Abstract
Tumor necrosis factor-α (TNFα) and transforming growth factor-β1 (TGFβ1) are potent peptide growth factors that are likely to play important roles in the development of interstitial pulmonary fibrosis (IPF). Previously we showed that TNFα and TGFβ1 are up-regulated in macrophages, epithelial and mesenchymal cells early after exposure to chrysotile asbestos, particularly at sites of fiber deposition in vivo. We also showed that TNFα receptor knockout mice are resistant to asbestos-induced fibrosis. Importantly, vectors that over-express TNFα cause inflammation and fibrogenesis along with increased TGFβ1 production in C57Bl/6 mice. Recently we reported that TNFα activates the extracellular regulated kinase pathway in fibroblasts leading to a 200–400% increase in TGFβ1 mRNA and protein. The mechanism of TNFα induction of TGFβ1 expression appears to be complex, involving both transcriptional and post-transcriptional mechanisms. In asbestos-exposed animals, this TGFβ1 is produced on alveolar surfaces in a latent form (controlled by binding of a latent associated peptide [LAP]) that must be activated for the TGFβ1 to bind to its receptors and induce its multiple biological effects. Thus, we recently reported that, in vitro, reactive oxygen species (ROS) derived from chrysotile and crocidolite asbestos activate TGFβ1 by oxidation of the LAP. Now, in preliminary findings, we have shown that over-expression of latent TGFβ1 prior to asbestos exposure of fibrogenic-resistant TNFα receptor knockout mice produces asbestos lesions with the same severity as seen in normal C57/Bl6 mice. This finding plus the demonstration of increased amounts of TGFβ1, increased Smad activation and amelioration of the developing disease by treating the mice with an anti-oxidant all support the concept that, in vivo, latent TGFβ1 is activated by asbestos-generated oxygen radicals and consequently mediates at least a component of the consequent fibrogenesis. Taken together, these findings support the postulate that TNFα controls fibrogenesis by regulating TGFβ1 expression and that one mechanism through which ROS induce lung fibrosis is by activating latent TGFβ 1.
INTRODUCTION
Interstitial pulmonary fibrosis (IPF) is a progressive and usually fatal disease. IPF can be caused by multiple factors including immune mechanisms, infectious agents and toxic gases and particles including asbestos (Morris and Brody, Citation1998). Symptoms of the disease may not appear for many years and a large proportion of cases remain idiopathic (Katzenstein and Myers, Citation1998). IPF is characterized by increased extracellular matrix production, varying degrees and types of inflammation, and proliferation of a variety of cells as the disease develops. IPF afflicts millions of individuals worldwide, yet there are no effective therapies available for any of the forms of IPF (Selman et al., Citation2004). A better understanding of the molecular mechanisms involved in initiation and progression of the disease may lead to new therapies.
Asbestosis displays the hallmarks of IPF (Brody, Citation1997; Mossman and Churg, Citation1998). We have been using brief exposure of rodents to inhaled chrysotile asbestos fibers as a model of IPF to study early molecular events important in initiating and sustaining the fibrogenic response. Our current work is focused on two potent peptide growth factors that appear to play key roles in the development of IPF. These are transforming growth factor-β1 (TGFβ1) and tumor necrosis factor-α (TNFα).
TGFβ1 is a potent inducer of extracellular matrix proteins, and up-regulation of TGFβ1 expression is a consistent feature of most fibrotic diseases. Indeed, patients with IPF have increased levels of TGFβ1 in their lungs and the levels correlate with severity of lung disease (Bartram and Speer, Citation2004). We showed that inhalation exposure of rats to asbestos produces increased expression of TGFβ1 and fibronectin at the sites of fiber deposition (Perdue and Brody, Citation1994) that correlates with elevated levels of extracellular matrix (ECM) as measured by morphometry (Chang et al., Citation1988; Coin et al., Citation1996). Importantly, over-expression of TGFβ1 in the lung via a recombinant adenovirus expressing constitutively active TGFβ1 is sufficient to cause the histopathological changes of pulmonary fibrosis in rats (Sime et al., Citation1997) and mice (Warshamana et al., Citation2002a and 2002b), while inhibitors of TGFβ1 activity such as neutralizing antibodies to TGFβ1 (Giri et al., Citation1993), TGFβ1 soluble receptor (Wang et al., Citation2002) and Smad 7 protein expression block the development of bleomycin-induced fibrogenesis in mice (Nakao et al., Citation1999). Thus, it is clear that the induction of TGFβ1 expression is an important step in the development of fibrosis. However, TGFβ1 is synthesized and secreted in a latent form as a high molecular weight complex with the latent associated peptide (LAP). Latent TGFβ1 must be converted to the mature, active form to bind to its receptors and induce its multiple biological effects.
Latent TGFβ1 can be activated by a number of mechanisms including proteolytic cleavage (Harpel et al., Citation1992), interaction with the integrin α Vβ 6 (Munger et al., Citation1999) or exposure to reactive oxygen species (Ehrhart et al., Citation1996). The factors mediating the expression and activation of TGFβ 1 in asbestos lesions are not all understood. It is our hypothesis that TNFα induces expression of TGFβ1 in cells of the lung (Sullivan et al., Citation2005) and that asbestos-induced reactive oxygen species activate latent TGFβ1 leading to increased expression of ECM and ultimately the scar that typifies asbestosis and IPF (Pociask et al., Citation2004).
TNFα has been shown to induce the expression of TGFβ1 in a variety of cell types including microglial cells (Chao et al., Citation1995), mature adipocytes (Samad et al., Citation1999), human proximal tubular cells (Phillips et al., Citation1996), and rat pulmonary artery endothelial cells (Phan et al., Citation1992) in vitro. We previously showed that TNFα rapidly up-regulates TGFβ1 and collagen mRNA expression in both primary mesenchymal and epithelial cells of the lung (Warshamana et al., Citation2001).
Importantly, Sime and colleagues showed that adenovirus-mediated expression of TNFα in lungs of normal rats induces TGFβ1 production and interstitial fibrogenesis (Sime et al., Citation1998). In another study, administration of anti-TNFα antibody reduced bleomycin-induced lung injury and decreased expression of TGFβ1 in the treated mice (Phan and Kunkel, Citation1992). Importantly, we showed that animals genetically engineered to have both TNFα receptors knocked out are resistant to the fibrogenic effects of asbestos (Liu and Brody, Citation2001), silica and bleomycin (Ortiz et al., Citation1999). There is also accumulating evidence from patients with IPF that TNFα is an important mediator of fibrogenesis. TNFα expression is increased in the lungs of these patients, and agents such as pirfenidone and etanercept with anti-TNFα properties have shown promise in treatment of some patients (Selman et al., Citation2004). Furthermore, TNFα polymorphisms indicative of high levels of TNFα expression correlate with an increased risk of developing IPF (Whyte et al., Citation2000; Maier et al., Citation2001; Yucesoy et al., Citation2001).
TNFα Induces Expression of TGFβ1 Through the ERK Pathway
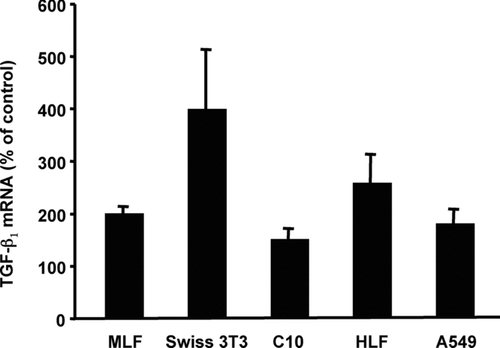
In a recent study (Sullivan et al., Citation2005), we showed that treatment of fibroblasts isolated from the lungs of adult C57 mice with recombinant murine TNF α (5 ng/ml) resulted in increased levels of TGFβ 1 mRNA within 2 hr, peaking at 200–400% of untreated controls after 12 hr, and persisting for 24 hr. The increase in TGFβ1 mRNA correlated with an increase in secretion of latent TGFβ1 as measured by ELISA (R&D Systems, Minneapolis, MN). We also found that cell lines derived from mouse embryo fibroblasts (Swiss 3T3) (Sullivan et al., Citation2005), mouse Type II alveolar epithelial cells (C10), human alveolar epithelial (A549) and human lung fibroblasts (HFL1) up-regulated their TGFβ1 expression in response to TNFα ().
FIG. 1 TNFα induces expression of TGFβ1 mRNA in lung fibroblasts and alveolar epithelial cells. Confluent monolayers of primary mouse lung fibroblasts (MLF), Swiss 3T3 fibroblasts, mouse alveolar epithelial cells (C10), human lung fibroblasts (HFL1) or human alveolar epithelial cells (A549) were serum-starved for 48 hr and then treated with TNFα (10 ng/ml) or media alone (untreated control). At the indicated times, total RNA was isolated and TGFβ1 mRNA was measured by quantitative real-time RT-PCR. The average CT calculated for TGFβ1 mRNA in unstimulated cells was 25.5, except for A549 cells that were 20.3. Relative quantitation was determined using the comparative CT method with data normalized to 36B4 riboprotein mRNA and calibrated to the average Δ CT of the corresponding untreated control. Data are represented as the mean ± SEM, relative units, with an n = 3.
The mitogen activated protein kinases (MAPKs), extracellular-stimulus regulated kinase 1 and 2 (ERK1/ERK2), c-jun N-terminal kinase (JNK), and p38 have been implicated in regulation of TGFβ1 expression (Grewal et al., Citation1999; Hamaguchi et al., Citation1999; Isono et al., Citation2000; Yue and Mulder, Citation2000) and TNFα is a potent activator of MAPKs (Wajant et al., Citation2003; Sullivan et al., Citation2005). To determine if MAPK are required for TNFα induction of TGFβ1, we used a panel of well-characterized pharmacological inhibitors of individual MAPKs. Both PD98059 and U0126, specific inhibitors of ERK1/ERK2 activation, significantly blocked TNFα -induced expression of TGFβ1 mRNA and protein in MLFs while inhibitors of JNK or p38 had little effect (Sullivan et al., Citation2005).
These results suggest a critical role for ERK1/ERK2 activation in TNFα -induced expression of TGFβ1. The specificity of the response to TNFα was demonstrated by treating fibroblasts isolated from lungs of mice that are genetically engineered to lack both TNFα receptor 1 and receptor 2 (Jackson Laboratory, Bar Harbor, ME) and showing that the ERK pathway was not activated and TGFβ1 expression was not increased in these cells (Sullivan et al., Citation2005). To determine if these results are biologically relevant, we over-expressed TNFα in the lungs of mice via a recombinant adenovirus carrying a cDNA for murine TNFα (AdTNF) and then examined ERK1/ERK2 activation and TGFβ1 expression by immunohistochemistry. We found significant increases in phosphorylated-ERK1/ERK2 (phosphorylation is indicative of activation) and TGFβ1 protein in the lungs of AdTNF-treated animals when compared to control adenovirus-treated animals. These results suggest that TNFα activates the ERK1/ERK2 pathway and up-regulates TGFβ1 in vivo.
TNFα Increases Transcription of the TGFβ1 Gene and Stabilizes TGFβ1 mRNA
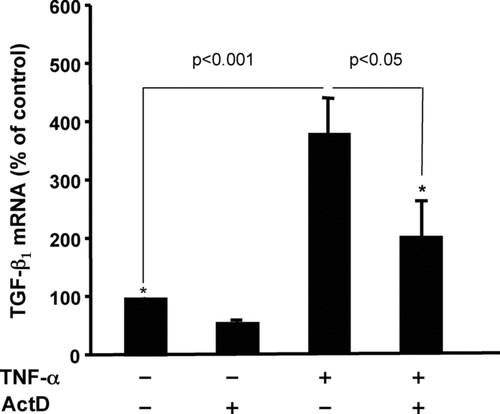
An increase in the steady-state levels of an mRNA could be the result of increased transcription and/or mRNA stabilization. Treatment of Swiss 3T3 fibroblasts with actinomycin D (ActD), an inhibitor of transcription, prior to addition of TNFα partially blocked the TNFα induction of TGFβ 1 suggesting that de novo transcription is required for maximal TNFα induction of TGFβ1 mRNA (). Nuclear runon transcription assays confirmed an increase in TGFβ1 transcription in response to TNFα (unpublished data). Chromatin immunoprecipitation assays revealed that the increase in TGFβ1 transcription was associated with recruitment of RNA polymerase II to the TGFβ1 gene (unpublished data). We also observed significant stabilization of the TGFβ1 mRNA in response to TNFα (Sullivan et al., Citation2005). In untreated cells, the half-life of TGFβ1 was ∼ 15 hr, whereas in TNFα -treated cells no degradation of the mRNA was detected after 24 hr exposure (Sullivan et al., Citation2005). This combination of transcriptional and post-transcriptional events leads to a 3–4-fold increase in steady-state TGFβ1 mRNA 6–12 hr after exposure to TNFα. The mechanisms involved in TNFα -induced TGFβ1 transcription and mRNA stabilization are currently under investigation.
FIG. 2 Inhibition of transcription partially blocks TNFα induction of TGFβ1 mRNA. Quiescent Swiss 3T3 fibroblasts were pre-treated with actinomycin D (5 μ M) or vehicle (DMSO) as control for 30 min followed by addition of 10 ng/ml TNFα for another 6 hr. Expression of TGFβ1 mRNA was determined by quantitative real-time RT-PCR as described in . Data are represented as the mean ± SEM, relative units, with an n = 3. An unpaired Student's t-test was used to assess the difference between the indicated groups.
Asbestos-Induced Reactive Oxygen Species Activate Latent TGFβ1
Deposition of asbestos fibers at the bronchiolar-alveolar duct bifurcations is associated with the induction of reactive oxygen species (ROS) and the release of TGFβ1. TGFβ1 is produced on alveolar surfaces in a latent form that must be activated to have a biological effect. We recently reported that ROS derived from chrysotile and crocidolite asbestos can activate TGFβ1 in vitro by oxidation of the latent associated peptide (Pociask et al., Citation2004). We also showed that over-expression of active TGFβ1 was sufficient to induce fibrosis in fibrogenic-resistant TNFα receptor knockout mice (Liu et al., Citation2001). To investigate the role of asbestos-induced ROS in the activation of TGFβ1 in vivo, TNFα receptor knockout mice were transduced with an adenovirus vector that over-expresses full-length latent TGFβ1 (adLTGF-β) or an empty vector control virus (MG3), and 7 d later exposed to chrysotile asbestos.
Mice over-expressing latent TGFβ1 and exposed to asbestos had significantly higher levels of active TGFβ1 in whole lung lysates measured by ELISA, greater amounts of Smad-2 phosphorylation assessed by immunohistochemistry and, importantly, more severe asbestos lesions than did mice receiving MG3 and asbestos or adLTGF-β alone. Latent TGFβ1 over-expressers treated with the anti-oxidant Tempol (4-hydroxy-2,2,6,6-tetramethylpiperidine-N-oxyl) prior to asbestos exposure demonstrated less active TGFβ1, lower levels of Smad-2 phosphorylation and consequently less severe asbestos lesions than untreated mice that were exposed to adLTGF-β and asbestos. Together these results suggest that latent TGFβ1 can be activated by asbestos-derived ROS at sites of fiber deposition and that TGFβ 1 expression plays a central role in the development of the fibrogenic lesions.
SUMMARY AND CONCLUSIONS
Exposure to asbestos fibers leads to a rapid increase in the expression of TNFα and TGFβ 1, as well as other cytokines, at the sites of fiber deposition followed by synthesis and deposition of ECM. It is clear that expression of TGFβ1 plays an important role in the pathogenesis of asbestosis and IPF. TNFα is also emerging as a key cytokine in the fibrogenic process. Our findings provide evidence that the fibrotic properties of TNFα may be mediated at least in part by its ability to up-regulate TGFβ1 through a complex mechanism involving the ERK-specific MAPK signaling pathway. TGFβ1 expression may be controlled at the levels of transcription, mRNA stability, mRNA translation, secretion of preformed protein, activation of the latent protein to its active form (Phillips et al., Citation1995, Citation1996, Citation1997; Morrisey et al., Citation2001) and inactivation by circulating proteins and ECM (Bartram and Speer, Citation2004).
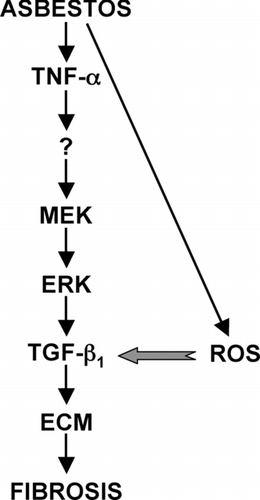
We showed that TNFα increases transcription of the TGFβ1 gene and stabilizes the transcript leading to a 2–4-fold increase in TGFβ1 expression (Sullivan et al., Citation2005). Asbestos-induced ROS provide a mechanism for activating this latent TGFβ1. Whether ROS activate latent TGFβ1 in vivo directly, as seen in vitro (Pociask et al., Citation2004), or through modulation of proteases that are known to activate latent TGFβ1 (Harpel et al., Citation1992) remains to be determined. Regardless, the increase in biologically active TGFβ1 at the alveolar surface may lead to autoinduction of TGFβ1 (Van Obberghen-Schilling et al., Citation1988) induction of other profibrotic factors such as CTGF (connective tissue growth factor) (Grotendorst, Citation1997; Duncanet al., Citation1999; Lasky and Brody, Citation2000) and synthesis and deposition of ECM (Bartram and Speer, Citation2004). Our current model is depicted in .
FIG. 3 Proposed role of TNFα in asbestos-induced fibrogenesis. TNFα is expressed by a variety of cell types in the lung early after exposure to asbestos. TNFα binding to its cellular receptors activates the MEK/ERK signaling pathway leading to increased TGFβ1 expression by mesenchymal and epithelial cells. Asbestos-induced ROS activate latent TGFβ1 that can then bind to its cellular receptors activating signaling pathways that lead to increased production of ECM, a hallmark of fibrosis. Whether TNFα directly up-regulates TGFβ1 or does so through a secondary mediator; elucidation of the cis- and trans-acting elements involved in TNFα up-regulation of TGFβ1; and the precise mechanism of ROS activation of latent TGFβ1 are currently under investigation in our laboratory.
TNFα has also been shown to antagonize TGFβ1-induced signaling (Verrecchia et al., Citation2000) suggesting that these two pleiotropic cytokines may tightly regulate each other to maintain homeostasis. Clearly, the regulatory mechanisms underlying TNFα induction of TGFβ1 expression and activation of the latent protein could be central to understanding the pathogenesis of asbestosis and IPF and may present new targets for therapeutic intervention.
Grant support is acknowledged from the National Institutes of Health (HL 60532 [ARB], ES06766 [ARB]).
REFERENCES
- Bartram U., Speer C. P. The role of transforming growth factor-β in lung development and disease. Chest 2004; 125: 754–765
- Brody A. R. Asbestos. Toxicology of the Respiratory System, R. R. Roth. Pergamon Press, New York 1997; 393–413
- Chang L. Y., Overby L. H., Brody A. R., Crapo J. D. Progressive lung cell reactions and extracellular matrix production after a brief exposure to asbestos. Am. J. Pathol. 1988; 131: 156–170
- Chao C. C., Hu S., Sheng W. S., Tsang M., Peterson P. K. Tumor necrosis factor-α mediates the release of bioactive transforming growth factor-β in murine microglial cell cultures. Clin. Immunol. Immunopathol. 1995; 77: 358–365
- Coin P. G., Osornio-Vargas A., Roggli V. L., Brody A. R. Pulmonary fibrogenesis after three consecutive inhalation exposures to chrysotile asbestos. Am. J. Respir. Crit. Care Med. 1996; 154: 1511–1519
- Duncan M. R., Frazier K. S., Abramson S., Williams S., Klapper H., Huang X., Grotendorst G. R. Connective tissue growth factor mediates transforming growth factor β -induced collagen synthesis: Down-regulation by cAMP. FASEB J 1999; 13: 1774–1786
- Ehrhart E. J., Gillette E. L., Barcellos-Hoff M. H. Immunohistochemical evidence of rapid extracellular matrix remodeling after iron-particle irradiation of mouse mammary gland. Radiat. Res. 1996; 145: 157–162
- Giri S. N., Hyde D. M., Hollinger M. A. Effect of antibody to transforming growth factor-β on bleomycin-induced accumulation of lung collagen in mice. Thorax 1993; 48: 959–966
- Grewal J. S., Mukhin Y. V., Garnovskaya M. N., Raymond J. R., Greene E. L. Serotonin 5-HT2A receptor induces TGF-β 1 expression in mesangial cells via ERK: Proliferative and fibrotic signals. Am. J. Physiol. 1999; 276: F922–930
- Grotendorst G. R. Connective tissue growth factor: A mediator of TGF-β action on fibroblasts. Cytokine Growth Factor Rev. 1997; 8: 171–179
- Hamaguchi A., Kim S., Izumi Y., Zhan Y., Yamanaka S., Iwao H. Contribution of extracellular signal-regulated kinase to angiotensin II-induced transforming growth factor-β 1 expression in vascular smooth muscle cells. Hypertension 1999; 34: 126–131
- Harpel J. G., Metz C. N., Kojima S., Rifkin D. B. Control of TGF-β activity: Latency vs. activation. Prog. Growth Factor Res. 1992; 4: 321–335
- Isono M., Cruz M. C., Chen S., Hong S. W., Ziyadeh F. N. Extracellular signal-regulated kinase mediates stimulation of TGF-β 1 and matrix by high glucose in mesangial cells. J. Am. Soc. Nephrol 2000; 11: 2222–2230
- Katzenstein A. L., Myers J. L. Idiopathic pulmonary fibrosis: Clinical relevance of pathologic classification. Am. J. Respir. Crit. Care Med. 1998; 157: 1301–1315
- Lasky J. A., Brody A. R. Interstitial fibrosis and growth factors. Environ. Health Perspect. 2000; 108(S4)751–762
- Liu J. Y., Brody A. R. Increased TGF-β 1 in the lungs of asbestos-exposed rats and mice: Reduced expression in TNFα receptor knockout mice. J. Environ. Pathol. Toxicol. Oncol. 2001; 20: 77–87
- Liu J. Y., Sime P. J., Wu T., Warshamana S., Pociask D., Tsai S. Y., Brody A. R. Transforming growth factor β1 over-expression in tumor necrosis factor-α receptor knockout mice induces fibroproliferative lung disease. Am. J. Respir. Cell Mol. Biol. 2001; 25: 3–7
- Maier L. A., Sawyer R. T., Bauer R. A., Kittle L. A., Lympany P., McGrath D., Dubois R., Daniloff E., Rose C. S., Newman L. S. High beryllium-stimulated TNFα is associated with the -308 TNFα promoter polymorphism and with clinical severity in chronic beryllium disease. Am. J. Respir. Crit. Care Med. 2001; 164: 1192–1199
- Morris G., Brody A. Molecular mechanisms of particle-induced lung disease. Environmental and Occupational Medicine, Third Edition, W. N. Rom. Lippincort-Raven, Philadelphia 1998; 305–333
- Morrisey K., Evans R. A., Wakefield L., Phillips A. O. Translational regulation of renal proximal tubular epithelial cell transforming growth factor-β 1 generation by insulin. Am. J. Pathol. 2001; 159: 1905–1915
- Mossman B. T., Churg A. Mechanisms in the pathogenesis of asbestosis and silicosis. Am. J. Respir. Crit. Care Med. 1998; 157: 1666–1680
- Munger J. S., Huang X., Kawakatsu H., Griffiths M. J., Dalton S. L., Wu J., Pittet J. F., Kaminski N., Garat C., Matthay M. A., Rifkin D. B., Sheppard D. The integrin α Vβ 6 binds and activates latent TGFβ 1: A mechanism for regulating pulmonary inflammation and fibrosis. Cell 1999; 96: 319–328
- Nakao A., Fujii M., Matsumura R., Kumano K., Saito Y., Miyazono K., Iwamoto I. Transient gene transfer and expression of Smad7 prevents bleomycin-induced lung fibrosis in mice. J. Clin. Invest. 1999; 104: 5–11
- Ortiz L. A., Lasky J., Lungarella G., Cavarra E., Martorana P., Banks W. A., Peschon J. J., Schmidts H. L., Brody A. R., Friedman M. Up-regulation of the p75 but not the p55 TNFα receptor mRNA after silica and bleomycin exposure and protection from lung injury in double receptor knockout mice. Am. J. Respir. Cell Mol. Biol. 1999; 20: 825–833
- Perdue T. D., Brody A. R. Distribution of transforming growth factor-β 1, fibronectin, and smooth muscle actin in asbestos-induced pulmonary fibrosis in rats. J. Histochem. Cytochem. 1994; 42: 1061–1070
- Phan S. H., Gharaee-Kermani M., McGarry B., Kunkel S. L., Wolber F. W. Regulation of rat pulmonary artery endothelial cell transforming growth factor-β production by IL-1β and tumor necrosis factor-α. J. Immunol. 1992; 149: 103–106
- Phan S. H., Kunkel S. L. Lung cytokine production in bleomycin-induced pulmonary fibrosis. Exp. Lung Res. 1992; 18: 29–43
- Phillips A. O., Steadman R., Topley N., Williams J. D. Elevated D-glucose concentrations modulate TGF-β 1 synthesis by human cultured renal proximal tubular cells: The permissive role of platelet-derived growth factor. Am. J. Pathol. 1995; 147: 362–374
- Phillips A. O., Topley N., Morrisey K., Williams J. D., Steadman R. Basic fibroblast growth factor stimulates the release of pre-formed TGF-β 1 from human proximal tubular cells in the absence of de-novo gene transcription of mRNA translation. Lab. Invest. 1997; 76: 591–600
- Phillips A. O., Topley N., Steadman R., Morrisey K., Williams J. D. Induction of TGF-β 1 synthesis in D-glucose primed human proximal tubular cells: Differential stimulation by the macrophage derived pro-inflammatory cytokines IL-1β and TNFα. Kidney Int. 1996; 50: 1546–1554
- Pociask D. A., Sime P. J., Brody A. R. Asbestos-derived reactive oxygen species activate TGF-β 1. Lab. Invest. 2004; 84: 1013–1023
- Samad F., Uysal K. T., Wiesbrock S. M., Pandey M., Hotamisligil G. S., Loskutoff D. J. Tumor necrosis factor-α is a key component in the obesity-linked elevation of plasminogen activator inhibitor 1. Proc. Natl. Acad. Sci. USA 1999; 96: 6902–6907
- Selman M., Thannickal V. J., Pardo A., Zisman D. A., Martinez F. J., Lynch J. P., 3rd. Idiopathic pulmonary fibrosis: Pathogenesis and therapeutic approaches. Drugs 2004; 64: 405–430
- Sime P. J., Marr R. A., Gauldie D., Xing Z., Hewlett B. R., Graham F. L., Gauldie J. Transfer of tumor necrosis factor-α to rat lung induces severe pulmonary inflammation and patchy interstitial fibrogenesis with induction of transforming growth factor-β 1 and myofibroblasts. Am. J. Pathol. 1998; 153: 825–832
- Sime P. J., Xing Z., Graham F. L., Csaky K. G., Gauldie J. Adenovector-mediated gene transfer of active transforming growth factor-β 1 induces prolonged severe fibrosis in rat lung. J. Clin. Invest. 1997; 100: 768–776
- Sullivan D. E., Ferris M., Pociask D., Brody A. R. Tumor necrosis factor-α induces transforming growth factor-β 1 expression in lung fibroblasts through the extracellular signal-regulated kinase pathway. Am. J. Respir. Cell Mol. Biol. 2005; 32: 342–349
- Van Obberghen-Schilling E., Roche N. S., Flanders K. C., Sporn M. B., Roberts A. B. Transforming growth factor-β 1 positively regulates its own expression in normal and transformed cells. J. Biol. Chem. 1988; 263: 7741–7746
- Verrecchia F., Pessah M., Atfi A., Mauviel A. Tumor necrosis factor-α inhibits transforming growth factor-β /Smad signaling in human dermal fibroblasts via AP-1 activation. J. Biol. Chem. 2000; 275: 30226–30231
- Wajant H., Pfizenmaier K., Scheurich P. Tumor necrosis factor signaling. Cell Death Differentiation 2003; 10: 45–65
- Wang Q., Hyde D. M., Gotwals P. J., Giri S. N. Effects of delayed treatment with transforming growth factor-β soluble receptor in a three-dose bleomycin model of lung fibrosis in hamsters. Exp. Lung Res. 2002; 28: 405–417
- Warshamana G. S., Corti M., Brody A. R. TNFα, PDGF, and TGFβ 1 expression by primary mouse bronchiolar-alveolar epithelial and mesenchymal cells: TNFα induces TGFβ 1. Exp. Mol. Pathol. 2001; 71: 13–33
- Warshamana G. S., Pociask D. A., Fisher K. J., Liu J. Y., Sime P. J., Brody A. R. Titration of non-replicating adenovirus as a vector for transducing active TGFβ 1 gene expression causing inflammation and fibrogenesis in the lungs of C57BL/6 mice. Int. J. Exp. Pathol. 2002a; 83: 183–201
- Warshamana G. S., Pociask D. A., Sime P., Schwartz D. A., Brody A. R. Susceptibility to asbestos-induced and transforming growth factor-β 1-induced fibroproliferative lung disease in two strains of mice. Am. J. Respir. Cell Mol. Biol. 2002b; 27: 705–713
- Whyte M., Hubbard R., Meliconi R., Whidborne M., Eaton V., Bingle C., Timms J., Duff G., Facchini A., Pacilli A., Fabbri M., Hall I., Britton J., Johnston I., Di Giovine F. Increased risk of fibrosing alveolitis associated with interleukin-1 receptor antagonist and tumor necrosis factor-α gene polymorphisms. Am. J. Respir. Crit. Care Med. 2000; 162: 755–758
- Yucesoy B., Vallyathan V., Landsittel D. P., Sharp D. S., Weston A., Burleson G. R., Simeonova P., McKinstry M., Luster M. I. Association of tumor necrosis factor-α and interleukin-1 gene polymorphisms with silicosis. Toxicol. Appl. Pharmacol. 2001; 172: 75–82
- Yue J., Mulder K. M. Requirement of Ras/MAPK pathway activation by transforming growth factor-β for transforming growth factor-β 1 production in a Smad-dependent pathway. J. Biol. Chem. 2000; 275: 30765–30773