Abstract
In 1981–1982, individuals in fourteen central and northwest provinces in Spain were affected by an illness that was eventually labeled toxic oil syndrome (TOS) by the World Health Organization. Thousands of individuals were diagnosed with, and 356 people eventually died from, the disease. The disease shares striking similarities with several autoimmune diseases, particularly eosinophilia-myalgia syndrome (EMS) and diffuse fasciitis with eosinophilia (DFE). As with many other autoimmune diseases, women were more severely affected than men and made up a significant portion of TOS-related deaths. While a number of etiologic agents were investigated, disease occurrence was found to be significantly associated with consumption of contaminated rapeseed oil produced by a particular refinery. Two compounds, 1,2-di-oleyl ester (DEPAP) and oleic anilide are considered to be biologically relevant contaminants that may contribute to disease development. Toxic oil syndrome was a three-phase disease with an initial non-necrotizing vasculitis in multiple organs. Suspected immune mechanisms in TOS include activation of T-cells, altered cytokine production, and several studies have associated disease severity with HLA-DR2 and polymorphisms in metabolism and immune response genes. While a number of animal models have been used to investigate the underlying immune mechanisms in TOS, only a few studies in rodents have demonstrated the classical symptoms of TOS. Biotransformation and oxidation of the parent compound(s) to reactive intermediates prior to induction of autoreactive pathways appears to be an important component of the disease process. These reactive intermediates could haptenate self-proteins and activate autoreactive T-cells, disrupt signal transduction, or induce apoptosis and necrosis to release abnormal forms of self-antigens. Although the TOS epidemic was limited to a discrete period of time, the origin of the contamination determined, and the spread of the disease halted by government intervention, the underlying immune mechanisms have yet to be elucidated.
INTRODUCTION
In 1981–1982, individuals in Madrid province and thirteen other central and northwest provinces in Spain were affected by an illness that was eventually labeled toxic oil syndrome (TOS) by the World Health Organization. In less than two years, 20,096 people were afflicted with, and 356 people (2% of all TOS victims) eventually died from, the new disease, with the majority of the cases (14,292; 71%) in Madrid province (Philen et al., Citation1997). Women, especially those under 40 years of age, were affected more severely than men, possibly due to higher exposure in the home (Abaitua Borda et al., Citation1998) or genetic/hormonal effects; 61% of the victims and 66% of the TOS-related deaths were women (Sanchez-Porro Valades et al., Citation2003). Although TOS was originally believed to be an allergic reaction (Tabuenca, Citation1981), the disease is strikingly similar to a number of autoimmune diseases, particularly eosinophilia-myalgia syndrome (EMS), and diffuse fasciitis with eosinophilia (DFE) (Kaufman and Krupp, Citation1995). TOS pathology includes an initiating vasculitis, eosinophilia in the acute phase, and sicca syndrome (the destruction of salivary and lacrimal glands by lymphocytes and plasma cells), neuropathy, scleroderma, Raynaud's phenomenon, and musculoskeletal inflammation in the chronic phase. More than 70% of TOS patients presented with eosinophilia, regardless of age or gender, but motor/sensory neuropathy and scleroderma were more common in women under 40 than in older women or men (Sanchez-Porro Valades et al., Citation2003).
Several potential etiologies were investigated, including infectious agents such as Legionella and Mycoplasma; biological vectors carried by pets; chemically or microbiologically contaminated fruits, vegetables and other dietary elements (Tabuenca, Citation1981; Philen et al., Citation1997; Gelpi et al., Citation2002); organophosphate pesticide exposure (Hard, Citation2000); and, vinyl chloride contamination from food containers (Vicario et al., Citation1982), before a link was made between the new disease and a specific type of cooking oil. The relationship between disease symptoms and consumption of the oil, and the subsequent readmission of many patients who had been treated and discharged in good condition yet continued to use the same oil, led to an official oil recall by the Spanish Ministry of Health and Consumer Affairs two months after the onset of the epidemic (Tabuenca, Citation1981; Posada de la Paz et al., Citation1996; Philen et al., Citation1997).
Chemical analysis of the case-associated oil identified brassicasterol, a marker for rapeseed oil, trace amounts of aniline, oleyl anilide (OA), and other fatty acid anilides and contaminants (Posada de la Paz et al., Citation1996). The toxin(s) appear to be stable since consumption of toxic oil one year after the main epidemic led to development of the disease (Posada de la Paz et al., Citation1989). The development of new cases of TOS in Spain followed a pattern that suggested a point source epidemic. There was a sharp rise in diagnosis of new cases initially (12,000 people diagnosed in six weeks), with a single peak during weeks 5–7, followed by a steady decline over the next 18 weeks, reflecting a short duration of exposure to a single agent and decline after the contaminated batch had been used (Gomez de la Camara et al., Citation1997; Philen et al., Citation1997). Cases clustered in families and households, while students, military personnel, and hospital personnel and patients, were not disproportionately affected, suggesting that exposure was related to the home (Tabuenca, Citation1981; Philen et al., Citation1997). All contaminated oils were traced to a batch of rapeseed oil refined at the Industria Trianera de Hidrogenacion (ITH) refinery and sold by RAELCO. To protect a major domestic export product, the Spanish government limited imported edible oils to those denatured for industrial use. However, evidence suggests that a number of companies imported rapeseed oil denatured with 2% aniline from France, refined the oil, mixed it with other oils, and fraudulently sold the product as pure olive oil through itinerant vendors (Posada de la Paz et al., Citation1996; Philen et al., Citation1997). Anilide-contaminated oils returned in the recall by a household in which at least one family member had developed the disease were classified “case” oils for future studies.
Clinical Features of Toxic Oil Syndrome
Toxic oil syndrome was a three phase, multisystem disease (Alonso-Ruiz et al., Citation1993; Kaufman et al., Citation1995; Sanchez-Porro Valades et al., Citation2003), with an interval between ingestion of contaminated oil and presentation of symptoms of four to ten days (Tabuenca, Citation1981; Philen et al., Citation1997). Subjective estimates of consumption by victims suggest that the degree of illness varied proportionately with the amount and frequency of intake (Tabuenca, Citation1981); however, this has not been validated through objective, quantitative studies. Criteria for diagnosis of TOS were proposed by the Spanish Clinical Commission (a component of the Ministry of Health and Consumer Affairs) in August 1981 (Philen et al., Citation1997). The initiating incident is believed to be non-necrotizing endothelial damage in vessels in multiple organs, a form of vasculitis. In the acute phase (0–2 months), patients exhibited endothelial lesions, endovasculitis, eosinophilia, high IgE levels, pulmonary infiltrates and edema, pneumonopathy, myalgia, fever, and a rash (Tabuenca, Citation1981; Alonso-Ruiz et al., Citation1993; Kaufman et al., Citation1995; Gomez de la Camara et al., Citation1997; Sanchez-Porro Valades et al., Citation2003). Aniline was detected in pleural effusion in approximately 50% of the victims (Tabuenca, Citation1981). Children under 15 years old (21.8% of TOS cases) (Gomez de la Camara et al., Citation1997) presented a pruriginous, polymorphic rash more frequently than adults (Philen et al., Citation1997). The primary cause of death for patients in the acute phase was respiratory failure (Gomez de la Camara et al., Citation1997; Sanchez-Porro Valades et al., Citation2003). Approximately 60% of all TOS patients progressed to the intermediate phase (2-4 months) that was characterized by eosinophilia, pulmonary hypertension, sicca syndrome, dermal infiltration/edema, myalgia, cachexia, liver disease, and hypertriglyceridemia; the primary causes of TOS related death during the intermediate phase were thromboembolism of the great arteries and pulmonary hypertension (Gomez de la Camara et al., Citation1997; Lahoz et al., Citation1997; Sanchez-Porro Valades et al., Citation2003). Early in the chronic phase (4 months–2 years) of the disease, patients developed sicca syndrome, scleroderma, pulmonary hypertension, neuropathy, and liver disease. Late in the chronic phase (> 2 years), musculoskeletal pain, pulmonary edema and hypertension, carpal tunnel syndrome, Raynaud's phenomenon, muscle cramps, psychologic disturbances, and hyperlipidemia were common. The primary causes of TOS-related death throughout the chronic phases were respiratory failure due to neurologic infection and pulmonary hypertension (Alonso-Ruiz et al., Citation1993; Gomez de la Camara et al., Citation1997; Sanchez-Porro Valades et al., Citation2003). The symptoms that were associated with the worst prognosis were liver disease and pulmonary hypertension. In 1981 and 1982, TOS ranked as the second and third leading cause of all deaths for women and men, respectively, in the official Spanish cohort of exposed individuals, behind cardiovascular disease. Two years after the start of the epidemic, the number of deaths, from any cause, among TOS victims was below the expected figure for the Spanish population, and in 1985, at the recommendation of the World Health Organization (WHO) TOS Study Scientific Committee, the census for the official TOS cohort (maintained by the Toxic Oil Syndrome Research Centre, CISAT) was closed. Between 1982 and 1995, TOS, in the chronic phase, continued to be the third leading cause of death for both men and women in the cohort. In people under 40 years of age, TOS was the leading cause of death from 1981 to 1995 (Sanchez-Porro Valades et al., Citation2003). A 1997 assessment by the Spanish court and Spanish National Institute of Social Security recognized 3,787 TOS victims (19% of the original cohort) as having some type of permanent disability (Gelpi et al., Citation2002).
Chemistry of Toxic Oil Syndrome
The initial chemical analysis identified OA as the primary contaminant and marker for case-associated oils, but the number of anilides and unidentified contaminants suggested that other compounds might also be involved in the mechanism of TOS (Aldridge, Citation1992; Posada de la Paz et al., Citation1996). A review of 70 chemical entities associated with mineral oil and the oil refinery process stream indicated that none of the common refinery products, additives or contaminants induced symptoms and pathology consistent with those of TOS (Hard, 2000). More recently, liquid chromatography/mass spectrometry allowed identification of anilino-propanediol derivatives that may potentially be associated with the disease, in particular 3-(N-phenylamino)-1,2-propanediol (PAP), and two of its esters, 3-oleyl-ester (MEPAP) and 1,2-di-oleyl ester (DEPAP). The OA and propanediols were formed by the reaction of aniline with the oleyl side chain fatty acids abundant in rapeseed oil. Fatty acid anilides were detected in all oils tested, including denatured unrefined oil shipped to the suspected source, oil refined at this source, and non-TOS oils illicitly processed at other refineries. However, the PAP derivatives were present only in TOS-associated oils, suggesting that PAP esters were the products of the refining process and were not formed spontaneously during storage. DEPAP was the dominant form and is now considered to be the most specific marker of case-relatedness and the most likely candidate as the etiological agent (Hill et al., Citation1995; Schurz et al., Citation1997; Ruiz-Mendez et al., Citation2001). However, oleic anilide is also considered to be a biologically relevant contaminant that may contribute to disease development alone or in conjunction with PAP and PAP esters.
Attempts to reproduce the toxic oil created at the ITH refinery have shown that the most critical factors are the length of storage time prior to refining, the temperature of distillation, and the use of stripping steam during distillation (Hill et al., Citation1995; Ruiz-Mendez et al., Citation2001). Under normal refining conditions, the formation of anilides increases with increased storage time prior to refining, but distillation with stripping steam reduces the concentration. PAP esters have been found in non-TOS denatured rapeseed oil from the epidemic and aniline-denatured canola oil after heating at 300°C for 4 hours. Similarly, at a distillation temperature of 300°C, and without steam stripping, formation of stable anilides increased 3–7 fold compared to the conventional distillation temperature of 250°C. Under the latter conditions, the distillate contained a higher oleic:linoelic anilide ratio and a high concentration of saturated fatty acids typical of TOS oils, but no stable PAP esters (Schurz et al., Citation1997; Ruiz-Mendez et al., Citation2001). Ruiz-Mendez et al. (Citation2001) proposed that a malfunction in the bleaching step may have necessitated an increase in the deodorization and distillation temperature to drive off the strong flavor, allowing increased transesterification of aniline, possibly exacerbated by reduced circulation from insufficient stripping steam. Other studies have indicated that heated anilides are more toxic to rats than their non-heated counterparts (Boor et al., Citation1991; Khan et al., Citation1991a and b), and that high cooking temperatures could oxidize the anilide derivatives in the oil (Weatherhill et al., Citation2003).
Immunologic Features of TOS
A distinguishing characteristic of autoimmune diseases is that the targets of the reactive lymphocytes are self-molecules, rather than foreign agents. Tolerance, the ability to distinguish foreign organisms and molecules from self (host) and to respond appropriately to protect the host, is maintained by the complex interactions of T-cells, B-cells, and antigen presenting cells. Loss of tolerance, leading to tissue destruction and organ damage, can result from insults, including chemical exposure, that block the deletion of autoreactive lymphocytes, modify self-molecules so that they are recognized as foreign, and/or alter the expression of critical mediator molecules or receptors. It is likely that autoimmune diseases are the result of multiple mechanisms working simultaneously (Rao and Richardson, Citation1999; Anderson and Halfler, Citation2000). The major risk factors proposed are genetic susceptibility, neuroendocrine influences, and environmental triggers (Kammuller et al., Citation1988). Two examples of such diseases are autoimmune lymphoproliferative syndrome (ALPS) and systemic lupus erythematosus (SLE). In ALPS, a congenital defect in the Fas/FasL genes leads to suppression of apoptosis of autoreactive lymphocytes and development of the disease (Straus et al., Citation1999). Procainamide and silica are two compounds that can induce SLE. Exposure to procainamide can lead to alteration of DNA conformation to a more immunogenic form and, subsequently, induction of anti-DNA antibodies (Thomas, Citation1986) while silica can upregulate macrophage secretion of IL-1 and TNFα, and surface Fc receptor and MHC II molecule expression to induce autoimmunity (Koeger et al., Citation1995). Women, particularly young, post-pubescent women, have a higher risk of developing autoimmune disease than men; 75% of those afflicted with autoimmune diseases are female (Jacobson et al., Citation1997). This is similar to the demographics of TOS; 60% of TOS cases were women and TOS was most lethal to women under 40 years old (Sanchez-Porro Valades et al., Citation2003). Vasculitis, a primary finding in TOS, is a hallmark of many autoimmune diseases, and may be initiated when reactive intermediates of xenobiotics chemically modify or damage endothelial cells, ultimately leading to chronic T-cell activation and destruction of endothelium by cellular or humoral effector mechanisms (Kammuller et al., Citation1988).
Analysis of sera from a cross-section of TOS patients (n = 98) in the acute phase of the disease showed no difference in IgG, IgM, IgA, chemical-specific IgG or IgE antibodies, GM-CSF, IL-4, IL-6, or TNF levels, compared to controls. However, soluble IL-2 receptor (sIL-2R) and eosinophil granule major basic protein (MBP) levels were significantly increased in TOS patients, and although not statistically significant, IgE and soluble CD23 (sCD23) levels were consistently increased 2–3 fold, compared to controls (Brostoff et al., Citation1982; Gallardo et al., Citation1994; Lahoz et al., Citation1997). Elevated IgE was confirmed in two additional studies that evaluated, respectively, 345 pediatric patients (Tabuenca, Citation1981) and 37 patients following discharge (Brostoff et al., Citation1982). Positive immunohistochemical localization of IL-4 was observed in the skin biopsies of TOS patients (Lahoz et al., Citation1997). Post-mortem, there was no difference in IL-1α, GM-CSF, or CD25 gene expression in lung tissue from TOS patients with pulmonary hypertension and edema (n = 26), compared to controls, in either the acute or chronic phases. Consistent with serum sCD23 (see above), expression of CD23 was elevated two fold in TOS patients. Both Th1 (IL-2 and IFNγ) and Th2 (IL-4 and IL-5) cytokines were up-regulated in TOS patients compared to controls. Further, among TOS cases, the Th1:Th2 ratio was altered such that Th2 cytokines were significantly elevated compared to Th1 cytokines; this effect was not observed among controls where there was no significant difference in expression between the two cytokine classes. Collectively, these reports suggest a T-cell dependent mechanism (del Pozo et al., Citation1997; Lahoz et al., Citation1997). Aberrant cytokine production has been associated with a number of autoimmune diseases, including vasculitis (Tomer et al., Citation1999), Graves disease, diabetes (Kallmann et al., Citation1997), systemic lupus erythematosus (SLE) (Gomez et al., Citation2004), arthritis (Ruschpler and Stiehl, Citation2002), and systemic sclerosis (SSc) (Mavalia et al., Citation1997). IL-2 and IFNγ participate in delayed-type hypersensitivity responses, which could help explain the rash and allergic-type symptoms noted early in the disease progression, when the Th1 cytokine levels were highest (del Pozo et al., Citation1997; Lahoz et al., Citation1997). In addition, IFNγ up-regulates class II major histocompatibility (MHC) expression in epithelial cells and other nonlymphoid tissue, facilitating antigen presentation (Zhang and Michael, Citation1990). Cytokine profile changes have been documented in other autoimmune diseases as the disease progresses. During the acute, inflammatory phase of graft vs. host (GVH) response in the vasculature, Th1 cytokines are preferentially detected; as the disease progresses to the chronic phase with endothelial lesions, Th2 cytokines are dominant. Two distinct subsets of autoreactive T-cells, differentiated by their cell receptor gene elements via CDR3 spectratype analysis, have been identified in the lymphocytic infiltrate and are believed to be responsible for the shift in cytokine secretion (Chen et al., Citation2001). Th2 cells can induce loss of tolerance through chronic lymphocyte stimulation and subsequent production of autoantibodies (Kammuller et al., Citation1988; Rao and Richardson, Citation1999). The symptoms of the intermediate and chronic phases of TOS are especially consistent with this paradigm. IL-5, an eosinophilopoietic factor, and eosinophil granule MBP, a mediator of eosinophil toxicity, could play a role in eosinophilia and eosinophil degranulation-related organ damage. In addition, increased IL-4 and sCD23 have been associated with increased IgE antibody production by B-cells (del Pozo et al., Citation1997; Lahoz et al., Citation1997). One postulated sequence of events in the pathogenesis of TOS is that T-cell activation leads to IL-4 and IL-5 secretion, driving increased IgE production and eosinophilia, respectively; degranulation of eosinophils deposits MBP in tissues (). These events are critical to the development of vasculitis and, ultimately, organ damage (del Pozo et al., Citation1997; Gelpi et al., Citation2002).
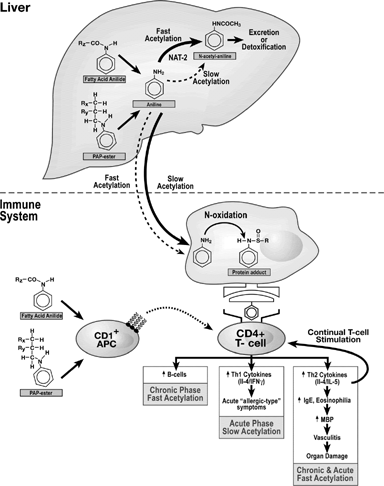
FIG. 1 Putative Sequence of Immunological Events Leading to Toxic Oil Syndrome. This figure summarizes the immune system pathways involved in developing the TOS disease state. In this model, aniline-coupled lipids are initially metabolized in the liver. The aniline moiety is either detoxified and excreted, or transported to the immune system. In immune cells, the aniline moiety may be oxidized to reactive metabolites that bind to self-proteins. These haptenated proteins may be presented as foreign to Th-cells, which secrete cytokines and initiate autoimmunity. An alternative hypothesis is that CD1+ antigen presenting cells recognize the aniline-coupled lipids directly and present them to T-cells. Solid arrows represent major pathways and dashed arrows represent alternative pathways. Adapted from Wulferink et al. (Citation2001), del Pozo et al., Citation1997, Lahoz et al. (Citation1997), Gelpi et al. (Citation2002), Bell et al. (Citation1996), and Berking et al. (Citation1998).
More than 40 autoimmune diseases have been associated with the human leukocyte antigen (HLA) type. Association with specific alleles in HLA Class I or II suggests that a disease may have an autoimmune etiology and that the immunogenic stimulus may be a specific HLA-binding peptide (Moller, Citation1998). Cardaba et al. (Citation1999) investigated the expression of HLA II genes in TOS. There was no HLA II association among survivors of the disease (n = 117), but 73% of the chronic phase victims for whom TOS was the major cause of death showed increased expression of HLA-DR2 in lung tissue. This is consistent with the work of Arnaiz-Vilena et al. (1982) who found elevated DR2 in TOS patients in the chronic phase who had experienced symptoms of atypical pneumonia during the acute phase. No other HLA antigens were found associated with TOS in these studies. Vicario et al. (Citation1982) reported an increase in HLA-DR3 and DR4 in female TOS patients, although others have questioned the results based on a lack of adequate controls (Gelpi et al., Citation2002).
Evaluation of genes for Phase I and Phase II metabolizing enzymes in TOS patients (n = 73) demonstrated an increase in defective alleles of arylamine N-acetyltransferase-2 (NAT2), a Phase II enzyme involved in biotransformation and acetylation of carcinogens and toxicants, but no association with genes encoding for cytochromes P450 1A1 (CYP1A1) and 2D6 (CYP2D6) and glutathione S-transferase M1 (GSTM1) was observed (Ladona et al., Citation2001). Polymorphisms in the acetyltransferase gene determine whether acetylation proceeds at a fast or slow rate. When chemicals are detoxified via N-acetylation, an NAT2 fast acetylator phenotype would be advantageous to protect the subject against development of neoplasia and other cell damage. By contrast, if the toxin is activated by O-acetylation, an NAT2 slow acetylator phenotype is advantageous (Hein, Citation2002). Cardaba et al. (Citation1999) concluded that TOS could be lethal in the acute phase, operating by Th1 mechanisms in individuals with the slow acetylator phenotype. In the chronic phase, TOS follows a Th2 paradigm in individuals with the fast acetylator phenotype, leading to autoimmune disease.
The haptoglobin alpha (Hp) gene is also implicated as a marker for risk factors for several inflammatory diseases and autoimmune disorders. The Hp protein binds free hemoglobin, acts as an anti-oxidant, and is involved in the acute phase immune response. The two alleles, Hp1 and Hp2, combine to produce six different expression phenotypes; the Hp2 allele is reported to have greater immune reactivity than the Hp1 allele. Proteomic analysis, including two-dimensional electrophoresis and mass spectrometry/peptide mass fingerprinting, of pooled human sera indicated differential expression of Hp isoforms in TOS patients, compared to controls. The most frequent control phenotype was Hp2-2, while cases demonstrated increased frequencies of the Hp2-1s and Hp1-1s phenotyes (Quero et al., Citation2004).
Experimental Studies of TOS
An animal model that reflects the disease endpoints associated with human TOS has yet to be developed, although a wide variety of different species (mouse, rat, guinea pig, chicken, dog, minipigs, monkeys, hamster, and rabbit) have been experimentally exposed to both case and reconstituted oils. Only a few studies have reported the classical symptoms of TOS in mice (eosinophilia and elevated IgE) and rats (lung edema, respiratory difficulties, splenomegaly). Possible explanations for the generally negative results in traditional animal models include: 1) TOS may be a uniquely human disease; 2) animals may have a lower sensitivity to toxic oils; and, 3) multiple chemicals, genetic factors, and biochemical alterations may be involved in disease development (Aldridge, Citation1992; Gelpi et al., Citation2002).
Since many autoimmune diseases require both genetic susceptibility and an environmental trigger, mice with genetic predisposition to developing autoimmune disease are commonly employed in TOS research. MHC class has been used to distinguish autoimmune disease-prone strains and establish a hierarchy in the strength of the immune response, with H-2s as the high responding mice, H-2b as the low responders and H-2a as nonresponders (Berking et al., Citation1998). MRL/lpr (H-2s) mice develop idiopathic autoimmune diseases and, due to a fatal deletion in the fas gene, are prone to a high degree of apoptosis (Hard, Citation2002). Koller et al. (Citation2001) administered case, reconstituted, and canola oils to MRL/lpr mice by oral gavage. Surprisingly, case oil suppressed the progression of glomerulonephritis. Serum IgE levels were reduced, and serum autoantibodies were increased, by all toxic oils tested compared to the naïve mice. Due to the high variance between mice, the early onset of spontaneous autoimmune disease typical of this strain, and the many positive responses observed in mice treated with the canola oil control, it was generally concluded that this model is not suitable for studying subtle immune function alterations induced by TOS (Koller et al., Citation2001, Citation2002; Weatherhill et al., Citation2003). A.SW mice, an H-2s strain that is susceptible to autoimmunity but requires environmental or infectious induction to develop the disease, failed to develop TOS symptoms. Autoantibody titers and serum levels of IgG1, IgG2, and IgE, were unaffected by treatment with case or reconstituted oils (Weatherhill et al., Citation2003).
Aniline derivatives that possess a functional group in the para position relative to the amino group on the molecule have been shown to induce allergy and autoimmune disease (Wulferink et al., Citation2001). The reactive aniline metabolite N-hydroxylaniline, the aniline-coupled lipids linoleic anilide and linolenic anilide, and a mixture of PAP esters all induced primary, T-cell dependent, lymph node cell proliferation, and an increase in the percentage of B-cells in the popliteal lymph node (PLN) assay in Balb/c mice (Wulferink et al., Citation2001). Nitrosobenzene, another reactive aniline metabolite, stimulated both primary and secondary PLN responses. Aniline, the non-protein reactive metabolites nitrobenzene, p-aminophenol, and N-acetyl-p-aminophenol, and the aniline-free lipids linoleic acid, linolenic acid, and triolein did not induce a response. Cultured bone marrow cells (BMC) metabolized aniline to produce a protein-reactive product that induced a primary PLN reaction in mice. Isolated T-cells from mice primed with nitrosobenzene recognized a BMC-produced aniline metabolite in culture and proliferated, suggesting antigen specificity (Wulferink et al., Citation2001). These authors proposed a prohapten-hapten model, in which aniline is a small molecular weight molecule that is not protein reactive. If the aniline moiety is cleaved from the aniline-lipid complex, however, the resulting metabolites undergo oxidation to reactive compounds such as nitrosobenzene. The reactive intermediates act as haptens and covalently bind the nucleophilic amino acid side chain of a protein that binds to MHC molecules. The altered self-protein is subsequently recognized as immunologically foreign and induces autoimmune disease via a T-cell response directed against altered self (). The same authors also offered a second hypothesis, that aniline-coupled lipids activate T-cells via non-classical pathways, in which CD1 molecules function as non-MHC antigen presenting cells to present the antigenic lipids to T-cells and initiate autoimmunity (Wulferink et al., Citation2001).
Capitalizing on the unique genetic traits of each H-2 class, Bell et al. developed a model with B10.S, C57BL/6 and A/J mice to examine the susceptibility factors associated with TOS (Bell, Citation1996; Bell et al., Citation1999). In both B10.S (H-2s, fast acetylator) and A/J (H-2a, slow acetylator) mice, OA was more toxic than linoleyl anlide and linoleic DEPAP. Linoleic DEPAP, a diester of PAP, induced cachexia, eosinophilia, and pulmonary congestion and disease in both strains. B10.S mice did not exhibit outward clinical symptoms of TOS in response to OA (Bell et al., Citation1992, Citation1999; Bell, Citation1996). However, serum immunoglobulin levels (IgE, IgG1, and IgM), IL-1β and IL-6 mRNA expression in spleen, the number of IgG and IgM positive splenocytes, and autoantibodies for denatured DNA, histones, and rheumatoid factor were elevated in this strain (Bell et al., Citation1992; Bell, Citation1996). Congestion and edema in the lungs, and splenomegaly were observed upon necropsy (Bell et al., Citation1992, Citation1999).In vitro OA-restimulation of splenic lymphocytes resulted in elevated secretion of IL-10 (Bell, Citation1996). Similarly, C57BL/6 (H-2b, fast acetylator) mice did not exhibit outward symptoms following treatment with OA. While serum IgE and IgM levels were elevated, mRNA expression of TNF-β and IL-1β were suppressed. Fifty to sixty percent of A/J mice died within 5 days, and another 20% died within two weeks of exposure to OA. This strain exhibited increased serum IgE, IgG, and IgM, and upregulation of IL-10, IL-1α, IL-6, and IFNγ mRNA in the spleen (Bell, Citation1996; Berking et al., Citation1998; Bell et al., Citation1999). The difference in the responses of the various strains of mice tested suggests a genetic component in susceptibility to TOS. Consistent with the symptoms presented in human TOS patients, the serologic and gene expression changes in all three strains suggest a Th2-mediated mechanism, with possible Th1 involvement in the acute phase, and a humoral immune response with polyclonal B-cell activation in the chronic phase () (Bell, Citation1996; Berking et al., Citation1998). It has been proposed that slow acetylator A/J mice model the acute phase of TOS, and process toxins through metabolic pathways that result in the rapid accumulation of reactive immunogenic metabolites (Bell et al., Citation2002). Fast acetylator mice, such as C57BL/6 and B10.S, may be able to eliminate some toxins by acetylation to stable products, the majority of which are quickly excreted. The continuous exposure to small amounts of remaining reactive metabolites can lead to the chronic phase condition (Bell et al., Citation1992, Citation2002; Bell, Citation1996; Berking et al., Citation1998).
Modulation of apoptosis and necrosis contributes to the autoimmune condition by either preventing the deletion of autoreactive lymphocytes (Rao and Richardson, Citation1999; Anderson and Halfler, Citation2000) or facilitating cell lysis and the release of abnormal forms of self-antigens (Gallardo et al., Citation1997; Lahoz et al., Citation1997). DEPAP induced both apoptosis and necrosis in human peripheral blood lymphocytes in vitro in a time- and dose-dependent fashion. Apoptosis preceded membrane damage in early cultures, while in late cultures, cytotoxicity was characterized by necrosis (Gallardo et al., Citation1997). PAP esters induced upregulation of c-fos, a key member of the AP-1 family, in rat lung fibroblasts; inhibition of phospholipase D mitigated this effect. PAP esters are structurally similar to phospholipids, and some products of phospholipid metabolism mediate nuclear factors, such as AP-1, that regulate apoptosis, cell proliferation and differentiation, and inflammation (Serrano-Mollar et al., Citation2004).
Summary
The true etiologic agent(s) of TOS and the mechanism(s) of action remain elusive, although many theories have been proffered. The prevailing models are not mutually exclusive and could, conceivably, operate simultaneously to induce autoimmunity. The higher incidence of TOS in women, the correlation with DR2 and HP in humans and H-2 in mice, and the putative association with a specific acetylator phenotype, suggest a genetic component. Cytokine profiling supports a T-cell mediated mechanism with genetic modification of soluble mediators of immunoregulation. Chronic lymphocyte stimulation could perpetuate the cycle of induction, damage and cell death. One commonality observed in TOS research is the biotransformation and oxidation of the parent compound(s) to reactive intermediates prior to induction of auto-reactive pathways. The reactive intermediates could haptenate self proteins and activate auto-reactive T-cells (Wulferink et al., Citation2001), destabilize cell membranes and disrupt signal transduction, or induce apoptosis and necrosis to release abnormal forms of self-antigens when cells lyse (Gallardo et al., Citation1997; Lahoz et al., Citation1997). Although the TOS epidemic was limited to a discrete period of time, the origin of the contamination determined, and the spread of the disease halted by government intervention in 1981, the issues surrounding the mechanism of action are still unresolved. More than 3000 people in Spain continue to suffer some type of permanent disability resulting from TOS. There is a strong clinical similarity between TOS and EMS, an autoimmune disease epidemic initiated by point source contamination of a dietary supplement (L-Trp) product. Elucidation of the etiology and toxicokinetics of TOS may contribute not only to improving the health status of victims of this, and other similar, diseases, but also to a knowledge base that could help prevent future food and drug contaminant induced epidemics.
Acknowledgements
We extend our sincere thanks to Dr. Robert Luebke and Dr. Scott Masten for their insightful critique of the manuscript.
REFERENCES
- Abaitua Borda I., Philen R. M., Posada de la Paz M., Gomez de la Camara A., Diez Ruiz-Navarro M., Gimenez Ribota O., Alvargonzalez Soldevilla J., Terracini B., Severiano Pena S., Fuentes Leal C., Kilbourne E. M. Toxic oil syndrome mortality: The first 13 years. Int. J. Epidemiol. 1998; 27: 1057–1063, [PUBMED], [INFOTRIEVE], [CSA]
- Aldridge W. N. The toxic oil syndrome (TOS, 1981): From the disease towards a toxicological understanding of its chemical aetiology and mechanism. Toxicol. Lett. 1992; 64/65: 59–70, [CROSSREF], [CROSSREF]
- Alonso-Ruiz A., Calabozo M., Perez-Ruiz F., Mancebo L. Toxic oil syndrome. A long-term follow-up of a cohort of 332 patients. Medicine (Baltimore) 1993; 72: 285–295
- Anderson D. E., Halfler D. A. Immune tolerance. Textbook of the Autoimmune Diseases, R. G. Lahita, N. Chiorazzi, W. H. Reeves. Lippincott Williams & Wilkins, Philadelphia 2000; pp. 69–79
- Arnaiz-Villena A., Vicario J. L., Serrano-Rios M., Bellas C., Mamposo F. Glomerular basement-membrane antibodies and HLA-DR2 in Spanish rapeseed oil disease. N. Engl. J. Med. 1982; 307: 1404–1405, [PUBMED], [INFOTRIEVE]
- Bell S. A. The toxic oil syndrome: An example of an exogenously induced autoimmune reaction. Mol. Biol. Rep. 1996; 23: 261–263, [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF], [CROSSREF]
- Bell S. A., Hobbs M. V., Rubin R. L. Isotype-restricted hyperimmunity in a murine model of the toxic oil syndrome. J. Immunol. 1992; 148: 3369–3376, [PUBMED], [INFOTRIEVE]
- Bell S. A., Kuntze I., Caputo A., Chatelain R. Strain-dependent in vitro and in vivo effects of oleic acid anilides on splenocytes and T-cells in a murine model of the toxic oil syndrome. Food Chem. Toxicol. 2002; 40: 19–24, [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF], [CROSSREF]
- Bell S. A., Sander C., Kuntze I., Chatelain R. The acute pathology of fatty acid anilides and linoleic diester of 3-phenylamino-1,2-propanediol in mice: Possible implication as aetiologic agents for the toxic oil syndrome. Arch Toxicol 1999; 73: 493–495, [PUBMED], [INFOTRIEVE], [CROSSREF], [CROSSREF]
- Berking C., Hobbs M. V., Chatelain R., Meurer M., Bell S. A. Strain-dependent cytokine profile and susceptibility to oleic acid anilide in a murine model of the toxic oil syndrome. Toxicol. Appl. Pharmacol. 1998; 148: 222–228, [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF], [CROSSREF]
- Boor P. J., Khan M. F., Kaphalia B. S., Jerrells T. R., Ansari G. A. Synergistic vascular toxicity and fatty acid anilides in the toxic oil syndrome. J. Am. Coll. Cardiol. 1991; 18: 1824–1828, [PUBMED], [INFOTRIEVE], [CSA]
- Brostoff J., Blanca M., Boulton P., Serrano S. Absence of specific IgE antibodies in toxic oil syndrome. The Lancet 1982; 277, [CROSSREF], [CROSSREF]
- Cardaba B., Enzendam J., Gallardo D., del Pozo V., Izquierdo M. M., Martin C., Cortegano M. I., Aceituno E., Rojo M., Arrieta I., Palomino P., Posada de la Paz M., Lahoz C. DR2 antigens are associated with severity of disease in toxic oil syndrome (TOS). Tiss. Antigens 1999; 55: 110–117, [CSA], [CROSSREF], [CROSSREF]
- Chen W., Thoburn C. J., Miura Y., Sommer M., Hruban R., Qian Z., Baldwin W., Hess A. D. Autoimmune-mediated vasculopathy. Clin. Immunol. 2001; 100: 57–70, [PUBMED], [INFOTRIEVE], [CROSSREF], [CROSSREF]
- del Pozo V., de Andres B., Gallardo S., Cardaba B., de Arruda-Chaves E., Cortegano M. I., Jurado A., Palomino P., Oliva H., Aguilera B., Posada M., Lahoz C. Cytokine mRNA expresion in lung tissue from toxic oil syndrome patients: A TH2 immunological mechanism. Toxicology 1997; 118: 61–70, [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF], [CROSSREF]
- Gallardo S., Cardaba B., del Pozo V., Belen d. A., Cortegano M. I., Jurado A., Tramon P., Palomino P., Lahoz C. Study of apoptosis in human lymphocytes by toxic substances implicated in toxic oil syndrome. Toxicology 1997; 118: 71–82, [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF], [CROSSREF]
- Gallardo S., del Pozo V., Cardaba B., de Andres B., Martin-Orozco E., Fernandez J. C., Tramon P., Posada de la Paz M., Abaitua Borda I., Palomino P., Lahoz C. Immunological basis of toxic oil syndrome (TOS). Toxicology 1994; 93: 289–299, [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF], [CROSSREF]
- Gelpi E., Posada de la Paz M., Terracini B., Abaitua I., Gomez de la Camara A., Kilbourne E. M., Lahoz C., Nemery B., Philen R. M., Soldevilla L., Tarkowski S. The Spanish toxic oil syndrome 20 years after its onset: A multidisciplinary review of scientific knowledge. Environmental Health Perspectives 2002; 110: 457–464, [PUBMED], [INFOTRIEVE], [CSA]
- Gomez D., Correa P. A., Gomez L. M., Cadena J., Molina J. F., Anaya J. M. TH1/TH2 cytokines in patients with systemic lupus erythematosus: Is tumor necrosis factor alpha protective?. Semin. Arthritis Rheum. 2004; 33: 404–413, [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF], [CROSSREF]
- Gomez de la Camara A., Abaitua Borda I., Posada de la Paz M. Toxiocologists versus toxicological disasters: Toxic oil syndrome, clinical aspects. Applied Toxicology: Approaches Through Basic Sciences, J. P. Seiler, E. Vilanova. Springer, Berlin Heidelberg 1997; pp. 31–40
- Hard G. C. Short-term adverse effects in humans of ingested mineral oils, their additives, and possible contaminants—a review. Human and Experimental Toxicology 2000; 19: 158–172, [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF], [CROSSREF]
- Hard G. C. A search for an animal model of the Spanish toxic oil syndrome. Food Chem. Toxicol. 2002; 40: 1551–1567, [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF], [CROSSREF]
- Hein D. W. Molecular genetics and function on NAT1 and NAT 2: Role in aromatic amine metabolism and carcinogenesis. Mutat. Res. 2002; 506–507: 65–77
- Hill R. H., Jr., Schurz H. H., Posada de la Paz M., Abaitua Borda I., Philen R. M., Kilbourne E. M., Head S. L., Bailey S. L., Driskell W. J., Barr J. R. Possible etiologic agents for toxic oil syndrome: Fatty acid esters of 3-(n-phenylamino)-1,2-propanediol. Arch. Environ. Contam. Toxicol. 1995; 28: 259–264, [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF], [CROSSREF]
- Jacobson D. L., Gange S. J., Rose N. R., Graham N. M. H. Epidemiology and estimated population burden of selected autoimmune diseases in the United States. Clin. Immunol. Immunopath. 1997; 84: 223–243, [CSA], [CROSSREF], [CROSSREF]
- Kallmann B. A., Huther M., Tubes M., Feldkamp J., Bertrams J., Gries F. A., Lampeter E. F., H., K. Systemic bias of cytokine production toward cell-mediated immune regulation in IDDM and toward humoral immunity in Graves' Disease. Diabetes 1997; 46: 237–243, [PUBMED], [INFOTRIEVE]
- Kammuller M. E., Bloksma N., Seinen W. Chemical-induced autoimmune reactions and Spanish toxic oil syndrome. Focus on hydantions and related compounds. Clin. Toxicol. 1988; 26: 157–174
- Kaufman L., Krupp L. Eosinophilia-myalgia syndrome, toxic-oil syndrome, and diffuse fasciitis with eosinophilia. Curr. Opin. Rheumatol. 1995; 7: 560–567, [PUBMED], [INFOTRIEVE], [CSA]
- Kaufman L. D., Izquierdo Martinez M., Serrano J. M., Gomez-Reino J. J. 12-Year follow-up study of epidemic Spanish toxic oil syndrome. J. Rheumatol. 1995; 22: 282–288, [PUBMED], [INFOTRIEVE]
- Khan M. F., Kaphalia B. S., Ansari G. A. Heated linoleic acid anilide reduces serum enzyme activities in rats. Res. Commun. Chem. Pathol. Pharmacol. 1991a; 73: 107–110, [PUBMED], [INFOTRIEVE]
- Khan M. F., Kaphalia B. S., Palafox A., Jerrells T. R., Ansari G. A. Heated linoleic acid anilide: Toxicity and relevance to toxic oil syndrome. Toxicology 1991b; 68: 143–155, [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF], [CROSSREF]
- Koeger A., Lang T., Alcaix D., Milleron B., Rozenberg S., Chaibi P., Arnaud J., Mayaud C., Camus J., Bourgeois P. Silica-associated connective tissue disease. A study of 24 cases. Medicine (Baltimore) 1995; 74: 221–237, [CSA], [CROSSREF], [CROSSREF]
- Koller L. D., Stang B. V., Hall J. A., Posada de la Paz M., Ruiz-Mendez M. V. Immunoglobulin and autoantibody responses in MRL/lpr mice treated with “toxic oils.”. Toxicology 2002; 178: 119–133, [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF], [CROSSREF]
- Koller L. D., Stang B. V., Posada de la Paz M., Ruiz-Mendez M. V. Pathology of “toxic oils” and selected metals in the MRL/lpr mouse. Toxicol. Pathol. 2001; 29: 630–638, [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF], [CROSSREF]
- Ladona M. G., Izquierdo Martinez M., Posada de la Paz M., de la Torre R., Ampurdanes C., Segura J., Sanz E. J. Pharmacogenetic profile of xenobiotic enzyme metabolism in survivors of the Spanish toxic oil syndrome. Environ. Health Perspect. 2001; 109: 369–375, [PUBMED], [INFOTRIEVE], [CSA]
- Lahoz C., del Pozo V., Gallardo S., Cardaba B., Jurado A., Cortegano M. I., del Amo A., Arrieta I., Palomino P. Immunological aspects of the toxic oil syndrome. Applied Toxicology: Approaches Through Basic Science, J. P. Seiler, E. Vilanova. Springer, Berlin Heidelberg 1997; pp. 63–73
- Mavalia C., Scalettei C., Romagnani P., Carossino A. M., Pignone A., Emmi L., Pupilli C., Pizzolo G., Maggi E., Romagnani S. Type 2 helper T-cell predominance and high CD30 expression in systemic sclerosis. Am. J. Pathol. 1997; 151: 1751–1758, [PUBMED], [INFOTRIEVE]
- Moller E. Mechanisms for induction of autoimmunity in humans. Acta Paediatr. Suppl. 1998; 424: 16–20, [PUBMED], [INFOTRIEVE], [CSA]
- Philen R. M., Posada de la Paz M., Hill R. H., Schurz H. H., Abaitua Borda I., Gomez de la Camara A., Kilbourne E. M. Epidemiology of the Toxic Oil Syndrome. Berlin Heidelberg 1997
- Posada de la Paz M., Abaitua Borda I., Kilbourne E. M., Tabuenca Oliver J., Diaz de Rojas F., Castro Garcia M. Late cases of toxic oil syndrome: Evidence that the aetiological agent persisted in oil stored for up to one year. Food Chem. Toxicol. 1989; 27: 517–521, [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF], [CROSSREF]
- Posada de la Paz M., Philen R. M., Abaitua Borda I., Sicilia Socias J. M., Gomez de la Camara A., Kilbourne E. M. Toxic oil syndrome: Traceback of the toxic oil and evidence for a point source epidemic. Food Chem. Toxicol. 1996; 34: 251–257, [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF], [CROSSREF]
- Quero C., Colome N., Prieto M. R., Carrascal M., Posada de la Paz M., Gelpi E., Abian J. Determination of protein markers in human serum: Analysis of protein expression in toxic oil syndrome studies. Proteomics 2004; 4: 303–315, [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF], [CROSSREF]
- Rao T., Richardson B. Environmentally-induced autoimmune diseases: Potential mechanisms. Environmental Health Perspectives 1999; 107: 737–742, [PUBMED], [INFOTRIEVE], [CSA]
- Ruiz-Mendez M., Posada de la Paz M., Abian J., Calaf R., Blount B., Castro-Molero N., Philen R. M., Gelpi E. Storage time and deodorization temperature influence the formation of aniline-derived compounds in denatured rapeseed oils. Food Chem. Toxicol. 2001; 39: 91–96, [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF], [CROSSREF]
- Ruschpler R., Stiehl P. Shift in TH1 (Il-2 and IFN-γ) and TH2 (Il-10 and Il-4) cytokine mRNA balance within two new histological main-types of rheumatoid arthritis (RA). Cell. Mol. Biol. 2002; 48: 285–293, [PUBMED], [INFOTRIEVE], [CSA]
- Sanchez-Porro Valades P., Posada de la Paz M., de Andres Copa P., Gimenez Ribota O., Abaitua Borda I. Toxic oil syndrome: Survival in the whole cohort between 1981 and 1995. J. Clin. Epidemiol. 2003; 56: 701–708, [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF], [CROSSREF]
- Schurz H. H., Hill R. H., Philen R. M., Posada de la Paz M., Abaitua Borda I., Kilbourne E. M., Bernert T., Needham L. L. Analytical measurements of products of aniline and triglycerides in oil samples associated with the toxic oil syndrome. Applied Toxicology: Approaches Through Basic Science, J. P. Seiler, E. Vilanova. Springer, Berlin Heidelberg 1997; pp. 53–63
- Serrano-Mollar A., Fernandez-Zabalegui L., Bulbena O., Gelpi E., Closa D. Induction of c-fos messenger RNA by 3-(N-phenylamino)-1,2-propanediol esters, compounds related to toxic oil syndrome. Chem.-Biol. Inteact. 2004; 149: 117–123, [CSA], [CROSSREF], [CROSSREF]
- Straus S., Sneller M., Lenardo M., Puck J., Strober W. An inheritied disorder of lymphocyte apoptosis: The autoimmune lymphoproliferative syndrome. Ann. Intern. Med. 1999; 130: 591–601, [PUBMED], [INFOTRIEVE], [CSA]
- Tabuenca J. M. Toxic-allergic syndrome caused by ingestion of rapeseed oil denatured with aniline. The Lancet 1981; 2: 567–568, [CROSSREF], [CROSSREF]
- Thomas T. Effects of lupus-inducing drugs on the b to z transition of synthetic DNA. Arthritis Rheum. 1986; 29: 638–645, [PUBMED], [INFOTRIEVE]
- Tomer Y., Barak V., Gilburd B., Shoenfeld Y. Cytokines in experimental autoimmune vasculitis: Evidence for a TH2 type response. Clin. Exp. Rheumatol. 1999; 17: 521–526, [PUBMED], [INFOTRIEVE], [CSA]
- Vicario J. L., Serrano-Rios M., San Andres F., Arnaiz-Villena A. HLA-DR3, DR4 increase in chronic stage of Spanish oil disease. The Lancet 1982; 276
- Weatherhill A. R., Stang B. V., O'Hara K., Koller L. D., Hall J. A. Investigating the onset of autoimmunity in As.W mice following treatment with ‘toxic oils.’. Toxicol. Lett. 2003; 136: 205–216, [CSA], [CROSSREF], [CROSSREF]
- Wulferink M., Gonzalez J., Goebel C., Gleichmann E. T-Cells ignore aniline, a prohapten, but respond to its reactive metabolites generated by phagocytes: Possible implications for the pathogenesis of toxic oil syndrome. Chem. Res. Toxicol. 2001; 14: 389–397, [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF], [CROSSREF]
- Zhang Z. Y., Michael J. G. Orally inducible immune unresponsiveness is abrogated by IFN-γ treatment. J. Immunol. 1990; 144: 4163–4165, [PUBMED], [INFOTRIEVE]