Abstract
Pre-existing or contributing risk factors, including genetic predisposition and environmental influences, are largely thought to play a crucial (though ill-elucidated) role in the development of autoimmunity. Trichloroethylene (TCE) is a widely used organic solvent, which has been suspected of increasing the prevalence of autoimmune diseases, e.g., lupus, following environmental contamination. Although few epidemiological data are available, several studies reported an accelerated and more severe disease in TCE-exposed autoimmunity-prone MRL+/+ mice. To test whether TCE can exert similar deleterious effects on organ-specific autoimmune diseases, non obese diabetic (NOD) mice were given 5 mg/ml TCE via the drinking water for 12 weeks. TCE administration induced a decrease in CD44+ splenic T-cells and CD45RBhigh, CD54+ blood and splenic T-cells. Conversely, the number of CD45RBlow splenocytes was increased. Interestingly, the progressive increase in serum TNF-α and IFN-γ levels normally seen with age in these mice was inhibited by TCE. There was also a relative lower incidence of histological changes in the pancreas of TCE-exposed NOD mice than in unexposed mice. Contrary to what has been found in systemic models of autoimmunity, TCE did not accelerate the diabetes of NOD mice and may have a protective effect. This finding suggests that comparative studies using different genetically related autoimmune-prone models are needed to investigate the role of xenobiotics in the precipitation of autoimmunity, particularly in sensitive populations.
INTRODUCTION
Trichloroethylene (TCE) is a volatile organic compound widely used as a solvent to remove grease from metal parts. It can also be found in many industrial and household products. Due to this widespread use, TCE is a common environmental contaminant listed as a hazardous air and drinking water pollutant by the U.S. Environmental Protection Agency. Extensive assessment has shown that exposure to TCE is associated with neurotoxicity, developmental toxicity, liver, kidney and endocrine toxicity, and several forms of cancer (ATSDR, 1997). Different P450 enzymes have been identified as playing a role in hepatic TCE metabolism. TCE is metabolized by the cytochrome P450 2E1 (CYP2E1) to several metabolites, i.e., chloral, chloral hydrate, trichloroacetic acid (TCA), and dichloroacetic acid (DCA) (Lash et al., Citation2000).
Only limited and somewhat conflicting data are available regarding the influence of TCE on the immune system. TCE and DCA were shown to suppress humoral and cellular immunity after exposure via the drinking water or via inhalation, with a greater susceptibility of female than male mice (Sanders et al., Citation1982). A 3-day exposure via the intraperitoneal route inhibited NK-cell activity, but not mitogen-induced lymphocyte proliferation in both rats and mice (Wright et al., Citation1991). In another experiment, CD-1 mice exposed to chloral hydrate for 14 or 90 days via the drinking water did not exhibit changes in either humoral or cellular immunity (Kaufmann et al., 1982).
In fact, the most recent focus was on autoimmunity following the observation that humans exposed to TCE via contaminated well water had significantly higher antinuclear antibody titers (Kilburn and Warshaw, 1992). Ten out of 362 exposed subjects had clinical signs of systemic lupus erythematosus or other connective tissue diseases. Subsequently, several epidemiological studies underscored the causative role of exposure to organic solvents, in particular TCE, in the development of systemic scleroderma, a rare autoimmune disease (Bovenzi et al., Citation1995; Nietert et al., Citation1998; Aryal et al., Citation2001; Garabrant et al., Citation2003).
A series of experimental studies have been conducted in MRL+/+ mice, an autoimmunity-prone strain that spontaneously develops a lupus-like disease. Khan et al. (Citation1995) showed a statistically significant increase in antinuclear, anti-ssDNA, anti-cardiolipin autoantibodies and serum immunoglobulin levels in MRL+/+ mice treated with TCE or DCA by intraperitoneal injections. Similar findings were obtained in MRL+/+ mice following exposure to TCE for up to 22 weeks via the drinking water (Gilbert et al., Citation1999). In contrast to d-penicillamine and mercuric chloride, TCE produced no immune changes in Brown-Norway rats, another autoimmunity-prone rodent strain (White et al., Citation2000).
Type-1 insulin-dependent diabetes mellitus (IDDM) is an organ-specific autoimmune disease characterized by the progressive destruction of the insulin-secreting β cells in the islets of Langerhans. The incidence and development of IDDM depend upon a variety of genetic and nongenetic factors, including environmental influences, such as chemicals, diet, and infections (Kukreja and Maclaren, 1999). The mechanism of IDDM has been extensively investigated in diabetes-prone rodents, particularly the nonobese diabetic (NOD) mouse. The cell-mediated destruction of β-cells is associated with increased expression of the pro-inflammatory cytokines interleukin-1 (IL-1) and tumor necrosis factor (TNF-α), and the type 1 T (TH1) cytokines interferon-γ (IFN-γ), IL-2, and IL-12 (Rabinovitch, Citation1998).
The present study was conducted to investigate the influence of TCE exposure via the drinking water for 12 weeks on the spontaneous development of the autoimmune diabetes in female NOD mice. The course of the diabetes was evaluated with standard parameters including glycemia, histological examinations, and immunological endpoints (including lymphocyte subset analysis of naive and activated T-cells and cytokines level analysis).
MATERIALS AND METHODS
Animals
Fifty-seven female NOD/Bom mice were purchased from M&B (Gannat, France) at the age of 8 weeks. They were group-housed in polycarbonate cages in controlled conditions (19 to 25°C, < 40% humidity, at least 8 air changes per day, 12-hour light cycle) with free access to diet and water. They were randomly assigned to experimental groups following 7 days of acclimatization.
Treatment
TCE was purchased from Aldrich (Milwaukee, WI) and Alkamuls EL-620 from Rhodia (Clamecy, France). TCE (at least 99% pure) was suspended in drinking water containing 1% of emulsifier (Alkamuls EL-620).
Mice were randomly assigned to the treated (n = 27) and control groups (n = 30). The treated group received TCE via the drinking water at the concentration of 5.0 mg/ml from 11 weeks of age. Ten per group were terminated at 15 weeks of age, 10 per group were terminated at 19 weeks of age, 7 treated and 10 control females were terminated at 23 weeks of age. Control mice were given drinking water containing 1% Alkamuls EL-620. The drinking water was changed every 3–4 days.
Clinical Examination
The animals were observed daily and weighed once weekly. Drinking water consumption was recorded twice a week for each cage of animals. Food consumption was recorded weekly for each cage throughout the study.
Blood Glucose Determination
Blood glucose analysis was performed every two weeks using a Lifescan One Touch Profile Meter (Issy les Moulineaux, France) with one drop of blood sampled from the tail of non fasted animals. A 1.0 g/l calibration standard solution was assayed at least every 15 samples. The threshold for positivity was set as the mean of the pretest values plus two standard deviations.
Sampling for Immunological Investigations
Blood was withdrawn from the retro-orbital sinus of mice under isoflurane anesthesia and collected in tubes without anticoagulant for cytokine analysis every 4 weeks. On the day of necropsy, blood was withdrawn by intracardiac puncture from mice following overnight fasting and deeply anesthetized under isoflurane. Samples were collected in tubes with EDTA for lymphocyte subset analysis. An additional sample was also collected in tubes without anticoagulant for cytokine assays. The serum samples were stored at −80°C. In addition, the spleen was removed aseptically and a fragment was sampled. Spleen cells were suspended in RPMI 1640 supplemented with 10% fetal bovine serum containing 100 IU/ml penicillin and 100 μ g/ml of streptomycin (Biowhittaker, Walkersville, MD) for lymphocyte subset analysis. A cell suspension of approximately 107 cell/ml was prepared for each animal.
T- and B-Cell Subset Analysis
Total white blood cells were counted and the absolute and relative lymphocyte numbers were determined in peripheral blood using an ADVIA 120 Hematology System (Bayer Corporation, Tarrytown, NY). All antibodies used for the lymphocyte subset analysis were purchased from BD Biosciences Pharmingen (San Diego, CA).
T- and B-Cells. Blood and splenic cells were stained with peridinin chlorophyll-a protein (PerCP)-conjugated anti-CD3 (clone 145-2C11) and either fluorescein isothiocyanate (FITC)-conjugated anti-CD45RB220 (clone RA3-6B2) or R-phycoerythrin (R-PE)-conjugated anti-CD4 (clone RM4-5) and FITC-conjugated anti-CD8 (clone 53-6.7).
Memory and Activated T-Cells. Blood and splenic cells were stained with either PerCP/cyanine dye (PerCP-CY5.5)-conjugated anti-CD4 (clone RM4-5) and FITC-conjugated anti-CD8 (clone 53-6.7) followed by either biotinylated anti-CD44 (clone IM7), anti-CD45RB (clone 16A) or anti-CD54 (clone 3E2). A streptavidin-phycoerythrin conjugate was used as second-step reagent for the biotinylated primary antibodies. Two populations of CD45RB+ CD4+ T-cells were observed. The population with a high or a low intensity of fluorescence was identified as the CD45RBhigh or CD45RBlow CD4+ T-subset, respectively.
The phenotypic analysis was performed using a FACScan (Becton-Dickinson, Franklin Lakes, NJ) following the lysis of the red blood cells. A staining with an appropriate Ig isotype control was performed.
Cytokine Level Analysis
IL-2, IL-4, IL-5, IFN-γ, and TNF-α serum levels were assayed using the BD TH1/TH2 cytokine Cytometric Bead Array (CBA) kit (BD Biosciences, San Diego, CA).
Necropsy and Histological Examination
Ten mice per group were sacrified by carbon dioxide inhalation and exsanguination after 4 or 8 weeks of treatment, and 7 treated plus 10 control mice after 12 weeks of treatment, i.e., at 15, 19, and 23 weeks of age, respectively. A full necropsy was performed on all animals. Histological examinations were performed following fixation of the tissues in formalin (Chimie Plus, Denice, France) and embedding in paraffin wax (Lambert Riviere, Pierre-Benite, France). Then, 4-μ m sections were cut and stained with hematoxylin-eosin prior to examination. Pancreas, lymphoreticular tissues, liver and lungs were submitted to histological examination.
Statistical Analysis
All results are expressed as mean ± SEM. Differences between the treated and control group were analyzed using a standard 2-tailed Student's t-test.
RESULTS
Mortality, Body Weight, Food and Water Consumption
The treated mice ingested between 118 and 190 μ g/kg/day of TCE based on mean body weights and water consumption. TCE concentrations were not verified, however, so the actual concentrations at the end of the 3–4-day period of use may have been reduced due to evaporation.
There were no mortality and no relevant clinical signs at any time point during the study. There were no treatment-related effects on the food intake or body weight gain.
Blood Glucose Levels
Animals with blood glucose levels exceeding 2.0 g/l on 2 successive occasions were considered positive. The progressively increasing incidence of mice with hyperglycemia in both the control and treated groups were consistent with data provided by the animal supplier. There were thus no treatment-related effects on the spontaneous course of IDDM in the TCE-exposed mice.
Lymphocyte Subset Analysis
Blood T- and B-Cells
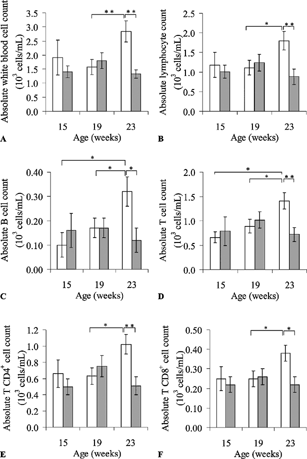
Statistically significant TCE-related reductions in the total white blood cell count and the numbers of total lymphocytes in the peripheral blood became evident at 23 weeks of age, but were not detected at 15 or 19 weeks of age (). The T-lymphocyte subsets (CD3+, CD4+, and CD8+) were principally affected. Inconsistent variations were noted at 15 and 19 weeks of age. Sharp increases in these parameters noted in the controls at 23 weeks of age, were not seen in the TCE-treated mice. The differences in total lymphocyte counts and T-lymphocyte subsets between the control and treated mice at 23 weeks of age were highly significant (p < 0.01 for total, T- and CD4+ T-lymphocytes counts; p < 0.05 for CD8+ T-lymphocytes counts). In the same way, there was also a TCE-related inhibition of B lymphocyte proliferation.
FIG 1. Lymphocyte subset analysis (absolute cell count in 103 cells/ml) in peripheral blood samples from TCE-treated and control mice: total white blood cell (A), lymphocyte (B), B (C), T (D), CD4+ T (E), CD8+ T (F) cell counts (103 cells/mL). Presented values are mean ± SEM. *p < 0.05; **p < 0.01.
Memory and Activated CD4+ T-Cells
There was a decrease in CD45RBhigh expression of splenic CD4+ T-lymphocytes (p < 0.05) in the TCE-treated mice at 15 weeks of age. CD45RBlow expression, on the other hand, was significantly higher in CD4+ T-lymphocytes from the blood (p < 0.01) and spleen (p < 0.05) of the treated mice at 23 weeks of age by comparison with the control mice (). In addition, there were significant decreases in CD44high (p < 0.05) and CD54 (p < 0.01) expression of spleen CD4+ T-lymphocytes in the treated mice at 19 weeks of age, and in CD54 expression of blood CD4+ T-lymphocytes at 23 weeks of age by comparison with the control mice (p < 0.05).
TABLE 1 Expression of markers of CD4+ T-cell activation (CD44, CD54 and CD45RB) on peripheral blood T-cells or splenocytes from female NOD mice (fluorescence intensity)
Memory and Activated CD8+ T-Cells
No changes in CD44, CD45RB and CD54 expression of CD8+ T-lymphocytes were observed either in the blood or the spleen of TCE-treated mice at 15 weeks of age. CD8+ Tlymphocytes from the spleen of treated mice at 19 weeks of age had a higher CD45RB expression (p < 0.05) than control ones (). In contrast to blood CD4+ T-lymphocytes, an up-regulation of CD44 expression was observed on CD8+ T-lymphocytes from treated mice at 23 weeks of age (p < 0.001). In addition, TCE-treatment induced a decrease in CD54 expression on blood CD8+ T-lymphocytes (p < 0.05).
TABLE 2 Expression of markers of CD8+ T-cell activation (CD44, CD54 and CD45RB) on peripheral blood T-cells or splenocytes from female MOD mice (fluorescence intensity)
Cytokine Levels
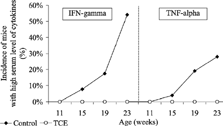
There were no treatment-related effects on serum IL-2, IL-4, and IL-5 levels. The number of serum samples from control NOD mice with a high serum level (concentration > 20 pg/ml) of the TH1 cytokines IFN-γ and TNF-α showed a regular progression throughout the study (). In contrast, there were no sera with high levels of IFN-γ and TNF-α in TCE-treated NOD mice.
FIG 2. Progression of the incidence of TCE-treated NOD mice with a high serum level of IFN-gamma and TNF-alpha compared with in control NOD mice from (concentration > 20 pg/ml).
Histological Examination
TCE-treated mice at 15 weeks of age had marginally more islets in the pancreas than controls (). Three out of 10 controls and one of seven TCE-treated mice at 23 weeks of age had an absence of islets in the pancreas. However, this result was not confirmed at 19 weeks of age. In addition, 2 of the 7 treated mice at 23 weeks of age had grade 3–4 lymphoid cell infiltration in the pancreas versus 4 of 10 control mice (moderate to severe). Although control mice showed more lymphoid hyperplasia in the thymus, mesenteric and axillary lymph nodes than TCE-treated mice at 15 weeks of age, there were no treatment-related effects in the lymphoreticular tissues. Apart from 1 control animal at 15 weeks and 2 at 23 weeks with hepatic inflammation and fibrosis, changes in liver and lungs were similar between control and treated mice.
Incidence of histopathological changes in the pancreas, thymus, mesenteric and axillary lymph nodes of TCE-treated and control female NOD mice
DISCUSSION
Even though the mechanisms leading to autoimmune diseases are not clearly elucidated, the role of preexisting risk factors including genetic predisposition and environmental influences is largely accepted. Infections, drugs and chemicals may be major environmental factors leading to the development of autoimmune diseases (Hess, Citation2002; Morahan and Morel, 2002). Amongst the chemicals suspected of inducing autoimmune diseases, there is some evidence supporting a link between several organic compounds (e.g. vinyl chloride, trichloroethylene and tetrachloroethylene) and various autoimmune disorders, such as systemic lupus erythematosus and scleroderma (Kilburn and Warshaw, 1992; Nietert et al., Citation1998).
TCE-induced acceleration of spontaneous autoimmune diseases was first demonstrated in MRL+/+ mice. Increases in antinuclear, anti-ssDNA, anti-cardiolipin autoantibodies and serum immunoglobulin levels were shown following intraperitoneal injections (Khan et al., Citation1995) or long-term exposure via the drinking water (Gilbert et al., Citation1999). Interestingly, TCE induced an increased expression of the activation markers CD44, CD45RBlow and CD54 on CD4+ T-lymphocytes from the spleen of MRL+/+ (). In addition, splenic CD4+ T-cells produced more IFN-γ and less IL-4 than control T-cells, suggesting a type 1 response (Griffin et al., Citation2000a, 2000b). It was concluded that TCE exposure of MRL+/+ mice is associated with the activation of pathogenic CD4+ TH1-cells (Gilbert et al., Citation1999; Griffin et al., Citation2000a).
TABLE 4 Reported role of the markers and effects of trichloroethylene administration on the spontaneous evolution of the autoimmune diseases in MRL+/+ and NOD mice
In different strains of mice, the CD45RBlow CD4+ T-cell subset was shown to exert regulatory effects. Inflammatory bowel disease was induced in immunodeficient mice by transfer of CD45RBhigh CD4+ T-cells, whereas the co-transfer of CD45RBlow CD4+ T-cells failed to induce the disease. The two subsets CD45RBlow and CD45RBhigh CD4+ T-cells were characterized as protective and pathogenic T-cells, respectively (Roncarolo and Levings, 2000). In NOD mice, CD45RBlow CD4+ T-lymphocytes can exert a protective and a facilitating role on the spontaneous development of IDDM. Their function seems to evolve from a protective to a pathogenic role as the disease progresses with a shift in the cytokine profile from TH0- and TH2-type to TH1-type responses (Shimada et al., Citation1996a). CD45RBlow CD4+ cells from young pre-diabetic NOD mice were found to be protective through the production of IL-4. CD45RBhigh CD4+ cells, on the other hand, secrete IFN-γ (Shimada et al., Citation1996b). CD45RBhigh CD4+ T-cells that are considered as a pathogenic subset in most autoimmune models, failed to induce diabetes in immunodeficient NOD-scid mice (Shimada et al., Citation1996b).
The intercellular adhesion molecule ICAM-1 (CD54) binds to lymphocyte function-associated antigen-1 (LFA-1) and macrophage-1 antigen (MAC-1). This interaction is involved in the activation of lymphocytes. The CD44 antigen is another cell adhesion receptor involved in T-lymphocyte activation (Haynes et al., Citation1989). CD44 levels increase upon activation of B lymphocytes, CD4+ T-cells, and CD8+ T-cells. Memory and activated T-cells can be recognized by their CD44high phenotype, whereas naive CD4+ T-cells express the CD44low phenotype (Dubey and Croft, 1996; Campbell et al., Citation2000). Activated cells that express CD44high and CD54 markers were also investigated in NOD mice. The prevention of IDDM was associated with a reduced ICAM-1 expression on CD4+ T-cells and a decreased number of memory CD44high CD4+ T-lymphocytes (Martin et al., Citation2001; Weiss et al., Citation2000). Additionally, there is an elevated production of IFN-γ and TNF-α by cells that infiltrate the pancreas. This correlates with the destruction of β islet cells and the progression of diabetes in NOD mice (Suk et al., Citation2001).
Under our experimental conditions, TCE at the concentration of 5 mg/ml in the drinking water did not accelerate IDDM in NOD mice. There was a statistically significant decrease in total lymphocyte, T-lymphocyte, CD4+ and CD8+ T-lymphocyte counts at the end of exposure in treated mice compared with the controls. In addition, TCE induced a down-regulation of markers of pathogenic T-cells. CD44 expression on CD4+ T-lymphocytes from the spleen, CD45RBhigh and CD54 expression on CD4+ T-lymphocytes from both the blood and spleen were significantly decreased (). Conversely, CD45RBlow expression on CD4+ T-lymphocytes from the spleen was up-regulated. There was an increase in CD44 and CD45RB expression on CD8+ T-lymphocytes that remains unexplained and there was a decrease in CD54 expression on CD8+ T-lymphocytes. At the same time, the progressive increase in TNF-α and IFN-γ production was inhibited by TCE exposure.
Our findings on activated T-cells and cytokine levels correlate with the lower incidence of histological changes in the pancreas of TCE-treated mice. Therefore, these results are in sharp disagreement with those obtained in MRL+/+ (Khan et al., Citation1995) or MRL-lpr/lpr (Kaneko et al., Citation2000) mice which develop a lupus-like autoimmune disease. TCE did not accelerate IDDM in NOD mice and seemed to have protective effect on the progression of the autoimmune process even though the onset of diabetes was not postponed.
Similar conflicting results between autoimmune models have already been obtained with mercuric chloride (HgCl2). In susceptible mice such as MRL+/+ and (NZB x NZW) F1 mice, HgCl2 induces immune activation through a TH2 immune response (Pollard et al., Citation1999). In NOD mice, on the first hand, treatment with mercury induced a strong TH2-like autoimmune response characterised by an increase in IgG1, IgG2a, IgG3, and IgE serum levels associated with high levels of autoantibodies against ssDNA and thyroglobulin, and renal immune complex deposits (Brenden et al., Citation2001). On the other hand, HgCl2 reduced the development of insulitis and delayed the onset of autoimmune diabetes in NOD mice with a down-regulation of IFN-γ. Exposures to mercury in (NZB × NZW) F1, MRL and NOD mice and comparisons of results obtained in TCE-treated NOD and MRL mice confirm that the hypothesis of the polarized TH1 and TH2 cell population is a simplistic view of autoimmunity and autoimmune responses to xenobiotics (Liew, Citation2002). The mechanisms involved in drug-induced autoimmune diseases might be more complex than previously thought with an important role of the timing and the duration of exposure and disease stage (O'Shea et al., Citation2002; Yadav and Sarvetnick, 2003).
It was demonstrated that TCE is metabolized by CYP2E1 to form protein adducts that are required for the induction of CD4+ T-cell defects in TCE-treated MRL+/+ mice (Griffin et al., Citation2000c; Halmes et al., Citation1996). The involvement of CYP2E1 in TCE metabolism was also demonstrated in the Fischer 334 rat kidney (Cummings et al., Citation2001) and in human liver (Lash et al., Citation2000). Sex, species, and strain differences in TCE metabolism may explain the variable susceptibility to TCE (Lash et al., Citation2000). Therefore, a different metabolism pathway of TCE in NOD mice might explain the conflicting results with respect to MRL+/+ mice. Unfortunately, there is no data in the literature to support or rule out this hypothesis.
A different effect of TCE on the dysregulation of apoptosis in NOD and MRL mice might also explain these conflicting results. Dysregulation of apoptosis is a common event in both tumorigenesis and autoimmune processes. Apoptosis can be altered by hypomethylation of several genes. TCA and DCA were recently shown to induce hypomethylation of the proto-oncogenes c-jun and c-myc, which participate in the control of cell proliferation and apoptosis in female B6C3F1 mice (Tao et al., Citation1998, 2000). This mechanism might explain the particular susceptibility of the MRL strain to TCE effects on apoptosis and the induction of autoimmunity. Hypomethylation of DNA was shown to be increased in the thymus and lymph nodes of MRL+/+ mice when compared with Fas-deficient MRL-lpr/lpr mice (Mizugaki et al., Citation1997). In Fas-deficient MRL-lpr/lpr mice, increased and accelerated autoimmune diseases are linked to the lpr (lymphoproliferation) gene and this accounts for the higher sensitivity of the MRL+/+ strain to altered apoptosis. Fas-deficient NOD mice failed to develop autoimmune diabetes when compared with control NOD mice (Savinov et al., Citation2003). TCE-treatment by altering apoptosis via hypomethylation of genes may significantly decrease or delay the autoimmune diabetes in NOD mice. Therefore, the difference in the susceptibility to altered apoptosis between MRL+/+ and NOD mice may be hypothesized to explain the conflicting effects of TCE.
Our results suggest that a similar interference in disease development via the Fas pathway may retard or prevent diabetes progression in NOD mice and accelerate the lupus-like syndrome in MRL+/+ mice. These results lend support to the hypothesis that individuals with several predisposing factors may have different level of risk to xenobiotic-induced or accelerated autoimmunity.
In view of the likely multiple mechanisms involved in the initiation and progression of autoimmune diseases, it is unlikely that the use of a single autoimmune-prone model could consistently generate reliable safety data for hazard identification. Further research is warranted to identify those individuals that may be at a higher risk of developing autoimmunity following exposure to xenobiotics. Comparative studies using different genetically related autoimmune-prone models, including organ-specific and systemic autoimmune diseases, transgenic and normal mice at different stages (acute vs chronic), are needed to investigate the role of xenobiotics in the precipitation of autoimmunity, particularly in sensitive populations.
REFERENCES
- Adorini L., Gregori S., Harrison L. C. Understanding autoimmune diabetes: Insights from mouse models. Trends Mol. Med. 2002; 8: 31–38
- ATSDR. Toxicological profile for trichloroethylene (Update). Public Health Service, U.S. Department of Health and Human Services. Agency for Toxic Substance Substances and Disease Registry, Atlanta, NC 1997
- Aryal B. K., Khuder S. A., Schaub E. A. Meta-analysis of systemic sclerosis and exposure to solvents. Am. J. Ind. Med. 2001; 40: 271–274
- Bovenzi M., Barbone F., Betta A., Tommasini M., Versini W. Scleroderma and occupational exposure. Scand. J. Work Environ. Health 1995; 21: 289–292
- Brenden N., Rabbani H., Abedi-Valugerdi M. Analysis of mercury-induced immune activation in nonobese diabetic (NOD) mice. Clin. Exp. Immunol. 2001; 125: 202–210
- Campbell S. B., Komata T., Kelso A. Differential effects of CD4 and CD8 engagement on the development of cytokine profiles of murine CD4+ and CD8+ T-lymphocytes. Immunology 2000; 99: 394–401
- Cooper G. S., Miller F. W., Germolec D. R. Occupational exposures and autoimmune diseases. Int. Immunopharmacol. 2002; 2: 303–313
- Cummings B. S., Parker J. C., Lash L. H. Cytochrome p450-dependent metabolism of trichloroethylene in rat kidney. Toxicol. Sci. 2001; 60: 11–19
- Dubey C., Croft M. Accessory molecule regulation of naive CD4 T-cell activation. Immunol. Res. 1996; 15: 114–125
- Garabrant D. H., Lacey J. V., Laing T. J., Gillespie B. W., Mayes M. D., Cooper B. C., Schottenfeld D. Scleroderma and solvent exposure among women. Am. J. Epidemiol. 2003; 157: 493–500
- Gilbert K. M., Griffin J. M., Pumford N. R. Trichloroethylene activates CD4+ T-cells: Potential role in an autoimmune response. Drug Metab. Rev. 1999; 31: 901–916
- Griffin J. M., Blossom S. J., Jackson S. K., Gilbert K. M., Pumford N. R. Trichloroethylene accelerates an autoimmune response by Th1 T-cell activation in MRL+/+ mice. Immunopharmacology 2000a; 46: 123–137
- Griffin J. M., Gilbert K. M., Lamps L. W., Pumford N. R. CD4+ T-cell activation and induction of autoimmune hepatitis following trichloroethylene treatment in MRL+/+ mice. Toxicol. Sci. 2000b; 57: 345–352
- Griffin J. M., Gilbert K. M., Pumford N. R. Inhibition of CYP2E1 reverses CD4+ T-cell alterations in trichloroethylene-treated MRL+/+ mice. Toxicol. Sci. 2000c; 54: 384–389
- Halmes N. C., McMillan D. C., Oatis J. E., Jr., Pumford N. R. Immunochemical detection of protein adducts in mice treated with trichloroethylene. Chem. Res. Toxicol. 1996; 9: 451–456
- Haynes B. F., Telen M. J., Hale L. P., Denning S. M. CD44—a molecule involved in leukocyte adherence and T-cell activation. Immunol. Today 1989; 10: 423–428
- Hess E V. Environmental chemicals and autoimmune disease: Cause and effect. Toxicology 2002; 181-182: 65–70
- Kaneko T., Saegusa M., Tasaka K., Sato A. Immunotoxicity of trichloroethylene: A study with MRL-lpr/lpr mice. J. Appl. Toxicol. 2000; 20: 471–475
- Kauffmann B. M., White K. L., Sanders V. M., Douglas K. A., Sain L. E., Borzelleca J. F., Munson A. E. Humoral and cell-mediated immune status in mice exposed to chloral hydrate. Environ. Health Perspect. 1982; 44: 147–151
- Khan M. F., Kaphalia B. S., Prabhakar B. S., Kanz M. F., Ansari G. A. Trichloroethene-induced autoimmune response in female MRL+/+ mice. Toxicol. Appl. Pharmacol. 1995; 134: 155–160
- Kilburn K. H., Warshaw R. H. Prevalence of symptoms of systemic lupus erythematosus (SLE) and of fluorescent antinuclear antibodies associated with chronic exposure to trichloroethylene and other chemicals in well water. Environ. Res. 1992; 57: 1–9
- Kukreja A., Maclaren N. K. Autoimmunity and diabetes. J. Clin. Endocrinol. Metab. 1999; 84: 4371–4378
- Lash L. H., Fisher J. W., Lipscomb J. C., Parker J. C. Metabolism of trichloroethylene. Environ. Health Perspect. 2000; 108: 177–200
- Liew F. Y. TH1 and TH2 cells: A historical perspective. Nat. Rev. Immunol. 2002; 2: 55–60
- Martin S., van den Engel N. K., Vinke A., Heidenthal E., Schulte B., Kolb H. Dominant role of intercellular adhesion molecule-1 in the pathogenesis of autoimmune diabetes in non-obese diabetic mice. J. Autoimmun. 2001; 17: 109–117
- Mizugaki M., Yamaguchi T., Ishiwata S., Shindo H., Hishinuma T., Nozaki S., Nose M. Alteration of DNA methylation levels in MRL lupus mice. Clin. Exp. Immunol. 1997; 110: 265–269
- Morahan G., Morel L. Genetics of autoimmune diseases in humans and animal models. Curr. Opin. Immunol. 2002; 14: 803–811
- Nietert P. J., Sutherland S. E., Silver R M., Pandey J. P., Knapp R. G., Hoel D. G., Dosemeci M. Is occupational organic solvent exposure a risk factor for scleroderma?. Arthritis Rheum. 1998; 41: 1111–1118
- O'Shea J. J., Ma A., Lipsky P. Cytokines and autoimmunity. Nat. Rev. Immunol. 2002; 2: 37–45
- Pollard K. M., Pearson D. L., Hultman P., Hildebrandt B., Kono D. H. Lupus-prone mice as models to study xenobiotic-induced acceleration of systemic autoimmunity. Environ. Health Perspect. 1999; 107: 729–735, suppl 5
- Rabinovitch A. An update on cytokines in the pathogenesis of insulin-dependent diabetes mellitus. Diabetes. Metab. Rev. 1998; 14: 129–151
- Roncarolo M. G., Levings M. K. The role of different subsets of T regulatory cells in controlling autoimmunity. Curr. Opin. Immunol. 2000; 12: 676–683
- Rosmalen J. G., van Ewijk W., Leenen P. J. T-cell education in autoimmune diabetes: Teachers and students. Trends Immunol. 2002; 23: 40–46
- Sanders V. M., Tucker A. N., White K. L., Kauffmann B. M., Hallett P., Carchman R. A., Borzelleca J. F., Munson A. E. Humoral and cell-mediated immune status in mice exposed to trichloroethylene in the drinking water. Toxicol. Appl. Pharmacol. 1982; 62: 358–368
- Savinov A. Y., Tcherepanov A., Green E. A., Flavell R. A., Chervonsky A. V. Contribution of Fas to diabetes development. Proc. Natl. Acad. Sci. USA 2003; 100: 628–632
- Shimada A., Charlton B., Rohane P., Taylor-Edwards C., Fathman C. G. Immune regulation in type 1 diabetes. J. Autoimmun. 1996a; 9: 263–269
- Shimada A., Rohane P., Fathman C. G., Charlton B. Pathogenic and protective roles of CD45RBlow CD4+ cells correlate with cytokine profiles in the spontaneously autoimmune diabetic mouse. Diabetes 1996b; 45: 71–78
- Suk K., Kim S., Kim Y. H., Kim K. A. Chang I., Yagita H., Shong M., Lee M. S. IFN-γ /TNF-α synergism as the final effector in autoimmune diabetes: A key role for STAT1/IFN regulatory factor-1 pathway in pancreatic β cell death. J. Immunol. 2001; 166: 4481–4489
- Tao L., Kramer P. M., Ge R., Pereira M. A. Effect of dichloroacetic acid and trichloroacetic acid on DNA methylation in liver and tumors of female B6C3F1 mice. Toxicol. Sci. 1998; 43: 139–144
- Tao L., Yang S., Xie M., Kramer P. M., Pereira M. A. Effect of trichloroethylene and its metabolites, dichloroacetic acid and trichloroacetic acid, on the methylation and expression of c-Jun and c-Myc protooncogenes in mouse liver: Prevention by methionine. Toxicol. Sci. 2000; 54: 399–407
- Weiss L., Slavin S., Reich S., Cohen P., Shuster S., Stern R., Okon E., Rubinstein A. M., Naor D. Induction of resistance to diabetes in non-obese diabetic mice by targeting CD44 with a specific monoclonal antibody. Proc. Natl. Acad. Sci. USA 2000; 97: 285–290
- White K. L., David D. W., Butterworth L. F., Klykken P. C. Assessment of autoimmunity-inducing potential using the Brown Norway rat challenge model. Toxicol. Lett. 2000; 112-113: 443–551
- Wright P. F., Thomas W. D., Stacey N. H. Effects of trichloroethylene on hepatic and splenic lymphocytotoxic activities in rodents. Toxicology 1991; 70: 231–242
- Yadav D., Sarvetnick N. Cytokines and autoimmunity: Redundancy defines their complex nature. Curr. Opin. Immunol. 2003; 15: 697–703