Abstract
This review discusses currently available methods for predicting B-cell epitopes on proteins. The use of animals for assessing protein immunogenicity is addressed primarily to highlight the differences in B- and T-cell epitope recognition between species. These differences have to be considered when interpreting potential B-cell epitopes identified by the methods addressed here. “In vitro alternatives” focuses on the strengths and limitations of peptide-based technologies. Three types of computer-based methods for identifying potential B-cell epitopes are discussed: (i) methods applying physico-chemical and structural propensity scales for predicting linear epitopes from the primary structure of a protein, (ii) comparative methods basing prediction upon amino acid sequence and structural similarities between antigenically known and unknown proteins, and (iii) a method combining structural features with a B-cell epitope motif database for predicting linear and conformational antigenic determinants. With respect to human safety, the usefulness of antibody-based tests is limited to comparative studies between an antigenically known protein and variants thereof. Similarly, computer-based methods using data mining can address similarities in B-cell epitope profiles between related proteins, if a proper cut off can be defined for the minimal amino acid sequence similarity required for obtaining an acceptable accuracy. Among the physico-chemical and structural scales, scales identifying in a protein hairpin and non-specific turns seem useful for predicting epitopes with a continuous primary binding site. When conformational epitopes have to be identified as well, a novel computer-based tool seems to be the most promising alternative to X-ray crystallography. However, both methods remain to be extensively evaluated and validated. Thus, promising tools for B-cell epitope identification have been developed. But, no validated method for B-cell epitope identification on antigenically unknown proteins is available yet.
Keywords :
INTRODUCTION
Therapeutic proteins of non-human and human origin have revolutionized the treatment of many diseases, and the prospect is that even more therapeutic proteins will become available for an increasingly wide range of indications in the near future.
One drawback of using proteins as drugs is the risk of triggering the immune response of the patient, typically measured by the presence of specific antibodies in circulation following drug administration. Therapeutic proteins that are phylogenetic distant from humans will trigger the highly organised and regulated innate and adaptive networks of cells, and soluble and membrane-associated molecules that have developed throughout evolution to protect man against offending agents and compounds (Stoy, Citation2001; Chaplin, Citation2003). To circumvent this problem, protein drugs of non-human origin were gradually replaced by native and recombinant human proteins, the general idea behind this strategy being that the immunologic response to these proteins, if any at all, would be down regulated by mechanisms involved in development and maintenance of tolerance to self-proteins. Two such mechanisms are known. T- and B-cells responding to self-proteins are eliminated in the thymus and bone marrow, respectively, by clonal deletion, receptor editing and functional silencing (central tolerance), while cells escaping into the periphery are controlled by other cells such as dendritic cells and regulatory CD25+CD4+ T-cells (peripheral tolerance) (Nossal, Citation1991; Sakaguchi et al., Citation2001; Miller, Citation2005).
Unfortunately, antibody production in humans against human proteins has been extensively documented for growth factors, receptors, antagonists, cytokines and hormones (Oberg et al., Citation1989; Peces et al., Citation1996; Zang et al., Citation2000; Schellekens, Citation2002b). In general, the frequency and levels of these antibodies are influenced by patient-related, treatment-related and drug-related factors (). Some of these products such as interferon-β and interleukin (IL)-2, IL-3-granulocyte macrophage colony stimulating factor (GM-CSF) fusion protein PIXY321, GM-CSF and tumour necrosis factor (TNF) receptor fusion protein were shown to induce antibodies in 88–95% of patients treated with these products (Schellekens, Citation2002b).
TABLE 1 Factors influencing the immunogenic potency of therapeutic proteins
The clinical implications of drug immunogenicity are severe, and include (1) impairment of treatment by reduction of the efficacy of the therapeutic protein, (2) autoimmunity if the exogenous drug triggers antibodies recognised by the patient's own endogenous protein, and (3) an allergic or anaphylactic response (Schellekens, Citation2002a). Thus, proper testing of therapeutic proteins for their capacity to induce immune responses must be a conditio sine qua none prior to administration.
The objective of this review is to establish a status on methods for preclinical assessment of the capacity of a therapeutic protein to trigger antibody responses. It is well known that B-cells cannot produce a proper antibody response to most antigens without the help of TH2 cells. However, identification and location of B-cell epitopes on proteins makes it possible to address the pathophysioloic and immunologic mechanisms involved in a number of diseases, such as auto-immune diseases and allergic reactions. Furthermore, T-cell tolerance is the most important mechanism for maintaining B-cell tolerance, but it may not be the only mechanism. There is evidence suggesting that B-cells can be subject to positive selection generated and maintained on the basis of their auto-reactivity. This would make these cells essential in promoting systemic auto-immunity (Chan et al., Citation1999; Hayakawa et al., Citation1999; Zouali, Citation2001). In addition, antigen dose and antigen patterns seem to play an important role in regulating B-cell responsiveness to self-antigens. Bachmann and Zinkernagel (Citation1996, Citation1997) demonstrated that a sufficient dose of self-antigen exhibited in a repetitive form (e.g., protein complexes) elicits a B-cell response in a T-cell-independent fashion, while monomeric self-antigens do not induce antibody production because the necessary T-cell help is not available. Recently, Hermeling et al. (Citation2006) reported data on interferon-α 2a suggesting that protein aggregates are able to break tolerance, especially if the conformation of the aggregated molecules is native-like. Whether or not this process also is T-cell independent is not clear yet.
TESTING ANTIBODY PRODUCTION USING ANIMAL MODELS
In general, assessing antibody responses to a protein using animal models will tell whether or not, and to what extent, the study protein will produce antibody, but it will not provide any detailed insight in the B-cell epitopes existing on the study protein. However, antibodies that are produced by these animals are often used for in vitro identification of potential B-cell epitopes, while information of antibody-binding sequences is incorporated in a number of computer-based mapping tools. Animal models have also been used to evaluate protein-engineered variants of a protein of interest in an effort to define the amino acids involved in antibody binding.
This paper intends to highlight the issues to be considered before adapting the obtained data to measurements for assuring human safety.
Animal Testing: Evaluation of Drug Immunogenicity Allows for Development of Drug-Specific Immunochemical Assays
It is common practice to assess the immunogenicity of therapeutic proteins in a variety of animal species, a lack of antibody response being an indication for low probability and a strong antibody response for a high probability of immunogenicity.
Considering the fact that the mammalian immune system has evolved to react promptly to foreign compounds, there is no reason to expect but an immune response to a therapeutic protein that is related to the phylogenetic distance between the receiving mammal and the protein drug. With respect to therapeutic proteins of human origin, animal testing for immunogenicity should be considered irrelevant, as animals will react with a vigorous immune response to this neo-protein in contrast to humans. In an effort to come around this issue, protocols have been developed for testing the immunogenicity of human therapeutic proteins in transgenic mice with immune tolerance to the native human protein that they express (Stewart et al., Citation1989; Ottesen et al., Citation1994; Palleroni et al., Citation1997; Hermeling et al., Citation2006).
The antibodies produced by these animal models allow for development of drug-specific immunoassays that could be useful in B-cell epitope mapping studies. However, the information on potential B-cell epitopes obtained by methods using these antibodies (e.g., peptide-based immunoscreening), or the information provided by them (e.g., computer-based data mining), has to be considered with care. Indeed, the antigenic information provided by antibodies of non-human origin may or may not be predictive for adverse effects in humans because species (including transgenic animal species), as well as different strains of the same species, may differ in antigen presentation, and T- and B-cell epitope recognition (Milich and Leroux-Roels, Citation2003). Thus, B- and T-cell epitopes that are recognized in mice, for example, may not be recognised in humans and vice versa.
While animal testing may be of limiting value for assessing the absolute immunogenic potential of a therapeutic protein, some animal models have been proven useful in predicting the relative immunogenicity of, for example, different forms of human proteins (Zwickl et al., Citation1991; Wierda et al., Citation2001).
Identification of B-Cell Epitopes by a Combination of Protein Engineering and Animal Experimentation: a Comparative Tool
Animal models have also been used to compare the immunogenicity of a study protein and variants thereof (Scandella et al., Citation1988; Keil and Wagner, Citation1989; Stewart et al., Citation1989; Alexander et al., Citation1992; Laroche et al., Citation2000; Meyer et al., Citation2000; Renukaradhya et al., Citation2002).
In these studies, amino acids critical for antibody-binding were identified in the context of the folded protein by preparing a large number of well-characterized variants of the parental protein by site-directed mutagenesis, and by testing them for immunological activity in a variety of antibody-binding assays and confirmed by animal experimentation.
For the sake of feasibility amino acids to be subjected to protein engineering were often selected after consultation of the data obtained by in vitro B-cell epitope identification, and by propensity scales addressing the physico-chemical characteristics of amino acids (e.g., accessibility, hydrophilicity, flexibility) and/or structural features (e.g., hairpin loops, non-specific turns). These methods are discussed later in this review.
Structural and functional integrity are key issues for this approach. There is ample evidence showing that modifications of the protein structure and function e.g., by glutaraldehyde, DMSO, urea or salt treatment, formulation or protein engineering may induce structural changes which have a significant but indirect (i.e., unrelated to changes in the amino acid sequence of B-cell epitopes) impact on antibody-binding and immunogenicity (Janssen et al., Citation1996; Andersson et al., Citation2001; Hermeling et al., Citation2004; Wu et al., Citation2004). The availability of well-established assays addressing these issues is a prerequisite for this approach to be successful.
The outcome of these B-cell epitope mapping studies is two-fold. First, amino acid positions within the study protein that are involved in antibody-binding and production are identified. When visualised on the three-dimensional structure of the parental protein, these amino acid positions often describe coherent patches of amino acids on the surface of the three-dimensional structure of the protein, which are related to antigenic determinants (). Second, the impact of specific amino acid exchanges is determined. As shown in , this impact can be positive, negative or neutral. This information can be used as guidance for subsequent epitope engineering aiming at improving the antigenic or immunogenic characteristics of the therapeutic.
FIG. 1 Epitope mapping by peptide scanning. State of the art protein engineering was performed on Bacillus subtilis serine protease. The targeted amino acids were selected on the basis of their accessibility (i.e., being surface exposed) and their unlikelihood to affect enzyme stability. Each amino acid position was replaced by any of the 19 remaining amino acids. This protease library was screened at two levels before further evaluation in rats. First, a functional assay allowed for identification of enzyme variants that were structurally and functionally comparable with the parent protein. Second, the impact of the mutations on the antigenicity (i.e., antibody-binding capacity) of the protein variants was assessed by ELISA. Rats were immunised subcutaneously on a weekly base for 15 weeks, and protease-specific serum IgG and IgE levels were detected by ELISA. Amino acid positions for which mutagenesis resulted in modification of the antigenic and immunogenic properties were localised on the X-ray crystallographic structure of the protease. Thus, two potential antigenic determinants were revealed.
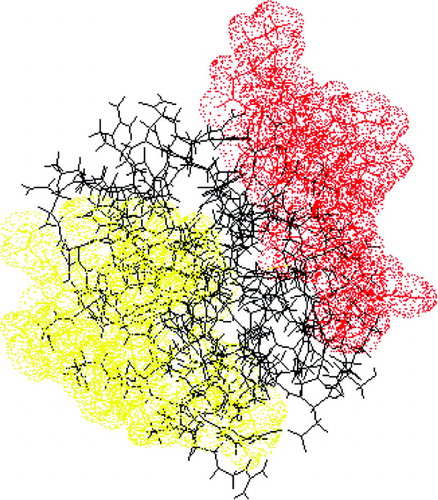
TABLE 2 The impact of specific amino acid substitutions on the antigenicity of Bacillus subtilis serine protease
In most cases the immuno-assays for assessing antigenicity are using antibodies of non-human origin. Thus, the obtained information on potential B-cell epitopes has to be considered with care when to be used for measurements to assure human safety. Occasionally (e.g., after clinical studies) though, human polyclonal sera are available for development of a human antibody binding assay. In these assays, reduced binding of the human antibodies to variants of the parental protein will provide relevant information of the existing human epitope areas on the protein. However, the occurrence of new epitope areas (i.e., introduced by protein engineering) with direct relevance for humans can only be assessed by new clinical studies.
B-CELL EPITOPE PREDICTION USING IN VITRO TECHNIQUES
The most direct way of identifying B-cell epitopes on a folded protein still is by X-ray crystallography of, for example, protein-monoclonal antibody complexes (Amit et al., Citation1986; Bentley, Citation1996; Mirza et al., Citation2000; Li et al., Citation2003). This approach is, however, very lengthy and resource demanding as it requires well-characterized monoclonal antibodies as well as the availability of pure protein-monoclonal antibody complexes.
A variety of relatively simple assays (e.g., direct ELISA, sandwich ELISA, competitive ELISA) for assessing the antigenicity (i.e., antibody-binding capacity) of a protein are available, but only a competitive test format (e.g., two monoclonal antibodies competing for the same protein, or two proteins competing for the same antibody) can give information about potential B-cell epitopes. However, this information is very general (i.e., not revealing specific antigenic amino acids), and mainly comparative.
In contrast to the ELISA-formats, immunoscreening of oligopeptides with antibody specific for the study protein may provide more detailed insight in the antibody-binding amino acid sequences of that protein. Methods using monoclonal and polyclonal antibodies have been used most frequently and will be discussed next. Not discussed is an exciting, but yet to be more substantiated, method using large repertoires of human Fab immunoglobulin (Ig) fragments to map B-cell epitopes on proteins. These Ig fragments were produced by molecular cloning and expression of Ig from activated human B-cells. Successful results have been reported for thyroid peroxidase, Bet v 1, and Humicola lanuginosa lipase (Nishikawa et al., Citation1996; Jakobsen et al., Citation2004).
B-Cell Epitope Mapping by Peptide Scanning: A Linear Solution to a Three-Dimensional Problem
A frequently used experimental method for investigating B-cell epitopes assesses specific binding of antibodies to sequential overlapping peptides encompassing the entire sequence of the study protein (). Eventually, alanine substitution of every single native amino acid of the antibody-binding peptides may help to identify amino acids in the reactive peptides that are critical for antibody binding (Elsayed et al., Citation1989; Burks et al., Citation1997; Stanley et al., Citation1997; Williams et al., Citation1998; Beezhold et al., Citation2001; Sorokin et al., Citation2002).
FIG. 2 Epitope mapping by peptide scanning. Based upon the amino acid structure of the study protein (primary structure) peptides of 10 to 15 amino acids of length covering the entire primary structure of the protein, are synthesised. These peptides will overlap typically with five amino acids. These peptides are immobilised e.g., on some sort of membrane (e.g., nitrocellulose) prior to incubation with specific serum, IgG or IgE. A potential test format could be any type of standard immunochemical assay, e.g., ELISA, dot-blot or line immunoassay. Following development of the membrane, specific peptides may be visualised by various degrees of staining.
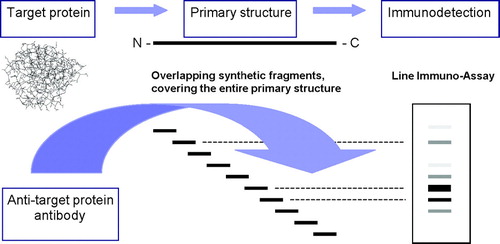
It is generally appreciated that epitopes identified by this method reveal some inherent limitations. Attempts to mimic protein epitopes by means of short synthetic peptides derived from the primary structure of the study protein are bound to select for linear epitopes. Conformational epitopes will be predicted only if they have a continuous interaction site that is long enough to allow for antibody binding, and which has the highest overall affinity for the antibody. Since protein surface analysis seem to suggest that most antigenic determinants are discontinuous (Barlow et al., Citation1986; Van Regenmortel and Pellequer, Citation1994; Bentley, Citation1996), peptide scanning may not provide the expected information on the overall antigenicity of the study protein.
B-Cell Epitope Mapping by Immuno-Competitive Screening of Phage Libraries Expressing Random Oligo-Peptides: A First Step Toward a Three-Dimensional Solution
Biopanning of phage-displayed random peptide libraries is a powerful technique for defining peptide structures that mimic natural epitopes. In contrast to other methods, epitope mapping by phage display screening involves specific monoclonal or polyclonal antibodies, but not necessarily the antigen (). The overall advantage of this technology is believed to be that it allows for identification of linear and conformational epitopes. In other words, antibodies raised against conformational epitopes will recognize their epitope also when it is presented as a continuous amino acid sequence.
FIG. 3 Epitope mapping by immuno-screening of phage libraries expressing random oligopeptides. A library of random oligo-peptides displayed on the surface of a phage as part of selected membrane proteins, are incubated with antibody specific to the study protein. Prior to this incubation, these antibodies were immobilised to a solid support, typically magnetic beads allowing for fast recovery of the beads. Phage expressing a peptide in a three-dimensional context that can be recognised by the immobilised antibodies with high enough an affinity will be bound and can be concentrated together with the magnetic beads. These phages are eluted by modifying the pH or ionic strength of the buffer, or by adding the study protein to the buffer used for binding (competitive immunoscreening). Subsequently, the recovered phages are subjected to at least two more rounds of selection. Finally, the DNA inserts expressed by the phages as part of a membrane protein are analysed to determine the amino acid sequence of the selected peptides. Eventually, the resulting peptide sequences can be assessed on the primary or three dimensional structure of the protein directly or after identification of specific motifs using sequence alignment.
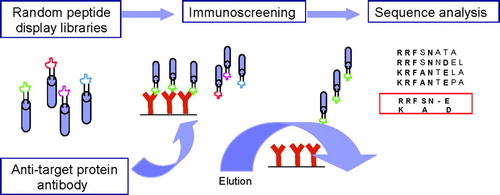
An important problem inherent to the phage display technology is background. Thus, it has to be considered that a considerable fraction of the peptides derived by using polyclonal antisera, but also monoclonal antibodies, may represent irrelevant amino acid sequences. Davies and co-workers (Citation1999) addressed this problem apparently successfully by applying a multiple sequence alignment algorithm (PILEUP) together with a matrix for scoring amino acid substitutions based upon physico-chemical properties to generate guide trees depicting relatedness of selected peptides. An elegant alternative approach was proposed by Mittag et al. (Citation2006). By competitive immunoscreening of phage-displayed random oligopeptide libraries, using the study protein as the elutriator in stead of salt or pH, the level of irrelevant phages was reduced from 90% to 10% of the total number of phages obtained by biopanning. In addition to the problems typically associated with the use of antibodies, it is generally appreciated that the information provided by this method is biased towards those regions of an epitope with the highest overall affinity for the antibody. Therefore, the identified amino acid sequence may not reflect the entire epitope (Van Regenmortel, Citation1998). The results of epitope mapping may also be biased by the method used to present the antigen to the antibody (Van Kampen et al., Citation2001). Identification of antibody-binding sequences by immunoscreening of phage-displayed oligopeptides is also complicated by biases imposed primarily by the viral morphogenesis process (Rodi et al., Citation2002). Finally, localization of the sequences in the three-dimensional structure of the target protein may not always be straightforward and is often subjective.
In spite of these biases, both linear and conformational epitopes have been located by this method in the three-dimensional structure of a number of proteins (Parhami-Seren et al., Citation1997; Rudolf et al., Citation1998; Williams et al., Citation1998; Davies et al., Citation1999; Ganglberger et al., Citation2000; Hantusch et al., Citation2004). Recently, Mittag et al. (Citation2006) implemented this technique for prediction of cross-reactivity and potential allergenicity of novel foods. Selection of phage-displayed peptide mimics by competitive immunoscreening with serum IgE from allergic patients in combination with computer-based mapping of the peptide mimics onto the surface of the three-dimensional allergen structure showed useful to investigate IgE epitope specificity in individual patients. Combined with information about conservation of epitopes on homologous proteins it may also be useful to predict potential cross-reactivity patterns and their clinical relevance. This tool may also support the development of patient-tailored reagents for immunotherapy and diagnostic tools (Shreffler et al., Citation2004; Vrtala et al., Citation2004).
BIO-INFORMATICS
Using one or a combination of the approaches described have, antigenic structures have been identified for a number of proteins. These data were used to develop a number of computer-based tools aiming at assessing the antigenicity of proteins whose epitopes had not been analyzed before.
These currently available methods can be divided into three groups: (i) methods applying physico-chemical and structural propensity scales for predicting linear epitopes from the primary structure of a protein, (ii) comparative methods basing prediction upon amino acid sequence and structural similarities between antigenically known and unknown proteins, and (iii) a method combining structural features with a B-cell epitope motif database for predicting linear and conformational antigenic determinants.
The common overall expectation for each of these methods is that they can predict antigenicity of any relevant protein.
Physico-Chemical and Structural Scales for Predicting Amino Acids Potentially Involved in Antibody-Binding from the Primary Structure of the Protein: Tossing the Coin
A number of tools have been developed for protein identification and analysis (http://www.expasy.org/tools/protscale-ref.html, Gasteiger et al. Citation2005), of which some have been assessed to predict B-cell epitopes from the primary amino acid sequence of the study protein. The low prediction efficacies revealed by analysis of the prediction algorithms strongly suggest though that no single propensity scale, with the exception of specific scales for addressing turns in proteins, is correlated with antigenicity ().
TABLE 3 Comparison of different prediction scales applied to proteins with known epitopes
There is considerable evidence that continuous antigenic sites are often located in or nearby turns of proteins (Mendz and Moore, Citation1985; Williamson et al., Citation1986; Laczko et al., Citation1992). Thus, three turn scales were developed using a three-dimensional database of high-resolution proteins extracted from the Brookhaven Protein Data Bank, corresponding to (1) helical turns, (2) hairpin turns and (3) non-specific turns, respectively. Sequently, the different scales were assessed on the studied protein by the PREDITOP package (Pellequer and Westhof, Citation1993; Pellequer et al., Citation1993). Interestingly, algorithms for predicting amino acid stretches defining in a protein structure hairpin turns and non-specific turns, but not helical turns, were shown to have a 70% prediction efficacy for antigenicity. This high reliability may be due to the fact that only a limited number of epitopes are predicted (low sensitivity), but with a high predictivity. The results also seem to indicate that these turns incorporate structural information which cannot be represented by a single physico-chemical parameter (Pellequer et al., Citation1994).
Jameson and Wolf (Citation1988) were the first to combine different physico-chemical parameters each representing a different aspect of protein architecture, i.e., surface accessibility, regional backbone flexibility and predicted secondary structure. This method was modified by Alix (Citation2000) who incorporated a new hydrophilic scale (Parker et al., Citation1986) and assigned a weight to each class of biophysical parameter. Subsequently, an “Antigenic Index” was computed from the individually profiles in the Predictive Estimation of Protein Linear Epitopes (PEOPLE) program. Recently, Odorico and Pelleques (Citation2003) developed the BEPITOPE program that adapts the propensity scales such that positive values in the hydrophobicity scale correspond to hydrophilic regions, and those in the flexibility scale with flexible regions, etc. Both programs indicated putative epitopes by concrete peaks, with each putative epitope containing a sequence of 10 (PEOPLE) or 15 (BEPITOPE) amino acid residues centred around each peak.
For each of these methods, these peaks were claimed to describe surface exposed regions which coincide with actual antigenic sites on representative proteins. While these specific programs are still waiting for being evaluated in terms of sensitivity and positive predictive value, an extensive evaluation was published by Saha and Raghava (Citation2004). They evaluated their method 〈http://www.imtech.res.in/raghava/bcepred〉, which combines flexibility, hydrophilicity, polarity and surface, on a data set consisting of 1029 non-redundant B-cell epitopes obtained from the Bcipep database. Based on their analysis, combining the four properties resulted in a disappointing maximum accuracy of 59% which was marginally, if at all, better than the accuracy of any single property (53-58%).
It is generally believed that algorithms based upon physico-chemical characteristics and structural features will address predominantly linear epitopes. However, conformational epitopes are occasionally identified. This is explained by the fact that the primary (contiguous) interaction sites described above may fall in a loop or protruding region of the protein. However, none of these methods can predict conformational epitopes lacking such a primary site, nor can they predict hidden antigenic amino acids brought into contact with paratope residues by specific antigen–antibody interactions (Alexander et al., Citation1992; Nair et al., Citation2002).
B-Cell Epitope Mapping by Sequence Similarities Using Computer-Aided Data Mining Systems: A Question of Defining Similarity
Alternative methods have based the prediction of B- and T-cell epitopes on similarities between primary amino acid sequences and three-dimensional structures of proteins with known and unknown antigenicity. In general, the strength of these systems is the coupling of secondary structure predictions with multiple alignments, performed on a sequence subset extracted following a similarity search, using tools such as BLAST, PSI-BLAST, SSEARCH, FASTA or PattInProt (e.g., http://pbil.ibcp.fr/NPSA; Combet et al., Citation2000; http://fermi.utmb.edu/SDAP/index.html; Ivanciuc et al., Citation2002). The basic principle of these approaches is that if two proteins share sufficient linear sequence similarity, they will also share three-dimensional structure, and therefore functional, homology.
The question of what represents a significant sequence similarity remains still unanswered. Related proteins may share only 25% amino acid identity, but cross-reactivity caused by proteins sharing epitopes is believed to be rare at less than 50% sequence similarity (Pearson, Citation2000; Aalberse et al., Citation2001). Aalberse (Citation2000) has suggested that at least 70% amino acid identity is required across the full length of the protein sequence. However, cross-reactivity between crustacean, mollusc and insect allergens has been confirmed by clinical tests using sera from patients sensitive to cockroach allergen. The overall sequence identity between these tropomyosins is 50–60%. (Santos et al., Citation1999; Leung and Chu, Citation2001). Furthermore, IgE cross-reactivity between the major birch pollen allergen Bet v 1, and Bet v 1 homologues of fruits (e.g., Pru av 1), nuts and vegetables (Gly m 4, Ara h 8) is well documented (Vieths et al., Citation2002; Mittag et al., Citation2006). Nevertheless, fruit and nut homologous of Bet v 1 reveal only 57–67% sequence identity with Bet v 1, while Bet v 1 homologous in vegetables are as low as 38-48% (Vieths et al., Citation2002).
Hileman and co-workers (Citation2002) compared the protein sequences of six insecticidal proteins, three common low allergenic food proteins and 50 corn proteins to allergens using the FAST algorithm and by searching for matches of contiguous identical stretches of 6, 7 and 8 amino acids of length. Using stretches of 8 amino acids, these authors found only seven corn proteins matching with an allergen. Six out of seven shared at least 50% identity to the matching allergen. Thus, the authors proposed to add an additional search for matches of 8 amino acids to the FASTA search, to add a margin of safety when assessing the potential allergenicity of a protein. They recognised however that additional work is required to evaluate specific threshold criteria (e.g., % of sequence similarity) to improve bioinformatics as a tool for prediction of allergenicity.
B-Cell Epitope Mapping Using a Computer-Based Method Using an Epitope Motif Database: Combining Physico-Chemical and Structural Features with Antigenic Signatures
With the exception of algorithms predicting hairpin loops and non-specific turns, propensity scales addressing physico-chemical characteristics and structural features of amino acid sequences have not been shown to be very useful as predictive tools for antigenicity. However, the association between antigenic amino acid stretches, and hydrophilic (i.e., surface exposed) amino acid stretches (Hopp and Woods, Citation1981; O'Lorcain et al., Citation1996; Alix, Citation2000); hairpin loops and non-specific loops (Pellequer et al., Citation1994) should not be ignored. Thus, collecting this sort of information should be considered as a first step towards the understanding of antigenicity.
Most antigenic determinants are believed to be discontinuous and thus defined by the protein structure (Barlow et al., Citation1986; Bentley, Citation1996). The relation between antigenicity and structure was further supported by comparative analysis of the three-dimensional structures of diverse allergens performed to understand the molecular basis for cross-reactivity (Aalberse et al., Citation2001; Furmonaviciene and Shakib, Citation2001; Vieths et al., Citation2002). Thus, the second step towards understanding antigenicity should involve structural information, e.g., information on the localisation of accessible amino acid patches and loops on the three-dimensional structure of the study protein.
Batori et al. (Citation2006) were the first to provide evidence suggesting that antigenicity of a protein is also defined by specific amino acid motifs and combinations of such motifs. These amino acid motifs (N = 1013) emerged from sequence alignment of well-characterised human, rat, mouse and rabbit IgG and IgE antibody-binding sequences of as yet 105 proteins, obtained from the literature and by competitive immunoscreening of phage libraries expressing random oligopeptides. Interestingly, the resulting motifs showed to be nor protein nor species specific. Therefore, the hypothesis was raised that these motifs could be used to identify potential B-cell epitopes on a protein that had not provided antibody-binding sequences to the established epitope motif database.
This hypothesis was assessed on chicken lysozyme Gal d 4, for which linear and conformational epitopes have been characterised (Amit et al., Citation1986; Bentley, Citation1996). To allow for objective identification, the epitope motif database was first incorporated in a computer-based epitope mapping tool (EMT) assessing the epitope motifs on a three-dimensional or linear structure of the protein of interest. The algorithm used by the EMT allowed for customer-defined thresholds for flexibility (i.e., distance between subsequent amino acids) and accessibility (i.e., relative surface accessibility [RSA]) of the amino acids to be included in the mapping.
The first evidence ever suggesting the existence of general antibody-binding motifs was provided by the high sensitivity and the high positive predictive value of the EMT, as well as the relevance of the structural associations suggested by the EMT on Gal d 4 (). Differential mapping revealed that both parameters were dependent on the minimum RSA of the amino acids included in the mapping. These results seem to be in line with previous observations (Hopp and Woods, Citation1981; O'Lorcain et al., Citation1996; Alix, 1999) indicating that the most exposed amino acids are most likely to be involved in antigenic determinants. However, the low sensitivity of the computer-based mapping when applied on these very exposed amino acids, and of hydrophilicity/hydrophobicity analysis using established prediction scales (Manavalan and Ponnuswamy, Citation1978; Janin, Citation1979; Hopp and Woods, Citation1981; Kyte and Doolittle, Citation1982; Rose et al., Citation1985), show that amino acids with little or no surface exposure can constitute a substantial fraction of an epitope. Presently, efforts are undertaken to define how much an amino acid can be buried into a three-dimensional protein structure and still be part of an antigenic site. Epitope mapping performed on antigenically known industrial enzymes (N = 8), environmental allergens (N = 4), food allergens (N = 7) and therapeutic proteins (N = 2) seems to suggest that a 40% minimum RSA is an acceptable lower cutoff, giving balanced sensitivity (80%) and positive predictive value (75–100%) (Data not shown). As yet, only Ara h 2 epitope 4 (Stanley et al., Citation1997) and the Bet v 1 epitope reported by Mirza et al. (Citation2000) were shown to require amino acids with at least 10 and 30% minimum RSA, respectively.
FIG. 4 B-cell epitope mapping of Gal d 4: the relation between sensitivity, predictive value and accessibility. The three dimensional structure of Gald d 4 (1hel.pdb) was subjected to epitope mapping using the EMT. Differential epitope mapping was performed by decreasing the minimum relative surface accessibility (RSA) from 70 to 10% and the sensitivity and positive predictive value was determined using known epitopes as gold standard (Amit et al., Citation1986; Bentley, Citation1996). Dependency of the sensitivity (♦) and positive predictive value (▪) on the minimum RSA of the amino acids included in the mapping. (C–E) The sensitivity of the EMT for each of the known epitopes as a function of the minimum RSA of the amino acids included in the mapping.
Thus, the EMT seems to accommodate the fact that epitopes are three-dimensional entities with various degrees of accessibility. This observation opens up for the exiting possibility that the EMT might have the potential of becoming a fast alternative to B-cell epitope mapping by X-ray crystallography on protein–antibody complexes. This approach is too lengthy and too expensive for a number of applications such as modification of the overall antigenicity of pharmaceutical proteins, and the development of vaccines and industrial enzymes. In general, optimizing the antigenic and immunogenic characteristics of such proteins requires knowledge of the key amino acid positions, their localization and their overall structural association, rather than the detailed structure of epitopes, for guiding epitope engineering. This information can be obtained rapidly by the EMT.
FIG. 5 Example of EMT guided epitope engineering. Step 1: The three-dimensional structure of the target protein was subjected to the EMT including amino acids with a minimum RSA of 50% (top) and 30% (bottom), respectively. Thus, amino acids potentially involved in antibody-binding are identified, as well as the probability by which they do (the epitope fingerprint). This information is put on the structure of the study protein, and will typically identify patches of amino acids (epitope areas). Step 2: The epitope information in combination with information provided by stability and performance (functionality) studies, an qualified selection of amino acid positions to be modified without loosing functionality, can be made. Using the EMT, the impact on the epitope finger print of each potential exchange in each of the selected positions can be evaluated before actually performing epitope engineering. Step 3: Protein variants are purified from the culture broth using a micro-purification procedure, and are screened in an immunochemical assay format (typically ELISA) to identify those variants with modified antigenicity. The figure shows the variation of the test when performed 96 micro-purified on the parental proteins (wt), on two epitope libraries covering the red epitopes and green epitope, respectively, in Step 1, as well as on a library obtained by random mutagenesis of surface exposed amino acids. Only the epitope engineered libraries showed a significantly different antigenicity profile as compared to the wt and randomly mutagenised library. Step 4: The antigenicity data are combined with data from stability and performance/functionality tests, and variants of interest are selected for in vivo evaluation of their immunogenicity.
The predictive efficacy of the EMT and existing prediction scales was compared after normalisation of the various scales in terms of sensitivity (). In agreement with previous data (), prediction scales based upon physicochemical characteristics defining the accessibility of amino acids in a three-dimensional structure proved to be poor predictors of the antigenic determinants on Gal d 4 for all normalization modes. Prediction scales based upon structural features, e.g., β -turns and flexibility, scored better as compared with the physico-chemical scales. The difference between the EMT and the structural prediction scales increased with decreasing sensitivity and increasing positive predictive value.
TABLE 4 Comparison of the EMT with existing prediction scales using Gal d 4 as a model
In order to make the EMT the tool of choice for prediction of potential antigenic sites on proteins of unknown antigenicity, a number of important issues remain to be addressed in detail. Decreasing the minimum RSA was shown to result in a decrease in predictive value of the EMT data. To what extent the observed drop is related to the identification of new potential epitopes or cryptic epitopes, or to increase in irrelevant noise is not fully clear yet. This issue is currently addressed by competitive immunoscreening of phage display libraries expressing random oligopeptides. The efficacy of EMT obviously relies on the quality of the 3D structure of the protein of interest. In particular, atomic surface accessibilities may vary significantly in different protein crystal forms. This issue is currently addressed using different X-ray crystallographic structures of Bet v 1. Preliminary data seem to indicate that the accessibility window is affected by the quality of the 3D structure used by the EMT.
Being a computer-based tool, the EMT can be used also for quick evaluation of engineering strategies (). Indeed, the amino acid in a target position in the 3D structure of a protein of interest can theoretically be exchanged with any of the remaining 19 amino acids. However, a number of the possible exchanges may introduce new or duplicate existing epitopes and/or may affect protein stability. To avoid these unwanted outcomes, the modified 3D structures can be subjected to the EMT to assess the impact of the mutation on the number of scored motifs, and to computer-based structural analysis to evaluate the effect of the mutation on stability and functionality. Thus, exchanges not affecting the score or increasing the number of motifs, and/or suspected to affect protein stability and functionality can be eliminated before the experimental work is initiated, if, for example, reducing the immunogenicity of a therapeutic protein is the objective.
CONCLUSION
Tools have been developed to identify B-cell epitopes on a study protein, but each of these methods seems to have strengths and limitations that have to be considered before translating data into measures assuring human safety ().
TABLE 5 Overview of the currently available methods for B-cell epitope identification
The in vitro alternative methods require the availability of specific serum and are therefore limited to assessment of cross-reactivity. No information is provided with regard to new epitopes. Another group of methods address the issue by predicting physico-chemical and structural features from the primary structure of the study protein. Obviously, these methods are limited to continuous epitopes and conformational epitopes with a primary binding core that has high enough an affinity to bind to specific antibody. Third, antigenicity can be addressed by comparing amino acid sequence and structural similarities. It is believed that proteins having a similar structure and at the same time share a substantial part of the amino acid sequences, share antigenic properties. The problem with this approach is that the minimal sequence similarity required to make this work still has to be defined. Finally, a computer-based epitope mapping tool was shown to identify, in the context of the folded protein, amino acid positions known to be involved in antibody binding. The high sensitivity and preliminary predictive value of the EMT on epitopes not included in the applied databases, as well as the relevance of the structural association between the amino acids describing the positive motifs, suggested the existence of amino acid motifs that are likely to be involved in antigenic determinants. Thus, the EMT has the potential of becoming a fast and simple alternative to X-ray crystallography for predicting structural antigenic determinants. The comparison with existing prediction scales demonstrated that information related to structural features, e.g., β -turns, could significantly improve the EMT.
Notes
2CI: confidence interval;
3RSA: relative surface accessibility.
REFERENCES
- Aalberse R. C. Structural biology of allergens. J. Allergy Clin. Immunol. 2000; 106: 228–238, [INFOTRIEVE], [CSA]
- Aalberse R. C., Akkerdaas J., van Ree R. Cross-reactivity of IgE antibodies to allergens. Allergy 2001; 56: 478–490, [INFOTRIEVE], [CSA], [CROSSREF]
- Alexander H., Alexander S., Getzoff E. D., Tainer J. A., Geysen H. M., Lerner R. A. Altering the antigenicity of proteins. Proc. Natl. Acad. Sci. USA 1992; 89: 3352–3356, [INFOTRIEVE], [CSA], [CROSSREF]
- Alix A. J. Predictive estimation of protein linear epitopes by using the program PEOPLE. Vaccine 2000; 18: 311–314, [CSA], [CROSSREF]
- Amit A. G., Mariuzza R. A., Phillips S. E., Poljak R. J. Threedimensional structure of an antigen-antibody complex at 2.8 Å resolution. Science 1986; 223: 747–753, [CSA]
- Andersson K., Choulier L., Hämäläinen M. D., van Regenmortel M. H., Altshuh D., Malmqvist M. Predicting the kinetics of peptide-antibody interactions using multivariate experimental design of sequence and chemical space. J. Mol. Recognition 2001; 14: 62–71, [CSA], [CROSSREF]
- Bachmann M. F., Zinkernagel R. M. The influence of virus structure on antibody responses and virus serotype formation. Immunol. Today 1996; 17: 553–558, [INFOTRIEVE], [CSA], [CROSSREF]
- Bachmann M. F., Zinkernagel R. M. Neutralizing antiviral B cell responses. Ann. Rev. Immunol. 1997; 15: 235–270, [CSA], [CROSSREF]
- Barlow D. J., Edwards M. S., Thornton J. M. Continuous and discontinuous protein antigenic determinants. Nature 1986; 322: 747–748, [INFOTRIEVE], [CSA], [CROSSREF]
- Batori V., Friis E. P., Nielsen H., Roggen E. L. An in silico method using an epitope motif database for predicting the location of antigenic determinants on proteins in a structural context. J. Mol. Recognition 2006; 19: 21–29, [CSA], [CROSSREF]
- Beezhold D. H., Hickey V. L., Sussman G. L. Mutational analysis of the IgE epitopes in the latex allergen Hev b 5. J. Allergy Clin. Immunol. 2001; 107: 1069–1076, [INFOTRIEVE], [CSA], [CROSSREF]
- Bentley G. H. Lysozymes: Model Enzymes in Biochemistry and Biology, S. P. Jollé. Birkhauser, Basel 1996; 301–319
- Bhaskaran R., Ponnuswamy P. K. Positional flexibilities of amino acid residues in globular proteins. Int. J. Peptide Prot. Res. 1988; 32: 242–255, [CSA]
- Burks A. W., Shin D., Cockrell G., Stanley J. S., Helm R. M., Bannon G. A. Mapping and mutational analysis of the IgE-binding epitopes on Ara h l, a legume vicilin protein and a major allergen in peanut hypersensitivity. Eur. J. Biochem. 1997; 245: 334–339, [INFOTRIEVE], [CSA], [CROSSREF]
- Chan O. T., Madaio M. P., Shlomchik M. J. B-cells are required for lupus nephritis in the polygenic Fas-intact MRL model of systemic autoimmunity. J. Immunol. 1999; 163: 3592–3596, [INFOTRIEVE], [CSA]
- Chaplin D. D. Overview of the immune response. J. Allergy Clin. Immunol. 2003; 111: S442–S459, [INFOTRIEVE], [CSA], [CROSSREF]
- Combet C., Blanchet C., Geourjon C., Deléage G. NPS×: Network protein. Trends Bioch. Sci. 2000; 25: 147–150, [CSA], [CROSSREF]
- Davies J. M., Scealy M., Cai Y., Whisstock J., Mackay I. R., Rowley M. J. Multiple alignement and sorting of peptides derived from phage-displayed random peptide libraries with polyclonal sera allows discrimination of relevant phagotopes. Mol. Immunol. 1999; 36: 659–667, [INFOTRIEVE], [CSA], [CROSSREF]
- De Weck A. L. Low Molecular weight antigens. The Antigens, M. Sela. Academic Press, New York 1974; Vol. 2: 99, 141–248
- Deleage G., Roux B. An algorithm for protein secondary structure prediction based on class prediction. Protein Eng. 1987; 1: 289–294, [INFOTRIEVE], [CSA]
- Elsayed S., Holen E., Dybendahl T. Synthetic allergenic epitopes from the amino-terminal regions of the major allergens of hazel and birch pollen. Int. Arch. Allergy Appl. Immunol. 1989; 89: 410–415, [INFOTRIEVE], [CSA]
- Furmonaviciene R., Shakib F. The molecular basis of allergenicity: Comparative analysis of the three dimensional structures of diverse allergens reveals a common structural motif. Molec. Pathol. 2001; 54: 155–159, [CSA], [CROSSREF]
- Ganglberger E., Grunberger K., Sponer B., Radauer C., Breiteneder H., Boltz-Nitulescu G., Scheiner O., Jensen-Jarolim E. Allergen mimotopes for 3-dimensional epitope search and induction of antibodies inhibiting human IgE. FASEB J. 2000; 14: 2177–2184, [INFOTRIEVE], [CSA], [CROSSREF]
- Gasteiger E., Hoogland C., Gattiker A., Duvaud S., Wilkins M. R., Appel R. D., Bairoch A. Protein identification and analtsis tools on the ExPASy Server. The Proteomics Protocols Handbook, J. M. Walker. Humana Press, Tonkawa, NJ 2005; 571–607
- Hantusch B., Krieger S., Untermayer E., Scholl I., Knittelfelder R., Flicker S., Spitzauer S., Valenta R., Boltz-Nitulesen G., Scheiner O., Jensen-Jarolim E. Mapping of conformational IgE epitopes on Phl p 5a by using mimotopes from a phage display library. J. Allergy Clin. Immunol. 2004; 114: 1294–1300, [INFOTRIEVE], [CSA], [CROSSREF]
- Hayakawa K., Asano M., Shinton S., Gui M., Allman D., Stewart C., Silver J., Hardy R. Positive selection of natural autoreactive B-cells. Science 1999; 285: 113–116, [INFOTRIEVE], [CSA], [CROSSREF]
- Hermeling S., Crommelin D. J., Schellekens H., Jiskoot W. Structure-immunogenicity relationships of therapeutic proteins. Pharmacol. Res. 2004; 21: 897–903, [CSA], [CROSSREF]
- Hermeling S., Schellekens H., Maas C., Gebbink M. F., Crommelin D. J., Jiskoot W. Antibody responses to aggregated human interferon-α 2a in wild type and transgenic tolerant mice depends on type and level of aggregation. J. Pharm. Sci. 2006; 95: 1084–1096, [INFOTRIEVE], [CSA], [CROSSREF]
- Hileman R. E., Silvanovitch A., Goodman R. E., Rice E. A., Holleschak G., Astwood J. D., Hefle S. L. Bioinformatic methods for allergenicity assessment using a comprehensive allergen database. Int. Arch. Allergy Immunol. 2002; 128: 280–291, [INFOTRIEVE], [CSA], [CROSSREF]
- Hopp T. P., Woods K. R. Prediction of protein antigenic determinants from amino acid sequences. Proc. Natl Acad. Sci. USA 1981; 78: 3824–3828, [INFOTRIEVE], [CSA], [CROSSREF]
- Ivanciuc O., Schein C. H., Braun W. Data mining of sequences and 3D structures of allergenic proteins. Bioinformatics 2002; 18: 1358–1364, [INFOTRIEVE], [CSA], [CROSSREF]
- Jakobsen C. G., Bodtger U., Kristensen P., Poulsen L. K., Roggen E. L. Isolation of high-affinity human IgE and IgG antibodies recognising Bet v 1 and Humicola lanuginosa lipase from combinatorial phage libraries. Mol. Immunol. 2004; 41: 941–953, [INFOTRIEVE], [CSA], [CROSSREF]
- Jameson B. A., Wolf H. The antigenic index: A novel algorithm for predicting antigenic determinants. CABIOS 1988; 4: 181–186, [INFOTRIEVE], [CSA]
- Janin J. Surface and inside volumes in globular proteins. Nature 1979; 277: 491–492, [INFOTRIEVE], [CSA], [CROSSREF]
- Janssen R., Wauben M. H., Tomassen J. Quaternary structure of a carrier protein influences antigenicity and immunogenicity of an inserted T-cell determinant. Intl. Immunol. 1996; 8: 829–845, [CSA]
- Keil W., Wagner R. R. Epitope mapping by deletion mutants and chimeras of two vesicular stomatitis virus glycoprotein genes expressed by a vaccinia virus vector. Virology 1989; 170: 392–407, [INFOTRIEVE], [CSA], [CROSSREF]
- Kyte J., Doolittle R. F. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 1982; 157: 105–132, [INFOTRIEVE], [CSA], [CROSSREF]
- Laczko I., Hollosi M., Ürge L., Ugen K., Weiner D. B., Mantsch H. H., Thurin J., Otvös L. J. Synthesis and conformational studies of N-glycosylated analogues of the HIV-1 principal neutralizing determinant. Biochemistry 1992; 31: 4282–4288, [INFOTRIEVE], [CSA], [CROSSREF]
- Laroche Y., Heymans S., Capaert S., De Cock F., Demarsin E., Collen D. Recombinant staphylokinase variants with reduced antigenicity due to elimination of B-lymphocyte epitopes. Blood 2000; 96: 1425–1430, [INFOTRIEVE], [CSA]
- Leung P. S., Chu K. H. cDNA cloning and molecular identification of the major oyster allergen from the Pacific oyster Crassostera gigas. Clin. Exp. Allergy 2001; 31: 1287–1294, [INFOTRIEVE], [CSA], [CROSSREF]
- Levitt M. Conformational preferences of amino acids in globular proteins. Biochemistry 1978; 17: 4277–4285, [INFOTRIEVE], [CSA], [CROSSREF]
- Li Y., Li H., Yang F., Smith-Gill S. J., Mariuzza R. A. X-ray snapshots of the maturation of an antibody response to a protein antigen. Nature Struct. Biol. 2003; 10: 482–488, [INFOTRIEVE], [CSA], [CROSSREF]
- Manavalan P., Ponnuswamy P. K. Hydrophobic character of amino acid residues in globular proteins. Nature 1978; 275: 673–674, [INFOTRIEVE], [CSA], [CROSSREF]
- Mendz G. L., Moore W. J. NMR studies of myelin basic protein. Conformation of a peptide that is an antigenic determinant for B-cell reactivity. Biochem. J. 1985; 229: 305–313, [INFOTRIEVE], [CSA]
- Meyer D. L., Schultz J., Lin Y., Henry A., Scanderson J., Jackson J. M., Goshorn S., Rees A. R., Graves S. S. Reduced antibody response to streptavidin through site-directed mutagenesis. Protein Sci. 2000; 10: 491–503, [CSA], [CROSSREF]
- Milich D. R., Leroux-Roels G. Immunogenetics of the response to HBsAg vaccination. Autoimmun. Rev. 2003; 2: 248–257, [INFOTRIEVE], [CSA], [CROSSREF]
- Miller J. F. Principles of immunological tolerance. Transfus. Med. Hemother. 2005; 32: 322–331, [CSA], [CROSSREF]
- Mirza O., Henriksen A., Ipsen H., Larsen N., Wissenbach M., Spangfort M., Gajhede M. Dominant epitopes and allergic cross-reactivity: Complex formation between a Fab fragment of amonoclonal murine IgG antibody and the major allergen from birch pollen Bet v 1. J. Immunol. 2000; 165: 331–338, [INFOTRIEVE], [CSA]
- Mittag D., Batori V., Neudecker P., Wiche R., Friis E. P., Ballmer-Weber B. K., Vieths S., Roggen E. L. A novel approach for investigation of specific and cross-reactive IgE epitopes on Bet v 1 and homologous food allergens in individual patients. Mol. Immunol. 2006; 43: 79–89, [CSA], [CROSSREF]
- Nair D. T., Singh K., Siddiqui Z., Niyak B. P., Rao K. V., Salunke D. M. Epitope recognition by diverse antibodies suggests conformational convergence in an antibody response. J. Immunol. 2002; 168: 2371–2382, [INFOTRIEVE], [CSA]
- Nishikawa T., Rapoport B., McLachlan S. The quest for the autoantibody immunodominant region on thyroid peroxidase: Guided mutagenesis based on a hypothetical three-dimensional model. Endocrinology 1996; 137: 1000–1006, [INFOTRIEVE], [CSA], [CROSSREF]
- Nossal G. J. Molecular and cellular aspects of immunologic tolerance. Eur. J. Biochem. 1991; 202: 729–737, [INFOTRIEVE], [CSA], [CROSSREF]
- Novartis Foundation. The Genetics of Auto-Immunity. 2005, ISBN: 0-470-02137-3
- Oberg K., Alm G., Magnusson A., Lundqvist G., Theodorsson E., Wide L., Wilander E. Treatment of malignant carcinoid tumors with recombinant intereron alfa-2b: Development of neutralizing interferon antibodies and possible loss of antitumor activity. J. Natl. Cancer Inst. 1989; 81: 531–535, [INFOTRIEVE], [CSA]
- Odorico M., Pelleques J.-L. BEPITOPE: predicting the location of continuous epitopes and patterns in proteins. J. Mol. Recog. 2003; 16: 20–22, [CSA]
- O'Lorcain P., Talebi A., Mulcohy G. B-cell epitope mapping within the MA° 16 antigenic sequence found in Eimeria acervulina merozoites and sporozoites. Vet. Parasitol. 1996; 66: 147–157, [INFOTRIEVE], [CSA]
- Ottesen J. L., Nilsson P., Jami, Weilguny D., Dührkop M., Bucchini D., Havelund S., Fogh J. M. The potential immunogenicity of human insulin and insulin analogues evaluated in a transgenic mouse model. Diabetologia 1994; 37: 1178–1185, [INFOTRIEVE], [CSA]
- Palleroni A. V., Aglione A., Labow M., Brunda M. J., Pestka S., Sinigaglia F., Garotta G., Alsenz J., Braun A. Interferon immunogenicity: Preclinical evaluation of interferon-α 2a. J. Interferon Res. 1997; 17: S23–S27, [CSA]
- Parhami-Seren B., Keel T., Reed G. L. Sequences of antigenic epitopes of Streptokinase identified via random peptide libraries displayed on phage. J. Mol. Biol. 1997; 271: 333–341, [INFOTRIEVE], [CSA], [CROSSREF]
- Parker J. M., Guo D., Hodges R. S. New hydrophilic scale derived from high-performance liquid chromatography peptide retention data: Correlation of predicted surface residues with antigenicity and X-ray-derived accessible sites. Biochemistry 1986; 25: 5426–5432, [CSA]
- Pearson W. R. Flexible sequence similarity searching with the FAST3 program package. Methods Mol. Biol. 2000; 132: 185–219, [INFOTRIEVE], [CSA]
- Peces R., de la Torre M., Alcazar R., Urra J. M. Antibodies against recombinant human erythropoietin in a patient with erythroietin-resistant anemia. N. Engl. J. Med. 1996; 335: 523–524, [INFOTRIEVE], [CSA], [CROSSREF]
- Pellequer J. L., Westhof E. PREDITOP: A program for antigenicity prediction. J. Mol. Graph. 1993; 11: 204–210, [INFOTRIEVE], [CSA], [CROSSREF]
- Pellequer J. L., Westhof E., Van Regenmortel M. H. Methods in Enzymology, J. J. Langone. Academic Press, London 1991; Vol. 23: 176
- Pellequer J. L., Westhof E., Van Regenmortel M. H. Correlation between the location of antigenic sites and the prediction of turns in proteins. Immunol. Lett. 1993; 36: 83–100, [INFOTRIEVE], [CSA], [CROSSREF]
- Pellequer J. L., Westhof E., Van Regenmortel M. H. Epitope predictions from the primary structure of proteins. Peptide Antigens, A Practical Approach, G. B. Wisdom. IRL Press, Oxford 1994; 7–25
- Renukaradhya G., Sinnathamby G., Seth A., Rajasekhar M., Shaila M. S. Mapping of B-cell epitopic sites and delineation of functional domains on the hemagglutinin-neuraminidase protein of peste des petits ruminants virus. Virus Res. 2002; 90: 171–185, [INFOTRIEVE], [CSA], [CROSSREF]
- Rodi D. J., Soares A. S., Makowski L. Quantitative assessment of peptide sequence diversity in M13 combinatorial peptide phage display libraries. J. Mol. Biol. 2002; 322: 1039–1052, [INFOTRIEVE], [CSA], [CROSSREF]
- Rose M. P., Geselowitz A. R., Lesser G. J., Lee R. H., Zehfus M. H. Hydrophobicity of amino acid residues in globular proteins. Science 1985; 229: 834–838, [INFOTRIEVE], [CSA]
- Rudolf M. P., Vogel M., Kricek F., Ruf C., Zurcher A. W., Reuschel R., Auer M., Miescher S., Stadler B. M. Epitope-specific antibody response to IgE by mimotope immunisation. J. Immunol. 1998; 160: 3315–3321, [INFOTRIEVE], [CSA]
- Saha S., Raghava G. P. BcePred: Prediction of continuous B-cell epitopes in antigenic sequences using physicochemical properties. ICARIS 2004; 2004: 197–204, [CSA]
- Sakaguchi S., Sakaguchi N., Shimizu J., Yamazaki S., Sakihama T., Itoh M., Kuniyasu Y., Nomura T., Toda M., Takahashi T. Immunologic tolerance maintained by CD25+CD4+ regulatory T-cells: Their common role in controlling autoimmunity, tumor immunity and transplantation tolerance. Immunol. Rev. 2001; 182: 18–32, [INFOTRIEVE], [CSA], [CROSSREF]
- Santos A. B., Chapman M. D., Aalberse R. C., Vailes L. D., Ferriani V. P., Oliver C., Rizzo M. C., Naspitz C. K., Arruda L. K. Cockroach allergens and asthma in Brasil: Identification of tropomyosin as a major allergen with potential cross-reactivity with mite and shrimp allergens. J. Allergy Clin. Immunol. 1999; 104: 329–337, [INFOTRIEVE], [CSA], [CROSSREF]
- Scandella D., DeGraaf M. S., Mattingly M., Roeder D., Timmons L., Fulcher C. A. Epitope mapping of human factor VIII inhibitor antibodies by deletion analysis of factor VIII fragments expressed in E. coli. Proc. Natl. Acad. Sci USA 1988; 85: 6152–6156, [INFOTRIEVE], [CSA], [CROSSREF]
- Schellekens H. Bio-equivalence and the immunogenicity of biopharmaceuticals. Nat. Rev. Drug Discov. 2002a; 1: 457–462, [INFOTRIEVE], [CSA], [CROSSREF]
- Schellekens H. Immunogenicity of therapeutic proteins: Clinical implications and future prospects. Clin. Ther. 2002b; 24: 1720–1740, [INFOTRIEVE], [CSA], [CROSSREF]
- Shreffler W. G., Beyer K., Chu T. H., Burks A. W., Sampson H. A. Microarray immunoassay: Association of clinical history, in vitro IgE function, and heterogeneity of allergenic peanut epitopes. J. Allergy Clin. Immunol. 2004; 113: 776–782, [INFOTRIEVE], [CSA], [CROSSREF]
- Sorokin A. V., Kasachinskaia E. L., Ivanova A. V., Kachko A. V., Neteskov S. V., Bukreyev A. A., Laktev V. B., Razumov L. A. Mapping of two dominant sites of VP35 of Marsburg virus. Viral Immunol. 2002; 15: 481–492, [INFOTRIEVE], [CSA], [CROSSREF]
- Stanley J. S., King N., Burks A. W., Huang S. K., Sampson H., Cockrell G., Helm R. M., West C. M., Bannon G. A. Identification and mutational analysis of the immunodominant IgE binding epitopes of the major peanut allergen Ara h 2. Arch. Biochem. Biophys. 1997; 342: 244–253, [INFOTRIEVE], [CSA], [CROSSREF]
- Stewart T. A., Hollingshead P. G., Pitts S. L., Chang R., Martin L. E., Oakley H. Transgenic mice as a model to test the immunogenicity of proteins altered by site-specific mutagenesis. Mol. Biol. Med. 1989; 6: 275–281, [INFOTRIEVE], [CSA]
- Stoy N. Macrophage biology and pathobiology in the evolution of immune responses: A functional analysis. Pathobiology 2001; 69: 179–211, [INFOTRIEVE], [CSA], [CROSSREF]
- Van Kampen V., Liebers V., Sander J., Chen Z., Baur X., Raulf-Heimsoth M., Falkenberg F. W. B-cell epitopes of the allergen Chi t 1.01: Peptide mapping of epitopes recognized by rabbit, murine, and human antibodies. Allergy 2001; 56: 118–125, [INFOTRIEVE], [CSA], [CROSSREF]
- van Regenmortel M. H. Mimotopes, continuous paratopes and hydropathic complementarity: Novel approximations in the description of immunochemical specificity. Dispers. Sci. Technol. 1998; 19: 1199–1219, [CSA]
- van Regenmortel M. H., Pellequer J. L. Predicting antigenic determinants in proteins: Looking for unidimensional solutions to the three-dimensional problem?. Peptide Res. 1994; 7: 224–228, [CSA]
- Vieths S., Scheurer S., Ballmer-Weber B. Current understanding of cross-reactivity of food allergens and pollen. Ann. N. Y. Acad. Sci. 2002; 964: 47–68, [INFOTRIEVE], [CSA]
- Vrtala S., Focke-Tejkl M., Swoboda I., Kraft D., Valenta R. Strategies for converting allergens into hypoallergenic vaccine candidates. Methods (Duluth) 2004; 32: 313–320, [CSA]
- Wedemeyer G. J., Patten P. A., Wang L. H., Schultz P. G., Stevens R. C. Structural insights into the evolution of an antibody combining site. Science 1997; 276: 1665–1669, [CSA], [CROSSREF]
- Welling G. W., Weijer W. J., Van der Zee R., Welling-Wester S. Prediction of sequential antigenic regions in proteins. FEBS Lett. 1985; 188: 215–218, [INFOTRIEVE], [CSA], [CROSSREF]
- Wierda D., Smith H. W., Zwickl C. M. Immunogenicity of biopharmaceuticals in laboratory animals. Toxicology 2001; 158: 71–74, [INFOTRIEVE], [CSA], [CROSSREF]
- Williams S. C., Badley R. A., Davis P. J., Puijk W. C., Meloen R. H. Identification of epitopes within beta-lactoglobulin recognised by polyclonal antibodies using phage display and PEPSCAN. J. Immunol. Meth. 1998; 213: 1–17, [CSA], [CROSSREF]
- Williamson M. P., Hall M. J., Handa B. K. 1H-NMR assignment and secondary structure of a herpes simplex virus glycoprotein D-1 antigenic domain. Eur. J. Biochem. 1986; 158: 527–536, [INFOTRIEVE], [CSA], [CROSSREF]
- Wu K. J., Wang C. Y., Lu H. K. Effect of glutaraldehyde on the humoral immunogenicity and structure of porcine dermal collagen membranes. Arch. Oral Biol. 2004; 49: 305–311, [INFOTRIEVE], [CSA], [CROSSREF]
- Zang Y. C., Yang D., Hong J., Tejada-Simon M. V., Rivera V. M., Zhang J. Z. Immunoregulation and blocking antibodies induced by interferon beta treatment in MS. Neurology 2000; 55: 397–404, [INFOTRIEVE], [CSA]
- Zimmerman J. M., Eliezer N., Simha R. The characterization of amino acid sequences in proteins by statistical methods. J. Theor. Biol. 1968; 21: 170–201, [INFOTRIEVE], [CSA], [CROSSREF]
- Zouali M. B-cell tolerance to self in systemic auto-immunity. Arch. Immunol. Ther. Exp. 2001; 49: 361–365, [CSA]
- Zwickl C. M., Cocke K. S., Tamura R. N., Holzhausen L. M., Brophy G. T., Bick P. H., Wierda D. Comparison of the immunogenicity of recombinant and pituitary human growth hormone in rhesus monkeys. Fundam. Appl. Toxicol. 1991; 16: 275–287, [INFOTRIEVE], [CSA], [CROSSREF]