Abstract
Relatively little is known regarding mechanisms of environmental exposures in the development of autoimmune disease. However, several environmental agents are implicated in triggering or accelerating systemic autoimmune disease, including mercury, iodine, vinyl chloride, certain pharmaceuticals, and crystalline silica. There is increasing epidemiological evidence supporting the hypothesis that occupational silica exposure is associated with a variety of systemic autoimmune diseases, including scleroderma (SSc), rheumatoid arthritis (RA), systemic lupus erythematosus (SLE), glomerulonephritis (GN) and small vessel vasculitis (SVV). However, there have been few mechanistic studies examining silica exposure and autoimmune disease initiation and progression. This review summarizes human epidemiology data linking silica exposure with systemic autoimmune disease, but focuses on possible mechanisms by which silica can lead to the development and progression of autoimmunity.
INTRODUCTION
An association between occupational exposures of inhaled particulates and autoimmunity was postulated as early as 1914, when Bramwell reported increased frequency of diffuse scleroderma in stone masons (Bramwell, Citation1914). This was soon after scientists Paul Ehrlich, Karl Landsteiner, and Clemens von Pirquet began describing the existence and pathologic effects of autoantibodies (reviewed in (Silverstein, Citation2001). Since that time, autoimmunity has been associated with a wide range of environmental and occupational exposures, including mercury, iodine, vinyl chloride, certain pharmaceuticals, and crystalline silica, but the mechanisms leading to autoimmunity have remained enigmatic. The list of diseases now classified as autoimmune continues to grow and without a better understanding of the mechanisms, it will be difficult to develop effective therapeutic strategies. Identification of new antibodies helps draw better connections between autoantibodies and specific diseases, and may provide clues to mechanisms connecting environmental exposures with autoimmunity. It is possible that from the growing list of autoantibodies against specific targets, clues will be provided to advance our knowledge of the mechanisms involved.
The demonstration of mercury-induced systemic autoimmunity provides an excellent example of how mechanisms can be discovered through the identification of antibody specificities. Anti-fibrillarin antibodies are commonly found in systemic autoimmune diseases such as scleroderma, and there is some data linking mercury exposure with scleroderma (Mayes, Citation1999). Exposure to mercury in mouse models leads to the production of antibodies to fibrillarin, which gives a clumpy nucleolar staining pattern on HEp-2 cells by indirect immunofluorescence (IIF) (Hultman et al., Citation1989). Ultimately, this led to the discovery that mercury may interact with fibrillarin so that its presentation to the immune system leads to anti-fibrillarin antibodies (Pollard et al., Citation1997).
Chemical alteration of self-antigen does not explain many exposure-associated autoimmune responses, however. Another interesting and plausible explanation has developed through the work of several laboratories studying the induction of apoptosis by the same substances, such as crystalline silica, that are associated with autoimmunity. This review will cover data suggesting a role for silica-induced apoptosis in the induction and progression of systemic autoimmune disease.
Crystalline Silica
Commonly known as quartz, crystalline silica is an abundant mineral found in rock, sand and soil. The highest exposures to silica occur in the dusty trades, such as mining, farming, foundry work, ceramic and pottery making, glass making, drilling and sandblasting. Silica is widely used in materials, such as absorbents, dessicants, catalysts, fillers, lubricants, thickening agents and paints. Prolonged or acute high exposures of silica can cause pulmonary inflammation and fibrosis, which is termed silicosis (Society, Citation1997). The potential to develop silicosis depends on the concentration of silica in the ambient air, the content of crystalline silica in inhaled dust, the size of the particles (under 1 μm being most fibrogenic) and the duration and amount of exposure (Hughes et al., Citation1982).
Silica occurs in crystalline and amorphous forms with the crystalline form considered more biologically active. Cutting, grinding and milling of silica results in the formation of fractured crystal, generating Si and Si-O radicals on its surface. Thus, freshly fractured crystalline silica may have surface properties that make it more reactive in the lung. Crystalline silica has been shown to have a negative charge on its surface which provides an increased potential to adsorb and interact with cellular components (Castranova and Vallyathan, 1996).
Silica particles of respirable size are deposited in the alveolar spaces of the lung. The particles are phagocytosed by alveolar macrophages, which are removed by the mucociliary movement of macrophages up the respiratory tract or drained to the lymphatics (Brody et al., Citation1982). However, at high concentrations it is reported that the mucociliary clearance of silica particles is impaired, resulting in an accumulation of silica-laden macrophages within the lymphatics and interstitial spaces of the lung (Brody et al., Citation1982). Consequently, these silica-laden macrophages are able to interact with other immune cells, possibly mediating many of the immune effects of silica, including systemic autoimmune disease.
Clinical Data: Silica Exposure and Autoimmune Disease
There are historical case reports linking silica exposure to several autoimmune diseases, but recently there have been several population based studies examining silica exposure as a risk factor for the development of autoimmune disease (Parks et al., Citation1999, 2002, 2003). Parks et al. reported a higher prevalence of systemic lupus erythematosus (SLE) in patients following silica exposure than in a control population (Parks et al., Citation2002). Further, they reported an association between the level of silica exposure and the development of SLE. These data suggest that silica exposure may promote the development of SLE in some individuals and that the development of disease may be more prevalent in highly exposed individuals. They further examined several common types of autoantibodies and found a strong association between silica exposure and anti-DNA and anti-Sm autoantibody levels. An association between silica exposure and anti-neutrophil cytoplasmic autoantibody (ANCA) formation has also been reported to be involved in small vessel vasculitis (Hogan et al., Citation2001). Therefore, it appears silica exposure results in the production of several specific types of autoantibodies.
Use of this epidemiologic data has provided clues about autoimmune mechanisms with silica exposure. Other reports showed associations between silica exposure and both RA and SSc, and subsequently for SLE (Caplan, Citation1953; Rodnan et al., Citation1967; Koeger et al., Citation1995). Many of the same antibodies are seen in both idiopathic SSc and silica-associated SSc, and they are very similar in clinical presentation (McHugh et al., Citation1994) (Rustin et al., Citation1990). Of particular interest is the association of anti-topoisomerase antibodies with both silica-associated SSc and diffuse idiopathic SSc with pulmonary involvement, suggesting some common link in their pathogenesis (McHugh et al., Citation1994). That link may be suggested in the recent data showing that topoisomerase I is over-expressed in scleroderma fibroblasts (Zhou et al., Citation2001), and is uniquely cleaved in apoptosis and metal-catalyzed redox reactions (Casciola-Rosen et al., Citation1997; Rosen and Casciola-Rosen, 1999) yielding targets for autoantibodies. At the same time, it has been shown that anti-topoisomerase antibody responses in scleroderma may be associated with specific HLA haplotypes, which would incorporate into a model of genetic susceptibility due to the ability to successfully present those antigens (Whyte et al., Citation1994).
Lymphocytes have been examined in regard to activation and immune abnormalities in patients with silicosis, and these same abnormalities may play a role in the progression of autoimmune disease. The number of T-helper cells was reported to be increased in the lymph nodes of the lung hilus in silicosis patients; however, the overall number of all classes of T-lymphocytes was decreased (Watanabe et al., Citation1987). Further, silicosis patients who were reported to have a higher incidence of autoimmune disease also had a significant decrease in B-, T- and NK-cells compared to healthy volunteers (Subra et al., Citation2001). However, a significant increase in the percentage of activated CD3+ T-cells was reported (Subra et al., Citation2001). It is not known whether the increase of activated T cells is due to chronic stimulation by silica and if this is occurring directly or indirectly through other immune cells or cytokines. Further, it is not known if the overall decrease in B, T, and NK cells is due to increased elimination of these cells or to altered levels of proliferation. Lymphopenia has been reported to induce autoimmune diseases experimentally through reduction of regulatory-T-cells that help control T-helper cell responses; therefore, this may provide an additional mechanism to describe the effects of silica on immune dysregulation (Shevach, Citation2000). These data show many consistencies with effects on lymphocytes seen in mouse models of silica exposure, which are described later.
While these data may provide clues to underlying mechanisms involved in silica-induced autoimmunity, there is relatively little data examining specific pathways of this process in animal models that may then be applied to therapeutic intervention. Mouse models have, however, been used extensively to study silica-induced apoptosis.
Silica and Apoptosis: Setting the Stage for Autoimmunity
Apoptosis can play several roles in the pathogenesis of systemic autoimmunity (Reviewed in Greidinger, Citation2001). First, patients and animal models that have defects in apoptosis are more susceptible to autoimmunity due to impaired tolerance mechanisms (Cohen and Eisenberg, 1991; Vaishnaw et al., 1999). Second, apoptosis of target cells may be a mechanism of immune complex-mediated target organ injury by autoimmune responses. However, there is also evidence that impaired clearance of apoptotic material could provide antigenic material to drive autoimmune responses. Mouse models prone to SLE-like disease have been shown to exhibit impaired clearance of apoptotic cells (Potter et al., Citation2003), and autoimmunity occurs in nonautoimmune prone mice when targeted gene knockouts impair apoptotic clearance (Hanayama et al., Citation2004). Work by Antony Rosen et al. demonstrated that intracellular antigens that are often the targets of autoantibodies may become immunogenic during apoptotic processes such as proteolytic cleavage (Casciola-Rosen et al., Citation1997; Rosen and Casciola-Rosen, 1999). Apoptosis also alters golgi antigens, which might explain the development of anti-golgi complex autoantibodies in autoimmune diseases such as Sjögren's Syndrome and SLE (Nozawa et al., Citation2002).
Cells of the lung efficiently clear most respired particulates without residual damage. Silica, however, leads to chronic inflammation and subsequent fibrosis, possibly due to disruption of alveolar macrophage (AM) function and excess production of pro-inflammatory cytokines (Davis et al., Citation1998). The adjuvant-like, broad immune activating effect of silica has long been recognized to have an effect on antibody production (Pernis and Paronetto, 1962). Therefore, exposure to silica is able to establish an immune activated/inflammatory environment.
In addition, silica is quite cytotoxic, causing both necrosis and apoptosis (Iyer et al., Citation1996; Leigh et al., Citation1997), but it seems unlikely that cytotoxicity alone would lead to a loss of tolerance since the ingestion of apoptotic bodies by phagocytes normally down-regulates production of inflammatory cytokines (Fadok et al., Citation1998). It has previously been reported that uptake of silica and subsequent apoptosis of alveolar macrophages appears to be mediated in part through scavenger receptor class A (SR-A) (Hamilton et al., Citation1996; Chao et al., Citation2001; Thibodeau et al., Citation2003). SR-A is characterized by broad ligand binding specificity and is a member of the pattern recognition receptor family. SR-A has been mainly studied for its role in atherosclerosis due to the binding of modified low density lipoproteins (Krieger, Citation1997), but silica has also been reported to bind this receptor (Kobzik, Citation1995). By blocking the binding of silica to SR-A using an antibody, Chao et al. reported that the induction of caspase 3 and subsequent apoptosis was inhibited (Chao et al., Citation2001). This could provide a mechanism by which silica might selectively delete SR-A expressing cells. It has been shown that silica appears to shift the population of human AM to a more immune active phenotype, through selective apoptosis of the normally immune suppressive AM population (Holian et al., Citation1997; Hamilton et al., Citation2001). The remaining macrophages are much better antigen presenting cells, expressing increased co-stimulatory molecules and cytokines (Hamilton et al., Citation2001), which would lead to activation rather than tolerance of T-cells. These immune active macrophages, or dendritic cells, may therefore be presenting excess apoptotic material (from apoptosis of normal macrophages and infiltrating neutrophils) to T-cells, leading to autoimmune induction. In an in vitro system, unstimulated macrophages were found to prevent immune responses to apoptotic material by competing with dendritic cells for uptake of apoptotic blebs, demonstrating the importance of the cell population silica is targeting for injury (Albert et al., Citation1998). Silica-induced apoptosis of alveolar macrophages leads to release and uptake of silica by other alveolar macrophages, producing a cyclical process of inflammation and cell death (Cooper et al., Citation2002). Therefore, silica induces a highly inflammatory state in the exposed lung, in addition to apoptosis of alveolar macrophages. In this environment, excess apoptosis and the inability to effectively clear the apoptotic cells could lead to excess presentation of self-antigens, therefore exacerbating an autoimmune response. This mechanism could be relevant not only to silica-associated autoimmunity, but also to other environmental exposures for which a common thread is the induction of apoptosis.
Consistent with this hypothesis, animals intravenously exposed to apoptotic cellular material develop autoantibodies (Mevorach et al., Citation1998). In addition, the presence of concentrated autoantigens has been reported within the blebs of apoptotic cells (Rosen and Casciola-Rosen, 1999). Further, lupus autoantigens are represented by structures that are chemically cleaved or modified during apoptosis and if this material is not removed by noninflammatory processes, apoptotic material could be presented by specialized antigen presenting cells to induce immune responses (Utz and Anderson, 1998; Ronchetti et al., Citation1999). Consequently, it appears apoptosis may play a central role in the development of autoimmune disease following silica exposure. Therefore, mechanisms of silica-induced apoptosis, including caspase activation, Fas/Fas ligand, TNF-α, and protein kinase Cδ are reviewed next.
Silica and Caspase Activation
The central effectors in apoptotic cell death involve the activation of a group of cysteine proteases known as caspases. Thirteen caspases have been identified and are named caspase-1 through -13. Two main pathways of caspase activation have been studied, the intrinsic and extrinsic pathways, both of which converge at the major effector caspase-3 (reviewed in (Boatright and Salvesen, 2003; Creagh et al., Citation2003).
The intrinsic pathway involves mitochondrial damage resulting in redistribution of cytochrome c from the mitochondria to the cytoplasm of the cell leading to the activation of caspases (Boatright and Salvesen, 2003). In addition to activation of caspases through mitochondrial release of cytochrome c, caspases can also be activated through cell surface receptors (Thorburn, Citation2004) The extrinsic pathway of caspase activation involves several tumor necrosis factor receptor (TNFR) family members, such as TNFR-1 and CD95 (Fas). These receptors have an intracellular signaling domain referred to as the death domain (DD) (Thorburn, Citation2004). Upon receptor engagement by TNF or Fas ligand, the DD is bound by another class of proteins referred to as adaptor proteins which can activate caspase-8 and -10. Through downstream events, caspase-8 and -10 lead to the cleavage and activation of caspase-3, resulting in apoptosis.
Silica has been reported to alter mitochondrial membrane permeability inducing release of procaspase-9, the production of the active p39 subunit of caspase-9, and cleavage and activation of caspase-3, thereby initiating apoptosis in MH-S cells (a BALB/c derived alveolar macrophage cell line) (Thibodeau et al., Citation2003). Using a general caspase inhibitor (z-VAD-fmk) or a caspase-9 specific inhibitor (z-LEHD-fmk), Thibodeau et al. were able to inhibit silica-induced activation of caspase-9 and -3 in MH-S cells (Thibodeau et al., Citation2003). They further reported that inhibition of the mitochondrial permeability pore by cyclosporin A partially decreased caspase-9 and -3 activation in silica exposed MH-S cells (Thibodeau et al., Citation2003). Therefore, activation of caspases by silica appears to be partially dependent on loss of mitochondrial integrity.
Borges et al. (Citation2002) have reported a role for apoptosis in silicosis by using caspase inhibitors in silica exposed BALB/c mice. Treatment with pan-caspase inhibitors reduced inflammation and collagen deposition in the lungs of BALB/c mice that received 20 mg of silica. Silica-induced apoptotic cells were also found in the draining lymph nodes of these silica-instilled BALB/c mice where they were able to interact with lymphocytes (Borges et al., Citation2002). They further reported that within the draining lymph nodes of these silica-treated mice, electron microscopy showed lymphocytes undergoing activation-induced cell death (Borges et al., Citation2002). Therefore, it appears apoptosis plays a role in silicosis and that silica-induced apoptotic cells are found outside of the lung, suggesting a systemic immune response.
As the caspase pathways for silica-induced apoptosis are clarified, it may become possible to explore the array of substrates for those caspases and determine whether antibodies to those substrate proteins are found in individuals with silica-associated autoimmunity. Although caspase cleavage is certainly not the only mechanism by which altered proteins could be presented to the immune system, it has been shown to be a factor in the production of anti-golgi antibodies (Nozawa et al., Citation2002). In a recent study, serum antibodies from silica-exposed NZM mice were shown to bind to apoptotic cells, in which the apoptosis had been triggered by silica (Pfau et al., Citation2004). To test whether any of those targets were exposed due to caspase activity, it was further shown that a broad-spectrum caspase inhibitor, Boc-D-FMK, inhibited both silica-induced apoptosis and the increased binding of serum autoantibodies from silica-exposed NZM mice to apoptotic cells (Pfau et al., Citation2004). Further study is needed to determine what the specific targets are and whether any of them are specific to apoptotic processes activated by silica.
Silica and Fas/FasL
Fas is a cell surface receptor protein involved in apoptosis which belongs to the TNF receptor family, and Fas abnormalities have been reported in human autoimmune diseases such as SLE and RA (reviewed in (Ricci-Vitiani et al., Citation2000; Rieux-Laucat et al., Citation2003; Siegel et al., Citation2003). Fas ligand (FasL) induces apoptosis by binding its membrane receptor Fas (Krammer, Citation2000). Outside of the thymus, most of the T-cell receptor-mediated apoptosis of T-cells, referred to as activation-induced cell death (AICD), is induced through the Fas pathway. Cytotoxic-T-lymphocytes also have the ability to use FasL to trigger apoptosis in Fas bearing cells, such as virus-infected cells or cancer cells. The importance of Fas in AICD is demonstrated by the lpr and gld strains of mice that have alterations in the Fas gene (Rieux-Laucat et al., Citation2003). Lpr and gld mice develop a lymphoproliferative disorder that resembles systemic lupus erythematosus. The MRL/lpr and gld mice have been studied with regard to silicosis; however, no studies have examined autoimmune responses following silica exposure in these models (Davis et al., Citation1998; Borges et al., Citation2002). In MRL/lpr mice, silica has been reported to result in increased prominent cellular infiltrates and fibrosis when compared to BALB/c or C3H mice which have normal levels of Fas (Davis et al., Citation1998). Borges et al. reported that silica-exposed T-cells from BALB.gld mice were resistant to AICD as compared to wild-type BALB/c mice suggesting that self-reactive T-cells may be able to escape apoptosis in this silicosis model (Borges et al., Citation2002). Further, silicosis patients with autoimmune disease have been reported to have elevated serum soluble Fas levels as compared to healthy volunteers (Otsuki et al., Citation1998; Tomokuni et al., Citation1999; Hamzaoui et al., Citation2003). Soluble Fas is produced as an alternative spliced product of the Fas gene that protects the cell from apoptosis by antagonization of the binding between membrane bound Fas and FasL. Therefore, apoptosis may play two distinct additional roles in silica-associated autoimmunity: first, it is possible that self-reactive immune cells may survive by escaping Fas mediated apoptosis, and second, silica may allow excess lymphocyte activation-induced cell death leading to immune abnormalities.
Silica and TNF-α
Tumor necrosis factor-α (TNF-α) is a potent cytokine mediator of inflammatory and immune functions (reviewed in (Wallach et al., Citation1999). TNF-α mediates a broad range of biological activities, but was originally characterized by its ability to induce apoptosis of tumor cells. TNF-α is predominately produced by macrophages and signals through TNF receptors which are found on nearly all cell types. There are 2 types of TNFR, type I (p55) and type II (p75). The type I receptor is found on most cells, while the type II receptor is found mainly on hematopoietic cells (Wallach et al., Citation1999). The TNFR has a death domain in the cytoplasmic tail which signals an adaptor protein named TRADD (TNF receptor-associated death domain). For apoptosis signaling, TRADD recruits FADD (Fas associated death domain), which in turn recruits and activates caspase-8 and -10.
TNF-α has been studied in regards to silica-induced fibrosis; however, the same role TNF-α plays in fibrosis may be relevant to silica-induced autoimmune disease. When macrophages are exposed to silica in vitro, enhanced TNF-α production occurs (Savici et al., Citation1994; Ortiz et al., Citation1999). Increases in TNF-α production activate an inflammatory cascade, which induces release of cytokines, including TNF-α, and activation of several cell signaling cascades (Pfeffer, Citation2003). Inhibition of TNF-α production or activity significantly reduces fibrosis, and addition of recombinant TNF-α increases collagen deposition in the lungs of mice (Piguet et al., Citation1990; Ortiz et al., Citation1999). Ortiz et al. (Citation1999) reported that silica-exposed C57Bl/6 and BALB/c mice acutely upregulated mRNA levels of TNF-α and the p75 TNFR and that these mice developed fibrosis. They also reported that silica-exposed TNFR knockout mice did not develop fibrosis as compared to wild-type mice. Thus, silica-induced lung injury involves TNF-α production and signaling by macrophages (Ortiz et al., Citation1999). TNF-α has been extensively studied as a pro-apoptotic cytokine; therefore, it is possible that silica-induced production of TNF-α by macrophages may lead to apoptosis and that apoptosis may be playing a role in lung injury. A 1.5-fold increase has recently been reported for TNF-α levels in lavage fluid from silica-exposed NZM mice 14 weeks following exposure (Brown et al., Citation2004). It appears silica is able to induce both acute and long-term production of TNF-α, which may provide an inflammatory environment with continual apoptosis of macrophages.
Silica and Protein Kinase Cδ
We have recently examined protein kinase Cδ (PKCδ), an additional pro-apoptotic protein, following silica exposure. PKCδ is a novel PKC family member that has been reported to play a role in the initiation of apoptosis in many cell types (Pongracz et al., Citation1999; Webb et al., Citation2000; Bertho et al., Citation2002; Lounsbury et al., Citation2002; Brodie and Blumberg, 2003; Shukla et al., Citation2003). PKCδ is activated by diacylglycerol (DAG)/phorbol esters in a calcium-independent manner. A variety of apoptotic stimuli in many different cellular systems have been shown to induce PKCδ activation and translocation. Furthermore, PKCδ is activated in spontaneous apoptosis of neutrophils, in H2O2-induced apoptosis, TNF-α-induced apoptosis, Fas-mediated apoptosis and asbestos-induced apoptosis of alveolar epithelial cells (Pongracz et al., Citation1999; Webb et al., Citation2000; Bertho et al; 2002; Lounsbury et al., Citation2002; Brodie and Blumberg Citation2003; Shukla et al., Citation2003).
A study by Shukla et al. reported a role for PKCδ in apoptosis of alveolar type II cells induced by exposure to asbestos (Shukla et al., Citation2003). They reported that asbestos activated PKCδ, leading to its translocation to the mitochondria, and inducing the release of cytochrome c and activation of caspase-9 (Shukla et al., Citation2003). Pre-treatment of these cells with rottlerin, reported by some as an inhibitor of PKCδ, prevented activation of caspase-9 and apoptosis of asbestos-exposed type II cells (Shukla et al., Citation2003). These results suggest that apoptosis of alveolar type II cells by asbestos is dependent on PKCδ, and similar mechanisms may be involved in silica-induced apoptosis.
Microarray analysis (complete table of results can be found at 〈http://www.umt.edu/cehs/imtx.htm〉) of AM recovered from NZM mice 14 weeks after silica exposure indicated increased expression of PKCδ that was confirmed by Western analysis (unpublished data). In addition, we recently found increases in expression of PKCδ and caspase-9 at 2 hours followed by increases in caspase-3 at 4 hours following silica treatment in bone marrow derived macrophages, suggesting that silica induces PKCδ upstream of caspase-9 thereby initiating the apoptotic process (unpublished data). Using rottlerin, in vivo, we significantly inhibited the exacerbation of systemic autoimmune disease by silica exposure in NZM mice (unpublished data). In addition, we observed decreased levels of autoantibodies to histone and decreased IgG and C3 deposition within the kidneys (unpublished data). These data are consistent with previous data and suggest that silica-induced apoptosis of alveolar macrophages plays a major role in the exacerbation of autoimmune disease in the NZM mouse model.
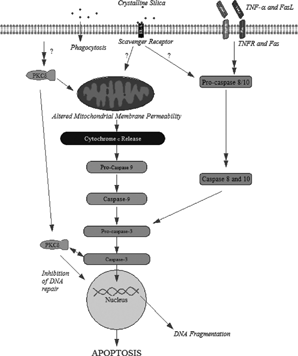
Abnormalities at many points in the process of apoptosis may be related to autoimmune disease pathogenesis. summarizes silica-induced signaling pathways involved in the induction of apoptosis. The induction of these apoptotic pathways by silica may lead to altered antigen, or to excess antigen thereby progressing the development of autoimmunity. More importantly, silica is inducing apoptosis within the alveolar macrophage, the very cell that should clear respired particles and apoptotic cells. Therefore, silica is not being cleared and additional macrophages may undergo apoptosis by the various signaling pathways leading to accumulation and impaired clearance of apoptotic material.
FIG. 1 Schematic of signal transduction pathways potentially activated by silica and leading to apoptosis.
Silica Exposure and Lymphocytes
As discussed previously, silica-induced apoptosis of alveolar macrophages may provide excess or modified antigen that is presented to lymphocytes thereby eliciting autoantibody production and T-cell-mediated responses. Further, the inflammatory response initiated by silica provides activation signals via cytokines to lymphocytes. Several studies have further examined silica's effects on lymphocytes using animal models.
Silica exposure has been reported to result in massive increases in lymph node size and sustained activation of distinct T-cell populations suggesting silica may be indirectly mitogenic for T-cells (Garn et al., Citation1997, 2000; Davis et al., Citation2000). Lymph nodes from silica exposed rats increased in weight up to 35-fold 52 weeks after silica exposure and the increase was due to an early influx of CD4+ T-cells and later influx of B-cells (Friedetzky et al., Citation1998). Six weeks following silica exposure, Friedetzky {et al}. reported finding accumulations of macrophages with ingested silica scattered throughout the lymphoid tissue and these macrophages were interacting with lymphocytes undergoing apoptosis (Friedetzky et al., Citation1998). Garn et al. further reported that T-cells from lymph nodes of silicotic rats had become activated with enhanced IFN-γ gene transcription, thereby resulting in macrophage activation (Garn et al., Citation1997). They further examined cytokine production from T-cells in silicotic rats and reported a shift towards production of the Th1 cytokines: IL-12, IL-18 and IFN-γ (Garn et al., Citation2000). Therefore, T-cells appear to migrate into the lymph nodes of silicotic animals and produce cytokines, thereby providing additional signals for immune activation.
Consistent with previous data, silica exposure in NZM mice resulted in migration of CD4+ T-helper cells and B1a B cells into a draining lymph node of the lung (Brown et al., Citation2004). The number of CD4+ T-helper cells was increased 3-fold in silica exposed mice as compared to saline-exposed mice (Brown et al., Citation2004). This large increase in T-helper cells was concurrent with a decrease in the number of CD4+CD25+ regulatory T-cells within the draining lymph node (Brown et al., Citation2004). It is possible that silica-induced apoptosis of pulmonary cells is providing antigen to macrophages or dendritic cells thereby inducing T-cell migration to regional lymph nodes.
Within the lymph nodes of silica exposed NZM mice, we reported a 6-fold increase in the number of B1a B cells (Brown et al., Citation2004). B1a B cells are very potent antigen presenting cells due to high expression levels of CD80 and CD86 (Mohan et al., Citation1998). Therefore, an increase in the number of B1a B-cells may allow for excess antigen presentation to T-helper cells thereby allowing peripheral tolerance to be broken.
Studies using T-cell deficient nude mice have suggested a role for T-lymphocytes in the migration of macrophages to the lung and in the termination of a neutrophil response following silica exposure (Hubbard, Citation1989). In this study, Hubbard reported that silica exposure in nude mice resulted in a massive influx of neutrophils that persisted over two months, whereas in the T-cell sufficient mice, the neutrophil response was short lived and followed by a migration of macrophages into the lung (Hubbard, Citation1989). This suggests that T-cells play a role in influencing the cell type within the lung of silica-exposed mice. However, the silica exposed nude mice did not develop fibrosis (Hubbard, Citation1989). These results suggest that T-cells may play a role in the migration of macrophages to the lung and that macrophages are playing a major role in the induction of the fibrotic process. A similar role for T-cells inducing macrophage migration to the lung may also be an important mechanism for the progression of autoimmune disease.
Lymphocytes appear to migrate to regional lymph nodes of silica-exposed animals, but antigen-presenting cell and T-cell interactions may also be occurring within the lung itself. The accumulation of lymphocytes has been reported within the alveolar spaces of the lung and lymphoid tissue of silica-treated C3H/HeN mice (Davis et al., Citation2001). These lymphocytes consisted primarily of CD4+ T-cells, but also numerous CD8+ T-cells, NK cells and γ δ T-cells (Davis et al., Citation2001). Further, they reported enhanced IFN-γ production and mRNA transcripts for IL-12 and IL-18, thereby suggesting a Th1 response in silica exposed mice (Davis et al., Citation2001). Using IFN-γ deficient C57Bl/6, Davis et al. (Citation2001) further reported less extensive silicosis than in wild-type mice. Lymphocytes are found within the lung of silica-exposed animals, but very little data exist as to their role in the progression of autoimmune disease.
Taken together, these data suggest that silica exposure results in T- and B-cell activation primarily within the draining lymph nodes of the lung. It appears that silica and apoptotic material-laden antigen presenting cells are migrating to these lymph nodes and presenting self-antigen to T-cells, thereby inducing autoimmune responses. The activation of T-cells can then lead to B cell activation and autoantibody production further inducing immune complex formation and complement activation.
Future Needs in Silica and Autoimmunity Studies
The challenge for future work in this area will be to build on existing data to design studies that will identify both the initiating mechanisms and the pathologic outcomes of silica-associated autoimmunity. These two goals are generally explored as distinct and unrelated events, but the unique sets of autoantibodies found in some exposure-associated autoimmune responses suggest that there may indeed be a relationship between initiation of autoimmune responses and disease pathology. That finding would be extremely important in its implications for the etiology of systemic autoimmune disease.
Human epidemiology data from Parks et al. (Citation2002) examined several common types of autoantibodies and found a strong association between silica exposure and anti-DNA and anti-Sm autoantibody levels. Further, Hogan et al. (Citation2001) reported an association between silica exposure and anti-neutrophil cytoplasmic autoantibody formation which is involved in small vessel vasculitis. Many common autoantibodies that are found in systemic autoimmune diseases were present in silica-exposed NZM mice including increases in the levels of anti-nuclear, anti-dsDNA and anti-histone autoantibodies (Brown et al., Citation2004). In addition, there was increased recognition of apoptotic cells by autoantibodies from silica exposed NZM mice (Pfau et al., Citation2004). It appears that silica exposure in NZM mice results in autoantibodies which recognize apoptotic cells and not live or necrotic cells, suggesting that during apoptosis self molecules are being uniquely altered to become antigenic (Pfau et al., Citation2004). Although no data exists examining possible unique autoantigens formed during silica-induced apoptosis, technologies such as probing cDNA expression libraries, and analysis of serum antibodies by multiplexed antigen-coated bead arrays, may make this data more and more attainable. Discovery of those targets will ultimately allow examination of whether the binding of the autoantibodies to their targets plays a role in disease pathogenesis.
It is quite possible that gene expression changes in tissues exposed to silica, particularly looking for changes in some of the common autoantigens or in the apoptotic and immune activation pathways, may provide clues for new targets. Anti-topoisomerase antibodies develop in both silica-associated SSc and diffuse idiopathic SSc with pulmonary involvement, suggesting some common link in their pathogenesis (McHugh et al., Citation1994). A clue to that link appeared in the recent cDNA microarray data showing that topoisomerase I is over-expressed in scleroderma fibroblasts (Zhou et al., Citation2001), suggesting that over-expression of this antigen led to autoimmune recognition. Since the target is expressed in fibroblasts, the question is raised whether binding of those antibodies might play a role in the activation of fibroblasts and their contribution to the fibrosis of scleroderma.
As noted earlier, the discovery of the possible involvement of PKCδ arose from examining gene expression changes in AM from silica-exposed NZM mice. From those studies, a few additional genes were reproducibly up-regulated or down-regulated and may serve to seed future research in this area. Those genes included those for macrophage migration inhibitory factor, thymosin β-10, Gc-group vitamin D binding protein, Api5, Bin1, and Tnfsf6 (Fas ligand). These are briefly described below in terms of their relevance to autoimmunity.
Macrophage Migration Inhibitory Factor
Macrophage migration inhibitory factor (MIF) is a cytokine with enzymatic properties that has been reported to act as a critical mediator in a number of inflammatory diseases including septic shock, rheumatoid arthritis, glomerulonephritis, inflammatory lung disease, delayed type hypersensitivity and cancer (Lue et al., 2002). MIF was originally described to activate macrophages and also inhibit their migration, and serve as a key regulator of innate immunity by suppressing the anti-inflammatory effects of glucocorticoids (Calandra and Roger, 2003). MIF has been associated with systemic lupus erythematosus (Foote et al., Citation2004), juvenile idiopathic arthritis (Donn et al., Citation2004), and several other autoimmune disorders (Barton et al., Citation2003; Calandra and Roger, 2003). MIF regulates cytokine secretion and the expression of receptors involved in innate immunity, inhibits p53 production and activates the MAP kinase and Jab-1 pathways (Lue et al., 2002). MIF has been found at elevated levels in serum, synovial fluid and macrophages from rheumatoid arthritis patients compared to a control population (Leech et al., Citation1999; Santos et al., Citation2001; Morand et al., Citation2002). In addition to rheumatoid arthritis, MIF has been reported to be upregulated in human glomerulonephritis and this correlates with leukocyte infiltration and histologic damage (Lan et al., Citation2000). Therefore, it is possible that the prolonged expression of MIF in the lungs of NZM mice is contributing to silica-induced autoimmunity through the recruitment of macrophages and T-cell accumulation in sites of inflammation and apoptosis.
Thymosin β-10 (Tmsb10)
Thymosin β-10 is a G-actin binding protein that prevents globular actin monomers from polymerizing spontaneously. However, in addition to regulating the rate of actin polymerization, thymosins regulate cell growth and differentiation. Cells overexpressing thymosin β -10 have a reduced growth rate, grow in a disorganized fashion and accelerate apoptosis (Hall, Citation1995). A mouse macrophage cell line (RAW264.7) overexpressing thymosin β-10 developed spontaneous apoptosis in 66.5% of the cells (Gutierrez-Pabello et al., Citation2002). Further, Mycobacterium bovis infection of bovine macrophages induced the upregulation thymosin β-10 leading to apoptosis (Gutierrez-Pabello et al., Citation2002). Our microarray data suggests that silica exposure upregulates thymosin β-10, which may be playing an additional role in the induction of apoptosis in alveolar macrophages.
Gc-Group Specific Component (Vitamin D Binding Protein) DBF
This protein is a multifunctional protein found on the surface of many cell types and secreted into many body fluids. It can associate with membrane-bound immunoglobulin on the surface of B-lymphocytes or with the Fc receptor of immunoglobulin on T-lymphocytes. Gumireddy {et al}. (2003) demonstrated that Gc could be involved in increasing propapoptotic caspases (3, 8, and 9) and inducing apoptosis in a murine macrophage model RAW 264.7. Polymorphisms within this gene have been linked to various human diseases, including Grave's disease, and COPD.
Api5
Api5 is a protein of approximately 25 kDa, conserved in evolution, and ubiquitously expressed. This protein has been shown to rescue serum-starved cell lines (Tewari et al., Citation1997) from apoptosis, and is thus considered a survival factor. This protein contains a leucine zipper motif, critical to its function, and interacts with FGF2. Up-regulation of this protein may preserve T-cell populations normally targeted for apoptosis, contributing to the development of autoimmunity in the silica model.
Bin1
This protein is a nucleocytoplasmic adaptor protein with some of the features of a tumor suppressor (Negorev et al., Citation1996). Its expression is frequently lost in human hepatocellular carcinomas. It plays an important role by interacting with c-myc and inducing caspase-independent apoptosis. Bin1 has several isoforms, suggesting varied tissue specific functions. It may be that overexpression of this gene in specific lung macrophage populations leads to increased programmed cell death and loss of immune regulation of the response to silica.
Tnfsf6 (Fas Ligand)
This protein is the ligand for Fas, which is critical in triggering apoptosis, especially in lymphocytes. Defects in Tnfsf6 may be related to some cases of SLE (Wu et al., Citation1996), or toxic epidermal necrolysis (TEN) (Viard et al., Citation1998). The Tnfsf6/fas-mediated apoptosis has been shown to have a role in the induction of peripheral tolerance (Simon et al., Citation2002). The down-regulation of Tnfsf6 could be an attractive model for loss of tolerance and the induction of autoimmunity.
SUMMARY
Silica exposure has been associated with several autoimmune diseases, however the mechanisms of initiation and progression are unknown. This review explored possible mechanisms of silica exposure in the development of systemic autoimmune disease. Many studies have examined the effects of silica on the alveolar macrophage and the development of fibrosis, but very few studies have applied these same mechanisms to the development of autoimmune disease. Our model suggests that inhalation of crystalline silica results in concurrent activation and apoptosis of the alveolar macrophage resulting in an environment of inflammation and apoptosis. This environment may provide excess antigen that is further ingested by activated macrophages or dendritic cells that are able to migrate to local lymph nodes. Within the lymph nodes, these antigen presenting cells, laden with apoptotic material, activate T-cells and B-cells thereby inducing an autoimmune response. The poor ability of the body to clear silica from the lung prolongs this cycle of macrophage apoptosis and lymphocyte activation, thereby progressing from an autoimmune response into a systemic autoimmune disease. Several techniques exist to further test this model, both at the level of initiation of autoimmunity and in understanding the disease pathogenesis. The study of silica-associated autoimmunity is critical due to on-going exposures to this and other inhaled particulates, but it also provides an excellent model to begin to understand the etiology of other environmental autoimmune disease processes.
Related Research Data
REFERENCES
- Albert M. L., Pearce S. F., Francisco L. M., Sauter B., Roy P., Silverstein R. L., Bhardwaj N. Immature dendritic cells phagocytose apoptotic cells via alphavbeta5 and CD36, and cross-present antigens to cytotoxic T-lymphocytes. J. Exp. Med. 1998; 188(7)1359–1368
- Barton A., Lamb R., Symmons D., Silman A., Thomson W., Worthington J., Donn R. Macrophage migration inhibitory factor (MIF) gene polymorphism is associated with susceptibility to but not severity of inflammatory polyarthritis. Genes Immun. 2003; 4(7)487–491
- Bertho N., Blancheteau V. M., Setterblad N., Laupeze B., Lord J. M., Drenou B., Amiot L., Charron D. J., Fauchet R., Mooney N. MHC class II-mediated apoptosis of mature dendritic cells proceeds by activation of the protein kinase C-delta isoenzyme. Int. Immunol 2002; 14(8)935–942
- Boatright K. M., Salvesen G. S. Mechanisms of caspase activation. Curr. Opin. Cell. Biol. 2003; 15(6)725–731
- Borges V. M., Lopes M. F., Falcao H., Leite-Junior J. H., Rocco P. R., Davidson W. F., Linden R., Zin W. A., DosReis G. A. Apoptosis underlies immunopathogenic mechanisms in acute silicosis. Amer. J. Respir. Cell. Mol. Biol 2002; 27(1)78–84
- Bramwell B. Diffuse scleroderma: Its frequency; its occurrence in stone masons; its treatment by fibrolysis. Edinburgh Med. J 1914; 12: 387
- Brodie C., Blumberg P. M. Regulation of cell apoptosis by protein kinase c delta. Apoptosis 2003; 8(1)19–27
- Brody A. R., Roe M. W., Evans J. N., Davis G. S. Deposition and translocation of inhaled silica in rats. Quantification of particle distribution, macrophage participation, and function. Lab. Invest 1982; 47(6)533–542
- Brown J. M., Pfau J. C., Holian A. Immunoglobulin and lymphocyte responses following silica exposure in New Zealand mixed mice. Inhal. Toxicol 2004; 16(3)133–139
- Calandra T., Roger T. Macrophage migration inhibitory factor: A regulator of innate immunity. Nat. Rev. Immunol 2003; 3(10)791–800
- Caplan A. Certain unusual radiological appearances in the chest of coal-miners suffering from rheumatoid arthritis. Thorax 1953; 8(1)29–37
- Casciola-Rosen L., Wigley F., Rosen A. Scleroderma autoantigens are uniquely fragmented by metal-catalyzed oxidation reactions: Implications for pathogenesis. J. Exp. Med 1997; 185(1)71–79
- Castranova V. D., Vallyathan V. Role of Surface Free Radicals in the Pathogenicity of Silicosis: Silica and Silica-Induced Lung Diseases. CRC Press, Florida 1996
- Chao S. K., Hamilton R. F., Pfau J. C., Holian A. Cell surface regulation of silica-induced apoptosis by the SR-A scavenger receptor in a murine lung macrophage cell line (MH-S). Toxicol. Appl. Pharmacol. 2001; 174(1)10–16
- Cohen P. L., Eisenberg R. A. Lpr and gld: Single gene models of systemic autoimmunity and lymphoproliferative disease. Annu. Rev. Immunol. 1991; 9: 243–269
- Cooper G. S., Miller F. W., Germolec D. R. Occupational exposures and autoimmune diseases. Int. Immunopharmacol. 2002; 2(2-3)303–13
- Creagh E. M., Conroy H., Martin S. J. Caspase-activation pathways in apoptosis and immunity. Immunol. Rev. 2003; 193: 10–21
- Davis G. S., Holmes C. E., Pfeiffer L. M., Hemenway D. R. Lymphocytes, lymphokines, and silicosis. J. Environ. Pathol. Toxicol. Oncol. 2001; 20: 53–65, Suppl 1
- Davis G. S., Leslie K. O., Hemenway D. R. Silicosis in mice: Effects of dose, time, and genetic strain. J. Environ. Pathol. Toxicol. Oncol. 1998; 17(2)81–97
- Davis G. S., Pfeiffer L. M., Hemenway D. R. Persistent overexpression of interleukin-1beta and tumor necrosis factor-alpha in murine silicosis. J. Environ. Pathol. Toxicol. Oncol. 1998; 17: 99–114
- Davis G. S., Pfeiffer L. M., Hemenway D. R. Interferon-gamma production by specific lung lymphocyte phenotypes in silicosis in mice. Am. J. Respir. Cell. Mol. Biol. 2000; 22(4)491–501
- Donn R., Alourfi Z., Zeggini E., Lamb R., Jury F., Lunt M., Meazza C., De Benedetti F., Thomson W., Ray D. A functional promoter haplotype of macrophage migration inhibitory factor is linked and associated with juvenile idiopathic arthritis. Arthritis Rheum 2004; 50(5)1604–1610
- Fadok V. A., Bratton D. L., Konowal A., Freed P. W., Westcott J. Y., Henson P. M. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J. Clin. Invest 1998; 101(4)890–898
- Foote A., Briganti E. M., Kipen Y., Santos L., Leech M., Morand E. F. Macrophage migration inhibitory factor in systemic lupus erythematosus. J. Rheumatol 2004; 31(2)268–273
- Friedetzky A., Garn H., Kirchner A., Gemsa D. Histopathological changes in enlarged thoracic lymph nodes during the development of silicosis in rats. Immunobiology 1998; 199(1)119–132
- Garn H., Friedetzky A., Davis G. S., Hemenway D. R., Gemsa D. T-lymphocyte activation in the enlarged thoracic lymph nodes of rats with silicosis. Am. J. Respir. Cell. Mol. Biol. 1997; 16(3)309–316
- Garn H., Friedetzky A., Kirchner A., Jager R., Gemsa D. Experimental silicosis: A shift to a preferential IFN-gamma-based Th1 response in thoracic lymph nodes. Am. J. Physiol. Lung. Cell. Mol. Physiol 2000; 278(6)L1221–1230
- Greidinger E. L. Apoptosis in lupus pathogenesis. Front. Biosci 2001; 6: D1392–1402
- Gumireddy K., Reddy C. D., Swamy N. Mitogen-activated protein kinase pathway mediates DBP-maf-induced apoptosis in RAW 264.7 macrophages. J. Cell. Biochem 2003; 90(1)87–96
- Gutierrez-Pabello J. A., McMurray D. N., Adams L. G. Upregulation of thymosin beta-10 by Mycobacterium bovis infection of bovine macrophages is associated with apoptosis. Infect. Immun 2002; 70(4)2121–2127
- Hall A. K. Thymosin beta-10 accelerates apoptosis. Cell. Mol. Biol. Res 1995; 41(3)167–180
- Hamilton R. F., Iyer L. L., Holian A. Asbestos induces apoptosis in human alveolar macrophages. Am. J. Physiol 1996; 271(5)L813–819, Pt 1
- Hamilton R. F., Jr., Pfau J. C., Marshall G. D., Holian A. Silica and PM1648 modify human alveolar macrophage antigen-presenting cell activity in vitro. J. Environ. Pathol. Toxicol. Oncol 2001; 20: 75–84, Suppl 1
- Hamzaoui A., Ammar J., Grairi H., Hamzaoui K. Expression of Fas antigen and Fas ligand in bronchoalveolar lavage from silicosis patients. Mediators Inflamm 2003; 12(4)209–214
- Hanayama R., Tanaka M., Miyasaka K., Aozasa K., Koike M., Uchiyama Y., Nagata S. Autoimmune disease and impaired uptake of apoptotic cells in MFG-E8-deficient mice. Science 2004; 304(5674)1147–1150
- Hogan S. L., Satterly K. K., Dooley M. A., Nachman P. H., Jennette J. C., Falk R. J. Silica exposure in anti-neutrophil cytoplasmic autoantibody-associated glomerulonephritis and lupus nephritis. J. Am. Soc. Nephrol 2001; 12(1)134–142
- Holian A., Uthman M. O., Goltsova T., Brown S. D., Hamilton R. F. Asbestos and silica-induced changes in human alveolar macrophage phenotype. Environ. Health Perspect 1997; 105(S5)1139–1142
- Hubbard A. K. Role for T lymphocytes in silica-induced pulmonary inflammation. Lab. Invest 1989; 61(1)46–52
- Hughes J. M., Jones R. N., Gilson J. C., Hammad Y. Y., Samimi B., Hendrick D. J., Turner-Warwick M., Doll N. J., Weill H. Determinants of progression in sandblasters' silicosis. Ann. Occup. Hyg 1982; 26(1-4)701–712
- Hultman P., Enestrom S., Pollard K. M., Tan E. M. Anti-fibrillarin autoantibodies in mercury-treated mice. Clin. Exp. Immunol 1989; 78(3)470–477
- Iyer R., Hamilton R. F., Li L., Holian A. Silica-induced apoptosis mediated via scavenger receptor in human alveolar macrophages. Toxicol. Appl. Pharmacol. 1996; 141: 84–92
- Kobzik L. Lung macrophage uptake of unopsonized environmental particulates. Role of scavenger-type receptors. J. Immunol 1995; 155(1)367–376
- Koeger A. C., Lang T., Alcaix D., Milleron B., Rozenberg S., Chaibi P., Arnaud J., Mayaud C., Camus J. P., Bourgeois P. Silica-associated connective tissue disease. A study of 24 cases. Medicine (Baltimore) 1995; 74(5)221–237
- Krammer P. H. CD95's deadly mission in the immune system. Nature 2000; 407(6805)789–795
- Krieger M. The other side of scavenger receptors: Pattern recognition for host defense. Curr Opin Lipidol 1997; 8(5)275–280
- Lan H. Y., Yang N., Nikolic-Paterson D. J., Yu X. Q., Mu W., Isbel N. M., Metz C. N., Bucala R., Atkins R. C. Expression of macrophage migration inhibitory factor in human glomerulonephritis. Kidney Int 2000; 57(2)499–509
- Leech M., Metz C., Hall P., Hutchinson P., Gianis K., Smith M., Weedon H., Holdsworth S. R., Bucala R., Morand E. F. Macrophage migration inhibitory factor in rheumatoid arthritis: Evidence of proinflammatory function and regulation by glucocorticoids. Arthritis Rheum 1999; 42(8)1601–1608
- Leigh J., Wang H. A. B., Peters M., Ruan X. Silica-induced apoptosis in aleolar and granulomatous cells in vivo. Environ. Health. Perspect 1997; 105(S5)1241–1245
- Lounsbury K. M., Stern M., Taatjes D., Jaken S., Mossman B. T. Increased localization and substrate activation of protein kinase C delta in lung epithelial cells following exposure to asbestos. Am. J. Pathol 2002; 160(6)1991–2000
- Lue H., Kleemann R., Calandra T., Roger T., Bernhagen J. Macrophage migration inhibitory factor (MIF): Mechanisms of action and role in disease. Microbes Infect 2002; 4(4)449–460
- Mayes M. D. Epidemiologic studies of environmental agents and systemic autoimmune diseases. Environ. Health Perspect 1999; 107: 743–748, Suppl 5
- McHugh N. J., Whyte J., Harvey G., Haustein U. F. Anti-topoisomerase I antibodies in silica-associated systemic sclerosis. A model for autoimmunity. Arthritis Rheum 1994; 37(8)1198–1205
- Mevorach D., Zhou J. L., Song X., Elkon K. B. Systemic exposure to irradiated apoptotic cells induces autoantibody production. J. Exp. Med 1998; 188(2)387–392
- Mohan C., Morel L., Yang P., Wakeland E. K. Accumulation of splenic B1a cells with potent antigen-presenting capability in NZM2410 lupus-prone mice. Arthritis Rheum 1998; 41(9)1652–1662
- Morand E. F., Leech M., Weedon H., Metz C., Bucala R., Smith M. D. Macrophage migration inhibitory factor in rheumatoid arthritis: Clinical correlations. Rheumatology (Oxford) 2002; 41(5)558–562
- Negorev D., Riethman H., Wechsler-Reya R., Sakamuro D., Prendergast G. C., Simon D. The Bin1 gene localizes to human chromosome 2q14 by PCR analysis of somatic cell hybrids and fluorescence in situ hybridization. Genomics 1996; 33(2)329–331
- Nozawa K., Casiano C. A., Hamel J. C., Molinaro C., Fritzler M. J., Chan E. K. Fragmentation of Golgi complex and Golgi autoantigens during apoptosis and necrosis. Arthritis Res 2002; 4(4)R3
- Ortiz L. A., Lasky J., Lungarella G., Cavarra E., Martorana P., Banks W. A., Peschon J. J., Schmidts H. L., Brody A. R., Friedman M. Upregulation of the p75 but not the p55 TNF-alpha receptor mRNA after silica and bleomycin exposure and protection from lung injury in double receptor knockout mice. Am. J. Respir. Cell. Mol. Biol 1999; 20(4)825–833
- Otsuki T., Sakaguchi H., Tomokuni A., Aikoh T., Matsuki T., Kawakami Y., Kusaka M., Ueki H., Kita S., Ueki A. Soluble Fas mRNA is dominantly expressed in cases with silicosis. Immunology 1998; 94: 258–262
- Parks C. G., Conrad K., Cooper G. S. Occupational exposure to crystalline silica and autoimmune disease. Environ. Health Perspect 1999; 107(S5)793–802
- Parks C. G., Cooper G. S., Nylander-French L. A., Sanderson W. T., Dement J. M., Cohen P. L., Dooley M. A., Treadwell E. L., St Clair E. W., Gilkeson G. S., Hoppin J. A., Savitz D. A. Occupational exposure to crystalline silica and risk of systemic lupus erythematosus: A population-based, case-control study in the southeastern United States. Arthritis Rheum 2002; 46(7)1840–1850
- Parks C. G., Cooper G. S., Nylander-French L. A., Storm J. F., Archer J. D. Assessing exposure to crystalline silica from farm work: A population-based study in the Southeastern United States. Ann. Epidemiol 2003; 13(5)385–392
- Pernis B., Paronetto F. Adjuvant effects of silica (tridymite) on antibody production. Proc. Soc. Exp. Biol. Med. 1962; 110: 390–392
- Pfau J. C., Brown J. M., Holian A. Silica-exposed mice generate autoantibodies to apoptotic cells. Toxicology 2004; 195(2-3)167–176
- Pfeffer K. Biological functions of tumor necrosis factor cytokines and their receptors. Cytokine Growth Factor Rev 2003; 14(3-4)185–191
- Piguet P. F., Collart M. A., Grau G. E., Sappino A. P., Vassalli P. Requirement of tumour necrosis factor for development of silica-induced pulmonary fibrosis. Nature 1990; 344(6263)245–247
- Pollard K. M., Lee D. K., Casiano C. A., Bluthner M., Johnston M. M., Tan E. M. The autoimmunity-inducing xenobiotic mercury interacts with the autoantigen fibrillarin and modifies its molecular and antigenic properties. J. Immunol 1997; 158(7)3521–3528
- Pongracz J., Webb P., Wang K., Deacon E., Lunn O. J., Lord J. M. Spontaneous neutrophil apoptosis involves caspase 3-mediated activation of protein kinase C-delta. J. Biol. Chem 1999; 274(52)37329–37334
- Potter P. K., Cortes-Hernandez J., Quartier P., Botto M., Walport M. J. Lupus-prone mice have an abnormal response to thioglycolate and an impaired clearance of apoptotic cells. J. Immunol 2003; 170(6)3223–3232
- Ricci-Vitiani L., Conticello C., Zeuner A., De Maria R. CD95/CD95L interactions and their role in autoimmunity. Apoptosis 2000; 5(5)419–424
- Rieux-Laucat F., Le Deist F., Fischer A. Autoimmune lymphoproliferative syndromes: Genetic defects of apoptosis pathways. Cell Death Differ 2003; 10(1)124–133
- Rodnan G. P., Benedek T. G., Medsger T. A., Jr., Cammarata R. J. The association of progressive systemic sclerosis (scleroderma) with coal miners' pneumoconiosis and other forms of silicosis. Ann. Intern. Med 1967; 66(2)323–334
- Ronchetti A., Iezzi G., Crosti M. C., Garancini M. P., Protti M. P., Bellone M. Role of antigen-presenting cells in cross-priming of cytotoxic T lymphocytes by apoptotic cells. J. Leukoc. Biol 1999; 66(2)247–251
- Rosen A., Casciola-Rosen L. Autoantigens as substrates for apoptotic proteases: Implications for the pathogenesis of systemic autoimmune disease. Cell Death Differ 1999; 6(1)6–12
- Rustin M. H., Bull H. A., Ziegler V., Mehlhorn J., Haustein U. F., Maddison P. J., James J., Dowd P. M. Silica-associated systemic sclerosis is clinically, serologically and immunologically indistinguishable from idiopathic systemic sclerosis. Br. J. Dermatol 1990; 123(6)725–734
- Santos L., Hall P., Metz C., Bucala R., Morand E. F. Role of macrophage migration inhibitory factor (MIF) in murine antigen-induced arthritis: Interaction with glucocorticoids. Clin. Exp. Immunol 2001; 123(2)309–314
- Savici D., He B., Geist L. J., Monick M. M., Hunninghake G. W. Silica increases tumor necrosis factor (TNF) production, in part, by upregulating the TNF promoter. Exp. Lung Res. 1994; 20(6)613–625
- Shevach E. M. Regulatory T-cells in autoimmmunity. Annu. Rev. Immunol 2000; 18: 423–449
- Shukla A., Stern M., Lounsbury K. M., Flanders T., Mossman B. T. Asbestos-induced apoptosis is protein kinase C delta-dependent. Am. J. Respir. Cell. Mol. Biol 2003; 29(2)198–205
- Siegel R. M., Muppidi J., Roberts M., Porter M., Wu Z. Death receptor signaling and autoimmunity. Immunol. Res 2003; 27(2-3)499–512
- Silverstein A. M. Autoimmunity versus horror autotoxicus: The struggle for recognition. Nat. Immunol. 2001; 2(4)279–281
- Simon A. K., Gallimore A., Jones E., Sawitzki B., Cerundolo V., Screaton G. R. Fas ligand breaks tolerance to self-antigens and induces tumor immunity mediated by antibodies. Cancer Cell 2002; 2(4)315–322
- Society A. T. Adverse effects of crystalline silica exposure. Am. J. Respir. Crit. Care Med. 1997; 155: 761–768
- Steenland K., Brown D. Mortality study of gold miners exposed to silica and nonasbestiform amphibole minerals: An update with 14 more years of follow-up. Am. J. Ind. Med 1995; 27(2)217–229
- Steenland K., Goldsmith D. F. Silica exposure and autoimmune diseases. Am. J. Ind. Med 1995; 28(5)603–608
- Subra J. F., Renier G., Reboul P., Tollis F., Boivinet R., Schwartz P., Chevailler A. Lymphopenia in occupational pulmonary silicosis with or without autoimmune disease. Clin. Exp. Immunol 2001; 126(3)540–544
- Tewari M., Yu M., Ross B., Dean C., Giordano A., Rubin R. AAC-11, a novel cDNA that inhibits apoptosis after growth factor withdrawal. Cancer Res 1997; 57(18)4063–4069
- Thibodeau M., Giardina C., Hubbard A. K. Silica-induced caspase activation in mouse alveolar macrophages is dependent upon mitochondrial integrity and aspartic proteolysis. Toxicol. Sci 2003; 76(1)91–101
- Thorburn A. Death receptor-induced cell killing. Cell Signal 2004; 16(2)139–144
- Tomokuni A., Otsuki T., Isozaki Y., Kita S., Ueki H., Kusaka M., Kishimoto T., Ueki A. Serum levels of soluble Fas ligand in patients with silicosis. Clin. Exp. Immunol. 1999; 118: 441–444
- Utz P. J., Anderson P. Posttranslational protein modifications, apoptosis, and the bypass of tolerance to autoantigens. Arthritis Rheum 1998; 41(7)1152–1160
- Vaishnaw A. K., Toubi E., Ohsako S., Drappa J., Buys S., Estrada J., Sitarz A., Zemel L., Chu J. L., Elkon K. B. The spectrum of apoptotic defects and clinical manifestations, including systemic lupus erythematosus, in humans with CD95 (Fas/APO-1) mutations. Arthritis Rheum 1999; 42(9)1833–1842
- Viard I., Wehrli P., Bullani R., Schneider P., Holler N., Salomon D., Hunziker T., Saurat J. H., Tschopp J., French L. E. Inhibition of toxic epidermal necrolysis by blockade of CD95 with human intravenous immunoglobulin. Science 1998; 282(5388)490–493
- Wallach D., Varfolomeev E. E., Malinin N. L., Goltsev Y. V., Kovalenko A. V., Boldin M. P. Tumor necrosis factor receptor and Fas signaling mechanisms. Annu. Rev. Immunol. 1999; 17: 331–367
- Watanabe S., Shirakami A., Takeichi T., Ohara T., Saito S. Alterations in lymphocyte subsets and serum immunoglobulin levels in patients with silicosis. J. Clin. Lab. Immunol 1987; 23(1)45–51
- Webb P. R., Wang K. Q., Scheel-Toellner D., Pongracz J., Salmon M., Lord J. M. Regulation of neutrophil apoptosis: A role for protein kinase C and phosphatidylinositol-3-kinase. Apoptosis 2000; 5(5)451–458
- Whyte J., Artlett C., Harvey G., Stephens C. O., Welsh K., Black C., Maddison P. J., McHugh N. J. HLA-DQB1 associations with anti-topoisomerase-1 antibodies in patients with systemic sclerosis and their first degree relatives. United Kingdom Systemic Sclerosis Study Group. J. Autoimmun 1994; 7(4)509–520
- Wu J., Wilson J., He J., Xiang L., Schur P. H., Mountz J. D. Fas ligand mutation in a patient with systemic lupus erythematosus and lymphoproliferative disease. J. Clin. Invest 1996; 98(5)1107–1113
- Zhou X., Tan F. K., Xiong M., Milewicz D. M., Feghali C. A., Fritzler M. J., Reveille J. D., Arnett F. C. Systemic sclerosis (scleroderma): Specific autoantigen genes are selectively overexpressed in scleroderma fibroblasts. J. Immunol 2001; 167(12)7126–7133