult T-acute lymphoblastic leukemia cells via AKT/mTOR signal pathway. Leuk Res. 2016;46:1–9.
- Xu J, Wang D, Ma H, et al. KHSRP combines transcriptional and posttranscriptional mechanisms to regulate monocytic differentiation. Blood Sci. 2022;4(3):103–115. doi: 10.1097/BS9.0000000000000122
- Kafer R, Schmidtke L, Schrick K, et al. The RNA-Binding protein KSRP modulates cytokine expression of CD4(+) T cells. J Immunol Res. 2019;2019:4726532.
- Zhang Y, Peng L, Hu T, et al. La-related protein 4B maintains murine MLL-AF9 leukemia stem cell self-renewal by regulating cell cycle progression. Exp Hematol. 2015;43(4):309–18 e2. doi: 10.1016/j.exphem.2014.12.003
- Corley M, Burns MC, Yeo GW. How RNA-Binding proteins interact with RNA: molecules and mechanisms. Mol Cell. 2020;78(1):9–29. doi: 10.1016/j.molcel.2020.03.011
- Meyer C, Garzia A, Mazzola M, et al. The TIA1 RNA-Binding protein family regulates EIF2AK2-mediated stress response and cell cycle progression. Mol Cell. 2018;69(4):622–35 e6. doi: 10.1016/j.molcel.2018.01.011
- Del Gatto-Konczak F, Bourgeois CF, Le Guiner C, et al. The RNA-binding protein TIA-1 is a novel mammalian splicing regulator acting through intron sequences adjacent to a 5’ splice site. Mol Cell Biol. 2000;20(17):6287–6299. doi: 10.1128/MCB.20.17.6287-6299.2000
- Forch P, Puig O, Martinez C, et al. The splicing regulator TIA-1 interacts with U1-C to promote U1 snRNP recruitment to 5’ splice sites. Embo J. 2002;21(24):6882–6892. doi: 10.1093/emboj/cdf668
- Gal-Mark N, Schwartz S, Ram O, et al. The pivotal roles of TIA proteins in 5’ splice-site selection of alu exons and across evolution. PLOS Genet. 2009;5(11):e1000717. doi: 10.1371/journal.pgen.1000717
- Wang Z, Kayikci M, Briese M, et al. iCLIP predicts the dual splicing effects of TIA-RNA interactions. PLOS Biol. 2010;8(10):e1000530. doi: 10.1371/journal.pbio.1000530
- Mukherjee N, Corcoran DL, Nusbaum JD, et al. Integrative regulatory mapping indicates that the RNA-binding protein HuR couples pre-mRNA processing and mRNA stability. Mol Cell. 2011;43(3):327–339. doi: 10.1016/j.molcel.2011.06.007
- Li Y, Estep JA, Karginov FV. Transcriptome-wide identification and validation of interactions between the miRNA machinery and HuR on mRNA targets. J Mol Biol. 2018;430(3):285–296. doi: 10.1016/j.jmb.2017.12.006
- Yoon JH, De S, Srikantan S, et al. PAR-CLIP analysis uncovers AUF1 impact on target RNA fate and genome integrity. Nat Commun. 2014;5(1):5248. doi: 10.1038/ncomms6248
- Myer VE, Steitz JA. Isolation and characterization of a novel, low abundance hnRNP protein: A0. RNA. 1995;1(2):171–182.
- Thibault PA, Ganesan A, Kalyaanamoorthy S, et al. hnRNP A/B proteins: an encyclopedic assessment of their roles in homeostasis and disease. Biology. 2021;10(8):712. doi: 10.3390/biology10080712
- Briata P, Bordo D, Puppo M, et al. Diverse roles of the nucleic acid-binding protein KHSRP in cell differentiation and disease. Wiley Interdiscip Rev RNA. 2016;7(2):227–240. doi: 10.1002/wrna.1327
- Briata P, Chen CY, Ramos A, et al. Functional and molecular insights into KSRP function in mRNA decay. Biochim Biophys Acta. 2013;1829(6–7):689–694. doi: 10.1016/j.bbagrm.2012.11.003
- Feracci M, Foot JN, Grellscheid SN, et al. Structural basis of RNA recognition and dimerization by the STAR proteins T-STAR and Sam68. Nat Commun. 2016;7(1):10355. doi: 10.1038/ncomms10355
- Duggimpudi S, Kloetgen A, Maney SK, et al. Transcriptome-wide analysis uncovers the targets of the RNA-binding protein MSI2 and effects of MSI2‘s RNA-binding activity on IL-6 signaling. J Biol Chem. 2018;293(40):15359–15369. doi: 10.1074/jbc.RA118.002243
- Kuspert M, Murakawa Y, Schaffler K, et al. LARP4B is an AU-rich sequence associated factor that promotes mRNA accumulation and translation. RNA. 2015;21(7):1294–1305. doi: 10.1261/rna.051441.115
- Maraia RJ, Mattijssen S, Cruz-Gallardo I, et al. The La and related RNA-binding proteins (LARPs): structures, functions, and evolving perspectives. Wiley Interdiscip Rev RNA. 2017;8(6). doi: 10.1002/wrna.1430
- Mao R, Yang R, Chen X, et al. Regnase-1, a rapid response ribonuclease regulating inflammation and stress responses. Cell Mol Immunol. 2017;14(5):412–422. doi: 10.1038/cmi.2016.70
- Mino T, Takeuchi O. Regnase-1 and Roquin regulate inflammatory mRNAs. Oncotarget. 2015;6(20):17869–17870. doi: 10.18632/oncotarget.4891
- Nwosu GO, Powell JA, Pitson SM. Targeting the integrated stress response in hematologic malignancies. Exp Hematol Oncol. 2022;11(1):94. doi: 10.1186/s40164-022-00348-0
- Sanduja S, Blanco FF, Dixon DA. The roles of TTP and BRF proteins in regulated mRNA decay. Wiley Interdiscip Rev RNA. 2011;2(1):42–57. doi: 10.1002/wrna.28
- Ma W, Zhen G, Xie W, et al. In vivo reconstitution finds multivalent RNA–RNA interactions as drivers of mesh-like condensates. Elife. 2021;10:10. doi: 10.7554/eLife.64252
- Ma W, Mayr C. A membraneless organelle associated with the endoplasmic reticulum enables 3‘UTR-Mediated protein-protein interactions. Cell. 2018;175(6):1492–506 e19. doi: 10.1016/j.cell.2018.10.007
- Newton K, Dixit VM. Signaling in innate immunity and inflammation. Cold Spring Harb Perspect Biol. 2012;4(3):a006049–a006049. doi: 10.1101/cshperspect.a006049
- Wang J, Erlacher M, Fernandez-Orth J. The role of inflammation in hematopoiesis and bone marrow failure: what can we learn from mouse models? Front Immunol. 2022;13:951937. doi: 10.3389/fimmu.2022.951937
- Ratajczak MZ, Kucia M. Hematopoiesis and innate immunity: an inseparable couple for good and bad times, bound together by an hormetic relationship. Leukemia. 2022;36(1):23–32. doi: 10.1038/s41375-021-01482-0
- Goncalves AC, Cortesao E, Oliveiros B, et al. Oxidative stress and mitochondrial dysfunction play a role in myelodysplastic syndrome development, diagnosis, and prognosis: a pilot study. Free Radic Res. 2015;49(9):1081–1094. doi: 10.3109/10715762.2015.1035268
- van Galen P, Mbong N, Kreso A, et al. Integrated stress response activity marks stem cells in normal hematopoiesis and leukemia. Cell Rep. 2018;25(5):1109–17 e5. doi: 10.1016/j.celrep.2018.10.021
- Follo MY, Finelli C, Mongiorgi S, et al. PKR is activated in MDS patients and its subcellular localization depends on disease severity. Leukemia. 2008;22(12):2267–2269. doi: 10.1038/leu.2008.122
- Blalock WL, Grimaldi C, Fala F, et al. PKR activity is required for acute leukemic cell maintenance and growth: a role for PKR-mediated phosphatase activity to regulate GSK-3 phosphorylation. J Cell Physiol. 2009;221(1):232–241. doi: 10.1002/jcp.21848
- Cheng X, Byrne M, Brown KD, et al. PKR inhibits the DNA damage response, and is associated with poor survival in AML and accelerated leukemia in NHD13 mice. Blood. 2015;126(13):1585–1594. doi: 10.1182/blood-2015-03-635227
- Piwocka K, Vejda S, Cotter TG, et al. Bcr-Abl reduces endoplasmic reticulum releasable calcium levels by a Bcl-2-independent mechanism and inhibits calcium-dependent apoptotic signaling. Blood. 2006;107(10):4003–4010. doi: 10.1182/blood-2005-04-1523
- Kusio-Kobialka M, Podszywalow-Bartnicka P, Peidis P, et al. The PERK-eIf2alpha phosphorylation arm is a pro-survival pathway of BCR-ABL signaling and confers resistance to imatinib treatment in chronic myeloid leukemia cells. Cell Cycle. 2012;11(21):4069–4078. doi: 10.4161/cc.22387
- Dudka W, Hoser G, Mondal SS, et al. Targeting integrated stress response with ISRIB combined with imatinib treatment attenuates RAS/RAF/MAPK and STAT5 signaling and eradicates chronic myeloid leukemia cells. BMC Cancer. 2022;22(1):1254. doi: 10.1186/s12885-022-10289-w
- Biancon G, Joshi P, Zimmer JT, et al. Precision analysis of mutant U2AF1 activity reveals deployment of stress granules in myeloid malignancies. Mol Cell. 2022;82(6):1107–22 e7. doi: 10.1016/j.molcel.2022.02.025
- Diaz-Munoz MD, Kiselev VY, Le Novere N, et al. Tia1 dependent regulation of mRNA subcellular location and translation controls p53 expression in B cells. Nat Commun. 2017;8(1):530. doi: 10.1038/s41467-017-00454-2
- Bonomo I, Assoni G, La Pietra V, et al. HuR modulation counteracts lipopolysaccharide response in murine macrophages. Dis Model Mech. 2023;16(3): doi: 10.1242/dmm.050120
- Hu X, Li J, Fu M, et al. The JAK/STAT signaling pathway: from bench to clinic. Signal Transduct Target Ther. 2021;6(1):402. doi: 10.1038/s41392-021-00791-1
- Zhang T, Ma C, Zhang Z, et al. NF-kappaB signaling in inflammation and cancer. MedComm. 2021;2(4):618–653. doi: 10.1002/mco2.104
- Kudinov AE, Karanicolas J, Golemis EA, et al. Musashi RNA-Binding proteins as cancer drivers and novel therapeutic targets. Clin Cancer Res. 2017;23(9):2143–2153. doi: 10.1158/1078-0432.CCR-16-2728
- Majeti R, Jamieson C, Pang WW, et al. Clonal expansion of Stem/Progenitor cells in cancer, fibrotic diseases, and atherosclerosis, and CD47 protection of pathogenic cells. Annu Rev Med. 2022;73(1):307–320. doi: 10.1146/annurev-med-042420-104436
- Leppek K, Schott J, Reitter S, et al. Roquin promotes constitutive mRNA decay via a conserved class of stem-loop recognition motifs. Cell. 2013;153(4):869–881. doi: 10.1016/j.cell.2013.04.016
- Hauer J, Fischer U, Borkhardt A. Toward prevention of childhood ALL by early-life immune training. Blood. 2021;138(16):1412–1428. doi: 10.1182/blood.2020009895
- Carbone A, Tripodo C, Carlo-Stella C, et al. The role of inflammation in lymphoma. Adv Exp Med Biol. 2014;816:315–333.
- Popovic B, Nicolet BP, Guislain A, et al. Time-dependent regulation of cytokine production by RNA binding proteins defines T cell effector function. Cell Rep. 2023;42(5):112419. doi: 10.1016/j.celrep.2023.112419
- Heeren JJ F-V. Post-transcriptional control of T-cell cytokine production: implications for cancer therapy. Immunology. 2021;164(1):57–72. doi: 10.1111/imm.13339
- Gillis P, Malter JS. The adenosine-uridine binding factor recognizes the AU-rich elements of cytokine, lymphokine, and oncogene mRnas. J Biol Chem. 1991;266(5):3172–3177. doi: 10.1016/S0021-9258(18)49970-X
- Curdy N, Lanvin O, Cadot S, et al. Stress granules in the post-transcriptional regulation of immune cells. Front Cell Dev Biol. 2020;8:611185. doi: 10.3389/fcell.2020.611185
- Naz S, Battu S, Khan RA, et al. Activation of integrated stress response pathway regulates IL-1beta production through posttranscriptional and translational reprogramming in macrophages. Eur J Immunol. 2019;49(2):277–289. doi: 10.1002/eji.201847513
- Paget M, Cadena C, Ahmad S, et al. Stress granules are shock absorbers that prevent excessive innate immune responses to dsRNA. Mol Cell. 2023;83(7):1180–96 e8. doi: 10.1016/j.molcel.2023.03.010
- Kong G, Dou Y, Xiao X, et al. Transgenic expression of a Mutant Ribonuclease Regnase-1 in T cells disturbs T cell development and functions. Front Immunol. 2021;12:682220. doi: 10.3389/fimmu.2021.682220
- Bhat N, Virgen-Slane R, Ramezani-Rad P, et al. Regnase-1 is essential for B cell homeostasis to prevent immunopathology. J Exp Med. 2021;218(5): doi: 10.1084/jem.20200971
- Moore MJ, Blachere NE, Fak JJ, et al. ZFP36 RNA-binding proteins restrain T cell activation and anti-viral immunity. Elife. 2018;7. doi: 10.7554/eLife.33057
- Ott G, Rosenwald A, Campo E. Understanding MYC-driven aggressive B-cell lymphomas: pathogenesis and classification. Blood. 2013;122(24):3884–3891. doi: 10.1182/blood-2013-05-498329
- Izquierdo JM, Majos N, Bonnal S, et al. Regulation of fas alternative splicing by antagonistic effects of TIA-1 and PTB on exon definition. Mol Cell. 2005;19(4):475–484. doi: 10.1016/j.molcel.2005.06.015
- Izquierdo JM. Hu antigen R (HuR) functions as an alternative pre-mRNA splicing regulator of fas apoptosis-promoting receptor on exon definition. J Biol Chem. 2008;283(27):19077–19084. doi: 10.1074/jbc.M800017200
- Villa-Morales M, Cobos MA, Gonzalez-Gugel E, et al. FAS system deregulation in T-cell lymphoblastic lymphoma. Cell Death Dis. 2014;5(3):e1110. doi: 10.1038/cddis.2014.83
- Razzaghi R, Agarwal S, Kotlov N, et al. Compromised counterselection by FAS creates an aggressive subtype of germinal center lymphoma. J Exp Med. 2021;218(3): doi: 10.1084/jem.20201173
- Diaz-Munoz MD, Bell SE, Fairfax K, et al. The RNA-binding protein HuR is essential for the B cell antibody response. Nat Immunol. 2015;16(4):415–425. doi: 10.1038/ni.3115
- Papadaki O, Milatos S, Grammenoudi S, et al. Control of thymic T cell maturation, deletion and egress by the RNA-binding protein HuR. J Immunol. 2009;182(11):6779–6788. doi: 10.4049/jimmunol.0900377
- Yu S, Tripod M, Atasoy U, et al. HuR plays a positive role to strengthen the signaling pathways of CD4(+) T cell activation and Th17 cell differentiation. J Immunol Res. 2021;2021:9937243. doi: 10.1155/2021/9937243
- Vivori C, Papasaikas P, Stadhouders R, et al. Dynamics of alternative splicing during somatic cell reprogramming reveals functions for RNA-binding proteins CPSF3, hnRNP UL1, and TIA1. Genome Biol. 2021;22(1):171. doi: 10.1186/s13059-021-02372-5
- Kurata M, Onishi I, Takahara T, et al. C/EBPbeta induces B-cell acute lymphoblastic leukemia and cooperates with BLNK mutations. Cancer Sci. 2021;112(12):4920–4930. doi: 10.1111/cas.15164
- Piva R, Pellegrino E, Mattioli M, et al. Functional validation of the anaplastic lymphoma kinase signature identifies CEBPB and BCL2A1 as critical target genes. J Clin Invest. 2006;116(12):3171–3182. doi: 10.1172/JCI29401
- Stelmach P, Trumpp A. Leukemic stem cells and therapy resistance in acute myeloid leukemia. Haematologica. 2023;108(2):353–366. doi: 10.3324/haematol.2022.280800
- Hubstenberger A, Courel M, Benard M, et al. P-Body purification reveals the condensation of repressed mRNA regulons. Mol Cell. 2017;68(1):144–57 e5. doi: 10.1016/j.molcel.2017.09.003
- Sanchez-Jimenez C, Ludena MD, Izquierdo JM. T-cell intracellular antigens function as tumor suppressor genes. Cell Death Dis. 2015;6(3):e1669. doi: 10.1038/cddis.2015.43
- Sung HM, Schott J, Boss P, et al. Stress-induced nuclear speckle reorganization is linked to activation of immediate early gene splicing. J Cell Bio. 2023;222(12): doi: 10.1083/jcb.202111151
- Galloway A, Saveliev A, Lukasiak S, et al. RNA-binding proteins ZFP36L1 and ZFP36L2 promote cell quiescence. Science. 2016;352(6284):453–459. doi: 10.1126/science.aad5978
- Kesarwani M, Kincaid Z, Gomaa A, et al. Targeting c-FOS and DUSP1 abrogates intrinsic resistance to tyrosine-kinase inhibitor therapy in BCR-ABL-induced leukemia. Nat Med. 2017;23(4):472–482. doi: 10.1038/nm.4310
- Reinhardt HC, Hasskamp P, Schmedding I, et al. DNA damage activates a spatially distinct late cytoplasmic cell-cycle checkpoint network controlled by MK2-mediated RNA stabilization. Mol Cell. 2010;40(1):34–49. doi: 10.1016/j.molcel.2010.09.018
- Kaloni D, Diepstraten ST, Strasser A, et al. BCL-2 protein family: attractive targets for cancer therapy. Apoptosis. 2023;28(1–2):20–38. doi: 10.1007/s10495-022-01780-7
- Stella S, Tirro E, Conte E, et al. Suppression of survivin induced by a BCR-ABL/JAK2/STAT3 pathway sensitizes imatinib-resistant CML cells to different cytotoxic drugs. Mol Cancer Ther. 2013;12(6):1085–1098. doi: 10.1158/1535-7163.MCT-12-0550
- Carter BZ, Mak PY, Mu H, et al. Combined targeting of BCL-2 and BCR-ABL tyrosine kinase eradicates chronic myeloid leukemia stem cells. Sci Transl Med. 2016;8(355):355ra117. doi: 10.1126/scitranslmed.aag1180
- Roberts AW, Wei AH, Huang DCS. BCL2 and MCL1 inhibitors for hematologic malignancies. Blood. 2021;138(13):1120–1136. doi: 10.1182/blood.2020006785
- Aichberger KJ, Mayerhofer M, Krauth MT, et al. Identification of mcl-1 as a BCR/ABL-dependent target in chronic myeloid leukemia (CML): evidence for cooperative antileukemic effects of imatinib and mcl-1 antisense oligonucleotides. Blood. 2005;105(8):3303–3311. doi: 10.1182/blood-2004-02-0749
- Donahue JM, Chang ET, Xiao L, et al. The RNA-binding protein HuR stabilizes survivin mRNA in human oesophageal epithelial cells. Biochem J. 2011;437(1):89–96. doi: 10.1042/BJ20110028
- Filippova N, Yang X, Wang Y, et al. The RNA-binding protein HuR promotes glioma growth and treatment resistance. Mol Cancer Res. 2011;9(5):648–659. doi: 10.1158/1541-7786.MCR-10-0325
- Abdelmohsen K, Pullmann R Jr., Lal A, et al. Phosphorylation of HuR by Chk2 regulates SIRT1 expression. Mol Cell. 2007;25(4):543–557. doi: 10.1016/j.molcel.2007.01.011
- Sasaki Y, Dehnad A, Fish S, et al. NOX4 regulates CCR2 and CCL2 mRNA stability in alcoholic liver disease. Sci Rep. 2017;7(1):46144. doi: 10.1038/srep46144
- Macanas-Pirard P, Quezada T, Navarrete L, et al. The CCL2/CCR2 axis affects transmigration and proliferation but not resistance to chemotherapy of acute myeloid leukemia cells. PLOS ONE. 2017;12(1):e0168888. doi: 10.1371/journal.pone.0168888
- Xiao X, Xu Q, Sun Y, et al. 5‑aza‑2‘‑deoxycytidine promotes migration of acute monocytic leukemia cells via activation of CCL2‑CCR2‑ERK signaling pathway. Mol Med Rep. 2017;16(2):1417–1424. doi: 10.3892/mmr.2017.6737
- Cappell KM, Kochenderfer JN. Long-term outcomes following CAR T cell therapy: what we know so far. Nat Rev Clin Oncol. 2023;20(6):359–371. doi: 10.1038/s41571-023-00754-1
- Wei J, Long L, Zheng W, et al. Targeting REGNASE-1 programs long-lived effector T cells for cancer therapy. Nature. 2019;576(7787):471–476. doi: 10.1038/s41586-019-1821-z
- Mai D, Johnson O, Reff J, et al. Combined disruption of T cell inflammatory regulators regnase-1 and roquin-1 enhances antitumor activity of engineered human T cells. Proc Natl Acad Sci U S A. 2023;120(12):e2218632120. doi: 10.1073/pnas.2218632120
- Zheng W, Wei J, Zebley CC, et al. Regnase-1 suppresses TCF-1+ precursor exhausted T-cell formation to limit CAR-T-cell responses against ALL. Blood. 2021;138(2):122–135. doi: 10.1182/blood.2020009309
- Liu Y, Li X, Zhang H, et al. HuR up-regulates cell surface PD-L1 via stabilizing CMTM6 transcript in cancer. Oncogene. 2021;40(12):2230–2242. doi: 10.1038/s41388-021-01689-6
- Cao X, Wang Y, Zhang W, et al. Targeting macrophages for enhancing CD47 blockade-elicited lymphoma clearance and overcoming tumor-induced immunosuppression. Blood. 2022;139(22):3290–3302. doi: 10.1182/blood.2021013901