Abstract
Pyridoxine, a B6 vitamin, is a co-factor in a variety of enzymatic reactions involved in intermediary metabolism. The effects of pyridoxine deficiency and supplementation on hematological profiles, lymphocyte function, and hepatic CYP1A1 and CYP2E1 were investigated in B6C3F1 mice fed a diet containing either 0 (i.e., pyridoxine-deficient diet, [PD]); or 7 mg pyridoxine-HCl/kg (i.e., control diet, [CD]) for 8 or 13 weeks followed by administration of 500 μg pyridoxine-HCl (IP) daily for either 2 (PD-S2 and CD-S2) or 3 (PD-S3 and CD-S3) consecutive days. Results demonstrated that erythrocyte aspartate aminotransferase activity coefficient (EAST-AC) values, which reflect host pyridoxine status, were significantly higher in PD mice than in CD mice, and dropped to control levels after supplementation. PD mice had significantly reduced weight gains, mean corpuscular volume (MCV), hemoglobin (HGB), and hematocrit (HCT) levels compared to CD mice after 8 and 13 weeks on the prescribed diet. In addition, PD mice had significantly lower circulating levels of total white blood cells, but higher red blood cell numbers after 8 weeks (compared to CD mice). Pyridoxine supplementation for 3 days restored HGB levels in PD mice to that of the unsupplemented CD controls; HCT, MCV and MCH levels were also increased in PD-S3 mice compared to their unsupplemented PD counterparts, but failed to reach comparable levels to those seen in mice fed a control diet. The pyridoxine-deficient diet also resulted in decreased mitogen stimulated T-lymphocyte proliferation after a 13-week feeding regimen and increased hepatic CYP1A1 activity that was reversed by pyridoxine supplementation. These studies demonstrate in a murine model that pyridoxine deficiency can cause multiple alterations that, in many cases, can be reversed by supplementation.
Introduction
Malnutrition and vitamin deficiency are health concerns in developing countries. Although the incidence of severe Vitamin B6 deficiency is rare, marginal to moderate vitamin deficiency is still prevalent, particularly in children and adolescents living in rural areas, pregnant women, the poor, and vegetarians. A major cause of Vitamin B6 deficiency is insufficient intake; deficiency can develop rapidly within a few weeks of consuming a diet low in Vitamin B6 (Schreurs et al., Citation1976; Kirksey et al., Citation1978; Leklem Citation1990; Vudhivai et al., Citation1991; Nontasut et al., Citation1996; Setiawan et al., Citation2000; Tada et al. Citation2002; Costello et al., Citation2003).
Vitamin B6 is a general term that refers to three primary forms, i.e., pyridoxine, pyridoxal, and pyridoxamine. Pyridoxine is converted to pyridoxal-5’-phosphate (PLP), a biologically active form involved in intermediary metabolism as a cofactor of enzymatic reactions in nucleotide and protein synthesis, cell proliferation, and maintenance of immune competence (Axelrod, Citation1981; Chaney, Citation1993; Rall and Meydani, Citation1993). The effects of pyridoxine deficiency in both human and experimental animals have been reported and include: delayed growth; seborrhea and pellagra-like dermatitis; peripheral neuropathy; impairment of both humoral and cell-mediated immunity, i.e., spleen and thymic atrophy, reductions in peripheral lymphocyte levels and decreased response to mitogens (Willis-Carr and St. Pierre, Citation1978; Axelrod, Citation1981; Gridley et al., Citation1987; Doke et al., Citation1998; Ladipo, Citation2000); development of microcytic anemia (Chandra, Citation1983; Ha et al., Citation1984; Gridley et al., Citation1987; Rall and Meydani, Citation1993; Frydas et al., Citation1999; Ronnenberg et al., Citation2000); and, increased tendency to develop infections (Rall and Meydani Citation1993; Holman, Citation1995). Studies on pyridoxine supplementation to improve pyridoxine-deficient status and to mitigate or repair toxicities associated with the deficient state have been reported (Cassel et al., Citation1978; Kwak et al., Citation2002; Chiang et al., Citation2005; Oshiro et al., Citation2005; Wolters et al., Citation2005).
Determination of plasma PLP and the urinary metabolite, 4-pyridoxic acid are routinely used for direct assessment of pyridoxine status (Bisp et al., Citation2002); however, indirect measurement of PLP-dependent enzyme activity is also common. Pyridoxal-5’-phosphate is a coenzyme of aspartate aminotransferase (also known as glutamic-oxaloacetic transaminase, which is present in both the cytoplasm and mitochondria of cells, particularly erythrocytes) and which is involved in the inter-conversion of amino acids and α-ketoacids by the transfer of amino groups (Holman, Citation1995). Therefore, in vitro measurements of erythrocyte aspartate aminotransferase activity, usually determined by spectrophotometry, reflect the activity afforded by pyridoxine available in erythrocytes. Pyridoxine deficiency decreases the activity of the enzyme with a concomitant induction in the activity coefficient; supplementation of pyridoxine increases the enzyme activity, resulting in a reduction of the activity coefficient (Raica and Sauberlich, Citation1964; Woodring and Storvick, Citation1970; Solomon and Hillman, Citation1978; Kwak et al., Citation2002). Thus, the measurement of erythrocyte aspartate aminotransferase activity coefficient (EAST-AC) has been frequently used to determine pyridoxine status in both humans and experimental animals (Woodring and Storvick, Citation1970; Pongpaew et al., Citation1993; Aramphongphan et al., Citation1994; Tovar et al., Citation1996; Sanchez, Citation1999; Chiang et al., Citation2005; Oshiro et al., Citation2005).
A diet deficient in pyridoxine has also been shown to cause reduction in the activities of a number of other PLP-dependent enzymes, e.g., alanine glyoxalate aminotransferase (Takada et al., Citation1984), serum glutamate-oxaloacetate transaminase (GOT), and glutamate-pyruvate transaminase (GPT) in the rat (Ruchirawat et al.,Citation1990). In addition, pyridoxine deficiency can result in alterations in PLP-independent enzymes (such as glyceraldehyde-3-phosphate dehydrogenase) and β-actin (Oka et al., Citation1993) for which modulation of gene expression by pyridoxine has been reviewed (Oka, Citation2001). It has also been reported that pyridoxine deficiency affects biotransformation and toxicity of chemicals including carcinogens such as N-nitrosodimethylamine (Ruchirawat et al., Citation1990), and leads to an increase in microsomal N-nitrosodimethylamine demethylase activity in rat liver and kidney, as well as NADPH cytochrome c reductase activity in pyridoxine-deficient rats (Aramphongphan et al., Citation1994).
Hepatic cytochrome P450 enzymes such as CYP1A1 and CYP2E1 are key enzymes involved in metabolic activation of many pro-carcinogens found in the environment, including N-nitrosodimethylamine (NDMA), polycyclic aromatic hydrocarbon (PAHs), and benzene. Cytochrome P4501A1 and 2E1 are expressed constitutively in the liver and extra- hepatic tissues, including kidneys, lung, lymphocytes, and bone marrow (Coon et al., Citation1996; Parkinson, Citation2001). Expression of CYP1A1 is constitutively negligible, but is markedly induced in a large number of tissues after induction by PAHs (Nebert et al., Citation2004). CYP2E1 is inducible by chemicals, including ethanol and acetone, (Smith, Citation1996; Gonzalez, Citation2007), as well as by other factors, such as nutritional status and starvation (Soh et al., Citation1996). Therefore, alterations of these enzymes are of great toxicological and carcinogenic relevance.
This study was undertaken to evaluate the effects of mild-to-moderate pyridoxine deficiency on hematological parameters, immune function, and alterations in host hepatic microsomal cytochrome P450 enzymes (CYP1A1 and CYP2E1). The effect of supplementation on the physiological/biochemical effects associated with the deficiency were also examined.
Materials and methods
Experimental animals and diet
The animal studies reported here were conducted at the Chulabhorn Research Institute Laboratory Animal Unit (Bangkok, Thailand) under the supervision of the Institute Animal Care and Use Committee (IACUC). This IACUC reviewed and approved all study protocols related to the use of animals to make sure that they adhered to internationally-accepted standards for the ethical use of animals in research.
All mice were maintained in the Chulabhorn Research Institute. Male weanling B6C3F1 mice (3-weeks-old at the initiation of the study) were housed individually in polycarbonate cages with wire mesh floor inserts. All mice were maintained under strict hygienic conditions in rooms with controlled temperature (24 ± 2°C), humidity (55 ± 10%), and light-dark cycle (12 hours: 12 hours). Food and water were provided ad libitum. Mice were acclimated for 2 weeks, and then randomly assigned to either a control diet (CD) or pyridoxine-deficient diet (PD) group. Average initial body weights did not significantly differ between the groups.
The control diet consisted of an agar-based semi-purified diet according to the American Institute of Nutrition formulation (AIN-93G), for which the recommended level of pyridoxine-HCl/kg is 7 mg (Reeves, Citation1997), while pyridoxine was completely omitted from the deficient diet ( 0 mg pyridoxine-HCl/kg diet). All ingredients were purchased from ICN (Costa Mesa, CA), except for the sucrose (Mitr-Phol, Supanburi, Thailand) and soybean oil (TIP Vegetable oil, Nonthaburi, Thailand). Food consumption was recorded daily and body weights were recorded twice weekly.
Experimental design
To study the development and progression of pyridoxine deficiency, separate groups of mice were fed control or pyridoxine-deficient diets for up to 14 weeks. Blood samples were collected weekly and pyridoxine status determined by erythrocyte aspartate aminotransferase activity coefficient (EAST-AC). The remaining animal groups were used to assess the effects from pyridoxine deficiency and supplementation. Mice were fed the test diets for 8 or 13 weeks and at the end of each feeding period, subgroups of mice in each respective diet regimen received 500 μg pyridoxine-HCl in normal saline daily via intraperitoneal (IP) injection (200 μl) for either 2 (CD-S2 and PD-S2) or 3 (CD-S3 and PD-S3) consecutive days. Administration by IP injection was selected rather than addition to food as it delivered a more accurate dose of pyridoxine hydrochloride that could be easily measured. Saline vehicle was administered (IP) to time-matched CD and PD mice that did not receive pyridoxine supplementation and mice were sacrificed 24 hours after the last injection. A separate set of naive animals was administered β-napthoflavone in corn oil (IP) at 24 mg/mouse for 2 days, or 5% acetone (v/v) in drinking water for three consecutive days before their livers were pooled and used for microsomal preparations (as positive controls for CYP1A1 and CYP2E1 studies, respectively).
Pyridoxine status
Pyridoxine status in all mice was monitored by tail-tip blood drawn into heparinized micro-hematocrit tubes. Samples were first centrifuged (10,000 × g) for 5 minutes to pack erythrocytes. A hemolysate was then prepared by mixing 30 μl of packed erythrocytes with 30 μl 0.85% (w/v) NaCl solution and 1. 14 ml 0.025% (w/v) saponin solution, incubated at 25°C for 30 minutes, and then centrifuged (1000 × g) to separate cell debris. The resulting supernatant was stored at -80°C until EAST-AC was analyzed.
Measurement of blood EAST-AC was carried out using the method of Changbumrung et al. (Citation1984). Briefly, 200 μl hemolysate was mixed with either 1.98 or 2. 0 ml (stimulated and unstimulated samples, respectively) of 50 mM/2.5 mM/150 mM triethanolamine-EDTA-aspartic acid buffer (BAA, pH 7.5). All tubes then received 50 μl of a 50 μg malate dehydrogenase/ml suspension (MDH; Roche Diagnostics, Indianapolis, IN) and 50 μl of 11.2 mM nicotinamide adenine dinucleotide (NADH; Roche). The stimulated sample then received 20 μl pyridoxal-5’- phosphate (PLP, 2 mg/ml in BAA; Sigma-Aldrich, St. Louis, MO), bringing the total volume of both stimulated and unstimulated samples to 2. 3 ml. Samples were incubated for 30 minutes (25°C) in the dark before 200 μl 0.2 M α-oxoglutaric acid (pH 6.8) was added to initiate the enzymatic reaction. The reduction of NADH over the first 10 minutes was measured at 340 nm in a UV 3100 spectrophotometer (Shimadzu, Kyoto, Japan). EAST-AC was then calculated by determining the ratio of stimulated (PLP added) to unstimulated sample activity.
Hematology and sample collection
After sacrifice by CO2 asphyxiation, blood was collected from each mouse by cardiac puncture. Samples (300 μl of whole blood) were transferred into tubes containing K3EDTA for complete blood counts and white blood cell differentiation in an automated blood cell analyzer (CELL-DYNE 3700, Abbott Laboratories, Abbott Park, IL). The remaining blood was transferred to heparinized tubes and centrifuged (10,000 × g for 5 minutes) to isolate plasma from packed cells. After blood collection, the liver was quickly harvested and transferred into ice-cold 0.9% NaCl for liver microsomal preparation. The spleen, kidneys, and thymus were also aseptically recovered and weighed.
Lymphocyte proliferation assay
Each spleen was rinsed in 1. 5 ml phosphate-buffered saline (PBS, pH 7.3) and then placed in a sterile petri-dish containing RPMI media (RPMI 1640 without phenol red and supplemented with 5% fetal bovine serum [FBS], 0.1% L-glutamine, 1% pyruvate, 1% non-essential amino acids, 0.2% gentamycin, and 1% penicillin/streptomycin [Gibco, Glen Cove, NY]). The organ was crushed through a metal mesh with the rubber end of a 3 ml syringe to generate a single-cell suspension of splenocytes. Six ml of the suspension was layered atop 5 ml Ficoll-Hypaque Plus (Amersham Bioscience, Manchester, England) in a 15 ml conical tube, and centrifuged at 400 × g (at 4°C) for 20 minutes. The mononuclear cell layer was removed and washed three times with supplemented RPMI media ( 2 ml each). The resultant lymphocyte pellet was resuspended in 2 ml supplemented RPMI, and cell number and viability were determined by hemocytometer counting and trypan blue exclusion, respectively. Cells were adjusted to 2 × 106 cells/ml for use in the proliferation assay. Concanavalin-A (ConA; Sigma) and lipopolysaccharide (LPS; Sigma) were used to stimulate T- and B-cell proliferation, respectively, using a method similar to that described by Ng and Zelikoff (Citation2008).
Prior to the actual study, dilutions (2–30 μg/ml) of ConA and LPS were tested to determine the appropriate stimulatory concentration (i.e., 10 μg/ml) for each mitogen. One hundred μl each of media and cell suspension (2 × 105 cells/well) was added to control wells of a flat-bottomed 96-well plate to determine basal (unstimulated) level proliferation. Background OD was determined from wells that received 200 μl media only. Other wells received 100 μl of cell suspension and an equal volume of the respective mitogen solutions. All samples were analyzed in triplicate. The cells were incubated at 37°C (with 5% CO2) for 48 hours, and then provided 20 μl freshly-prepared MTT (methylthiazolyldiphenyltetrazolium; Sigma, 5 mg/ml in PBS) and incubated for an additional 4 hours. A 50 μl aliquot of 10% sodium dodecyl sulfate (SDS; in 0.01 N HCl) was then added to stop the reaction and the cells were incubated overnight. The absorbance in each well was then assessed at 600 nm in an automatic microtiter plate reader (Molecular Devices, Sunnyvale, CA). The stimulation index (SI) was calculated based upon the following equation:
Microsome preparation
Harvested livers were perfused with ice-cold physiological saline (0.9% NaCl, [w/v]), weighed, and then homogenized in 3 vol of 0.01 M potassium phosphate/1.15% (w/v) potassium chloride buffer (pH 7.4), at a ratio of 1 g of liver to 3 ml buffer. The homogenate was centrifuged at 9,000 × g for 30 minutes (at 4°C) and the supernatant (S9 fraction) then transferred to a fresh ultracentrifuge tube for additional centrifugation for 1 hour at 105,000 × g (at 4°C). The microsomal pellet was then isolated from the cytosolic-supernatant and resuspended in 0.01 M potassium phosphate. Microsomal protein concentration was measured using a Bio-Rad protein assay kit (Bio-Rad, Hercules, CA), based on the method of Bradford (Bradford, Citation1976). All materials were stored at -80°C until needed.
Immunoblotting of hepatic CYP1A1 and CYP2E1
Hepatic microsomal protein (30 μg for CYP1A1 or 5 μg for CYP2E1) was denatured with SDS sample buffer (250 mM Tris-HCl [pH 6.8], 2% 2-mercaptoethanol, 4% SDS, 20% glycerol, and 0.02% bromophenol blue), boiled for 5 minutes, loaded to 10% (w/v) SDS-polyacrylamide gel (Towbin et al., Citation1979), and then electrophoresed (Mini-Protein III, Bio-Rad). The gel contents were then transferred electrophoretically to a Hybond™-P PVDF-membrane (Amersham). The membrane was placed (for 2 hours) in blocking solution (5% non-fat skimmed milk in 0.1% Tween-20 in Tris-buffer saline [TBST, pH 7.6]) and then incubated overnight (at 4°C) with primary rabbit anti-rat CYP1A1 or CYP2E1 polyclonal antibody (Chemicon International, Inc., Temecula, CA) at 1:500 and 1:20,000 dilutions (in blocking solution), respectively. After several washes in 0.1% TBST to remove non-adherent primary antibody, the membranes were incubated with horseradish peroxidase-conjugated goat anti-rabbit IgG secondary antibody (Chemicon; 1:10,000 dilution in 0.1% Tris-buffer saline [pH 7.6]) for 1 hour at room temperature. A chemiluminscence reaction using the ECL Plus kit (Amersham) was carried out followed by exposure to Hyperfilm ECL™ (Amersham). Imaging of all specific protein bands and band intensities were performed using a GS-710 Calibrated-Imaging densitometer and Quantity One Quantitation software (Bio-Rad).
Although housekeeping proteins (like glyceraldehyde-3-phosphate dehydrogenase [GAPDH] and β-actin) have been used as loading controls in Western blot analyses, they were not employed in these studies. This decision was based on the fact that pyridoxine deficiency caused an increase in hepatic microsomal β-actin protein expression in preliminary studies (data not shown). These effects were similar to those in other investigations. where it was reported that the expression of several mRNAs, including those for β-actin and GAPDH, was increased during pyridoxine deficiency (Oka et. al., Citation1993). As such, because the use of housekeeping proteins was not practical here, careful attention was instead paid to ensuring that all microsomal samples were prepared to the same protein concentration assuring consistency in protein loading between the individual gels.
CYP1A1 activity (Ethoxyresorufin O-deethylase [EROD] assay)
Measurement of EROD activity was performed in a 96-well microplate in which the incubation mixture in each well contained 10 μg of hepatic microsomal protein, 2 nM 7-ethoxyresorufin, and 67 mM phosphate buffer (pH 7.4), in a total volume of 200 μl. The enzymatic reaction was initiated by adding 10 mM nicotinamide adenine dinucleotide phosphate (NADPH) into the incubation mixture. EROD activity was measured as resorufin formation by the rate of fluorescence change by time; fluorescence was measured over a 5 minute period (at 30°C) at a 530 nm excitation and a 595 nm emission in a Spectra MAX-GeminiXS plate reader (Molecular Devices). The amount of resorufin formed was calculated against a calibration curve of resorufin which ranged from 2.5–20 pmol. EROD activity was expressed as pmol resorufin formed/mg microsomal protein/minute.
CYP2E1 activity (p-Nitrophenol Hydroxylase [p-NPH] activity)
Hydroxylation of p-nitrophenol to p-nitrocatechol was measured using spectrophotometry (Reinke and Moyer, Citation1985). Briefly, a mixture ( 1 ml) consisting of 0. 4 mg microsomes, 0.1 M potassium phosphate buffer (pH 7.4), 1 mM p-nitrophenol (Sigma), and water (to volume) was pre-incubated at 37°C for 3 minutes prior to addition of 10 μl 100 mM NADPH (Sigma). After 10 minutes, the reaction was terminated by addition of 200 μl 1.5 N perchloric acid (Merck, KGaA, Darmstad, Germany). The precipitated proteins were then removed by centrifugation for 10 minutes at 3,000 × g. The post- precipitation sample was then diluted 10:1 with 0. 1 ml 10 N NaOH, and the absorbance of p-nitrocatechol measured at 546 nm in a UV3100 spectrophotometer (Shimadzu, Kyoto, Japan). CYP2E1-specific p-NPH activity was expressed as nanomoles p-nitrocatechol formed per minute per mg protein, using an extinction coefficient of 10.28 mM−1 • cm−1.
Statistical analysis
All values were expressed as mean ± SE. Statistical analyses were performed using one-way analysis of variance (ANOVA) followed by post-hoc testing when appropriate using Fisher’s LSD or Kruskal–Wallis followed by the Mann–Whitney U-test using SPSS v15.0 software (SPSS, Inc., Chicago, IL). Differences were considered statistically significant at p-values < 0.05; differences with a p-value < 0.001 were considered highly significant.
Results
Effect of pyridoxine deficiency on mouse growth and food consumption
The development and progression of pyridoxine deficiency status was determined by EAST-AC in peripheral red blood cells collected weekly in male B6C3F1 mice, fed either a control (CD) or pyridoxine-deficient (PD) diet for 14 weeks (). EAST-AC levels in the deficient mice were significantly higher than those in time-matched CD mice (p < 0.001) within one week of the start of the diets (1.28 vs. 1.02, respectively). Highly significantly differences in EAST-AC levels became apparent about 5 weeks into the feeding period and persisted throughout the exposure period; in contrast the EAST-AC levels in the CD group did not exceed a value of 1.2 throughout the entire experimental period.
Figure 1. Time-course of pyridoxine status. The development and progression of pyridoxine status was determined as erythrocyte aspartate aminotransferase activity coefficients (EAST-AC) in control (CD, n = 6) and pyridoxine-deficient mice (PD, n = 5) over a 14-week feeding period. Values are expressed as mean ± SE per each experimental group. *Significantly different from the control mice at p < 0.001.
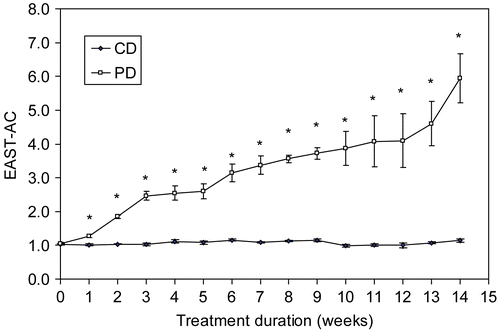
The effect of pyridoxine deficiency and supplementation on EAST-AC values was also examined in mice fed the deficient diet for 8 or 13 weeks (). By the end of each feeding period, PD mice had significantly greater EAST-AC values compared with time-matched CD control mice and these values increased with duration of feeding (2.3- and 5-fold at 8 and 13 weeks, respectively). After feeding for 8 weeks, daily supplementation for 2 or 3 days resulted in a significant lowering of EAST-AC values amongst the PD-S2 and PD-S3 mice compared to time-matched PD mice. In fact, resultant levels in the supplemented mice were comparable to those measured in the time-matched CD mice. In addition, no significant differences were observed between PD-S2 and CD-S2 or PD-S3 and CD-S3 mice. At 13 weeks, EAST-AC values in supplemented PD mice (i.e., PD-S2 and PD-S3) were significantly reduced relative to their unsupplemented counterparts (PD), but still remained somewhat elevated (albeit, not statistically significant) compared to those measured in the unsupplemented CD counterparts (1.37 vs. 1.15, p = 0.42).
Table 1. Effects of pyridoxine deficiency on erythrocyte-aspartate aminotransferase activity coefficient (EAST-AC) in B6C3F1 mice.
At the start of the experiment, the body weight range of experimental animals was 19-23 g, with no statistically significant differences within or between groups. After 5 weeks of feeding (), the body weight of PD mice was significantly lower than that of CD mice (25.6 vs. 27.8 g, p < 0.05). Although PD mice presented signs of reduced appetite (i.e., biting but not eating food and scattering food at the bottom of the cage), daily food consumption by these animals was not significantly different from that of the CD mice (6.7 vs. 6.8 g of food/day, respectively). Some animals in both treatment groups developed abdominal alopecia and after 13 weeks of feeding, some mice presented with tail scaling; however, no morbidity or mortality was observed over the entire course of the study. Pyridoxine supplementation for either 2 or 3 days had no significant effects on either CD or PD mouse body weights.
Figure 2. Effects of pyridoxine deficiency on growth. Body weights of control (CD) and pyridoxine-deficient (PD) mice, with or without supplementation of pyridoxine-HCl (500 μg, IP, daily for 2 [S2] or 3 consecutive days [S3]) were compared at the end of the 8 and 13 weeks feeding periods. Values are expressed as mean ± SE per each experimental group from 0–8 weeks (n = 13) and from 9–13 weeks (n = 6). *Significantly different from time-matched controls (CD) at p < 0.001.
![Figure 2. Effects of pyridoxine deficiency on growth. Body weights of control (CD) and pyridoxine-deficient (PD) mice, with or without supplementation of pyridoxine-HCl (500 μg, IP, daily for 2 [S2] or 3 consecutive days [S3]) were compared at the end of the 8 and 13 weeks feeding periods. Values are expressed as mean ± SE per each experimental group from 0–8 weeks (n = 13) and from 9–13 weeks (n = 6). *Significantly different from time-matched controls (CD) at p < 0.001.](/cms/asset/3cb2b68c-aa52-4e69-a8b9-5956ecc7ad8e/iimt_a_408559_f0002_b.gif)
After 8 weeks of feeding, relative liver weights (liver/ body weight ratio) of PD mice were not significantly different from those of the CD mice (0.042 vs. 0.040, ). Pyridoxine supplementation alone did not affect these values in CD mice, but caused relative liver weights to increase in the PD mice. Relative spleen weights were significantly greater in the PD mice, either with or without supplementation, when compared to their respective CD counterparts (p < 0.001), A significant increase in the absolute spleen weights was also observed (data not shown). After 13 weeks on their respective diets, relative liver and spleen weights of PD mice were all significantly greater (p < 0.001) than their respective CD counterparts (). No significant differences in relative kidney or thymus weights were observed amongst or between the treatment groups over the course of the study.
Figure 3. Effect of pyridoxine deficiency on relative organ weights. Mice were fed either a control (CD) or pyridoxine-deficient diet (PD) for 8 (Panel A; n = 7/experimental group) or 13 weeks (Panel B; n = 6/experimental group), with or without supplementation of pyridoxine-HCl (500 μg, IP, daily for 2 [S2] or 3 [S3]) consecutive days at the end of the feeding periods. Values are expressed as mean ± SE. Asterisks indicate significant differences from time-matched CD counterparts at p < 0.05 (*) and p < 0.001 (**). #Significantly different from unsupplemented PD counterparts, p < 0.001.
![Figure 3. Effect of pyridoxine deficiency on relative organ weights. Mice were fed either a control (CD) or pyridoxine-deficient diet (PD) for 8 (Panel A; n = 7/experimental group) or 13 weeks (Panel B; n = 6/experimental group), with or without supplementation of pyridoxine-HCl (500 μg, IP, daily for 2 [S2] or 3 [S3]) consecutive days at the end of the feeding periods. Values are expressed as mean ± SE. Asterisks indicate significant differences from time-matched CD counterparts at p < 0.05 (*) and p < 0.001 (**). #Significantly different from unsupplemented PD counterparts, p < 0.001.](/cms/asset/c35bf15a-5b70-48e4-b197-c2a717bdc7e4/iimt_a_408559_f0003_b.gif)
Effects on hematological profiles
As shown in , PD mice fed the deficient diet for 8 weeks exhibited hematotoxicity as demonstrated by significantly decreased (~37%) total numbers of peripheral white blood cells (WBC), but significantly increased (~7%) red blood cell numbers (RBC) compared to the CD mice. In addition, several RBC parameters, including mean corpuscular volume (MCV), mean corpuscular hemoglobin (MCM), as well as hemoglobin (HGB), and hematocrit (HCT) levels were significantly lower than those observed in CD mice (by 19, 12, and 13%, respectively). Moreover, all mice fed the deficient diet presented with significantly increased red cell distribution width (RDW) compared to either CD mice or their supplemented CD counterparts. Supplementation of pyridoxine for up to 3 days had no effect on any hematological parameters in mice fed the control diet (CD-S3). In contrast, supplementation of the deficient mice for three days (PD-S3) partially restored total WBC numbers (increased by ~22%).
Table 2. Hematological effects of pyridoxine deficiency in mice fed control (CD) or deficient (PD) diets for 8 weeks.
While still below those of the CD mice, total WBC values were significantly higher than those in the unsupplemented PD group. In addition, pyridoxine supplementation of the PD mice for three days led to a significant restoration of HGB values, reaching levels comparable to those observed in the CD group. Hematocrit, MCV, and MCH levels were also significantly increased after three days of supplementation relative to those values observed in the unsupplemented PD groups (8.2, 4.2 and 4.3%, respectively, p < 0.05); however, values were still significantly lower than those measured in the unsupplemented CD mice. The percentages of neutrophils, lymphocytes, monocytes, eosinophils, basophils, MCHC, or platelet (PLT) numbers were not significantly different amongst or between treatment groups.
After 13 weeks (), the deficient diet resulted in significantly lower levels of HGB, HCT, MCV, and MCH in PD mice compared to CD mice (18, 22, 28, and 24%, respectively). In contrast, MCHC and RDW were significantly higher in the PD group compared to CD mice (5.5 and 56.2%, respectively). Trends in the hematological profile after 13 weeks on the diet were similar to those observed after 8 weeks except for WBC numbers, which were unaffected by the longer treatment.
Table 3. Hematological effects of pyridoxine deficiency in mice fed control (CD) or deficient (PD) diets for 13 weeks.
Supplementation of mice fed a PD diet for 13-weeks with pyridoxine for three days restored HGB levels to normal levels. Although after 3 days of pyridoxine supplementation, HCT and MCV levels were significantly higher (by about 13.4 and 5.2%, respectively) than those in the unsupplemented PD group (PD-S3 vs. PD), they failed to reach control levels. Although platelet count (PLT) was only slightly higher in PD mice compared to their control counterparts (p = 0.42), supplementation of this group with pyridoxine resulted in significantly increased PLT counts and RDW compared to either unsupplemented control mice or supplementation-matched CD mice. Neither diet nor supplementation alone had any effect on total WBC numbers, the percentage of lymphocytes, neutrophils, eosinophils, basophils, or red blood cells, or on platelet number.
Effects on lymphocyte proliferation
Immunomodulation associated with pyridoxine deficiency and/or supplementation was determined, in part, by mitogen-induced proliferation of splenic lymphocytes. Neither pyridoxine deficiency nor supplementation had any effect on unstimulated lymphocyte proliferation for both feeding regimens (data not shown). After 8 weeks on the selected diet (), splenic T- and B-lymphocyte proliferation in response to either ConA or LPS, respectively, was unaffected by vitamin deficiency. Alternatively, increased treatment time, i.e., 13-weeks (), resulted in significant differences in T-lymphocyte proliferation between the groups. While pyridoxine deficiency by itself had no significant effect on lymphocyte proliferation (compared to controls), supplemented mice on a deficient diet demonstrated a significant reduction in proliferation compared to the supplement-matched control mice (i.e., PD-S2 vs. CD-S2, p < 0.05; PD-S3 vs. CD-S3, p < 0.05). Alternatively, no significant effects on LPS-stimulated B-lymphocyte proliferation were observed amongst or between the groups.
Figure 4. Effect of pyridoxine deficiency on lymphocyte proliferation. Splenic lymphocytes were stimulated with the T- or B-cell mitogens, concanavalin A (ConA) or lipopolysaccharide (LPS), respectively. Mice were fed a control (CD) or pyridoxine-deficient diet (PD) for 8 weeks (Panel A; n = 7/experimental group) or 13 weeks (Panel B; n = 6/experimental group), with or without pyridoxine supplementation (pyridoxine-HCl, 500 μg [IP] daily for 2 [S2] or 3 [S3] consecutive days at the end of the feeding period). Values are expressed as mean ± SE. a,bSignificantly different from supplementation-matched control mice at p < 0.05.
![Figure 4. Effect of pyridoxine deficiency on lymphocyte proliferation. Splenic lymphocytes were stimulated with the T- or B-cell mitogens, concanavalin A (ConA) or lipopolysaccharide (LPS), respectively. Mice were fed a control (CD) or pyridoxine-deficient diet (PD) for 8 weeks (Panel A; n = 7/experimental group) or 13 weeks (Panel B; n = 6/experimental group), with or without pyridoxine supplementation (pyridoxine-HCl, 500 μg [IP] daily for 2 [S2] or 3 [S3] consecutive days at the end of the feeding period). Values are expressed as mean ± SE. a,bSignificantly different from supplementation-matched control mice at p < 0.05.](/cms/asset/f089a38f-b265-4407-97d2-38bfcf0ba0f0/iimt_a_408559_f0004_b.gif)
Effect on hepatic CYP450 enzymes
Immunoblotting of CYP1A1 and CYP2E1
Both CYP1A1 and CYP2E1 protein expressions were analyzed by Western blot using hepatic microsomal samples prepared from individual mice and from livers pooled from three animals after 8 and 13 weeks of the diets respectively.
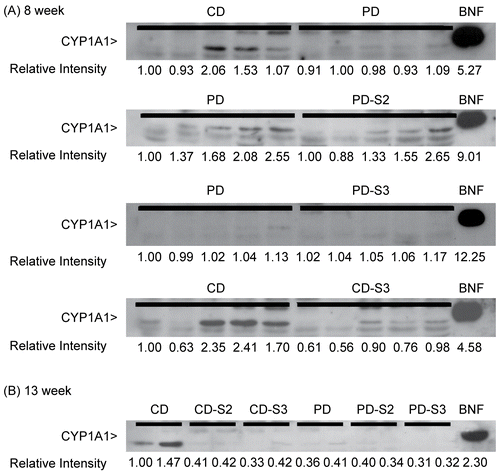
Although considerably large amounts of protein had been added to each lane (30 μg), CYP1A1 protein bands () displayed relatively weak intensities compared to those generated by liver microsomes treated with β-napthoflavone (BNF), a known inducer of CYP1A1. Protein band intensity was measured as the relative intensity in relation to that of the first lane of each blot (i.e., relative band intensity of 1.0). Despite intra-sample variation in CYP1A1 (particularly, between those bands in the 8 week group prepared from individual mice (). Diet intake for either 8 () or 13 weeks () appeared to have no significant effects on CYP1A1 protein levels.
Figure 5. Immunoblot of CYP1A1 in liver microsomes of mice fed a control (CD) or pyridoxine-deficient (PD) diet for 8 and 13 weeks. Panel A: Each lane represents microsomal samples prepared from an individual mouse. Five animals/group of control mice (CD), control mice with 3 days pyridoxine supplementation (CD-S3), pyridoxine-deficient mice (PD), and pyridoxine-deficient mice with 2 or 3 days’ supplementation (PD-S2 or PD-S3) after 8 weeks on the respective diets. Panel B: Each lane represents a microsomal sample pooled from the livers of three control mice (CD), controls with 2 or 3 days supplementation (CD-S2, CD-S3), pyridoxine-deficient mice (PD), and pyridoxine-deficient animals with 2 or 3 days’ supplementation (PD-S2 or PD-S3) for 13 weeks on the respective diets. Each lane was loaded with 30 μg of protein. Liver microsomes from β-napthoflavone (BNF, 2.5 μg per lane) treated mice were used as positive control. The level of CYP1A1 expression was measured as protein band intensity using a densitometer. The relative intensities of the first lane are arbitrarily assigned a value of 1.0 and intensities of the protein band in other lanes are expressed relative to that of the first lanes.
CYP2E1 was clearly and consistently detected by immunoblotting (); protein band intensities were analyzed using densitometry and presented as the relative intensity compared to those for control mice (CD) in the first lane. Levels of CYP2E1 protein in microsomal samples prepared from individual PD mice fed a pyridoxine-deficient diet for 8 weeks were slightly higher than those observed in CD mice (; the representative samples from two mice/treatment group are shown here). In contrast, a similar trend was not observed in the PD group fed a deficient diet for 13 weeks in which microsomal samples were prepared from livers pooled from three animals ().
Figure 6. Immunoblot of CYP2E1 in liver microsomes of mice fed a control (CD) or pyridoxine-deficient (PD) diet for 8 or 13 weeks. Panel A: The repernsentative samples from two individual mice/treatment group are shown here. Control mice (CD), control mice with 2 or 3 days supplementation (CD-S2, CD-S3), pyridoxine-deficient mice (PD), and pyridoxine-deficient mice with 2 or 3 d supplementation (PD-S2 or PD-S3) were compared after 8 weeks on the respective diets. Panel B: Each lane contained microsomal samples prepared from livers pooled from 3 individual mice from each dietary treatment group, i.e., control mice (CD), control mice supplemented with pyridoxine for 2 or 3 days (CD-S2, CD-S3), pyridoxine-deficient mice (PD), and pyridoxine-deficient mice supplemented with pyridoxine for 2 or 3 days (PD-S2 or PD-S3) after 13 weeks on the respective diets. Each lane was loaded with 5 μg of protein. Microsomes from acetone-treated mice (5 μg protein) were used as a positive control. The level of CYP2E1 expression was measured as protein band intensity using a densitometer. The relative intensities of controls (CD) in the first lanes are arbitrarily assigned a value of 1.0 and protein band intensities in other lanes are expressed relative to those of the controls.

CYP1A1 and CYP2E1 activity
The effects of pyridoxine deficiency and subsequent supplementation after 8 and 13 weeks on hepatic CYP1A1 and CYP2E1 activities were determined as EROD and p-NPH, respectively. Activity was measured in animals after 8 and 13 weeks using either individual or pooled livers, respectively. At 8 weeks (), EROD activity catalyzed by CYP1A1 in PD mice was significantly higher than that observed in CD mice (p < 0.05). Supplementation with pyridoxine in PD mice for 2 days (PD-S2) decreased CYP1A1 activity, though not significantly. Supplementation for 3 days significantly decreased CYP1A1 activity compared to PD and their supplemented CD-S3 counterparts. Supplementation of pyridoxine in CD mice for 2 or 3 consecutive days (CD-S2 and CD-S3, respectively) in the present study resulted in a slight increase in CYP1A1 activity compared to their CD counterpart, though this was not statistically significant. At 13 weeks (), neither pyridoxine deficiency nor supplementation had any effects on CYP1A1 activities in either control or pyridoxine-deficient groups.
Table 4. Effects of pyridoxine deficiency on CYP1A1 and CYP2E1 activities in liver microsomes of mice fed control or deficient diets for 8 weeks.
Table 5. Effects of pyridoxine deficiency on CYP1A1 and CYP2E1 activity in liver microsomes in mice fed control or deficient diet for 13 weeks.
CYP2E1 activity, expressed as formation of p-nitrophenol hydroxylase (p-NPH), in PD mice after 8 weeks on the deficient diet were slightly higher than those observed in the CD mice, but the differences were not statistically significant (). Subsequent supplementation of pyridoxine in PD mice for 2 or 3 consecutive days (PD-S2 and PD-S3, respectively) did not significantly alter cytochrome P450 2E1 activity, although there was an increasing trend.
After 13 weeks on the respective diets, CYP2E1 activity was unchanged in any of the treatment groups. However, as observed in the 8 weeks study, activity in the PD mice was slightly elevated compared to CD mice, p = 0.33 (). Moreover, CYP2E1 activity in PD mice after 13 weeks on the deficient diet were lower than that of mice fed a deficient diet for 8 weeks (p = 0.50). Compared to the PD counterparts, CYP2E1 activity increased slightly in pyridoxine-deficient mice supplemented with pyridoxine (PD-S2, p = 0.33 and PD-S3, p = 0.16); however, a similar effect was not observed in the supplemented control mice (CD-S2 and CD-S3).
Discussion and conclusion
The results of this study show that dietary restriction of pyridoxine can lead to increases in blood EAST-AC values in juvenile male B6C3F1 mice. Such values have been widely used for determination of pyridoxine status in humans (Raica and Sauberlich, Citation1964; Kwak et al., Citation2002; Oshiro et al., Citation2005) and other laboratory animals (Aramphongphan et al., Citation1994). The minimal activity coefficient value used as an indicator of pyridoxine deficiency in humans ranges from 2.0 to 3.0 (Schreurs et al., Citation1976; Changbumrung et al., Citation1985; Vudhivai et al., Citation1991) and is about 1.6 in rats (Aramphongphan et al., Citation1994). The minimum value for indicating pyridoxine deficiency in mice was calculated from the mean EAST-AC value plus 2.5 times the standard deviation (Heller et al., Citation1973; Changbamrung et al., Citation1985). In this study, EAST-AC values for both adult male and female B6C3F1 mice fed a control diet, averaged 1.04. Thus, EAST-AC values greater than 1.29 were considered here as an indicator of pyridoxine deficiency.
After 8 weeks on the deficient diet, the EAST-AC levels of PD mice were substantially higher than those observed in the CD mice. Moreover, pyridoxine-deficient mice presented with a slightly lower weight gain than control mice, beginning at about 2 weeks on the deficient diet. Thirteen weeks on the diet resulted in higher EAST-AC levels (5.37) than those seen in the 8-week group, reflecting a more severe pyridoxine deficiency. Although plasma-PLP is the most commonly used index for determining pyridoxine status, EAST-AC has been reported to be a very sensitive indicator (Dirige and Beaton, 1969) and requires less blood than the plasma-PLP assay. Thus, EAST-AC measurements were deemed a valid tool for evaluating pyridoxine nutritional status in the mouse model used here.
The general effects of pyridoxine deficiency were also investigated in this study. Pyridoxine-deficient mice displayed growth retardation after 2 weeks, which became significantly depressed after 5 weeks, of dietary treatment. In other studies, C57BL/6 mice fed a pyridoxine-deficient diet plus 4-deoxypyridoxine in the drinking water for 10 weeks ceased to gain weight; these mice also demonstrated reduced food consumption after 6 weeks. In addition, these pyridoxine-deficient mice had rough fur, denuding of the snout, and developed skin irritation after 21 weeks (Ha et al., Citation1984). Studies in rats have indicated an earlier onset of pyridoxine deficiency than that seen in mice.
This study clearly demonstrates that food intake in mice was not significantly affected by pyridoxine deficiency as reported earlier for rats provided a pyridoxine-deficient diet for 8-10 weeks. In that case, rats lost weight, consumed less food, and levels of serum transaminase dropped (Goswami and Coniglio, Citation1966). The absence of these overt morphological characteristics could suggest that male B6C3F1 mice might not be as sensitive to pyridoxine deficiency as other rodent models, particularly rats.
Relative liver weights were increased in PD mice after 13-weeks on the diet, an outcome similar to that observed in another study that demonstrated liver hypertrophy (Goswami and Coniglio, Citation1966) and liver hyperplasia in pyridoxine-deficient rats (Reynolds, Citation1986, Ruchirawat et al., Citation1990). In fact, PD mice that received vitamin supplementations for 2 or 3 subsequent days (PD-S2 and PD-S3, respectively) had even higher relative liver weights compared to the unsupplemented PD counterparts. Goswami and Coniglio (Citation1966) reported that administration of pyridoxine-HCl (50 μg/day for 6 days) to deficient rats resulted in almost complete reversal of the adverse effects (i.e. morphological signs, growth inhibition, and increased liver weights). However, the short supplementation periods used in the current study (i.e., 500 μg/day for 3 days) were likely of insufficient duration to correct liver weight abnormalities.
In contrast to the lack of effects observed here, several studies have reported that pyridoxine deficiency affects lymphoid organ weights. For example, male rats fed a pyridoxine-deficient diet and 0. 1 mg deoxypyridoxine HCl/ ml water for 2 weeks demonstrated thymic atrophy (Cassel et al., Citation1978). In another study, female C57BL/6 mice fed a pyridoxine-deficient diet for 21 weeks also had severe thymic atrophy (Ha et al., Citation1984), while female BALB/C mice fed a pyridoxine-deficient diet for 4 weeks demonstrated a significant reduction in thymus and spleen weights (Doke et al., Citation1998). The present study yielded different results. Although thymic size in PD mice here was somewhat smaller than that for CD mice, this change likely reflected the overall lack of body growth rather than PD-induced effects on the thymus itself. In addition, relative spleen weight was increased in pyridoxine-deficient mice. Discrepancies between the studies could be explained (at least in part) by the extent of pyridoxine deficiency produced in the animals. The study here was designed to induce mild to moderate pyridoxine deficiency while the other investigations brought about a more severe deficiency state.
As observed in other studies (Cartwright, Citation1947; Foy and Kondi, Citation1958; Shen et al., Citation1964; Shen and Ko, Citation1967), pyridoxine deficiency in this study induced anemia. While the total red blood cell (RBC) count and RBC distribution width (RDW) were increased in PD mice, hemoglobin (HGB) level, packed red cell volume (hematocrit [HCT]), mean corpuscular volume (MCV), and mean corpuscular hemoglobin (MCH) were all reduced in the deficient animals. Moreover, the longer the mice were maintained on the pyridoxine-deficient diets (without subsequent supplementation), the greater the effects on these specific anemic indices.
Studies in rats have demonstrated that there are several important mechanisms underlying the development of pyridoxine deficiency-induced anemia. These include: (1) defective heme synthesis; (2) impaired erythropoiesis; and, (3) increased RBC destruction in the circulation due to shortened RBC survival time (Foy and Kondi, Citation1958; Shen et al., Citation1964; Shen and Ko, Citation1967). The anemia observed in the current study markedly responded to pyridoxine-HCl treatment, i.e., there was a fairly quick restoration of HGB, HCT, and MCV towards normal levels, even though the degree of restoration was not uniform (i.e., restorative pattern was HGB > HCT > MCV). The reasons for these differences remain to be determined. However, as HGB synthesis requires the coordinated production of heme and globin, a deficiency of pyridoxine could act to impair overall HGB levels. As HGB was observed in this study to be one of the most sensitive indicators for disease pathogenesis, it is not surprising that pyridoxine supplementation would affect this RBC parameter to the greatest extent.
In general, the effects of pyridoxine supplementation in B6C3F1 mice reflected those reported by other investigators (Woodring and Storvick, Citation1970; Chiang et al., Citation2005), i.e., the extent of any restorative change was related to: the degree of anemia prior to supplementation; the concentration of the supplement; and, the route of administration. For example, Mirone and Jackson (Citation1959) reported that a daily injection of 50 μg pyridoxine hydrochloride to pyridoxine-deficient mice for 20 days restored growth and hematological indices to normal values. Although daily administration of 500 μg was higher than previously reported, a lack of complete restoration may have been due to the short period of supplementation.
Blood cell types other than RBC were also shown in this study to be affected in PD mice. For example, mice fed a pyridoxine-deficient diet for 8 weeks displayed significantly lower peripheral white blood cell (WBC) counts compared to controls. However, this effect was no longer apparent in mice fed a deficient diet for 13 weeks. That WBC numbers were reduced in the PD mice was not unexpected. Investigations with rats given 4-deoxypyridoxine (a pyridoxine antagonist) for 2 weeks also demonstrated altered WBC profiles (Cassel et al., Citation1978). Similarly, dietary pyridoxine deficiency has been shown to cause a severe depletion of thymic lymphocytes in the rat (Willis-Carr and St. Pierre, Citation1978), as well as peripheral blood leucopenia in mice (Mirone and Jackson Citation1959).
Apart from changes in numbers of non-erythrocyte cell types, the effect of pyridoxine deficiency on lymphocyte proliferation was also examined to demonstrate the effect (if any) of moderate pyridoxine deficiency. Earlier findings in rats (Willis-Carr and St. Pierre, Citation1978; Trakatellis et al., Citation1992) reported that pyridoxine-deficient diets gave rise to significant decreases in splenic T-lymphocyte proliferation in response to mitogen stimulation. In contrast, ConA- stimulated T-lymphocyte proliferation in PD mice in this study was only slightly below control levels. However, PD mice supplemented with pyridoxine HCl for either 2 or 3 days had significantly lower ConA-stimulated T-lymphocyte proliferation compared to their CD counterpart (see ).
The effects of pyridoxine deficiency on xenobiotic-metabolizing enzymes were also investigated by measuring the protein expression and activity of CYP1A1 and CYP2E1. In these studies, basal expression of hepatic CYP1A1 in control B6C3F1 mice in the present study was very low compared to that in mice treated with the known inducer of CYP1A1, β-napthoflavone (BNF), which is similar to that reported for mice in other investigations (Crawford et al., Citation1997; Nebert et al., Citation2004). Moderate deficiency resulting from intake of a diet lacking pyridoxine for 8 weeks had no effect on hepatic CYP1A1 protein expression.
In contrast to CYP1A1 protein expression, hepatic CYP1A1 activity in pyridoxine-deficient mice (PD) was significantly greater (1.3X; 44.35 vs. 36.66) than in control (CD) mice after 8 weeks on the respective diets (see ). Subsequent supplementation with pyridoxine in deficient mice for 2 days (PD-S2) lowered hepatic CYP1A1 activities; these dropped to control levels in mice supplemented with pyridoxine for an additional day (PD-S3). However, hepatic CYP1A1 protein expression and activity were unaffected in mice fed the vitamin deficient diet for 13 weeks. The disparity observed between mice fed a diet for 8 or 13 weeks in CYP1A1 activity could be due to exacerbation of the deficiency state as a result of prolonged feeding of the deficient diet. The exact mechanism by which pyridoxine deficiency acts to alter hepatic CYP1A1 activity is unclear and requires further study.
For CYP2E1, differences in CYP2E1 activity among treatment groups were not statistically significant. However, there was an increasing trend similar to that observed by Aramphongphan et al. (Citation1994) who reported that F344 rats fed a pyridoxine-deficient diet demonstrated a significant increase in hepatic and renal N-nitrosodimethylamine (NDMA)-demethylase activity (at 4 and 200 mM NDMA) after 3 weeks, and further increased up to 8 weeks. In this study, the levels of hepatic microsomal CYP2E1 protein expression and activities in mice fed a pyridoxine-deficient diet for 8 weeks were slightly higher than those of the control mice, but not statistically significant. However, hepatic CYP2E1 protein expression and activity were unaffected in mice fed the vitamin deficient diet for 13 weeks. Cytochrome P4502E1 is responsible for demethylation in the metabolism of NDMA at 4 mM (Hong et al., Citation1989; Bellec et al., Citation1996) and renal NDMA-demethylase activity was inhibited by a monoclonal antibody prepared against rat P4502E1 (Hong et al., Citation1989).
The increasing trend in CYP2E1 activity was also observed in deficient animals receiving pyridoxine supplementation, although the increases were not statistically significant. This contrasted with results observed in rats where daily supplementation with pyridoxine hydrochloride (500 μg, IP) for 2 days led to a significant reduction of NDMA-demethylase activity to normal levels (Aramphongphan et al., Citation1994). Regulation of CYP2E1 by the liver as well as by extra-hepatic organs (Powley and Carlson, Citation2000) is highly complex and can take place at the transcriptional, post-transcriptional, translational, or post-translational levels (Hukkanen, Citation2000). CYP2E1 can be induced by a variety of environmental toxicants including benzene (Gonzalez-Jesso et al., 2003). In this study, the levels of hepatic microsomal CYP2E1 protein expression in mice fed pyridoxine-deficient diet at 8 and 13 weeks were unaffected.
In summary, depletion of dietary pyridoxine for 8 or 13 weeks can induce: (1) a mild-to-moderate pyridoxine deficiency that is accompanied by growth retardation; (2) hematotoxic effects which increases with the degree of deficiency, particularly those associated with a microcytic anemia; (3) changes in WBC numbers and selected immune functions; and, (4) increased hepatic CYP1A1 activity in the absence of effects on protein expression. Although some aspects of deficiency can be reversed (to some extent) by short-term pyridoxine supplementation, many of the biochemical and/or physiological effects related to pyridoxine deficiency in mice seem to require a longer duration of supplementation for complete restoration. Even though further studies are required to understand the mechanisms by which pyridoxine deficiency may bring about hematotoxic alterations, the studies here provide some indication that a mild to moderate pyridoxine-deficient stage in B6C3F1 mice presented itself with immune alterations. Further investigation is also required to determine the influence of pyridoxine deficiency on hepatic CYP1A1 and the possible role of hepatic CYP1A1 in modulating the effect of pyridoxine deficiency.
Acknowledgments
The research described herein was supported by the Chulabhorn Research Institute and a grant from the Center of Excellence on Environmental Health, Toxicology and Management of Toxic Chemicals (ETM) under Science & Technology Postgraduate Education and Research Development Office (PERDO) of Ministry of Education.
Declaration of interest: The authors declare that there are no conflicts of interest in the design and conducting of this study. The authors alone are responsible for the content and writing of the paper.
References
- Aramphongphan, A., Navasumrit, P., Kuroiwa, Y., Yoshida, T., Frank, N., and Ruchirawat, M. 1994. Effects of pyridoxine deficiency on the metabolism of N-nitrosodimethylamine in the rat. J. Nutr. Sci. Vitaminol. (Tokyo) 40:105–113.
- Axelrod, A. E. 1981. Role of the B-vitamins in the immune response. Adv. Exp. Med. Biol. 135:93–106.
- Bellec, G., Dreano, Y., Lozach, P., Mdnez, J. F., and Berthou, F. 1996. Cytochrome P450 metabolic dealkylation of nine N-nitrosodialkylamines by human liver microsomes. Carcinogenesis 17:2029–2034.
- Bisp, M. R., Bor, M. V., Heinsvig, E. M., Kall, M. A., and Nexo, E. 2002. Determination of Vitamin B6 vitamers and pyridoxic acid in plasma: Development and evaluation of a high-performance liquid chromatographic assay. Anal. Biochem. 305:82–89.
- Bradford, M. M. 1976. A rapid and sensitive method for quantitation of microgram quantities of protein utilizing the principle of protein- dye-binding. Anal. Biochem. 72:248–254.
- Cartwright, G. E. 1947. Dietary factors concerned in erythropoiesis. Blood 2:111–153.
- Cassel, S., Robson, L., and Rosse, C. 1978. The effects of Vitamin B6 deficiency on the bone marrow of the rat. Anat. Rec. 191:47–53.
- Chandra, R. K. 1983. Nutrition and immune responses. Can. J. Physiol. Pharmacol. 61:290–294.
- Chaney, S. G. 1993. Principles of nutrition II: Micronutrients. In: Biochemistry with Clinical Correlations, 3rd Edition (Devlin, T. M., Ed.), Wiley-Liss, Inc. Press: New York, pp. 1126–1131.
- Changbumrung, S., Poshakrishana, P., Vudhivai, N., Hongtong, K., Pongpaew, P., and Migasena, P. 1984. Measurements of B1, B2, B6 status in children and their mothers attending a well-baby clinic in Bangkok. Int. J. Vitam. Nutr. Res. 54:149–159.
- Changbumrung, S., Schelp, F. P., Hongtong, K., Buavatana, T., Supawan, V., and Migasena, P. 1985. Pyridoxine status in preschool children in northeast Thailand: A community survey. Am. J. Clin. Nutr. 41:770–775.
- Chiang, E. P., Selhub, J., Bagley, P. J., Dallal, G., and Roubenoff, R. 2005. Pyridoxine supplementation corrects Vitamin B6 deficiency but does not improve inflammation in patients with rheumatoid arthritis. Arthritis Res. Ther. 7:R1404–1411.
- Coon, M. J., Vaz, A. D., and Bestervelt, L. L. 1996. Cytochrome P450 2: Peroxidative reactions of diversozymes. FASEB J. 10:428–434.
- Costello, A. M., and Osrin, D. 2003. Micronutrient status during pregnancy and outcomes for newborn infants in developing countries. J. Nutr. 133(S2):1757S–1764S.
- Crawford, R. B., Holsapple, M. P., and Kaminski, N. E. 1997. Leukocyte activation induces aryl hydrocarbon receptor up-regulation, DNA binding, and increased CYP1A1 expression in the absence of exogenous ligand. Mol. Pharmacol. 52:921–927.
- Dirige, O. V., and Beaton, J. R. 1969. Factors affecting Vitamin B6 requirement in the rat as determined by erythrocyte transaminase activity. J. Nutr. 97:109–116.
- Doke, S., Inagaki, N., Hayakawa, T., and Tsuge, H. 1998. Effects of Vitamin B6 deficiency on cytokine levels and lymphocytes in mice. Biosci. Biotechnol. Biochem. 62:1008–1010.
- Foy, H., and Kondi, A. 1958. Hypochromic anemias of the tropics associated with pyridoxine and nicotinic acid deficiency. Blood 13:1054–1062.
- Frydas, S., Papaioanou, N., Vlemmas, I., Theodoridis, I., Anogiannakis, G., Vacalis, D., Trakatellis, A., Barbacane, R. C., Reale, M., and Conti, P. 1999. Vitamin B6-deficient diet plus 4-deoxypyridoxine (4-DPD) reduces the inflammatory response induced by T. spiralis in diaphragm, masseter, and heart muscle tissue of mice. Mol. Cell. Biochem. 197:79–85.
- Gonzalez, F. J. 2007. CYP2E1 Drug Metab. Dispos. 35:1–8.
- González-Jasso, E., López, T., Lucas, D., Berthou, F., Manno, M., Ortega, A., and Albores, A. 2003. CYP2E1 regulation by benzene and other small organic chemicals in rat liver and peripheral lymphocytes. Toxicol. Lett. 144:55–67.
- Goswami, A., and Coniglio J. G. 1966. Effect of pyridoxine deficiency on the metabolism of linoleic acid in the rat. J. Nutr. 89:210–216.
- Gridley, D. S., Stickney, D. R., Nutter, R. L., Slater, J. M., and Shultz, T. D. 1987. Suppression of tumor growth and enhancement of immune status with high levels of dietary Vitamin B6 in Balb/c mice. J. Natl. Cancer Inst. 78:951–959.
- Ha, C., Kerkvliet, N. I., and Miller, L. T. 1984. The effect of Vitamin B6 deficiency on host susceptibility to Moloney sarcoma virus-induced tumor growth in mice. J. Nutr. 114:938–948.
- Heller, S. R., Salkeld, M., and Korner, W. F. 1973. Vitamin B6 status in pregnancy. Am. J. Clin. Nutr. 26:1339–1348.
- Hong, J., Pan, J., Ning, S., and Yang, C. 1989. Molecular basis for the sex-related difference in renal N-nitrosodimethylamine demethylase in C3H/HeJ mice. Cancer Res. 49:2973–2979.
- Holman P. 1995. Pyridoxine-Vitamin B6. J. Austral. Coll. Nutr. Environ. Med. 14:5–16.
- Hukkanen, J. 2000. Xenobiotic-metabolizing cytochrome P450 enzymes in human lung. Acta Univ. Oul. D 621:1–68.
- Kirksey, A., Keaton, K., Abernathy, R. P., and Greger, J. L. 1978. Vitamin B6 nutritional status of a group of female adolescents. Am. J. Clin. Nutr. 31:946–954.
- Kwak, H. K., Hansen, C. M., Leklem, J. E., Hardin, K., and Shultz, T. D. 2002. Improved Vitamin B6 status is positively related to lymphocyte proliferation in young women consuming a controlled diet. J. Nutr. 132:3308–3313.
- Ladipo, O. A. 2000. Nutrition in pregnancy: Mineral and vitamin supplements. Am. J. Clin. Nutr. 72(S1):280S–290S.
- Leklem, J. E. 1990. Vitamin B6: A status report. J. Nutr. 120(S11):1503–1507.
- Mirone, L., and Jakson, C. D. 1959. The development and cure of pyridoxine deficiency symptoms in weanling mice. J. Nutr. 67:167–179.
- Nebert D. W., Dalton T. P., Okey A. B., and Gonzalez, F. J. 2004. Role of aryl hydrocarbon receptor-mediated induction of the CYP1 enzymes in environmental toxicity and cancer. J. Biol. Chem. 279:23847–23850.
- Ng, S. P., and Zelikoff, J. T. 2008. The effects of prenatal exposure of mice to cigarette smoke on offspring immune parameters. J. Toxicol. Environ. Health 71:445–453.
- Nontasut, P., Changbumrung, S., Muennoo, C., Hongthong, K., Vudhivai, N., Sanguankiat, S., and Yaemput, S. 1996. Vitamin B1, B2, and B6 deficiency in primary school children infected with hookworm. Southeast Asian J. Trop. Med. Publ. Health 27:47–50.
- Oka, T., Komori, N., Kuwahata, M., Sassa, T., Suzuki, I., Okada, M., and Natori, Y. 1993. Vitamin B6 deficiency causes activation of RNA polymerase and general enhancement of gene expression in rat liver. FEBS Lett. 331:162–164.
- Oka, T. 2001 Modulation of gene expression by Vitamin B6. Nutr. Res. Rev. 14:257–265.
- Oshiro, M., Nonoyama, K., Oliveira, R. A., and Barretto, O. C. 2005. Red cell aspartate aminotransferase saturation with oral pyridoxine intake. Sao Paulo Med. J. 123:54–57.
- Parkinson, A. 2001. Biotransformation of xenobiotics. In: Casarett & Doull’s Toxicology: The Basic Science of Poison, Sixth Edition (Klaassen, C.D., Ed), McGraw-Hill: New York, pp. 133–224.
- Pongpaew, P., Tungtrongchitr, R., Radomyos, P., Vudhivai, N., Phonrat, B., Himman-ngan, T., Supawan, V., Tawprasert, S., Migasena, P., and Schelp, F. P. 1993. Parasitic infection and socio-demographic characteristics of urban construction site workers. Southeast Asian J. Trop. Med. Publ. Health 24:573–576.
- Powley, M. W., and Carlson, G. P. 2000. Cytochromes P450 involved with benzene metabolism in hepatic and pulmonary microsomes. J. Biochem. Mol. Toxicol. 14:303–309.
- Rall, L. C., and Meydani, S. N. 1993. Vitamin B6 and immune competence. Nutr. Rev. 51:217–225.
- Raica, N., Jr. and Sauberlich, H. E. 1964. Blood cell transaminase activity in human Vitamin B6 deficiency. Am. J. Clin. Nutr. 15:67–72.
- Reeves, P. G. 1997. Components of the AIN-93 diets as improvements in the AIN-76A diet. J. Nutr. 127(5S):838S–841S.
- Reinke, L. A., and Moyer M. J. 1985. p-Nitrophenol hydroxylation. A microsomal oxidation which is highly inducible by ethanol. Drug Metab. Disp. 13:548–552.
- Reynolds, R. D. 1986. Vitamin B6 deficiency and carcinogenesis. Adv. Exp. Med. Biol. 206:339–347.
- Ronnenberg, A. G., Goldman, M. B., Aitken, I. W., and Xu, X. 2000. Anemia and deficiencies of folate and Vitamin B6 are common and vary with season in Chinese women of childbearing age. J. Nutr. 130:2703–2710.
- Ruchirawat, M., Navasumrit, P., Aramphongphan, A., Mahathanatrakul, W., and Frank, N. 1990. Alterations in dimethylnitrosamine-induced lethality and acute hepatoxicity in rats during dietary thiamin, riboflavin and pyridoxine deficiency. J. Cancer Res. Clin. Oncol. 116:559–603.
- Sanchez, D. J., Murphy, M. M., Bosch-Sabater, J., and Fernandez-Ballart, J. 1999. Enzymic evaluation of thiamin, riboflavin, and pyridoxine status of parturient mothers and their newborn infants in a Mediterranean area of Spain. Eur. J. Clin. Nutr. 53:27–38.
- Schreurs, W. H., Migasena, P., Pongpaew, P., Vudhivai, N., and Schelp, F.P. 1976. The Vitamin B1, B2, and B6 status of school children in two resettlement areas in Northeast Thailand. Southeast Asian J. Trop. Med. Publ. Health 7:586–590.
- Setiawan, B., Giraud, D. W., and Driskell, J. A. 2000. Vitamin B6 inadequacy is prevalent in rural and urban Indonesian children. J. Nutr. 130:553–558.
- Shen, S. C., Wong.P. Y., and Oguro, M. 1964. Experimental production of pyridoxine deficiency anemia in rats. Blood 23:679–687.
- Shen, S. C., and Ko, R. L. 1967. Mechanism of the anemia of pyridoxine deficiency in the rat. Blood 30:425–441.
- Smith, M. T. 1996. The mechanism of benzene-induced leukemia: A hypothesis and speculations on the causes of leukemia. Environ. Health Perspect. 104(S6):1219–1225.
- Soh, Y., Rhee, H. M., Sohn, D. H., and Song, B. J. 1996. Immunological detection of CYP2E1 in fresh rat lymphocytes and its pretranslational induction by fasting. Biochem. Biophys. Res. Commun 227:541–546.
- Solomon, L. R., and Hillman, R. S. 1978. Vitamin B6 metabolism in human red cells. I. Variations in normal subjects. Enzyme 23:262–273.
- Tada, Y., Keiwkarnka, B., Pancharuniti, N., and Chamroonsawasdi, K. 2002. Nutritional status of the preschool children of the Klong Toey slum, Bangkok. Southeast Asian J. Trop. Med. Publ. Health 33:628–637.
- Takada, Y., Mori, T., and Noguchi, T. 1984. The effect of Vitamin B6 deficiency on alanine: glyoxylate aminotransferase isoenzymes in rat liver. Arch. Biochem. Biophys. 229:1–6.
- Towbin, H., Staehelin, T., and Gordon, J. 1979. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: Procedure and some applications. Proc. Natl. Acad. Sci. USA 76:4350–4354.
- Tovar, A. R., Torres, N., Halhali, A., and Bourges, H. 1996. Riboflavin and pyridoxine status in a group of pregnant Mexican women. Arch. Med. Res. 27:195–200.
- Trakatellis, A., Dimitriadou, A., Exindari, M., Scountzou, J., Koliakos, G., Christodoulou, D., Malissiovas, N., Antoniadis, A., and Polyzoni, T. 1992. Effect of pyridoxine deficiency on immunological phenomena. Post-grad. Med. J. 68(S1):S70–S77.
- Vudhivai, N., Ali, A., Pongpaew, P., Changbumrung, S., Vorasanta, S., Kwanbujan, K., Charoenlarp, P., Migasena, P., and Schelp, F. P. 1991. Vitamin B1, B2, and B6 status of vegetarians. J. Med. Assoc. Thai. 74:465–470.
- Willis-Carr, J. I., and St. Pierre, R. L. 1978. Effects of Vitamin B6 deficiency on thymic epithelial cells and T-lymphocyte differentiation. J. Immunol. 120:1153–1159.
- Wolters, M., Hermann, S., and Hahn, A. 2005. Effect of multivitamin supplementation on the homocysteine and methylmalonic acid blood concentrations in women over the age of 60 years. Eur. J. Nutr. 44:183–192.
- Woodring, M. J., and Storvick, C. A. 1970. Effect of pyridoxine supplementation on glutamic-pyruvic transaminase and in vitro stimulation in erythrocytes of normal women. Am. J. Clin. Nutr. 23:1385–1395.