
ABSTRACT
The aim of this study was to compare the circular transcriptome of divergent tissues in order to understand: i) the presence of circular RNAs (circRNAs) that are not exonic circRNAs, i.e. originated from backsplicing involving known exons and, ii) the origin of artificial circRNA (artif_circRNA), i.e. circRNA not generated in-vivo. CircRNA identification is mostly an in-silico process, and the analysis of data from the BovReg project (https://www.bovreg.eu/) provided an opportunity to explore new ways to identify reliable circRNAs. By considering 117 tissue samples, we characterized 23,926 exonic circRNAs, 337 circRNAs from 273 introns (191 ciRNAs, 146 intron circles), 108 circRNAs from small non-coding genes and nearly 36.6K circRNAs classified as other_circRNAs. Furthermore, for 63 of those samples we analysed in parallel data from total-RNAseq (ribosomal RNAs depleted prior to library preparation) with paired mRNAseq (library prepared with poly(A)-selected RNAs). The high number of circRNAs detected in mRNAseq, and the significant number of novel circRNAs, mainly other_circRNAs, led us to consider all circRNAs detected in mRNAseq as artificial. This study provided evidence of 189 false entries in the list of exonic circRNAs: 103 artif_circRNAs identified by total RNAseq/mRNAseq comparison using two circRNA tools, 26 probable artif_circRNAs, and 65 identified by deep annotation analysis. Extensive benchmarking was performed (including analyses with CIRI2 and CIRCexplorer-2) and confirmed 94% of the 23,737 reliable exonic circRNAs. Moreover, this study demonstrates the effectiveness of a panel of highly expressed exonic circRNAs (5–8%) in analysing the tissue specificity of the bovine circular transcriptome.
Introduction
The current reference genome for cattle (ARS-UCD1.2) is highly contiguous, complete and accurate [Citation1]. The protein coding transcriptome has been well characterized, for multiple different tissues and cell types [Citation2–4]. In contrast, little is known about how other RNA species are expressed in cattle tissues. In recent years, with the development of improved RNA sequencing methods and bioinformatics tools, to capture and characterize multiple RNA species, circular RNAs (circRNAs), with a closed covalent structure, have emerged as a fascinating new class of RNA molecules. The first two types of circRNAs described in 2012–2013 [Citation5–8] are now well described and their origin better understood (reviewed in [Citation9,Citation10]). They are likely to be a natural by-product of the splicing process [Citation11,Citation12] as other non-co-linear transcripts [Citation13,Citation14]. During splicing of linear primary transcripts (pre-mRNA), introns (non-coding regions) are spliced out in the form of lariat intronic RNA and exons are spliced together. Classically, a splicing event ligates the 5’ donor site located near the end of the upstream exon (i.e. in the intron on the 3’ side of the exon) with the 3’acceptor site located near the 5’ side of the downstream exon. The first type of circRNAs is generated by a specific splicing event, known as backsplicing, which results from the splicing of a downstream splice donor to an upstream splice acceptor. For example, at the circular junction we can see the ligation of exon3 end to exon2 start (see M&M_Adoc-1). This backsplicing (BS) leads to an exonic circRNA, which in the vast majority of cases contains only exonic sequences [Citation5,Citation15,Citation16]. The genesis of the second type of circRNAs is completely independent of a backsplicing event. Intronic circRNAs contain only intronic sequences and are by-products of classic splicing. The best-known and best-described intronic circRNAs are derived from lariat intronic RNA when intronic lariats escape degradation due to failure of intron debranching. The residue of intronic lariats can become circular RNA precursors to provide ciRNAs or lariat derived circRNAs [Citation8,Citation17,Citation18]. In addition to these ciRNAs, intron circles resulting from circularization of the entire intron have also been described [Citation17–19] and their genesis is beginning to be understood [Citation20].
Detection of circRNAs is performed using sequence data from total RNA libraries after depletion of ribosomal RNAs (total-RNAseq) [Citation21]. The identification of a circRNA is always based on the documentation of reads containing the circular junction. Numerous bioinformatic tools are currently available to accurately identify a high number of circRNAs while minimizing the number of false positives [Citation9,Citation22]. It is important to note that the criteria for managing this balance can vary significantly. A first approach leads to a tool retaining only circRNAs, which meet very precise annotation criteria. The most popular is CIRCexplorer [Citation23], which retains only circRNAs resulting from BS between two known exons (we reserve the term ‘exonic circRNA’ for circRNAs corresponding to this definition) and some putative intronic circRNAs. A second approach to identifying circRNAs is to retain only those suspected of originating from backsplicing; i.e. when the two parts of the canonical splicing motif are found on either side of the interval defined by the circRNA coordinates. This requirement ensures the identification of circRNAs originating from BS and may lead to the identification of circRNAs originating from BS involving unannotated exons. The most popular is CIRI2 [Citation24], which delivers a list of unannotated circRNAs. CIRI2 does not differentiate between exonic circRNAs and putative exonic circRNAs when the putative BS does not involve known exons. An alternative approach is to use a non-restrictive pipeline for the discovery of new types of circRNAs. Liu et al. [Citation25,Citation26] defined candidate interior circRNAs as those originating from single introns, exons, intergenic regions, and pairs of adjacent introns or adjacent exons (without regular backsplicing). For these interior circRNAs, the presence of repeated sequences appears to be the key to their genesis [Citation26]. To define sub-exonic circRNAs, only circRNAs are retained when both coordinates of the circular junction are located within a single exon [Citation27,Citation28] even though the class of sub-exonic circRNAs could also be considered as a subclass of interior circRNAs. Several interior circRNAs have been validated using experimental data [Citation25,Citation26]. The main feature of the interior/sub-exonic circRNAs is that more than one circRNA is detected from the same genomic locus [Citation25–28].
To manage the balance between true and false circRNAs, it is necessary to have a better understanding of the genesis of false circRNAs. Differentiating between a circRNA observed within a dataset as either generated in-vivo or being an artificial circRNA (not generated in-vivo) is difficult but necessary [Citation22,Citation29]. Nielsen et al. points to small circRNAs as particularly suspect [Citation22]. In our previous study [Citation27] we identified several circRNA clusters with unannotated exon boundaries along bovine chromosomes, likely reflecting the presence of sequence/assembly/annotation problems in these regions. The presence of falsely mapped inverted sequences in the genome assembly potentially leads to mapping of reads from the linear transcript as artificial reads spanning the circular junction. Although no circRNA generated in-vivo is expected in mRNAseq data (library prepared with poly(A)-selected RNAs) [Citation21,Citation22], these in-silico generated artificial circRNA annotations (in-silico artif_circRNAs) would be found in both mRNAseq and total-RNAseq. An analysis including total-RNAseq and mRNAseq prepared from the same sample would be informative to resolve this. A recent study examined two samples to establish the background of the circRNA identification process, but only for exonic and intronic circRNAs [Citation17]. One study has used mRNAseq data to conduct classical circRNAs analyses [Citation30] without indicating that these datasets are a priori unsuitable for characterizing circRNAs [Citation9]. In 2015, Lu et al. reported a comparison performed in rice between the circRNAs detected in mRNAseq and in poly(A)-depleted samples [Citation31]. The total numbers of detected circRNAs in mRNAseq was slightly higher than those in poly(A)-depleted samples. In 2023, Ma et al. [Citation21] suggested that non-specific binding of circRNAs with oligo(dT)-beads explained the presence of circRNAs in their mRNAseq data. In their study, a high fraction of reads were not mapped to the rice reference genome (55% in mRNAseq and 93.4% in poly(A)-depleted), indicating that the data quality may have been low. To resolve this conflict of opinion about the circRNAs present in mRNAseq datasets, we suggested comparing the circRNA content of mRNAseq with that of total-RNAseq from the same samples. This pairwise comparison method has the advantage of providing equal opportunity for any in-vivo-generated circRNA to be present in both mRNAseq and total-RNAseq datasets. We noticed there is a similar conflict of opinion regarding datasets generated after RNase-R treatment. This enzyme is used to eliminate the majority of linear transcripts and increase the concentration of circRNAs [Citation22]. Some authors consider circRNAs detected only after treatment to be low-expressed circRNAs [Citation5], while others do not consider them as reliable circRNAs [Citation32].
One of the aims of the European BovReg project (https://www.bovreg.eu/) was to generate a map of functionally active regulatory and structural elements in the bovine genome using a diverse catalogue of at least 26 tissue types collected from individuals of both sexes and from divergent breeds/crosses (117 samples in total) [Citation4,Citation33]. The data generated by BovReg provided an interesting opportunity to explore some aspects of the circRNA transcriptome in cattle because the transcriptome sequencing was performed in two ways: mRNAseq and total-RNAseq. The respective datasets were generated in very similar conditions to minimize any batch effects, and paired mRNAseq and total-RNAseq datasets were available for a subset tissues from the same animals. We performed the characterization of circRNAs using two bovine annotations (Ensembl and a new annotation generated by the BovReg project) and 117 samples obtained from 26 tissues, across 3 populations of cattle, with a first objective to understand the presence of non-exonic circRNAs. For this purpose, we also looked at a subset of 63 samples with available high-quality paired mRNAseq and total-RNAseq data. In a previous study performed on bovine, ovine and porcine tissues [Citation27], we had already obtained an indication of a large proportion (40% to 80%) of non-exonic/intronic circRNAs in bovine and ovine tissues. In this current study, we again could only annotate 40% of the highlighted circRNAs as exonic circRNAs in spite of a more comprehensive transcriptome annotation. With the availability of paired datasets, we had the chance to further explore mRNAseq-based output with respect to circRNAs. By performing these analyses, however, we did not expect to fully resolve the question that arose regarding the low proportion of circRNAs annotated as exonic circRNA. In this study, firstly we aimed: i) to understand the presence of non-exonic circRNAs in the cattle transcriptome, ii) to understand the origin of artificial circRNA, i.e. circRNA not generated in-vivo. Finally, we were able to build the first reliable catalogue of bovine circRNAs. Our second objective was to perform a comparison between the circular transcriptome (by considering only a small number of reliable circRNAs) of divergent tissues. Our results reflect the diversity of the circular transcriptome in cattle and provide a resource for comparative analysis across cattle populations and between species.
Materials and methods
Animals, samples and datasets
The six animals chosen for the sample collection originated from three populations kept in different environments representing different ages and sexes. Holstein Friesian calves from Belgium (neonatal: male calf 24 days and female calf 22 days), Kinsella composite juveniles from Canada (bullock 217 days and heifer 210 days) and Charolais x Holstein F2 cow and bull from Germany (adult: bull 18 months and cow 3 years, 7 months and 13 days).
All details of the animals are available in [Citation4]. Details of tissue sampling and storage, and RNA extraction, quality and integrity assessment are described in [Citation4]. All samples were sequenced in two ways: mRNAseq and total-RNAseq libraries were generated, quantified and sequenced by the GIGA Genomics platform (University of Liège, Belgium). mRNAseq libraries were built using the ‘TruSeq Stranded mRNA Library Prep’ kit (Illumina) following the protocol provided by the manufacturer. Total RNA libraries were built using the ‘TruSeq Stranded Total RNA Library Prep Gold’ kit (Illumina) following the protocol provided by the manufacturer. The Illumina NovaSeq 6000 instrument was used for sequencing, with a paired-end (PE) protocol (2 × 150bp).
For circRNA characterization, we considered a batch of 117 datasets obtained by total-RNAseq. Among the 26 tissues represented in this batch, 11 and 4 tissues were represented by 6 and 5 datasets, respectively, see Atab-1. A sub-batch of 63 samples were chosen for the comparison of total-RNAseq (63T) and mRNAseq (63 m). Among the 11 tissues represented in this batch, 8 and 3 tissues were represented by 6 and 5 datasets, respectively, see Atab-1.
Circular RNA detection and characterization
The RNAseq reads were mapped to the bovine genome reference assembly ARS-UCD1.2 (GCA_002263795.2) using the rapid splice-aware read mapper Spliced Transcripts Alignment to a Reference (STAR, version 2.7.10a [Citation34]). We selected the single-end alignments mode of STAR (STAR-SE) mapping mates of each pair independently. STAR was used with the previously proposed parameters [Citation35] that enable highlighting chimeric reads with only two mapped segments and with a minimal size for the smallest mapped segment of 15 bp.
Our approach to characterize circRNAs is described in (see M&M_Adoc-1–4 for details). The chosen circRNA tool, CD (CircDetector [Citation27,Citation36]), works in two steps. The initial step involves identifying reads that contain a circular junction, referred to as circular chimeric reads (CCRs), and generating two output files (). In the main CD_detection output file, detection.bed, CD reports a list of all circular RNAs and their associated number of CCRs, each circular RNA being defined by the coordinates of the circular junction (chromosome:start-end|strand). When CD is used with a gtf_file containing exon features (Ensembl v-105 and a new annotation generated by the BovReg project were used), the second module of CD is able to annotate certain circRNAs (, see also M&M_Adoc −2 & −3 for details). For instance, CD can identify circRNAs resulting from backsplicing events and provide a list of putative exonic circRNAs. It also identifies the two exons involved in the backsplicing and their respective parental gene (). In this study, we defined other_circRNAs as those not included in any of the three retained lists (). By creating this category, we set these circRNAs aside with the de-facto suspicion that some of them are artif_circRNAs, to be examined in detail in this study. We performed manual curation of each of the three retained output files to identify exonic circRNAs (associated with the use of blue colour in the figures), lariat-derived circRNAs (black/yellow), intron circles (black/pink), and sub-exonic circRNAs from small non-coding genes (snc) (black/green). For example, we rejected a potential exonic circRNA candidate, which would have resulted from backsplicing between two exons from different parental genes (Ensembl_gene A and Ensembl_gene B; or MSTRG_gene X and MSTRG_geneY). CircRNAs that were rejected during manual curation (symbolized by a trashcan in ) were not added to the other_circRNAs list (orange in the figures).
Figure 1. Analytical pipeline used to characterize circRNAs.
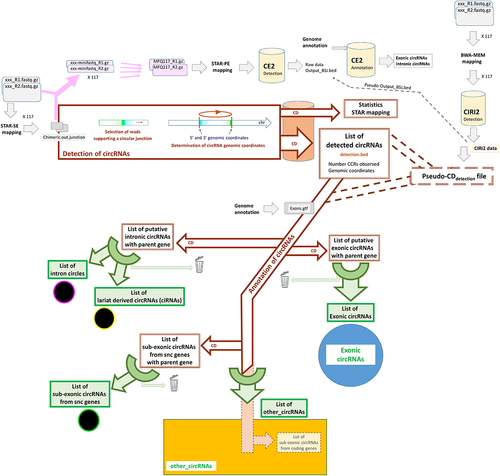
Annotating a large number of circRNA lists has never been performed previously as an individual task. This step was performed with pseudo-CDdetection output files using in particular a compilation of all detection.bed files from all datasets (, M&M_Adoc-3).
In addition, detection of circRNAs was also performed with CIRI2 [Citation24] (see ). Unlike CD and most other circRNA discovery tools, CIRI2 works from paired ends alignments. It requires these alignments to be performed by BWA-MEM (here we used the version 0.7.17-r1188) [Citation37]. CIRI2 implements multiple filtering strategies to eliminate false positives, including splice site analysis. CIRI2 (version 2.0.6.) was used with default parameters and only circRNAs detected by two reads (the maximum threshold option provided by the programme) containing a circular junction were retained. We have chosen to use CIRI2 without an annotation file and therefore the output file does not include any information about the parental gene. This output file contains the number of reads spanning the circular junction. The annotation of CIRI-circRNAs was performed by using CD with a pseudo-CDdetection output file. We used the BovReg annotation for classifying exonic and intronic circRNAs. For exonic circRNAs, we did not add a manual verification process; as a result, we identified only putative-exonic circRNAs.
Since the Chimeric.out.junction files derived from the STAR-SE alignments contain the coordinates of all reads spanning a circular junction, we suggested to use these lists to generate mini-fastq files (, all details in Atab-2). For the filtration step we used the Python3 script Fastaq (version 3.17.0, https://github.com/sanger-pathogens/Fastaq). These mini-fastq files represent an in-silico enrichment of reads spanning a circular junction and can be used with any circRNA detection tool. We built 117 paired mini-fastq files (R1 and R2) (doi:10.57745/IUJ40P) and a pair of MFQ117 files (, above in pink). The pool of chimeric reads (MFQ117 files) was mapped to the bovine genome using STAR in paired-end alignment mode. CircRNAs were detected with CIRCexplorer-2 (CE2) [Citation23] and a first output file was obtained (Output_BSJ.bed). The second module of CE2 allowed to perform the annotation using a gtf file derived from Ensembl v110 and led to the identification of exonic and intronic circRNAs (). For circRNAs identified by CIRI2, we were able to build a pseudo Output_BSJ.bed to annotate them.
Three circRNA tools were employed in this study. Although the authors of CIRI2 and CE2 utilize the term BSJ (backsplicing junction), we will refer to reads spanning a circular junction as CCR. None of the three tools utilized in this study identified exonic circRNAs exclusively. Circular RNAs are considered to be ‘validated’ if at least five CCRs are observed. Otherwise, they are simply considered to be ‘detected’.
To complete the benchmarking of circRNAs, we used lists of bovine circRNAs previously published by including genomic coordinates. More specifically, we used the list of exonic circRNAs characterized by CE2+CIRI2 and the list of intronic circRNAs characterized by CD published in 2021 (reported in Tables Suppl_list_1 and Suppl_list_3 of [Citation27] respectively).
Analyses relative to exonic sequences
The BovReg annotation consisted of a gtf_file defining 683,396 distinct exons (average length = 1,628 nt and median length = 226 nt). Only 235,049 were previously described by Ensembl v105 (average length = 308 nt and median length = 139 nt).
To perform what we call a minimal_annotation of exonic circRNAs, we built two sub-lists (Left_exons and Right_exons) from the list of all BovReg exons. To constitute the list of Left_exons, we selected exons according to their unique first genomic coordinate (M&M_Adoc-5) keeping only the exon with the smallest size in case of multiple exons with the same first coordinate (M&M_Adoc-6A). For the list of Right_exons, we only filtered for unique second coordinates (M&M_Adoc-6A). We retained a list of 636,307 distinct exons (582,688 in the list of Left_exons and 456,432 in the list of Right_exons). To perform a manual and ‘minimal’ annotation of exonic circRNAs, it is necessary to identify the two exons involved in backsplicing using these two lists (M&M_Adoc-6B). The list of Left_exons is used to identify the upstream exon (or ‘acceptor exon’) involved in the backsplicing when the parental gene is located on the forward strand and the downstream exon (or ‘donor exon’) when the parental gene is on reverse strand (see M&M_Adoc-1).
We used several tools available on the Galaxy platform proposed by Sigenae [Citation38] in particular to perform bedintersect (http://bedtools.readthedocs.io/en/latest/content/bedtools-suite.html). To examine the exonic sequence content of the genomic interval defined by the circRNA, we applied a 90% exon overlap threshold on the same strand. This allowed us to conclude that the circRNA interval contained what we then call a quasi-full exon. Bedintersect was also used to analyse the localization of the points defined by the two genomic coordinates of a circRNA, both inside and outside of an exon. The search was performed using the BovReg list of 683,396 exons, considering both strands. For each circRNA, two genomic intervals were defined: the first interval contains the 30 nucleotides downstream of the 5’ coordinate, and the second interval contains the 30 nucleotides upstream of the 3’ coordinate.
Statistical analyses
All the statistical analyses were carried out using R (v.4.0.2) (https://www.r-project.org/). Significant differences between circRNA proportions from contingency tables were identified with the Pearson’s Chi-squared test (chisq.test function from R STAT package v.4.0.2). A p-value less than .05 was considered as statistically significant.
Hierarchical clustering and principal component analyses
The hierarchical clustering analyses (HCA) were performed on the Galaxy platform proposed by Sigenae [Citation38]. All clusters were done with the ‘ward’ agglomeration method as suggested by developers [Citation39] and using Pearson’s correlations as distance. The principal component analyses (PCA) were also performed on this platform, with the function PCAFactoMineR, using the FactoMineR package. Data was transformed by the normalization module available on the Galaxy Platform. For HCA, the log-binary (binary log (expr +0.0001)) and standard score (standard score; mean = 0 - sd = 1) methods were used, while for PCA only the standard score method was used. These tools are part of a set of statistical tools made available by members of the BIOS4BIOL group (‘Normalization’, ‘Summary statistics’, ‘Hierarchical clustering’ and ‘PCAFactoMineR’) (see https://github.com/Bios4Biol).
For clustering, 96 samples were retained (see Atab-1). To avoid introducing a tissue represented by a single dataset, we selected 15 tissues, where samples were available for the two youngest (neonatal) and at least three of the older animals (juvenile or adult). In addition, we considered five tissues, where samples were available for the two young animals. For the PCA analysis of samples related to reproduction and hormonal function, 19 samples were considered from pituitary gland, adrenal gland, ovary, testis, uterus, and uterus-horn. For more details, see Atab-1.
Results
Characterization of 61,083 circRNAs less than 40% of them being exonic circRNAs
For circRNA characterization, we retained 117 samples sequenced with total-RNAseq from 26 distinct tissues providing a total of 10,052 million reads (150 bp) across all the samples. Three tissues samples from neonate animals (jejunum-female, rumen-male, pancreas-male) were sequenced at high depth (called ‘XL sequencing’). Two sequencing datasets were available for the cerebral cortex sample from the juvenile castrated male. 86% of reads mapped unambiguously to the bovine reference genome. At least 37 million uniquely mapped reads were obtained for each sample. For the three datasets with XL sequencing, 395, 410 and 432 million reads were available, while for the 114 other datasets we observed an average of 77 million reads that were uniquely mapped (all details are available in Atab-1). We did not observe any outlier samples, with a poor mappability, and all 117 datasets were considered for further analyses.
We started with the exhaustive list of circRNAs present in at least one sample. Rare circularization events were excluded by only retaining circRNAs detected by five reads containing the circular junction (CCRs for circular chimeric reads). Several studies have demonstrated the value of excluding such events [Citation32,Citation40]. The 117 output files generated by CD [Citation27,Citation36] were concatenated, resulting in the detection of 66,299 circRNAs. After discarding circRNAs with genomic size less than 70 nucleotides 61,083 circRNAs were retained.
Annotation with CD was performed using the list of 61,083 circRNAs as a single pseudo-sample using the two bovine annotations. All selected output files were manually inspected (, see also M&M_Adoc-3). We were aware that the list of circRNAs provided by CD can include false entries and we chose to retain only three sub-lists created by CD (see materials and methods, and ) and put the others in a list of other_circRNA. To create this list, we deducted from the initial list of 61,083 circRNAs the following: 24359 putative exonic circRNAs (CD BovReg annotation), 373 putative intronic circRNAs (CD BovReg annotation), and 108 sub-exonic circRNAs from snc genes (CD Ensembl annotation). Therefore, we retained 36,215 circRNAs as other_circRNAs for further analysis (1).
Figure 2. Overview of circRNAs detected in the 117 samples considered.
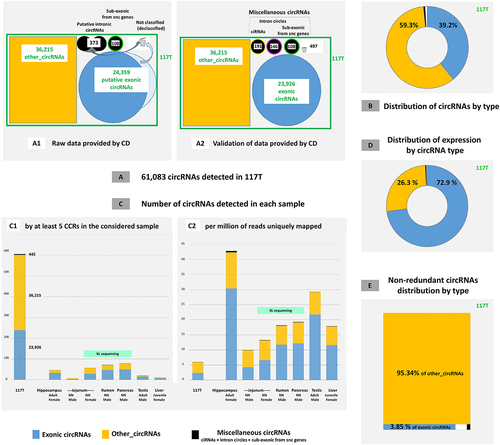
The analysis of the list of putative exonic circRNAs led to the characterization of 23,926 exonic circRNAs (i.e. resulting from backsplicing (BS) involving known exons; 2). More precisely, we were only able to annotate 20K circRNAs as exonic circRNAs using the Ensembl annotation and to add 4K circRNAs to the list of exonic circRNAs using the BovReg annotation. They are originating from approximately 8K parental genes (Res_Adoc-1). Furthermore, we observed 2K exonic circRNAs from a backsplicing between an exon with an Ensembl ID and a novel (MSTRG) exon in the BovReg annotation. From the sub-list of putative intronic circRNAs provided by CD, our analyses featured the annotation of 191 ciRNAs (147 introns concerned from 146 genes, SList-1) and 146 intron circles (126 introns concerned from 124 genes). We have grouped circRNAs that do not fit into the two main categories (exonic circRNAs and other_circRNAs) into ‘miscellaneous circRNAs’ (). We found ciRNAs, intron circles and sub-exonic circRNAs from snc genes. In these 117 samples considered, 39.2% of circRNAs are exonic circRNA and we classified 59.3% as other_circRNAs ().
Analysis of circRNA diversity observed in 117 bovine datasets (total-RNAseq)
In the full pseudo-sample (117T), we observed 61,083 circRNAs () with an average scaled read count of 6 circRNAs per million of uniquely mapped reads (). The landscape of the 117T merged list is very different from the 117 individual samples (). We observed less circRNAs per million of uniquely mapped reads in 117T than in each individual sample (): however, while the number mapped reads add up across the samples, the number of distinct circRNAs does not due to redundancy of circRNA identification in the different samples. The seven examples in show the great diversity of these 117 individual datasets. Three samples had very deep sequencing (XL sequencing), and this led to the identification of more circRNAs, including predominantly more of the other_circRNAs type ().
We observed that 6,982 circRNAs were detected in a single sample and were not confirmed in any other samples, not even when applying a threshold of a single CCR. It is hardly surprising to find the three samples that benefited from XL sequencing were among the samples with the highest proportion of circRNA not found in any other sample. Among the 6,982 non-redundant circRNAs detected in 117 datasets, only 268 were exonic circRNAs, i.e. we have 95.3% of other_circRNAs (). In other words, more than 18% (6,714 out of 36,215) of the other_circRNAs were detected by only five CCRs and in only one sample.
Brief description of CD-other_circRNAs
When we looked at the size of the genomic interval defined by the two genomic coordinates of an other_circRNA, we noted that 22.9% of other_circRNAs defined small genomic intervals (see Res_Adoc-2). When we looked at the exon content of the genomic interval defined by the two boundaries of the other_circRNAs, we found that 71.2% did not contain a quasi-full exon (90% of an exon, see MM section) (see Res_Adoc-3). Nevertheless, 62.4% of the other_circRNAs have their two genomic boundaries in exonic sequences (see Res_Adoc-3, and M&M_Adoc-4).
Among the 61,083 circRNAs, we identified 487 from the mitochondrial genome, all included in other-circRNAs. Other_circRNAs from the mitochondrial genome are more often smaller than other_circRNAs detected on the nuclear chromosomes (data reported in Res_Adoc-2 showed a statistically significant difference). As in our previous study [Citation27], we identified several clusters of other_circRNAs along the chromosomes, likely reflecting the presence of sequence/assembly/annotation problems in these regions. We identified 3,187 circRNAs (including 3,159 other_circRNAs) clustered in a region (BTA-27: 6.21–6.23Mb) known to contain the Defensin gene. Other_circRNAs from the Defensin region are less often of small size than other_circRNAs detected on other chromosomes (data reported in Res_Adoc-2 showed a statistically significant difference). The assembly of this region is thought to be difficult due to a substantial number of copies of the same or very similar sequences. In addition, it is assumed that bovine individuals differ in the number of Defensin gene copies. As such, this region is clearly a candidate to provide in-silico artif_circRNAs.
Brief description of circRNA expression
The number of circular junction reads associated with the detection of a circRNA is commonly used to quantify the expression of that circRNA (corrected for the number of uniquely mapped reads in the dataset). To determine the expression of each circRNA in each sample, an inventory integrating statistic relative to each sample was obtained from a second run (117X) of CD, but without a threshold on the number of CCRs (see also M&M_Adoc-3). At expression level, we noted a clear dominant impact of exonic circRNAs, since they are responsible for 72.9% of the global expression of circRNAs in our datasets (). This result is in contrast to the 39.2% of circRNAs annotated as exonic circRNAs (). Intronic circRNAs are responsible for 0.37% of the global expression of circRNAs (). The expression of intronic circRNAs, ciRNA-ATXN2L, which was found to be the dominating ciRNA in pigs [Citation28], was very low in most samples in the current analysis of bovine tissues, similar to a previous report [Citation36].
Although the expression of an exonic circRNA varied between 0 and 30, we defined ‘notable expression’ as expression above 0.05. We observed that 95.5% (22,846/23,926) of exonic circRNAs had a notable expression in at least one sample but only 5.3% (1,268/23,926) had a notable expression on average across all 117 samples. For example, seven exonic circRNAs were identified in the region of SMARCA5 (ENSBTAG0000000003399), but only one has a notable expression on average across all 117 samples. This exonic circRNA (17:14349781–14350241|-) has a notable expression in each of these 117 samples (SList-3) and the second highest average expression across the 117 samples. We remarked also an exonic circRNA with expression restricted to a single tissue (heart). This circRNA (2:18153915–18180018|+) originates from a region very poorly annotated in Ensembl, probably harbouring the TTN region. Nevertheless, the list of exonic circRNAs (Ext_Atab-2) from this region is long but it is unique with this tissue specificity (only detected in neonates Belgian animals).
Parallel analyses performed in total-RNAseq and mRnaseq reveals artificial circRNAs
The availability of bovine paired datasets was a good opportunity to perform a circRNA detection in total-RNAseq and mRNAseq in parallel. We put together a new dataset of 63 samples from 11 tissues (see Atab-1) with high-quality mRNA and total-RNA data available. Even though we avoided including samples from XL sequencing for total-RNAseq, the number of reads available for total-RNAseq was higher than for mRNAseq.
The 63 total-RNAseq dataset (63T, ) identified over 35,000 circRNAs, while the 63 mRNAseq dataset (63 m) identified 4,579 circRNAs (). The high number of circRNAs detected in 63 m, which represents more than 10% of the number detected in 63T, was completely inconsistent with an expected background. Indeed, we expected to find circRNAs existing in-vivo but resulting from non-specific binding to oligo (dT) beads and in-silico artif_circRNAs. Moreover, these two possible types of circRNAs present in mRNAseq were expected in total-RNAseq. Upon examining the 4,579 circRNAs from 63 m, it was observed that 63.4% (2,901 out of 4,579) had not been previously identified, i.e. in 117T (). Consequently, all circRNAs detected in mRNAseq were deemed unreliable and artificial. Additionally, it is important to determine the source(s) of these artif_circRNAs.
Figure 3. Analysis of circRNAs detected in mRNAseq.
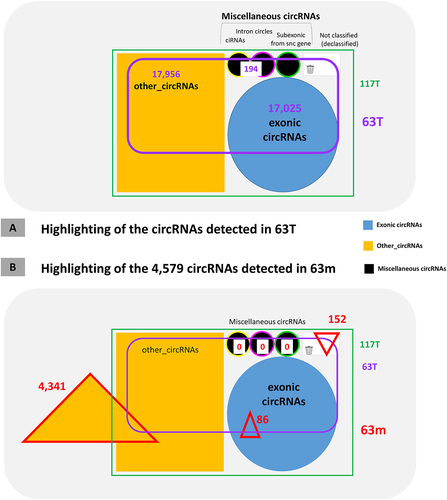
In mRNAseq datasets (63 m), we did not detect any intronic circles, ciRNAs, or sub-exonic circRNAs from snc genes. In addition, no AS-exonic circRNA was detected in the 63 m (13 and 20 were detected in 63T and 117T, respectively) and the eight aberrant-exonic circRNAs reported in Table S3 and detected in 63T were also absent in the 63 m (). We have no reason to suspect these different types of circRNAs as unreliable.
In the 63 m dataset, we identified 86 exonic circRNAs from the list of circRNAs found in 117T (). The level of artif_circRNAs is very, very low among exonic circRNAs. We cannot be as assertive among intronic circRNAs or sub-exonic from snc, as the samples were much poorer.
In the 63 m dataset, we retained 4,341 circRNAs as other_circRNAs but 2,812 (64.8%) have never been detected in total-RNAseq. We detected a higher proportion of other_circRNAs with a small genomic size (70–159 nt) in 63 m than in 63T (data reported in Res_Adoc-2 showed a statistically significant difference). This observation is certainly related to the higher proportion of other_circRNA that we could have classified as sub-exonic from multi-exonic genes (both genomic coordinates are located in the same exon, nuclear chromosomes and mitochondrial genome, Ensembl annotation, see Res_Adoc-4A). The fact that we observed the same number of these sub-exonic circRNAs per million uniquely mapped reads in 63 m and 63T (Res_Adoc-4B) does not support their reliability, as their genesis seems to be automatic or mechanical.
In an attempt to get a clearer picture of the reliability of other_circRNAs, we proposed to focus on three regions (see also Res_Adoc-5). In the albumin gene region, sample 63T is as informative as 117T for other_circRNAs. Most of the other_circRNAs identified in this region (57/59) have both genomic coordinates in exonic sequences (in the same exon (sub-exonic) or in two exons). Since the five new other_circRNAs detected in 63 m have the same feature (5/5), it was difficult to conclude that the 32 other_circRNAs identified only by total-RNAseq were reliable. The analysis of the other_circRNAs from the mitochondrial genome detected in 63 m led us to consider them all unreliable (see Res_Adoc-5). This is not surprising because the characteristics of other_circRNAs from the mitochondrial genome are very close to those of sub-exonic circRNAs from multi-exonic genes. In contrast to, the statistics about the other_circRNAs detected in the Defensin gene region (Res_Adoc-5) suggest that a very large proportion of them are reliable. Undeniably, this 63 m/63T comparative study casts doubt on the reliability of at least a large proportion of the other_circRNAs.
Complementary analyses performed with CIRI2
Using CIRI2 [Citation24] on the 117 total-RNAseq datasets 58,373 CIRI-circRNAs were detected with at least two reads spanning the circular junction. Even though we consider this too low a threshold, it is the upper limit for initial analysis implemented in the CIRI2 program. For example, 350 CIRI-circRNAs were identified in the Defensin region. Among the 23,926 exonic circRNAs identified by CD 20,531 were also identified as CIRI-circRNAs (). The overall confirmation rate for CD-exonic circRNAs is 85.8%, but only 2/27 for exonic circRNAs from the Defensin region. We also identified 2,305 other-circRNAs identified by CD among the CIRI-circRNAs (). The confirmation rate for CD-other_circRNAs is 6.4% (only 17/3,160 for other_circRNAs from the Defensin region). When the annotation of these 58,373 CIRI-circRNAs was performed with CD 48,310 putative exonic circRNAs were suggested. Among the 18 putative miscellaneous circRNAs, we retained three ciRNAs, nine intron circles 3 sub-exonic circRNAs from snc genes (). These data further defined a set of 10,102 CIRI-other_circRNAs (). Of these 10,081 circRNAs, only 111 (1.1%) had a small genomic size (135–160 nt) (Res_Adoc-2). Furthermore, none of them was from the mitochondrial genome. When we continued the comparisons of the features of these CIRI-other_circRNAs (see for details Res_Adoc-3), we were able to conclude that the other_circRNAs identified by CIRI2 were not the same as those identified by CD. These observations are not very surprising as the design of these two bioinformatic tools is different.
Figure 4. Analysis of circRNAs detected by CIRI2.
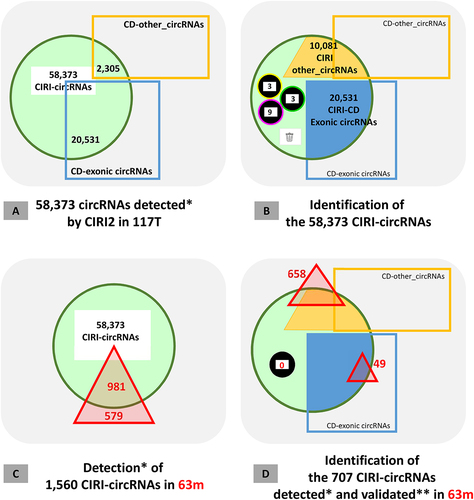
Subsequently, we performed a new detection of circRNAs in the 63 m dataset using CIRI2. Out of the 1,560 CIRI-circRNAs detected, 579 were not present in the list of 58,373 CIRI-circRNAs detected in 117T (). The detection in 63 m of these previously undescribed CIRI-circRNAs (37.1%) confirmed that all circRNAs detected in mRNAseq can be considered artificial. However, we only kept the 707 CIRI-circRNAs that were detected with at least five reads spanning the circular junction, which were validated as artificial circRNAs. Among them, we recognized 49 CD-CIRI exonic circRNAs from the list established on 117T by CD. We can conclude that the list of CIRI-circRNAs detected in mRNAseq included no more than 4% of the validated exonic circRNAs. These 49 circRNAs which were previously annotated as exonic circRNAs are now considered as artif_circRNAs, since they were detected in mRNAseq. Similar to CD, CIRI2 can also identify 63 circRNAs that were not detected in 63T. For example, 6 out of 49 CD-CIRI exonic circRNAs were identified.
Refinement of the list of exonic circRNAs
Identification of 103 artificial circRNAs among the list of exonic circRNAs
We identified 86 and 49 exonic circRNAs as artificial circRNAs from the analyses of 63T/63 m by CD and CIRI2, respectively. Since 32 were identified by the two approaches (see Atab-3), we suggest that 103 circRNAs previously annotated as exonic circRNAs are artif_circRNAs.
When we examined backsplicing falsely identified at the origin of these 103 artif_circRNAs, we found 2/5 from two Ensembl exons (42), 2/5 from two MSTRG exons (40) and 1/5 from mixed pairs (21). These observations showed a statistically significant difference with the observations made on the list of 23,926 exonic circRNAs where 77% of backsplicing involved a pair of Ensembl exons. (Res_Adoc-1, chisq_test with p-value <2.2 10−16).
Among this list of 103 artif_circRNAs, we find the circRNA with the highest average expression across all of the 117 samples (2:18153915–18180018|+). This circRNA was actually only detected in the two neonatal animals, which were also the two Belgian animals. It could be an artif_circRNA generated by differences in this genomic region (TTN), specific for these two animals of the same genetic origin (Holstein Friesian). Nevertheless, it is surprising that CD and CIRI2 detected it in 117T, while only CIRI2 detected it in 63 m. Among the set of 103 artif_circRNAs, we also noticed the presence of a cluster of 11 circRNAs from a region on BTA23 (28.52–28.72 Mb) containing a part of the major histocompatibility complex (MHC) class I genes. These 11 circRNAs were ‘linked’ by exon(s) identified as involved in (false) backsplicing and originating from the same MSTRG gene. This cluster included the circRNA with the highest average expression across the 117 samples, but was in fact only expressed in the tissues of neonatal animals. In the same region, 63 other_circRNAs were characterized in 63T, but 54 were invalidated by the analysis of 63 m. Moreover, 26 novel other_circRNAs were detected in 63 m.
Fine annotation of exonic circRNAs reveals some artificial annotations
Using two separate exon lists (Left_exons and Right_exons), a second annotation called minimal_annotation was performed for each of the 23,926 exonic circRNAs. In this way, we identified the two exons involved in each backsplicing, and when alternatives were possible, only the smaller exon was considered, regardless of the name of the parental gene (for details see the Materials and Methods section and M&M_Adoc-6). This minimal_annotation led to the characterization of a larger fraction of exonic circRNAs annotated with an Ensembl exon and an MSTRG exon compared to the classical CD-based circRNA annotation (Res_Adoc-1). Only 30,831 different exons (4.8%) out of the 683,396 described exons of the bovine genome (it would be more correct to take into account only the 636,307 considered for the minimal_annotation) were involved in the generation of exonic circRNAs.
We can describe the group of bovine exons involved in backsplicing by a mean size of 188 bp and a median size of 133 bp. These exons appear to be larger than those characterized as involved in exonic circRNAs in human HEK cells (160–165 bp for the mean size [Citation41]) or in exonic circRNAs from porcine testis (148 bp for the mean size [Citation17]). We detected 48 circRNAs annotated with two overlapping exons among the 23,926 exonic circRNAs, however, these two exons cannot be associated in the same transcript. Thus, these 48 circRNAs are not true in-vivo exonic circRNAs. Among them, we found 23 of the 27 circRNAs identified as exonic circRNAs from the Defensin gene. Our analysis also led to the identification of 1,025 single exon circRNAs (see the M&M_Adoc-5). The average length of these exons is 605 bp. This size is consistent with the one observed for a single-exon circRNAs from porcine testis (647 nt [Citation17]) or human HEK cells (709 nt [Citation41]). Among the list of 1,025 single exon circRNAs, we noted that seven were originating from the same parental gene (ENSBTAG00000006907, Nebulin, NEB), which is in itself suspicious. Moreover, we noted that four of them were detected in 63 m by CD, and, thus, were already suspected to be artif_circRNAs. An eighth exonic circRNAs from the same region seemed suspicious, since it involved a BS between two of the same exons. We suggested not retaining these eight circRNAs from the Nebulin region as exonic circRNA. Since single exon circRNAs range in size from 76 to 6,723 nt, we can suspect that exons larger than 7 kb are likely too large to be involved in backsplicing. When the list of 23,926 exonic circRNAs was examined in regard of the size of both exons involved in the backsplicing, we decided to not retain as exonic circRNA nine circRNAs involving at least one very large (15–38 kb) exon. All are MSTRG exons from the BovReg annotation.
This deep exon-based annotation allowed the identification of 48 (overlapping exons) + 8 circRNAs (many single exon circRNAs from the same gene) + 9 (very large exon involved), i.e. 65 circRNAs that were initially described as exonic circRNAs but share an annotation casting doubt on their true in-vivo existence.
Discovery of 26 exonic circRNAs that were very suspicious
When the list of 23,926 exonic circRNAs was examined with respect to the size of the genomic region defined by their two genomic coordinates, we found 26 circRNAs that defined a region of up to 500 kb (0.1%). Among them, we did not find any exonic circRNAs identified as the result of backsplicing between two Ensembl exons. The first with this feature defines a region of 483 kb. In addition, in CD-other_circRNAs we identified 380 circRNAs with this feature (1%) and CIRI2 considers only circRNAs defining a genomic interval < 200 kb. We considered these 26 circRNAs previously annotated as exonic circRNAs too suspicious to be reliable circRNAs, the probability that they are artif_circRNAs is very high.
The list of exonic circRNAs included 189 false entries
In addition to the 103 artif_circRNAs highlighted the analyses of 63T/63 m by CD and CIRI2, and to the 26 probable artif_circRNAs highlighted by the examination of the size of the genomic interval defining the circRNA, the process of fine annotation led to the highlighting of 65 artificial annotations. As a result, the list of exonic circRNAs was purified from 189 units and only 23,737 exonic circRNAs were considered for further analyses. The list of the 189 discarded exonic circRNAs is provided in Atab-3 (and in Ext_Atab-4).
Bovine circular transcriptome
For these analyses, we first considered the 117 samples and then a group of 15 tissues for which samples were available from the two young animals and at least three juvenile or old animals. We detected an average of 5,329 exonic circRNAs with non-null expression in each of the 117 samples (), but only 1,711 exonic circRNAs () with a notable expression. When we looked at the individual sample scale, ‘the number of exonic circRNAs with non-null expression’ () showed less homogeneity per tissue than ‘the number of exonic circRNAs with notable expression’ (). Considering these two criteria for the number of expressed circRNAs, we observed a similar ranking for the 5 or 6 animals in only two tissues out of 15 (cerebellum and spleen) (). Regarding the testis with two samples, we noted that the numbers of expressed circular exonic RNAs evaluated by the two criteria () are concordant and conclude that this number decreases with age in bovine. These results are consistent with our previous work characterizing circular exonic RNAs in tissues from three livestock species [Citation27]. The cerebellum sample from the juvenile female showed the highest mean values for these two criteria ().
Figure 5. Analyses of the possible presence of 23,737 exonic circRNAs in bovine tissues/samples.
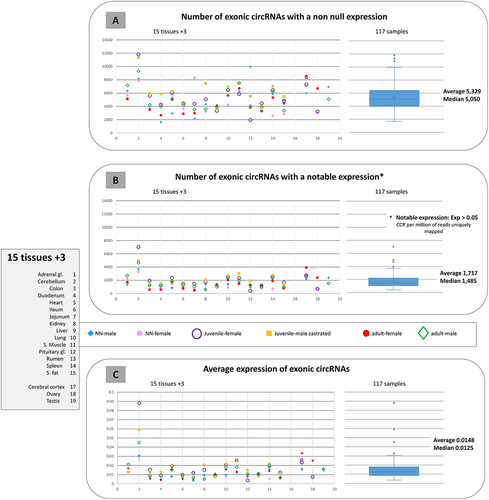
For each tissue sample, the average expression level across each of the 23,737 exonic circRNAs () was calculated. Among the four samples showing the highest expression level of the 117 samples, three were from the cerebellum (). The cerebellum differs from the other 14 tissues by the highest mean expression values per tissue (). The cerebellum was also distinguished by the variability in expression level that exists between samples ().
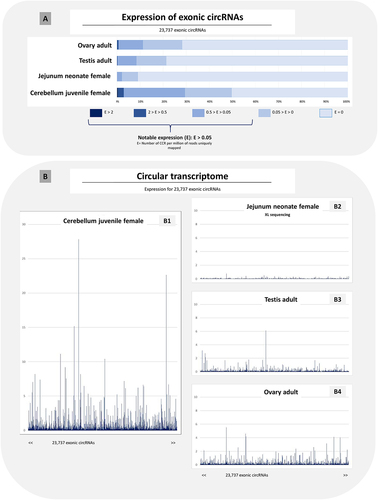
For the three criteria considered (), we observed that the XL sequencing, applied to three samples, did not affect the results. For these three criteria, the cerebellum from the juvenile female presented always the highest expression levels. This sample is also undoubtedly the sample with the most diverse circular transcriptome among the 117 samples considered here (). In contrast, the circular transcriptome can be described as poor in terms of diversity, complexity and expression level for digestive tissues. One of the ‘poorest’ circular transcriptomes considered here is that of the jejunum from the neonatal male (). Intermediate to the two extreme transcriptomes for cerebellum (rich) and jejunum (poor), are e.g. the testis and the ovary of adult animals (, B4).
Figure 6. Expression analysis of 23,737 exonic circRNAs in four samples.
Benchmarking of circRNAs
The initial objective is to benchmark the list of exonic (and intronic) circRNAs validated by CD. When utilizing two circRNA detection tools, it is logical to apply the validation threshold to only one of the two tools. The pool of chimeric reads obtained after STAR-SE mapping of 117 total RNA-seq datasets (MFQ117) was utilized to perform a novel circRNA detection by CE2. All circRNAs that were detected with at least one read spanning the circular junction were retained. In the CE2 list, we identified 61 out of 191 ciRNAs that had been validated by CD and 12 out of 146 intron circles that had been validated by CD. Thus, our findings demonstrate that 20,289 of the 23,737 exonic circRNAs validated by CD and considered to be reliable were detected by CE2. A total of 20,431 exonic circRNAs validated by CD were identified among the circRNAs detected by CIRI2. It is noteworthy that 8,726 exonic circRNAs validated by CD had also been validated by CE2+CIRI2 in the study of circRNAs from three bovine tissues. Moreover, we note that 8,726 exonic circRNA validated by CD had also been validated by CE2+CIRI2 in the study of circRNAs from three bovine tissues [Citation27]. All details of this benchmarking are shown on . In short, of the 23,737 exonic circRNAs validated by CD, only 1,453 were ever observed exclusively by CD, and 8,621 were identified by the four methods. These observed scores of 6.1% and 36.3% appeared somewhat reversed among the 189 unreliable exonic circRNAs, with 35.4% and 13.8%, respectively. (2). All available annotations for the 23,926 exonic circRNAs validated by CD in 117T (23,737 reliable + 189 unreliable) were reported in Ext_Atab −3 & −4, including, where possible, the official circRNA name according to the nomenclature proposed by Chen et al. [Citation42].
Figure 7. Benchmarking of 23,737 reliable exonic circRNAs and additional exonic circRNAs.
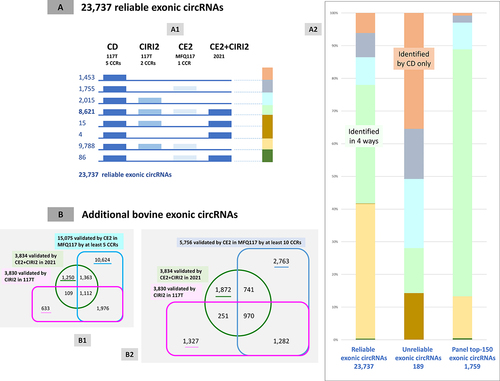
Our second objective was to propose an additional list of exonic circRNAs, only validated by CE2 in MFQ117 (Ensembl v110) or/and by CIRI2 in 117T (Ensembl v110) or/and by CE2+CIRI2 identified in 2021 (Ensembl v101) [Citation27]. Those validated by CIRI2 with at least 5 CCRs number 3,830 and those validated by CE2+CIRI2 in 2021 number 3,834 (). CE2 validated with a minimum of 5 CCR’s 15,075 number () and with a minimum of 10 CCR’s number 5,756 (). In light of the fact that the MFQ117 analysis indicates that the five CCRs may have originated from five distinct samples, it can be argued that a threshold of 10 CCRs is a more appropriate choice. The presence of these 9,206 circRNAs was analysed among the lists of circRNAs validated in 63 m by CD or by CIRI2. Five and twenty-one were detected, respectively, leading to the characterization of 23 additional exonic circRNAs that were subsequently deemed unreliable. The list of additional bovine exonic circRNAs includes now 9,183 units (Ext_Atab-5) and the list of unreliable exonic circRNAs 212 (Ext_Atab-4).
To highlight the tissue specificity of the circular transcriptome by using a small panel of exonic circRNAs
The circular transcriptome is very complex and as such, it was important to determine if it is possible to reduce the complexity to better identify tissue specificities. We analysed the tissue specificity of the circular transcriptome by considering a panel of reliable exonic circRNAs. To avoid evaluating a tissue by a single dataset, we selected 15 tissues where samples were available for the two youngest and at least three of the oldest animals. In addition, we considered the five tissues where samples were available for the two young animals. To this end, we performed hierarchical cluster analysis (HCA). The ideal result would be to find a clustering of the circular transcriptomes by tissues. Three HCAs were performed, with (1) 96 samples (15 tissues with 5/6 animals + 5 tissues with only young animals), (2) 56 samples (15 tissues with only juvenile or old animals), (3) 40 samples (20 tissues with only young animals). Although we explored 23,737 exonic circRNAs in 117 samples, only 386 to 6,995 had a notable expression in a given sample, and three samples were sequenced at a higher depth with XL sequencing. We wanted to prevent circRNAs with very low expression from becoming the discriminators. To construct an exonic circRNA tissue evaluation panel we included in the respective list of circRNA those samples which were the top-150 exonic circRNAs ranked according to their expression level in any of the 116 samples (we did not include circRNAs data obtained from the second total-RNAseq from the cerebral cortex of the juvenile castrated male). This method resulted in a panel of 1,749 exonic circRNAs (list available in Ext_Atab-6). The distribution of the 1,749 circRNAs in this panel appeared to be different from that in the original list (). However, we proposed that this was not a significant issue, particularly given that the top-150 panel comprises only 0.8% of circRNAs that were previously identified exclusively by CD (in contrast to 6.1% for the 23,737 reliable exonic circRNAs) and 75.5% that were identified through four different methods (in comparison to 36.3%) ().
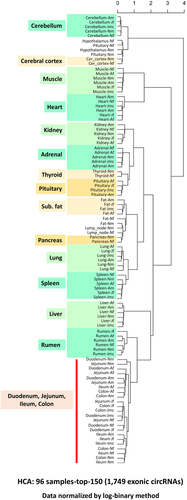
We began the HCA by considering the expression of 1,749 exonic circRNAs in 96 samples. With normalization performed using the log-binary method (), we observed tissue-wise clustering of all samples for nine tissues within the group of 15 tissues (cerebellum, muscle, heart, kidney, adrenal gland, lung, spleen, liver, and rumen). When we considered only the oldest animals, we noted clustering of two additional tissues where the two youngest animals were still available (fat and pituitary gland). In addition, the two samples from the youngest animals clustered together for thyroid, pancreas and cerebral cortex (tissues where samples from the oldest animals were not available). As the age of animals had an effect on the clustering pattern, we proposed to analyse separately young and juvenile/old animals. The clustering using the log-binary method considering only the 56 samples from the oldest animals and only 40 samples from the youngest animals were consistent with results observed on HCAs built with 96 samples (Res_Adoc-7).
Figure 8. Hierarchical clustering analysis (HCA).
Several panels increasing or decreasing the number of top exonic circRNAs ranked according to their expression were created with the expectation of improving the clustering (increasing the number of tissues where all samples clustered according to tissue). HCAs (96, 56, and 40 samples) were constructed with data normalized by the log-binary and standard score methods. Differences from the respective reference results (HCA obtained with top-150) were observed, but they were mainly negative differences (see Res_Adoc-7). No clear improvement was observed regardless of the normalization method used. These analyses were inconclusive for the four digestive tissues (duodenum, jejunum, ileum, and colon), which did not show a tissue- or organ-specific clustering pattern. In addition, we often observed a degradation of the clustering quality, especially when clustering small groups of samples (56 and 40).
These analyses showed that the top-100 and −150 panels are the most efficient, whatever the normalization method used, and even we considered only a subset of the 116 initial samples. The top-150 with 1,749 exonic circRNAs (7.4% of reliable exonic circRNAs) can be considered as a reference. We emphasized that this panel included the most highly expressed exonic circRNAs (top-150 for each of 116 samples). The lists of exonic circRNAs constituting the top-100 to top-250 panels are available in Ext_Atab-6.
Analysis of reproductive tissues
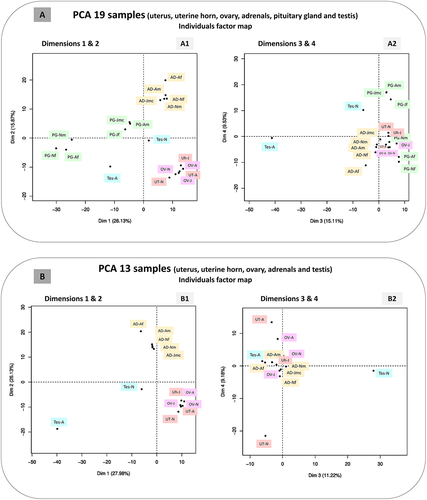
To analyse the circular transcriptome of reproductive tissues, we performed a PCA on the expression of circRNAs from the reference panel (1,749 exonic circRNAs, panel top-150) in these tissues (uterus, uterine horn, testis, and ovary). In addition, we considered the adrenal and pituitary gland samples. Initially, we considered these 6 tissues and 19 individual samples in total (). The first two and the first four PC dimensions explained 42.00% and 66.64% of the variance, respectively. The first dimensions allowed us to separate the samples from the pituitary gland into two groups (). We found that these groups did not reflect the age or the sex of the animals sampled. The most interesting element was probably that the testis of the adult animal appeared as an outlier in the dim-3 (). Since we were not convinced that the consideration of the pituitary gland was informative, a second PCA was performed with 5 tissues and 13 individual samples (). The performance of this PCA was better than the previous one, as the first two and first four dimensions explained 54.11% and 71.51% of the variance, respectively. The dimension-1 allowed the individualization of the sample from adult testis (Tes-A on ). The first dimensions allowed us to separate the samples into two groups and two individual samples (the two testis) (1). The first group included all female reproductive tissues (uterus, uterus horn, and ovary). The second group included all samples from the adrenal gland. The dimensions-3 and -4 showed a proximity between the testis of the adult animal and the adrenal gland samples of both adult animals (male and female) ().
Figure 9. Principal component analyses (PCA).
Discussion
In this study, we performed a circRNA characterization in bovine tissues with total-RNAseq data generated in a standardized manner for the BovReg project. We avoided the agglomeration of other available datasets to minimize batch effects that make interpretation of results difficult [Citation27]. This is because composition of the circRNA catalogue depends not only on the sample considered but also on the tissue collection and preservation method, RNA isolation and sequencing library preparation protocols, and data analysis pipeline. We chose to perform a main detection with CD and to use two other circRNA tools (CIRI2 and CE2) to obtain additional information. In all cases, we never included sporadic circularization events by applying a validation threshold. We also used the list of bovine exonic circRNAs previously characterized by CE2+CIRI2 in 2021 [Citation27] to complete the benchmarking. Most importantly, only 6.1% of the 23,737 circRNA exons validated by CD and considered reliable were observed exclusively by CD. This score could have been lower had CIRI2 detection of circRNAs been conducted without a minimum number of CCRs. Furthermore, it is crucial to highlight that the resource data utilized to annotate exonic circRNAs was more extensive for CD analyses than for the three other methods. Nevertheless, we think that CD is unable to characterize a significant proportion of exonic circRNAs. Therefore, in addition to the initial list of 23,926 circRNAs considered in this study, we propose a second list of 9,183 exonic circRNAs identified by other methods. Unfortunately, it is not possible to consider them for expression analyses.
What was more surprising than the number or list of exonic circRNAs, was the low proportion of bovine circRNAs validated by CD that can be annotated as exonic circRNAs, already observed in 2021 [Citation27]. The low percentage of exonic circRNAs (40%) observed in this study was far lower than the value we observed in pigs [Citation27,Citation28], though this value seems to vary depending on the tissue or the origin of the datasets [Citation27]. The use of the new transcriptome annotation allowed a 191% increase in the number of identified exons, but did not allow a clear improvement in the percentage of circRNAs annotated as exonic circRNAs compared to our previous study [Citation27]. However, of these exonic circRNAs, there are very few that have only been validated in one dataset, and as such they are likely to be more ‘reliable’ than the other_circRNAs where the majority are only validated in one dataset. The diversity in the population of other_circRNAs and the number of datasets considered led to a very low average percentage of exonic circRNAs. The exonic circRNAs characterized here with CD are likely to be reliable circRNAs, based on the criteria defined by Chuang et al. in [Citation43] while the other_circRNAs may not be.
The analyses conducted here showed that there were signals of circularization events in the reads obtained from mRNAseq, but that these were often never observed in total-RNAseq reads (CD and CIRI2 analyses). Moreover, these circRNAs are very rarely exonic circRNAs, even with CIRI2. This shows that Lv et al. [Citation30] had not worked on mRNAseq data as they reported. The essential feature of the artificial circularization events detected in mRNAseq seems to be that they are not reproducible. We were somewhat surprised that not all artif_circRNAs belonged to the other_circRNA category, since at least 103 artif_circRNAs were detected among those annotated as exonic circRNAs. Conversely, the other_circRNA category did not contain only artif_circRNAs. Among the ‘reliable’ other_circRNAs, we found most of the circRNAs identified in the Defensin genomic region.
As previously described [Citation27], clusters of other_circRNAs were characterized in several genomic regions known to be incompletely sequenced and incompletely assembled. We hypothesized that the presence of inverted regions in the assembled genome led to the mapping of some reads as artificial CCRs and to the identification of in-silico artif_circRNAs. We could have the same consequences in regions with gene clusters with segments of high homology, creating opportunities for misalignment. Therefore, we were not surprised to highlight more artif_circRNAs than real circRNAs in the MHC region. At the beginning of this study, we also thought that the Defensin region would be a good example of a region producing in-silico artif_circRNAs [Citation27]. Analysis of the other_circRNAs present in 63T and 63 m clearly showed that the other_circRNAs identified in the Defensin region seem ‘reliable’ circRNAs. It is possible that their identification as exonic circRNAs failed due to small gaps or errors at the boundaries of the exons. However, the statistics do not support this simple explanation. We showed that this region is capable of producing circRNAs (over 3,000), which seem reliable because they were not detected in mRNAseq data, but only four have been identified as exonic circRNAs. This region is particularly difficult to understand, undoubtedly due to a mixture of problems (e.g. sequencing/assembling, highly homologous genes with copy number variations between individuals, and non-poly(A) transcripts).
The consideration of mRNAseq in addition of total-RNAseq led to the identification of 103 + 26 artificial circRNAs among the list of exonic circRNAs. We can propose several hypotheses to explain these backsplicing falsely identified (): (1) the existence of inverted genomic sequences in the assembly. (2) The existence of genomic sequences with high similarity in the reference genome (gene family organized in clusters). (3) The presence of small regions in the genome of the affected animals with inverted genomic sequences or with chromosomal rearrangements. (4) Confusion with the identification of transcripts resulting from trans-splicing. (5) Possible template switching during reverse transcription in the library preparation process. The first three could be assimilated to in-silico circularization, and the fourth is due to an in-vivo event, but it is not a circularization event [Citation13,Citation29]. The third hypothesis may be illustrated by the artif_circRNA (2:18153915–18180018|+) detected in the TTN region, but only in the two Belgian animals. It is difficult to imagine that the hypothetical fifth event (in-vitro) would reproducibly lead to a circular junction identifiable as an exonic circRNA. We noted that the consideration of exons novelty annotated by BovReg increased the risk to annotate an (artificial) circRNA as an exonic circRNA. In addition, we were aware to take a risk by accepting circRNAs with backsplicing between a mixed pair of exons (Ensembl/MSTRG).
Figure 10. List and characteristics of different events leading to the formation of a circular junction.
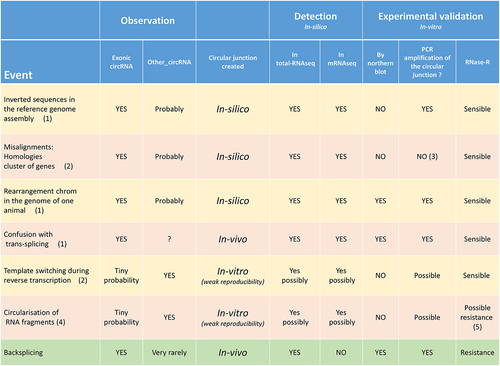
Based on the results of this study, we are convinced that the circularization in-vitro of RNA fragments during RNA preparation prior to sequencing is possible. The analyses conducted in this study revealed that a significant proportion of circRNAs identified in mRNAseq data were not detected in total-RNAseq data (63.4% for CD and 37.1% for CIRI2 analyses). These statistics would have been higher if sporadic circularization events had not been eliminated from our analyses. Moreover, we noticed that a similar high proportion of new circRNAs is often observed in datasets generated after RNase-R [Citation5,Citation32]. For example, Gruhl et al. [Citation32] had to eliminate 75% of the circRNAs that were detected in RNase-R-treated samples but not in untreated samples. We believe that the partial digestion of linear RNAs by RNase-R contributes to an increase in the number of RNA fragments. This may be one of the reasons for the large number of other_circRNAs detected in 117T. The sub-exonic circRNAs originating from one exon of a multi-exonic gene, the circRNAs with their two genomic coordinates in two different exons of the same gene, and circRNAs from the mitochondrial genome are candidates to be artificial circRNAs with an in-vivo genesis. The common feature of sub-exonic circRNAs and circRNAs from the mitochondrial genome is that they originate from genes that are abundantly transcribed in the considered tissue [Citation27]. We believe that the main feature of an artificial circRNAs obtained in-vitro is that this type of events is only weakly reproducible (to nucleotide precision) (). This could happen e.g. via template switching during reverse transcription in the library preparation process [Citation13,Citation44]. This in-vitro event does not lead to a circularization but only to the formation of a junction in the cDNA of junction that resembles to a circular junction. A genuine source of in-vitro circularization could be RNA fragments containing specific sequences that promote the formation of a double-stranded RNA with its two ends. The abundance of the initial linear transcript and treatments leading to RNA fragmentation probably increases the impact of such event. This is also the mechanism proposed by Liu et al. [Citation26] for the genesis of interior circRNAs. Template switching may also be favored by the abundance of RNA fragments.n the , we reported features of artificial circRNAs in comparison of exonic circRNAs. In addition to artificial circRNAs generated in-silico during the alignment process, in-vitro generation of artificial circRNAs should be considered. We noted that a new method has emerged recently to differentiate exonic circRNAs and other non-co-linear transcripts (fusion, trans-splicing) [Citation45,Citation46]. Northern blotting is an interesting technique for revealing the circular configuration of RNA, but is rarely used [Citation44,Citation47,Citation48]. A PCR amplification of the circular junction region as well as a test for resistance to RNase-R are often used to validate a circRNA [Citation43,Citation44,Citation47,Citation48]. We believe that only circular junctions generated in-silico after misalignment cannot be amplified by PCR whereas some in-vitro artificial circRNAs might pass these tests. We were not surprised to find in the literature that a significant fraction of non-exonic circRNAs detected by different tools could pass these validation tests [Citation29]. Among the 1,516 circRNAs considered by Vromman et al. [Citation49], we found in the lists published by the authors at least 172 ‘other_circRNAs’ that were validated by the three methods (qPCR, resistance to RNase-R and amplicon sequencing). Moreover, we identified 22 out of 39 sub-exonic circRNAs (circRNAs with both genomic coordinates in the same exon) from coding genes that were tested and were validated by the three methods. Using an approach focused on the RPGRorf15 locus, Apelbaum et al. [Citation50] confirmed the existence of several interior/sub-exonic circRNAs formed by back-fusion of linear parts (exonic and intronic) of the RPGRorf15 pre-mRNAs. Further verification is required, but the current study suggests that the main feature of (sub-exonic) in-vitro artificial circRNAs may be the multiplicity of circRNAs from the same locus [Citation27]. This is an easily detectable feature for highly expressed genes, but some circRNA tools tend to erase this feature. Another more surprising example is circRNAs from the mitochondrial genome, which, according to this study, are very likely to be artificial circRNAs [Citation51].
This study showed that the number of bovine introns involved in intron circles was close to that involved in the production of lariat-derived circRNAs (ciRNAs), 126 and 147, respectively (Ext_Atab-7 & −8). These observations are in line with those recently made in humans [Citation52]. This is not what has been observed previously in pigs, but that study involved only a very specific dataset [Citation53]. Two genes are able to produce the two types of intronic circRNAs from distinct introns (ENSBTAG00000001888, MED13L and ENSBTAG00000032087, ATXN2L) but we found that they are not able to produce exonic circRNAs. The number of parental genes for intronic circRNAs (268) is significantly lower compared to exonic circRNAs (8 to 8.5K).
From the 117 tissue samples, we analysed, we found that the cerebellum was the tissue with the highest number of distinct exonic circRNAs in cattle. A similar result was observed in pigs [Citation54]. We also found that the testis sampled from an adult animal could not be distinguished from the other tissues by the number of expressed exonic circRNAs. This result is consistent with comparisons made in pigs [Citation27,Citation54]. This non-distinct clustering status of testis compared to other tissues with respect to the number of exonic circRNAs is somewhat surprising, as testis is highly transcriptionally active and is the tissue in which the highest number of protein-coding genes are expressed [Citation55,Citation56]. Testicular exonic circRNAs seemed to be very tissue-specific, as demonstrated by the outlier status in the PCA analysis. These PCAs also showed that for the circular transcriptome there was some proximity between the adult adrenal and the adult testis and a large distance between these two tissues and the uterus, ovary and adrenal of non-pubertal animals. The circular transcriptome of the adrenal and testes is likely to be more affected by steroid synthesis than that of the ovary in bovine. However, this conclusion is probably due to the ovary sample used, which was taken from a cow 3 weeks after parturition, a period insufficient to observe normal ovarian function.
We proposed the creation of a new type of dataset (mini-fastq) that allows the characterization of circRNAs with less than 2% of the reads. It will also allow the rapid generation of comparative data, since this type of dataset can be analysed with most of the circRNA detection tools and with new criteria to validate circRNAs. It will also enable the update of the characterized circRNAs when a new version of the reference genome becomes available for the species in question. By analysing MFQ117 with CE2 we have demonstrated the ease and efficiency associated with using this type of dataset.
The 1,749 exonic circRNAs in the top-150 panel exhibit a distribution that may appear surprising. This panel comprises only 24% of exonic circRNAs that were not identified by CE2+CIRI2 in 2021, whereas the initial list of 23,937 included 49% (). It is also noteworthy that in this study, we characterized 86.1% (8,887 validated by CD and 1,956 validated by CE2 and/or CIRI2 out of 12,588) of the exonic circRNAs that had been previously characterized in analyses performed with CE2+CIRI2 in a smaller subset of tissues (muscle/liver/testis [Citation27]). The three tissues muscle/liver/testis are not the richest in terms of exonic circRNAs (neither in number nor in expression), but their contribution is significant in terms of diversity.
The overall tissue specificity of the circular transcriptome observed by hierarchical clustering analyses was very high for 8 tissues of 15 considered (kidney, cerebellum, muscle, heart, liver, lung, spleen, and adrenal gland). The 9th tissue for which we observed tissue-wise clustering of all samples was the rumen, but only when the data were normalized by the log-binary method. Clustering was biologically meaningful for two further tissues (fat and pituitary gland), if young animals are excluded from the analysis. No tissue specificity for the circular transcriptome was observed for four digestive tissues. Indeed, these five digestive tissues (duodenum, ileum, jejunum and colon) were the most resistant to clustering in the analyses of the 56 individual samples. These observations are not significantly different from those made in sheep considering only linear transcripts, which also showed that digestive tissues clustered poorly [Citation57]. The results of the HCA constructed using the top-100 panel or the top-150 panel of expressed circRNA appear to be the most robust, yet construct with a panel containing 4.8% or 7.4% of the exonic circRNAs identified in these samples. It is quite surprising that we cannot improve the results of these HCA. However, these results again show that it is efficient to focus on highly expressed exonic circRNAs [Citation27,Citation32].
Conclusion
One of our goals was to establish an exhaustive and reliable catalogue of the circular transcriptome in bovine tissues. It should be emphasized that this objective was achieved first and foremost thanks to the diversity of the tissues samples selected and the quality of the data analysed. This study compared circRNAs present in 117 samples with total-RNAseq and mRNAseq data, This study compared circRNAs present in 117 samples with total-RNAseq and mRNAseq data always excluding sporadic circularization events. Using this method, we confirmed the existence of several types of reliable circRNAs, including exonic circRNAs, ciRNA, intron circles, and sub-exonic circRNAs from snc genes. This study also identified a large number of circRNAs that are not generated in-vivo. The analysis of circRNA in mRNAseq datasets provided clear evidence that sub-exonic circRNAs from coding genes (introduced in [Citation27]) are artefacts, while sub-exonic circRNAs from small non-coding genes (introduced in [Citation28]) are not. Several hypotheses have been proposed to explain the presence of artif_circ RNAs in any RNAseq datasets. The most innovative are those related to in-silico and in-vitro factors. The possibility of in-vitro circularization of RNA fragments underlines the significance of the quality and integrity of the RNA source for the elaboration of datasets considered in circRNA studies. Our analysis leads us to recommend focusing on exonic circRNAs for tissue comparisons, such as those performed in this study of the bovine circular transcriptome for the BovReg project. By choosing to work with a large number of tissues from six very different animals, we did not expect to obtain particularly spectacular results on the tissue specificity of the bovine transcriptome. Nevertheless, to show that 5% to 7% of reliable circRNAs are sufficient to produce a comprehensive analysis is a major result. Finally, we expect that this study will lead to better integration and visibility of the bovine circular transcriptome in multi-species analyses. We proposed the creation of a novel type of dataset that would facilitate the generation of comparative data for circRNA analyses in a timely manner.
Atab.pdf
Download PDF (1.9 MB)Res_Adoc.pdf
Download PDF (2.8 MB)MM_Adoc.pdf
Download PDF (2.4 MB)Acknowledgments
Annie Robic acknowledges INRAE (more precisely the Animal Genetics and the M2I divisions) and the FBN (more precisely the Institute of Genome Biology), which supported her research stay in FBN in 2022–2023. We thank Dr Sylvain Foissac and Dr Laurence Liaubet (GenPhySE) for indirectly enriching this study through their insightful discussions.
Data availability statement
All data obtained concerning exonic and intronic circRNAs are available in several tables (all Ext_Atab) deposited in Dataverse repository (doi: 10.57745/XORQHK). The list of other_circRNAs is not available, as we were unable to distinguish between reliable and unreliable other_circRNAs. List of exons generated by the BovReg consortium and used in this study is available on upon request from CC. Datasets generated by the BovReg consortium and analysed during the current study are listed in Atab-1. We built 117 paired mini-fastq files (R1 and R2) to provide all sequences allowing a global and rapid characterization of circular RNAs present in bovine tissues (doi: 10.57745/IUJ40P).
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/15476286.2024.2375090
Additional information
Funding
References
- Rosen BD, Bickhart DM, Schnabel RD, et al. De novo assembly of the cattle reference genome with single-molecule sequencing. Gigascience. 2020;9(3). doi: 10.1093/gigascience/giaa021
- Goszczynski DE, Halstead MM, Islas-Trejo AD, et al. Transcription initiation mapping in 31 bovine tissues reveals complex promoter activity, pervasive transcription, and tissue-specific promoter usage. Genome Res. 2021;31(4):732–744. doi: 10.1101/gr.267336.120
- Ross EM, Sanjana H, Nguyen LT, et al. Extensive variation in gene expression is revealed in 13 fertility-related genes using RNA-Seq, ISO-Seq, and CAGE-Seq from Brahman Cattle. Front Genet. 2022;13:784663. doi: 10.3389/fgene.2022.784663
- Salavati M, Clark R, Becker D, et al. Improving the annotation of the cattle genome by annotating transcription start sites in a diverse set of tissues and populations using cap analysis gene expression sequencing. G3: Genes, Genomes, Genet. 2023;13(8):G3 13. doi: 10.1093/g3journal/jkad108
- Jeck WR, Sorrentino JA, Wang K, et al. Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA. 2013;19(2):141–157. doi: 10.1261/rna.035667.112
- Memczak S, Jens M, Elefsinioti A, et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature. 2013;495(7441):333–338. doi: 10.1038/nature11928
- Salzman J, Gawad C, Wang PL, et al. Circular RNAs are the predominant transcript isoform from hundreds of human genes in diverse cell types. PLOS ONE. 2012;7(2):e30733. doi: 10.1371/journal.pone.0030733
- Zhang Y, Zhang XO, Chen T, et al. Circular intronic long noncoding RNAs. Mol Cell. 2013;51(6):792–806. doi: 10.1016/j.molcel.2013.08.017
- Kristensen LS, Andersen MS, Stagsted LVW, et al. The biogenesis, biology and characterization of circular RNAs. Nat Rev Genet. 2019;20(11):675–691. doi: 10.1038/s41576-019-0158-7
- Liu CX, Chen LL. Circular RNAs: characterization, cellular roles, and applications. Cell. 2022;185(13):2390. doi: 10.1016/j.cell.2022.06.001
- Wilusz JE. Circular RNAs: unexpected outputs of many protein-coding genes. RNA Biol. 2017;14(8):1007–1017. doi: 10.1080/15476286.2016.1227905
- Yang L, Wilusz JE, Chen LL. Biogenesis and regulatory roles of circular RNAs. Annu Rev Cell Dev Biol. 2022;38(1):263–289. doi: 10.1146/annurev-cellbio-120420-125117
- Horiuchi T, Aigaki T. Alternative trans-splicing: a novel mode of pre-mRNA processing. Biol Cell. 2006;98(2):135–140. doi: 10.1042/BC20050002
- Chuang TJ, Chen YJ, Chen CY, et al. Integrative transcriptome sequencing reveals extensive alternative trans-splicing and cis-backsplicing in human cells. Nucleic Acids Res. 2018;46(7):3671–3691. doi: 10.1093/nar/gky032
- Dubois J, Sczakiel G. The human TRAM1 locus expresses circular RNAs. Sci Rep. 2021;11(1):22114. doi: 10.1038/s41598-021-01548-0
- Rahimi K, Veno MT, Dupont DM, et al. Nanopore sequencing of brain-derived full-length circRnas reveals circRNA-specific exon usage, intron retention and microexons. Nat Commun. 2021;12(1):4825. doi: 10.1038/s41467-021-24975-z
- Robic A, Faraut T, Djebali S, et al. Analysis of pig transcriptomes suggests a global regulation mechanism enabling temporary bursts of circular RNAs. RNA Biol. 2019;16(9):1190–1204. doi: 10.1080/15476286.2019.1621621
- Talhouarne GJ, Gall JG. Lariat intronic RNAs in the cytoplasm of Xenopus tropicalis oocytes. RNA. 2014;20(9):1476–1487. doi: 10.1261/rna.045781.114
- Taggart AJ, Lin CL, Shrestha B, et al. Large-scale analysis of branchpoint usage across species and cell lines. Genome Res. 2017;27(4):639–649. doi: 10.1101/gr.202820.115
- Ares M Jr., Igel H, Katzman S, et al. Intron lariat spliceosomes convert lariats to true circles: implications for intron transposition. Genes Dev. 2024;38(7–8):322–335. doi: 10.1101/gad.351764.124
- Ma XK, Zhai SN, Yang L. Approaches and challenges in genome-wide circular RNA identification and quantification. Trends Genet. 2023;39(12):897–907. doi: 10.1016/j.tig.2023.09.006
- Nielsen AF, Bindereif A, Bozzoni I, et al. Best practice standards for circular RNA research. Nat Methods. 2022;19(10):1208–1220. doi: 10.1038/s41592-022-01487-2
- Ma XK, Xue W, Chen LL, et al. CIRCexplorer pipelines for circRNA annotation and quantification from non-polyadenylated RNA-seq datasets. Methods. 2021;196:3–10. doi: 10.1016/j.ymeth.2021.02.008
- Gao Y, Zhang J, Zhao F. Circular RNA identification based on multiple seed matching. Brief Bioinform. 2018;19(5):803–810. doi: 10.1093/bib/bbx014
- Liu X, Frost J, Bowcock A, et al. Canonical and interior circular RNAs function as competing endogenous RNAs in Psoriatic Skin. Int J Mol Sci. 2021;22(10):5182. doi: 10.3390/ijms22105182
- Liu X, Hu Z, Zhou J, et al. Interior circular RNA. RNA Biol. 2020;17(1):87–97. doi: 10.1080/15476286.2019.1669391
- Robic A, Cerutti C, Kühn C, et al. Comparative analysis of the circular transcriptome in muscle, liver and testis in three livestock species. Front Genet. 2021;12:665153. doi: 10.3389/fgene.2021.665153
- Robic A, Demars J, Kühn C. In-depth analysis reveals production of circular RNAs from non-coding sequences. Cells. 2020;9(8):1806. doi: 10.3390/cells9081806
- Yu CY, Liu HJ, Hung LY, et al. Is an observed non-co-linear RNA product spliced in trans, in cis or just in vitro? Nucleic Acids Res. 2014;42(14):9410–9423. doi: 10.1093/nar/gku643
- Lv X, Chen W, Sun W, et al. Expression profile analysis to identify circular RNA expression signatures in hair follicle of Hu sheep lambskin. Genomics. 2020;112(6):4454–4462. doi: 10.1016/j.ygeno.2020.07.046
- Lu T, Cui L, Zhou Y, et al. Transcriptome-wide investigation of circular RNAs in rice. RNA. 2015;21(12):2076–2087. doi: 10.1261/rna.052282.115
- Gruhl F, Janich P, Kaessmann H, et al. Circular RNA repertoires are associated with evolutionarily young transposable elements. Elife. 2021;10. doi: 10.7554/eLife.67991
- Moreira GCM, Dupont S, Becker D, et al. Multi-dimensional functional annotation of the bovine genome for the BovReg project. In: Proceedings of 12th World Congress on Genetics Applied to Livestock Production (WCGALP), Rotterdam, the Netherlands; 2022. p. 2261–2264.
- Dobin A, Davis CA, Schlesinger F, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29(1):15–21. doi: 10.1093/bioinformatics/bts635
- Cheng J, Metge F, Dieterich C. Specific identification and quantification of circular RNAs from sequencing data. Bioinformatics. 2016;32(7):1094–1096. doi: 10.1093/bioinformatics/btv656
- Robic A, Cerutti C, Demars J, et al. From the comparative study of a circRNA originating from an mammalian ATXN2L intron to understanding the genesis of intron lariat-derived circRNAs. Biochim Et Biophys Acta (BBA) - Gene Regul Mechanisms. 2022;1865(4):194815. doi: 10.1016/j.bbagrm.2022.194815
- Li H. Toward better understanding of artifacts in variant calling from high-coverage samples. Bioinformatics. 2014;30(20):2843–2851. doi: 10.1093/bioinformatics/btu356
- SIGENAE. Available from: http://www.sigenae.org/
- HCA-Galaxy-tutorial. Available from: http://genoweb.toulouse.inra.fr/~formation/CATIBIOS4BIOL_stats/Learning_clustering_current.pdf
- Xu C, Zhang J. Mammalian circular RNAs result largely from splicing errors. Cell Rep. 2021;36(4):109439. doi: 10.1016/j.celrep.2021.109439
- Ragan C, Goodall GJ, Shirokikh NE, et al. Insights into the biogenesis and potential functions of exonic circular RNA. Sci Rep. 2019;9(1):2048. doi: 10.1038/s41598-018-37037-0
- Chen LL, Bindereif A, Bozzoni I, et al. A guide to naming eukaryotic circular RNAs. Nat Cell Biol. 2023;25(1):1–5. doi: 10.1038/s41556-022-01066-9
- Chuang TJ, Chiang TW, Chen CY. Assessing the impacts of various factors on circular RNA reliability. Life Sci Alliance. 2023;6(5):e202201793. doi: 10.26508/lsa.202201793
- Dodbele S, Mutlu N, Wilusz JE. Best practices to ensure robust investigation of circular RNAs: pitfalls and tips. EMBO Rep. 2021;22(3):e52072. doi: 10.15252/embr.202052072
- Chuang TJ, Wu CS, Chen CY, et al. NCLscan: accurate identification of non-co-linear transcripts (fusion, trans-splicing and circular RNA) with a good balance between sensitivity and precision. Nucleic Acids Res. 2016;44(3):e29. doi: 10.1093/nar/gkv1013
- Chen YC, Chen CY, Chiang TW, et al. Detecting intragenic trans-splicing events from non-co-linearly spliced junctions by hybrid sequencing. Nucleic Acids Res. 2023;51(15):7777–7797. doi: 10.1093/nar/gkad623
- Schneider T, Schreiner S, Preusser C, et al. Northern blot analysis of circular RNAs. Methods Mol Biol. 2018;1724:119–133.
- Mi Z, Zhongqiang C, Caiyun J, et al. Circular RNA detection methods: A minireview. Talanta. 2022;238:123066. doi: 10.1016/j.talanta.2021.123066
- Vromman M, Anckaert J, Bortoluzzi S, et al. Large-scale benchmarking of circRNA detection tools reveals large differences in sensitivity but not in precision. Nat Methods. 2023;20(8):1159–1169. doi: 10.1038/s41592-023-01944-6
- Appelbaum T, Aguirre GD, Beltran WA. Identification of circular RNAs hosted by the RPGR ORF15 genomic locus. RNA Biol. 2023;20(1):31–47. doi: 10.1080/15476286.2022.2159165
- Wu Z, Sun H, Wang C, et al. Mitochondrial genome-derived circRNA mc-COX2 functions as an oncogene in chronic lymphocytic leukemia. Mol Ther Nucleic Acids. 2020;20:801–811. doi: 10.1016/j.omtn.2020.04.017
- Rasmussen AM, Okholm TLH, Knudsen M, et al. Circular stable intronic RNAs possess distinct biological features and are deregulated in bladder cancer. NAR Cancer. 2023;5(3):zcad041. doi: 10.1093/narcan/zcad041
- Robic A, Kühn C. Beyond back splicing, a still poorly explored world: non-canonical circular RNAs. Genes (Basel). 2020;11(9):1111. doi: 10.3390/genes11091111
- Jin L, Tang Q, Hu S, et al. A pig BodyMap transcriptome reveals diverse tissue physiologies and evolutionary dynamics of transcription. Nat Commun. 2021;12(1):3715. doi: 10.1038/s41467-021-23560-8
- Soumillon M, Necsulea A, Weier M, et al. Cellular source and mechanisms of high transcriptome complexity in the mammalian testis. Cell Rep. 2013;3(6):2179–2190. doi: 10.1016/j.celrep.2013.05.031
- Yang W, Zhao F, Chen M, et al. Identification and characterization of male reproduction-related genes in pig (sus scrofa) using transcriptome analysis. BMC Genomics. 2020;21(1):381. doi: 10.1186/s12864-020-06790-w
- Clark EL, Bush SJ, McCulloch MEB, et al. A high resolution atlas of gene expression in the domestic sheep (Ovis aries). PLOS Genet. 2017;13(9):e1006997. doi: 10.1371/journal.pgen.1006997