Abstract
The therapeutic uses of immunostimulatory agents are generally in the treatments of infections or cancer. The traditional example of vaccination is one form of immunostimulation used in the prevention of pathogenic infections or cancer (e.g., human papillomavirus vaccine). Recombinant cytokines are increasingly used to stimulate immune system function. For example, interferon-α (IFNα) and interleukin (IL)-2 have been used to treat chronic hepatitis C virus infection and metastatic melanoma, respectively. In contrast, monoclonal antibodies are used to target malignant cells for elimination via antibody-dependent cytotoxicity mechanisms or apoptosis, including the anti-CD20 monoclonal antibody rituximab and the anti-CD56 monoclonal antibody alemtuzumab used in the treatment of B-cell malignancies, and the anti-erb2 receptor antibody trastuzumab used in the treatment of breast cancer. Finally, immunostimulation may develop via modulation of pathways involved in immune system regulation. For example, the anti-CD28 monoclonal antibody TGN1412 was developed as an agonist of regulatory T-cells for treatment of T-cell-mediated chronic inflammatory diseases or leukemias. A panel was convened to discuss potential toxicities associated with immunostimulation. At the Immunotoxicology IV meeting in 2006, a panel, moderated by Dr. Robert House (Dynport Vaccine Co., Frederick, MD), included Drs. Gary Burleson (Burleson Research Technologies, Inc., Raleigh, NC), Kenneth Hastings (US FDA, Center for Drug Evaluation and Research [CDER], Rockville, MD), Barbara Mounho (Amgen, Thousand Oaks, CA), Rafael Ponce (ZymoGenetics, Inc., Seattle, WA), Mark Wing (Huntington Life Sciences, Cambridgeshire, United Kingdom), Lauren Black (Navigators Consulting, Sparks, NV) and Anne Pilaro (US FDA, CDER, Rockville, MD). This paper reviews the major identified toxicities associated with immunostimulation, including the acute phase response, cell and tissue abnormalities/injury, cytokine release/cytokine storm, tumor lysis syndrome, vascular leak, and autoimmunity that were discussed by this panel.
INTRODUCTION
Systemic administration of immunostimulatory cytokines and antibody-based therapies has transformed the treatment of a number of life-threatening infections and cancers. For example, durable and complete response of malignant melanoma is observed in a subset of patients treated with recombinant IL-2 (aldesleukin) (reviewed in Atkins et al., Citation1999; O'Day and Boasberg, Citation2006) and a sustained virologic response is observed in the majority of patients treated with interferon-α -based therapies (reviewed in Thomas, Citation2006). The success of both of these therapies and that of other immunostimulatory agents is limited by observed toxicities associated with immune system activation, which may lead to (sub-optimal) dose reduction or treatment withdrawl. These toxicities appear to largely arise from either a primary or secondary cytokine imbalance associated with administration of the agent systemically and at supraphysiological concentrations. In this context, some of the toxicities associated with an induced immunostimulation may be predicted with an understanding of the mechanisms of immunostimulation.
Although the pathophysiological mechanisms underlying many aspects of immunostimu-lation-related toxicities are not established, the clinical experience with these agents has allowed identification of a number of observed toxicities and syndromes, including the acute phase response, immune system-mediated cell and tissue injury, cytokine release/cytokine storm, tumor lysis syndrome, vascular (capillary) leak syndrome, and autoimmunity, as summarized in . These toxicities may be understood in the context of current knowledge regarding the interaction of various cytokines and immune cells as modifiers of physiologic response in tissues, as depicted in . As toxicologists, it is imperative to remain cognizant of the value of animal models for predicting human responses for this class of agents, which was also addressed by this panel. Those interested in additional detail are referred to a recent review (Gribble et al., Citation2007).
FIG. 1 Overview of immune response, pharmacologic stimulation and potential pathways to toxicities. During the normal course of the immune response, innate and adaptive arms of the immune system are activated to clear infections. Immune stimulation of NK cells, macrophages, dendritic cells, and T-lymphocytes by therapeutic agonists administered systemically at supra-physiological doses enhances these “normal” processes leading to the various syndromes described. The potential pathological consequences of such therapies may be understood in the context of the pathways affected by treatment. A simplified representation of the various cell types and modulators leading to these syndromes and toxicities are depicted. Abbreviations: Mø, macrophage; DC, dendritic cell; NK, natural killer cell; B, B-lymphocyte; CD4 T, helper T-lymphocyte; CD8 T and CTL, cytotoxic T-lymphocyte; FcR, Fc receptor; TLR, Toll-like receptor; IL, interleukin; TNF, tumor necrosis factor; IFN, interferon (Figure used with permission from Gribble et al., Citation2007).
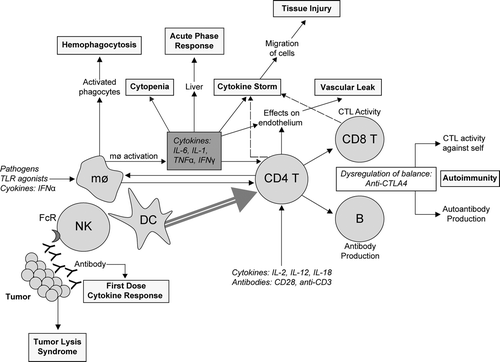
TABLE 1 Toxicities associated with immunostimulatory therapeutics (modified from Gribble et al., Citation2007)
Acute Phase Response
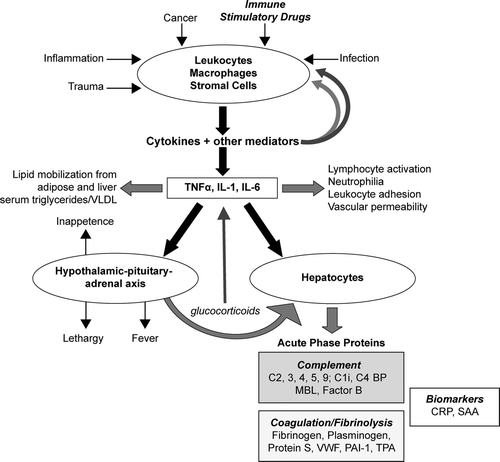
The acute phase response () is a series of reactions initiated in response to a variety of acute and chronic inflammatory conditions that limits tissue damage, reduces infection, and activates tissue repair (reviewed in Fey et al., Citation1994). The acute phase response is characterized by leukocytosis, fever, alterations in the metabolism of many organs, and changes in plasma concentrations of numerous acute-phase proteins, that are defined by plasma concentration changes of at least 25% during inflammatory disorder. Positive acute-phase proteins include fibrinogen, serum amyloid A, albumin, C-reactive protein. Negative acute-phase proteins include albumin, transferrin, and insulin growth factor I. The acute phase response is stimulated by inflammation-associated cytokines including interleukin (IL)-6, IL-1β, tumor necrosis factor-α (TNFα), interferon-γ (IFN-γ), and transforming growth factor β (TGF-β). The development of the acute phase response is identified by an elevation in body temperature, evaluation of peripheral blood counts/flow cytometric immunophenotyping, and monitoring of plasma acute phase proteins including C-reactive protein, fibrinogen, albumin, and others. Animal models are generally predictive of an acute phase response in humans.
FIG. 2 Overview of the acute phase response. Leukocytes, macrophages and stromal cells, among others, respond to certain stimuli by production of cytokines and other soluble mediators of inflammation. These mediators exert both positive and negative feedback on the inflammation process. Red arrows indicate stimulatory actions; blue arrows indicate inhibitory actions. The acute phase response (APR) is initiated when pro-inflammatory cytokines TNFα, IL-1, and IL-6 reach levels sufficient to activate a systemic response. The primary actions of APR involve the liver, the hematopoietic system, and the hypothalamic-pituitary-adrenal axis. Hepatocytes respond to these cytokines, primarily through alteration of gene transcription, to increase production and secretion of positive acute phase proteins. A select set of the approximately 40 different proteins involved in the APR is shown corresponding to major biomarkers and proteins that specifically affect complement and coagulation and fibrinolysis pathways. Observation of patient presentation, including measurement of biomarkers, provides a useful basis for monitoring development and progression of the acute phase response. Abbreviations: C1i, C1 inhibitor; MBL, mannose binding lectin; C4 BP, C4-binding protein; PAI-1, Plasminogen Activator Inhibitor-1; TPA, Tissue Plasminogen Activator; VWF, von Willebrand Factor; VLDL, very low density lipoprotein; CRP, C-reactive protein; SAA, serum amyloid A. (Figure used with permission from Gribble et al., Citation2007).
Cell and Tissue Injury
Various pathological changes have been associated with therapeutic immunostimulation, particularly with cytokines. As a rule, the observed effects are specific to the stimulated pathway and consistent with known physiological activity of each cytokine. For example, altered bone marrow production has been observed including both stimulation (e.g., IL-6-associated mega-karyocytopoiesis; Baatout, Citation1996) and inhibition (e.g., IFNα -associated bone marrow suppres-sion; Means and Krantz, Citation1996). Many cytokines affect leukocyte cell trafficking/infiltration and tissue inflammation associated with the expression of chemokines and cellular adhesion mole-cules leading to tissue homing.
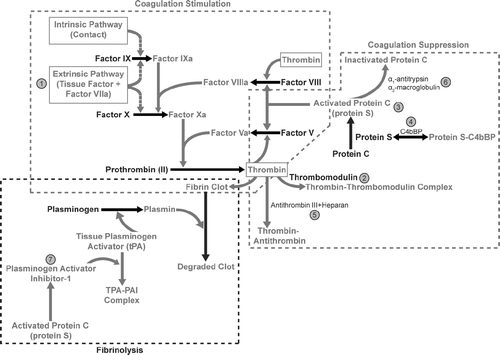
There is a broad literature describing the effects of immunostimulation on altered coagulation () that arises both indirectly from the acute phase response-mediated shift in hepatic protein production that results in decreased coagulation factor production and directly from cytokine-mediated alterations in coagulation factor activity. The net effect of inflammation is to create a pro-coagulant condition that, if severe enough, is associated with disseminated intravascular coagulation (Levi et al., Citation2003).
FIG. 3 Effects of inflammation on coagulation. Inflammation, cytokine release, and activation of platelets and leukocytes contribute to a systemic stimulation of coagulation, impairment in anticoagulant mechanisms, and alterations in mechanisms that remove fibrin deposition. Those components leading to a pro-coagulant state, or that increase clot stability are depicted in red letters and arrows. Taken together, inflammation tends to create a pro-coagulant state that is monitorable with evaluation of the activated partial thromboplastin time and prothrombin time. Those components that suppress coagulation or contribute to clot degradation are depicted in blue letters and arrows. Significant points of interaction are enumerated as follows: (1) IL-6, TNFα, C-reactive protein, IL-1, complement, complement activation, and endotoxin induce tissue factor on endothelial cells, monocytes and macrophages that stimulate the extrinsic pathway of coagulation. In addition, activated platelets release CD40 ligand, which increases tissue factor synthesis and inflammatory cytokines; (2) TNFα, IL-1β, IL-6, endotoxin, or neutrophil elastase decrease thrombomodulin transcription or activity, thereby contributing to a pro-coagulant condition; (3) TNF-α, IL-1β, and endotoxin decrease protein C receptor transcription and thereby contribute to a pro-coagulant state by removing key controls over thrombin generation; (4) IL-6 and IL-1β up-regulate C4-binding protein expression, which can be inhibited by TNFα. In addition, IL-6-mediated up-regulation of protein S production can be inhibited by TNF-α and IL-1; (5) TNF-α and IL-1β decrease glycosaminoglycans (heparin-like cofactors) necessary for optimal antithrombin activity; (6) α1-antitrypsin and α2-macroglobulin bind and inactivate protein C, thereby contributing to a pro-coagulant condition; and, (7) IL-6 increases production of new platelets, which are more thrombogenic than older platelets. Endotoxin, IL-8, and various chemokines increase platelet activation, thus providing a suitable membrane surface to support coagulation, and platelet aggregation, and thereby contributing to a pro-coagulant condition. TNFα, IL-1β increase plasminogen activator inhibitor (PAI)-1 and decrease free tissue plasminogen activator (tPA) thereby decreasing fibrin removal and increasing microvascular thrombosis. (Figure used with permission from Gribble et al., Citation2007).
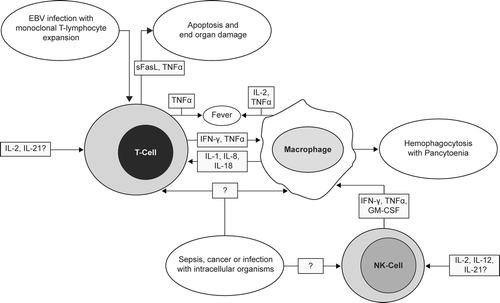
Increased cell destruction is one goal of therapeutic immunostimulation when the target is a tumor cell. However, immune system stimulation may be associated with non-specific cell destruction syndromes including macrophage activation syndrome () causing hemolytic anemia and thrombocytopenia, which are generally reversible upon termination of therapy (Fujiwara et al., Citation1993). Tissue-specific effects are observed from immunostimulation including thyroid atrophy, lymph node or splenic remodeling, and hyperbilirubinema and cholestasis. Dyslipidemia is another outcome of immunostimulation, with identified roles for a number of different cytokines in lipid metabolism (Khovidhunkit et al., Citation2004).
FIG. 4 Mechanisms underlying cytokine-mediated hemophagocytic lympohistiocytosis (HLH) and macrophage activation syndrome (MAS). Underlying chronic infection, cancer, or sepsis may activate innate and adaptive immune responses, resulting in the expression of a number of inflammatory cytokines that induce macrophages to phagocytize normal red blood cells, platelets, and other cells, contributing to pancytopenia. An acquired hemophagocytic syndrome may also develop upon administration of therapeutic immunostimulatory agents. Abbreviations: EBV, Epstein-Barr virus; sFasL, soluble Fas ligand; IL, interleukin; TNF, tumor necrosis factor; IFN, interferon; GM-CSF, granulocyte-macrophage colony-stimulating factor. (Figure used with permission from Gribble et al., Citation2007 as adapted from Fisman, Citation2000 and Arico et al., Citation2001).
Monitoring for cell and tissue injury is conducted with knowledge of the cytokine pathways affected by the therapeutic agent and includes peripheral blood differential cell counts or flow cytometric immunophenotyping (including the evaluation of activation markers), coagulation screening (activated partial thromboplastin time, D-dimers, thrombin-antithrombin complexes), serum chemistry (including conjugated and/or unconjugated bilirubin), and tissue pathology/immunohistochemistry (and possibly tissue-based flow cytometry). In general, animal models are generally predictive of effects in humans for many of these toxicities.
Cytokine Release/Cytokine Storm
A systemic cytokine release may develop in response to an intense inflammatory signal or antigen load that overwhelms immunomodulatory controls (Ertel et al., Citation1991; Panelli et al., Citation2004). The precise mechanisms underlying systemic cytokine release remain unclear, but are likely related to the uncontrolled activation of immune cells, mostly macrophages and T-cells, that results in the release of inflammatory mediators. The systemic and local presence of these molecules and the associated inflammation damages tissues and organs, and can result in organ failure and death if left untreated. Anti-inflammatory agents such as steroids help control this response. The recent response of healthy human volunteers to TGN1412 exemplifies the systemic nature and successful therapeutic management of this syndrome (Suntharalingam et al., Citation2006). Analysis of serum cytokines provides a primary means of monitoring the development and resolution of this syndrome, with supportive evidence provided by clinical signs/symptoms, peripheral blood differential cell counts, and flow cytometric analyses. Preclinical models in non-human primates of TGN1412 were not predictive of the cytokine release syndrome (with the associated clinical sequelae), although it appears that context and the animal model may be important. Unfortunately, interspecies comparisons of systemic cytokine release responses are lacking.
Tumor Lysis Syndrome
Tumor lysis syndrome is a potentially life-threatening metabolic imbalance produced by the rapid breakdown of a tumor in response to anti-cancer treatment (Cope, Citation2004). As the contents of necrosing cells are released into circulation, the body's ability to maintain homeostasis is overwhelmed leading to a variety of metabolic abnormalities (including hyperuricemia, hyperkalemia, hyperphosphotemia, hypocalcemia and uremia), often associated with renal failure. Tumor lysis syndrome has been most frequently observed in Burkitt lymphoma, T-cell acute lymphoblastic leukemia, acute lymphocytic leukemia and high-grade non-Hodgkin's lymphoma, and other cancers. Patient risk factors include having a tumor with a high proliferative rate, high sensitivity to chemotherapy, and/or large size, baseline decreased kidney function and elevated LDH. Tumor lysis syndrome may be observed following treatment with induction chemotherapy or monoclonal antibody therapy including rituximab (Jabr, Citation2005) and campath (Wing et al., Citation1996). Given that most nonclinical safety programs are conducted in normal animals, nonclinical models are generally not useful as predictive models.
Vascular (Capillary) Leak Syndrome
Vascular leak syndrome is characterized by an increase in vascular permeability and extravasation of fluids and proteins, resulting in interstitial edema and organ failure (e.g., Fishel et al., Citation2003). Manifestations of vascular leak syndrome include fluid retention and peripheral edema (including the abdominal cavity, connective tissues, lungs, and around the heart) and an increased body weight. In its most severe form, vascular leak results in pulmonary and cardiovascular failure. Vascular leak syndrome is the dose limiting toxicity of recombinant IL-2 therapy (Baluna and Vitetta, Citation1997). Development of vascular leak may be identified by hypotension, serum chemistry alterations (including hemoconcentration and hypoalbuminemia without albuminuria), clinical observations (generalized edema) and body weight. Interspecies comparisons are lacking, but animal models are reported to have a low sensitivity for this syndrome.
Autoimmunity
Autoimmunity results from the loss of “self” tolerance leading to an immune attack against our own tissues and organs associated with activation of B- and T-cells (). Though the precise mechanisms underlying development of autoimmunity are still subject to clearer definition, the end result is tissue and organ damage resulting from an immune response against the body's own proteins. Autoimmunity has been associated with CTLA-4 therapy as case reports of SLE, Type-1 diabetes, hepatitis, and dermatitis (Attia et al., Citation2005; Blansfield et al., Citation2005; Sanderson et al., Citation2005), and with IFN-α therapy as case reports of retinal abnormal-ities, thyroid disease, autoimmune thrombocytopenic purpura, SLE, Guillain-Barre Syndrome (e.g., Blanco et al., Citation2001; Seckin et al., Citation2004; Solans et al., Citation2004; Niewold and Swedler, Citation2005). Therapeutic administration of these agents to animals models do not appear to be predictive of autoimmunity in humans.
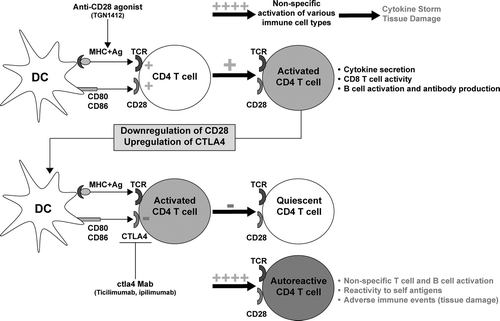
FIG. 5 T-Cell co-stimulation and balance of immune stimulation and tolerance. CD4 T-cells express members of the CD28 family (CD28, ICOS, OX40) that are needed for the efficient initiation and generation of a CD4 T-cell response that affects cellular (CD8 T-lymphocyte) and humoral (B lymphocyte) immunity. Superagonistic antibodies against CD28 (TGN1412) on T- and other immune cells provide a non-specific immune stimulation leading to a cytokine storm and inflammation. In contrast, activated T-cells are down-regulated by other members of the family, including CTLA4 and PD-1 to keep the immune response in check after clearance of infection. Antibodies that block this down-regulation (i.e., ipilimumab, ticilimumab) induce sustained T-cell activation and breakdown of self-tolerance, leading to reaction against self-antigens and autoimmunity. Abbreviations: DC, dendritic cell; MHC, major histocompatability complex, Ag; antigen; TCR, T-cell receptor; Mab, monoclonal antibody. (Figure used with permission from Gribble et al., Citation2007).
Predictive Value of Animal Models
The preceding review highlights an opportunity for toxicologists to develop and characterize improved animal models, in vitro assays, or in silico models for predicting toxicities associated with immune system activation. In particular, there is a pressing need to develop methods for predicting tumor lysis syndrome, autoimmunity, and systemic cytokine release responses, which are inadequately assessed using currently available models. In contrast, it appears that current animal models are generally adequate or good predictors of the acute phase response, cell and tissue injury, or vascular leak, with appropriate accounting of scaling differences for dose/pharmacokinetics and adjustments for interspecies differences in binding affinity, target distribution and other factors.
CONCLUSIONS
Recent clinical toxicities among healthy volunteers treated with TGN1412 demonstrate the complexity and challenge associated with therapeutic immunostimulation. Such experiences also provide us with an opportunity to be introspective regarding the value of current animal models for predicting the wide range of potential toxicities associated with immune system stimulation in humans. Moreover, such experiences must reinforce our mindfulness in transitioning any novel therapeutic agent from preclinical models to humans, which must be based in the full breadth of our scientific experience, and our responsibilities to continuously challenge our own assumptions.
Related Research Data
REFERENCES
- Arico M., Danesino C., Pende D., Moretta L. Pathogenesis of haemophagocytic lymphohistiocytosis. Br. J. Haematol. 2001; 114(4)761–769
- Atkins M. B., Lotze M. T., Dutcher J. P., Fisher R. I., Weiss G., Margolin K., Abrams J., Sznol M., Parkinson D., Hawkins M., Paradise C., Kunkel L., Rosenberg S. A. High-dose recombinant interleukin 2 therapy for patients with metastatic melanoma: Analysis of 270 patients treated between 1985 and 1993. J. Clin. Oncol. 1999; 17: 2105–2116
- Attia P., Phan G. Q., Maker A. V., Robinson M. R., Quezado M. M., Yang J. C., Sherry R. M., Topalian S. L., Kammula U. S., Royal R. E., Restifo N. P., Haworth L. R., Levy C., Mavroukakis S. A., Nichol G., Yellin M. J., Rosenberg S. A. Autoimmunity correlates with tumor regression in patients with metastatic melanoma treated with anti-cytotoxic T-lymphocyte antigen-4. J. Clin. Oncol. 2005; 23: 6043–6053
- Baatout S. Interleukin-6 and megakaryocytopoiesis: An update. Ann. Hematol. 1996; 73: 157–162
- Baluna R., Vitetta E. S. Vascular leak syndrome: A side effect of immunotherapy. Immunopharmacology 1997; 37: 117–132
- Berlex. 2004, Campath package insert
- Blanco P., Palucka A. K., Gill M., Pascual V., Banchereau J. Induction of dendritic cell differentiation by IFN-α in systemic lupus erythematosus. Science 2001; 294: 1540–1543
- Blansfield J. A., Beck K. E., Tran K., Yang J. C., Hughes M. S., Kammula U. S., Royal R. E., Topalian S. L., Haworth L. R., Levy C., Rosenberg S. A., Sherry R. M. Cytotoxic T-lymphocyte-associated antigen-4 blockage can induce autoimmune hypophysitis in patients with metastatic melanoma and renal cancer. J. Immunother. 2005; 28: 59–598
- Capuron L., Hauser P., Hinze-Selch D., Miller A. H., Neveu P. J. Treatment of cytokine-induced depression. Brain Behav. Immun. 2002; 16: 575–580
- Chapman P. B., Lester T. J., Casper E. S., Gabrilove J. L., Wong G. Y., Kempin S. J., Gold P. J., Welt S., Warren R. S., Starnes H. F., Sherwin S. A., Old L. J., Oettgen H. F. Clinical pharmacology of recombinant human tumor necrosis factor in patients with advanced cancer. J. Clin. Oncol. 1987; 5: 1942–1951
- Cheson B. D. Rituximab: Clinical development and future directions. Expert Opin. Biol. Ther. 2002; 2: 97–110
- Chiron. 2005, PROLEUKIN package insert
- Cope D. Tumor lysis syndrome. Clin. J. Oncol. Nurs. 2004; 8: 415–416
- Ertel W., Morrison M. H., Wang P., Ba Z. F., Ayala A., Chaudry I. H. The complex pattern of cytokines in sepsis. Association between prostaglandins, cachectin, and interleukins. Ann. Surg. 1991; 214: 141–148
- Feinberg B., Kurzrock R., Talpaz M., Blick M., Saks S., Gutterman J. U. A Phase I trial of intravenously-administered recombinant tumor necrosis factor-α in cancer patients. J. Clin. Oncol. 1988; 6: 1328–1334
- Ferran C., Sheehan K., Dy M., Schreiber R., Merite S., Landais P., Noel L. H., Grau G., Bluestone J., Bach J. F., et al. Cytokine-related syndrome following injection of anti-CD3 monoclonal antibody: Further evidence for transient in vivo T-cell activation. Eur. J. Immunol. 1990; 20: 509–515
- Fey G. H., Hocke G. M., Wilson D. R., Ripperger J. A., Juan S. C., Cui M. Z., Darlington G. J. Cytokines and the acute phase response of the liver. The Liver: Biology and Pathobiology, Third Ed., I. M. Arias, I. M. Boyer, N. Fausto. Raven Press, New York 1994; 113–137
- First M. R., Schroeder T. J., Hariharan S. OKT3-induced cytokine-release syndrome: renal effects (cytokine nephropathy). Transplant. Proc. 1993; 25(2 Suppl. 1)25–26
- Fishel R. S., Are C., Barbul A. Vessel injury and capillary leak. Crit. Care Med. 2003; 31: S502–511
- Fisman D. N. Hemophagocytic syndromes and infection. Emerg. Infect. Dis. 2000; 6(6)601–608
- Fujiwara F., Hibi S., Imashuku S. Hypercytokinemia in hemophagocytic syndrome. Am. J. Pediatr. Hematol. Oncol. 1993; 15: 92–98
- Gaston R. S., Deierhoi M. H., Patterson T., Prasthofer E., Julian B. A., Barber W. H., Laskow D. A., Diethelm A. G., Curtis J. J. OKT3 first-dose reaction: Association with T-cell subsets and cytokine release. Kidney Int. 1991; 39: 141–148
- Genentech. 2004, Rituxan package insert
- Gribble E. J., Sivakuar P. V., Ponce R. A., Hughes S. D. Toxicity as a result of immunostimulation by biologics. Expert Opin. Drug Metab. Toxicol. 2007; 3: 209–234
- Hodi F. S., Mihm M. C., Soiffer R. J., Haluska F. G., Butler M., Seiden M. V., Davis T., Henry-Spires R., MacRae S., Willman A., Padera R., Jaklitsch M. T., Shankar S., Chen T. C., Korman A., Allison J. P., Dranoff G. Biologic activity of cytotoxic T lymphocyte-associated antigen 4 antibody blockade in previously vaccinated metastatic melanoma and ovarian carcinoma patients. Proc. Natl. Acad. Sci. USA 2003; 100: 4712–4717
- Jabr F. I. Acute tumor lysis syndrome induced by rituximab in diffuse large B-cell lymphoma. Int. J. Hematol. 2005; 82: 312–314
- Khovidhunkit W., Kim M. S., Memon R. A., Shigenaga J. K., Moser A. H., Feingold K. R., Grunfeld C. Effects of infection and inflammation on lipid and lipoprotein metabolism: Mechanisms and consequences to the host. J. Lipid Res. 2004; 45: 1169–1196
- Levi M., Keller T. T., van Gorp E., ten Cate H. Infection and inflammation and the coagulation system. Cardiovasc. Res. 2003; 60: 26–39
- Means R. T., Jr., Krantz S. B. Inhibition of human erythroid colony-forming units by interferons alpha and beta: Differing mechanisms despite shared receptor. Exp. Hematol. 1996; 24: 204–208
- Moreau T., Coles A., Wing M., Isaacs J., Hale G., Waldmann H., Compston A. Transient increase in symptoms associated with cytokine release in patients with multiple sclerosis. Brain 1996; 119: 225–237
- Muggia F. M., Brown T. D., Goodman P. J., Macdonald J. S., Hersh E. M., Fleming T. R., Leichman L. High incidence of coagualopathy in Phase II studies of recombinant tumor necrosis factor in advanced pancreatic and gastric cancers. Anticancer Drugs 1992; 3: 211–217
- Niewold T. B., Swedler W. I. Systemic lupus erythematosus arising during interferon-alpha therapy for cryoglobulinemic vasculitis associated with hepatitis C. Clin. Rheumatol. 2005; 24: 178–181
- O'Day S., Boasberg P. Management of metastatic melanoma 2005. Surg. Oncol. Clin. N. Am. 2006; 15: 419–437
- Ortho. 2001, Orthoclone OKT3 package insert
- Panelli M. C., White R., Foster M., Martin B., Wang E., Smith K., Marincola F. M. Forecasting the cytokine storm following systemic interleukin (IL)-2 administration. J. Transl. Med. 2004; 2: 17
- Phan G. Q., Yang J. C., Sherry R. M., Hwu P., Topalian S. L., Schwartzentruber D. J., Restifo N. P., Haworth L. R., Seipp C. A., Freezer L. J., Morton K. E., Mavroukakis S. A., Duray P. H., Steinberg S. M., Allison J. P., Davis T. A., Rosenberg S. A. Cancer regression and autoimmunity induced by cytotoxic T lymphocyte-associated antigen 4 blockade in patients with metastatic melanoma. Proc. Natl. Acad. Sci. USA 2003; 100: 8372–8377
- Ribas A., Camacho L. H., Lopez-Berestein G., Pavlov D., Bulanhagui C. A., Millham R., Comin-Anduix B., Reuben J. M., Seja E., Parker C. A., Sharma A., Glaspy J. A., Gomez-Navarro J. Antitumor activity in melanoma and anti-self responses in a Phase I trial with the anti-cytotoxic T-lymphocyte-associated antigen 4 monoclonal antibody CP-675,206. J. Clin. Oncol. 2005; 23: 8968–8977
- Roche. 2003, Roferon-A package insert
- Roche. 2004, PEGASYS package insert
- Sanderson K., Scotland R., Lee P., Liu D., Groshen S., Snively J., Sian S., Nichol G., Davis T., Keler T., Yellin M., Weber J. Autoimmunity in a Phase I trial of a fully human anti-cytotoxic T-lymphocyte antigen-4 monoclonal antibody with multiple melanoma peptides and Montanide ISA 51 for patients with resected stages III and IV melanoma. J. Clin. Oncol. 2005; 23: 741–750
- Schering. 2005, PEG-Intron package insert
- Seckin D., Durusoy C., Sahin S. Concomitant vitiligo and psoriasis in a patient treated with interferon alfa-2a for chronic hepatitis B infection. Pediatr. Dermatol. 2004; 21: 577–579
- Siegel J. P., Puri R. K. Interleukin-2 toxicity. J. Clin. Oncol. 1991; 9: 694–704
- Solans R., Bosch J. A., Esteban I., Vilardell M. Systemic sclerosis developing in association with the use of interferon alpha therapy for chronic viral hepatitis. Clin. Exp. Rheumatol. 2004; 22: 625–628
- Suntharalingam G., Perry M. R., Ward S., Brett S. J., Castello-Cortes A., Brunner M. D., Panoskaltsis N. Cytokine storm in a Phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. New Engl. J. Med. 2006; 355: 1018–1028
- Thomas D. L. Options for treatment of hepatitis C in HIV-infected persons. J. Hepatol. 2006; 44(S1)S40–43
- Vial T., Descotes J. Clinical toxicity of interleukin-2. Drug Safety 1992; 7: 417–433
- Wing M. G., Moreau T., Greenwood J., Smith R. M., Hale G., Isaacs J., Waldmann H., Lachmann P. J., Compston A. Mechanism of first-dose cytokine-release syndrome by CAMPATH 1-H: Involvement of CD16 (FcγRIII) and CD11a/CD18 (LFA-1) on NK cells. J. Clin. Invest. 1996; 98: 2819–2826
- Winkler U., Jensen M., Manzke O., Schulz H., Diehl V., Engert A. Cytokine-release syndrome in patients with B-cell chronic lymphocytic leukemia and high lymphocyte counts after treatment with an anti-CD20 monoclonal antibody (rituximab, IDEC-C2B8). Blood 1999; 94: 2217–2224