
Abstract
Current advances in the study of cutaneous adverse drug reactions can be attributed to the recent understanding that the skin is both a metabolically and immunologically competent organ. The ability of the skin to serve as a protective barrier with limited drug biotransformation ability, yet highly active immune function, has provided insights into its biological capability. While the immune response of the skin to drugs is vastly different from that of the liver due to evolutionary conditioning, it frequently occurs in response to various drug classes and manifests as a spectrum of hypersensitivity reactions. The skin is a common site of adverse and idiosyncratic drug reactions; drug-specific T-cells, as well as involvement of an innate immune response, appear to be key mechanistic drivers in such scenarios. Association of other factors such as human leukocyte antigen (HLA) polymorphisms may play a significant role for particular drugs. This review aims to integrate emerging findings into proposed mechanisms of drug metabolism and immunity in the skin that are likely responsible for rashes and other local allergic responses. These unique biological aspects of the skin, and their translation into implications for drug development and the use of animal models, will be discussed.
Introduction
Recent advances in the knowledge of skin immunology and enzymatic ability have reshaped our understanding of the skin and its ability to respond to drugs and other foreign materials. Current evidence strongly suggests that most skin rashes are immune-mediated. In many cases, the immune response appears to be triggered by a chemically reactive drug or a reactive metabolite. Though the skin has limited ability to metabolize drugs (sulfotransferase and acetyltransferase have the greatest activity with only limited cytochrome P450 activity), the immune activity is quite high. Animal models are an essential tool to test hypotheses, and have enabled an understanding of the relationship between the metabolic capability and immunology of the skin. One of the few animal models of drug-induced skin rash is due to nevirapine in rats, which will be described in this review. This model highlights many of the unique physiologic features of the skin and demonstrates what chemical species is responsible for the rash, namely a reactive sulfate metabolite formed in the skin. However, other drugs that cause skin rashes in humans do not generally cause skin rashes in animals; the dominant response appears to be immune tolerance. Therefore, it may be possible to produce other animal models of drug-induced skin rash by impairing immune tolerance.
Drugs can induce a wide variety of skin reactions. Maculopapular exanthema (MPE) and erythema multiforme (EM) are common drug-induced skin rashes. Severe cutaneous adverse reactions (SCAR) are life-threatening drug hypersensitivity responses, carrying high mortality and morbidity. SCARs include drug-induced hypersensitivity syndrome (DIHS), drug reaction with eosinophilia and systemic symptoms (DRESS), Stevens-Johnson syndrome (SJS) and Toxic Epidermal Necrolysis (TEN) (Duong et al. Citation2017). Most reactions are likely allergic in nature, and thus hard to evaluate with animals in the non-clinical stage. The following further describes these clinically important types of skin reactions.
MPE is characterized by various erythema or papular eruptions, which are diffusely distributed on the body (Fernandez et al. Citation2009). Erythema is relatively homogeneous and divided into epidermal and dermal types. In the lesion, infiltration of lymphocytes is observed. MPE often occurs from 1–2 w following drug administration, and can be due to diverse drugs such as antibiotics (e.g. penicillin and sulfa drugs), antiepileptics (carbamazepine), allopurinol, or non-steroidal anti-inflammatory drugs (NSAID). This is a mild type of skin reaction and often resolves on stopping the medication.
EM is a skin disorder with a multiform of target lesions (usually three layers) with no or mild mucosal enanthema, and is due to diverse causes, including herpes simplex virus and various drugs (Lamoreux et al. Citation2006). EM can be divided into EM minor (in which lesions are limited to the face and four limbs) and EM major (with multiple lesions and fever). EM major leads to erythroderma by fusion of the lesions and may involve internal organ failure. EM is likely a delayed-type hypersensitivity reaction, and T-cell infiltration into the skin is often observed.
DIHS is often diagnosed by high-fever, maculopapular rash, involvement of at least one internal organ (mainly liver), blood abnormalities (lymphocytosis, eosinophilia, atypical lympho-cyte), lymphadenopathy, and human herpesvirus six reactivation (Shiohara et al. Citation2006). DRESS show similar symptoms, but have different diagnostic criteria such as no viral re-activation (Lally and Turner Citation2007). DIHS develops from 2–6 week after drug administration, and is due to limited drugs (i.e. antiepileptics, allopurinol, sulfa drugs, mexiletine, and minocycline), and requires hospitalization, with outcomes of high mortality (10%) and increases of the chemokine TARC/CCL17 in blood.
SJS/TEN represents the most severe forms of skin reactions (Duong et al. Citation2017). SJS/TEN show symptoms of wide-spread blistering exanthema, high fever, and, in SJS, mucosal injury. SJS/TEN are thought to be similar disorders classified by skin detachment/blistering among areas of the epidermis: < 10% of body surface area (BSA); SJS, 10%–30% of BSA; SJS/TEN overlap, and ≥ 30% of BSA:TEN. These reactions are very rare (1–5 cases/yr/1 million subjects) and develop within several days to weeks after initiating the drug. Patients suffering from SJS/TEN are hospitalized, and mortality rates are relatively high (SJS: 1–5%, TEN: 20–30%). Even after recovery, patients may suffer sequela, such as vision loss due to pseudo-membrane formation (Duong et al. Citation2017).
The lymphocyte transformation test (LTT, also drug-induced lymphocyte stimulation test) is one of the clinical tests used for investigating the suspected drugs. It detects proliferation of circulating drug-specific memory T-cells, and its sensitivity is relatively high (60–70%), particularly for DIHS, and MPE, but not for TEN (Pichler and Tilch Citation2004). As its sensitivity is high but specificity is not, positive results mean this drug is probably causative, but negative results do not imply exclusion of the drug as the causal agent.
Immune responses of the skin to drugs
The adaptive immune response: Overview
The immune system is quite complex, and while there are similarities between immune responses in the skin and other organs, there are also differences. A major difference between the immune response in the skin versus the liver is that the dominant immune response in the liver is immune tolerance. This is illustrated by the difference in the ease of transplanting liver and skin (Calne et al. Citation1969). Many cells are involved in dermal immune responses, including αβ and γδ T-cells, natural killer (NK) cells, B-cells, mast cells, and macrophages (Di Meglio et al. Citation2011). In addition, Langerhans cells (which express langerin) are a unique antigen-presenting cell (APC) in the skin. The APC are essential for initiation of an adaptive immune response. Originally, APC were thought to be responsible for initiating most immune responses in the skin. However, as it then been found that there are other cells that express langerin, including dermal dendritic cells (DC) (Clausen and Stoitzner Citation2015). Such DC appear to be required for T-cell-mediated immune responses in the skin.
Co-stimulatory and co-inhibitory pathways signal simultaneously to regulate whether antigen exposure will lead to T-cell activation. Danger signals released as a result of cell stress due to direct drug toxicity are an important stimulus for the up-regulation of co-stimulatory ligands on APC (Gallucci and Matzinger Citation2001). Similarly, it has been shown that dysregulated co-inhibitory receptor signaling can propagate autoimmunity (Chuang et al. Citation2005), and that blocking these pathways (such as PD-1 and CTLA-4) in cancer models can significantly enhance functional T-cell responses (Kouki et al. Citation2000; Hodi et al. Citation2010).
As discussed above, drug exposure can result in an array of cutaneous hypersensitivity reactions that vary in severity. Mild-to-moderate conditions, such as MPE, involve the preferential activation of perforin-producing CD4 T-cells, and in the case of β-lactam reactions, T-helper (TH)-22 cells play a dominant role (Castrejon et al. Citation2010; Whitaker et al. Citation2011; Sullivan et al. Citation2018). In contrast, urticaria is mediated by plasma cell-generated IgE antibodies, and pustular exanthema is mediated by neutrophils (Pichler Citation2003).
Various SCAR develop more rapidly and severely after the first episode in recurrent patients, suggesting that the pathogenesis of SCAR involves an immune memory response. The skin lesions of SCAR show infiltration of immune cells, in which, SJS and TEN have predominately CD8 T-cells and NK cells (Nassif et al. Citation2004; Wei et al. Citation2012; Yun et al. Citation2013; Cho et al. Citation2014). CD8 T-cells (also known as cytotoxic T-cells; CTL) have been shown to produce cytokines (e.g. interleukin [IL]-15 and IL-2), chemokines, and cytotoxic proteins (e.g. granulysin and granzyme B), to induce cutaneous inflammation and kill keratinocytes in SJS/TEN (Chung et al. Citation2008; Su et al. Citation2017). With the exception of abacavir, certain drugs can activate CD4 and CD8 T-cells in all hypersensitive patients (Pichler Citation2003; Adam et al. Citation2012; Lucas et al. Citation2015).
Proposed mechanisms for drug-induced hypersensitivity reactions in the skin
Three models have been proposed to examine the antigenicity of small-molecule drugs (Chung et al. Citation2016). The first is the hapten hypothesis, which goes back many years to the work of Landsteiner and Jacobs (Citation1935) who found that small molecules did not generally induce an immune response unless they covalently bound to proteins and thus formed an altered or “foreign” protein. However, foreign proteins do not provoke much of an immune response in the absence of activation of APC. This led to the second hypothesis, namely the danger hypothesis proposed by Matzinger (Citation1998), which stated that unless something was causing damage and triggered an innate sense of danger, the immune system ignored it. This damage could lead to production of danger-associated molecular pattern (DAMP) molecules that activate APC. Therefore, the hapten and danger hypotheses are complementary.
The third hypothesis stems from a concept of interaction of drugs with immune receptors that do not involve a reactive metabolite and includes two models: the pharmacological inter-action (p–i) model and the altered peptide repertoire model (Pichler Citation2002). The p–i model proposes that a drug or its metabolite directly and non-covalently binds to the human leukocyte antigen (HLA) molecule on APC in a region exposed to T-cells. In this model, antigen processing (generation of drug-self peptide complex) in APC can be bypassed. For example, carbamazepine was shown to bind the HLA-B*15:02 protein and induce CTL-mediated keratinocyte lysis, supporting this model (Wei et al. Citation2012). The altered peptide repertoire model, although distinct from the p–i model, is closely related and therefore discussed along with the p–i model. It postulates that the drug strongly binds to the cleft of a HLA molecule and alters the repertoire of the self-peptides presented by the HLA protein to T-cells. The anti-HIV drug abacavir was suggested to bind strongly to its associated HLA-B*57:01 protein and change the bound self-peptide (Illing et al. Citation2012).
Activation of the immune system based on any of these three hypotheses involves the classical sensitization and elicitation mechanism by which drugs presented by APC sensitize TH cells first, and then CTL are activated to induce keratinocyte damage through recognition of HLA-class I-drug complex. However recently, the heterologous immunity model was raised (White et al. Citation2015). This hypothesis proposes that virus-specific T-cells may cross-react with drug-bound peptide-HLA complexes to initiate the immune response. As intrinsic T-cells against viruses can react with the drug complex, the sensitization process may be skipped in this model. This may explain the occasional rapid onset of a rash on first exposure to a drug.
Other mechanistic hypotheses include mitochondrial injury, unfolded protein response, and the inflammogen hypotheses, which may or may not involve a reactive metabolite (Cho and Uetrecht Citation2017). These effects could directly cause idiosyncratic drug reactions (IDR) or lead to the production of various DAMP that promote an immune response.
Although the mechanistic progress has been striking in this area, several obstacles still exist for the prediction of skin toxicity/adverse reactions. First, knowledge of pathophysiological mechanisms is still scarce to explain the diverse types of adverse skin reactions. To solve this problem, greater mechanistic understanding is clearly necessary. Second, most adverse skin reactions are thought to be immune- or metabolism-mediated (at least partly), and thus, human-specific. Therefore, it is difficult to predict skin reactions using animal models, and thus proper in vitro or transgenic animal models are warranted based on mechanisms. Third, the predictability of the currently used LTT is insufficient to find a true causative drug. New diagnostic methods are warranted considering the mechanisms.
Molecular interaction between drugs and HLA/TCR in SJS and TEN
As HLA is the most polymorphic gene in the human genome, and has the ability to present antigens to activate T-cells, HLA has been proposed to be the risk factor of SCAR. The previous studies of Chung et al. (Citation2004) and Hung et al. (Citation2005) first discovered the strong HLA genetic predisposition to SCAR in Han Chinese, which included (1) HLA-B*15:02 and carbamazepine-, or oxcarbazepine-induced SJS/TEN (Chung et al. Citation2004; Chen et al. Citation2017), (2) HLA-B*58:01 and allopurinol-induced SCAR (Hung et al. Citation2005), and (3) HLA-A*31:01 and carbamazepine-induced DRESS and maculopapular eruption (Tate et al. Citation2006). These genetic associations were validated in different populations around the world (Lonjou et al. Citation2008; Tassaneeyakul et al. Citation2009; McCormack et al. Citation2011; Genin et al. Citation2014). In addition, there are increasing reports showing that drug hypersensitivity has strong HLA genetic predisposition, for example, abacavir hypersensitivity and HLA-B*57:01 (Mallal et al. Citation2002), and dapsone hypersensitivity and HLA-B*13:01. (Zhang et al. Citation2013). shows examples of HLA types reported to be associated with specific drug-induced SCARs in the ethnic populations (Kaniwa and Saito Citation2013; Chung et al. Citation2016). Based on these scientific reports, HLA markers are described in drug labels in several countries. displays a description of HLA marker types in carbamazepine labels.
Table 1. Examples of associated HLA types with SCARs.
Table 2. Description of HLA marker types in carbamazepine labels.
In addition to the immune determinant HLA genes, cytochrome P 2C9 (CYP2C9) variants causing defects of drug metabolism were shown to be strongly associated with pheny-toin-induced SCAR (Chung and Hung Citation2014). The defects of drug metabolism or clearance could also be due to renal deficiency in allopurinol-SCAR patients, and both genetic deficiency and underlying disease can lead to the accumulation of drug antigens (Chung and Hung Citation2014; Chung et al. Citation2015; Yang et al. Citation2015; Ng et al. Citation2016). The delayed drug clearance was associated with increased serum levels of granulysin, and high mortality within SCAR patients (Chung et al. Citation2015; Yang et al. Citation2015; Ng et al. Citation2016). As the strength of association is strong, numerous genetic markers have been applied in clinics to prescreen patients and identify genes that may indicate a predisposition to SCAR (Mallal et al. Citation2008; Chen et al. Citation2011; Pan et al. Citation2017).
The HLA and CYP predisposition observed in drug hypersensitivity are not only genetic markers, but play immunotoxicologic functional roles in the pathogenesis of SCAR. By applying mass spectrometry, Yang et al. (Citation2007) reported that there was no detection of specific carbamazepine-modified peptides with HLA-B*15:02 protein. Wei et al. (Citation2012) reported that endogenous peptide-loaded HLA-B*15:02 molecules presented carbamazepine to CTL without involvement of intracellular drug metabolism or antigen processing. The HLA-B*15:02-peptide-β2-microglobulin protein complex exhibited binding affinity toward chemicals with 5-carboxa-mide on the tricyclic ring, such as carbamazepine and oxcarbazepine. HLA-B75 family members share a structure similar to that of HLA-B*15:02, and could also present carbamazepine to activate CTL cells. Three residues (Asn63, Ile95, and Leu156) in the peptide-binding groove of HLA-B*15:02 were involved in carbamazepine presentation and CTL activation. In particular, Asn63 shared by members of the B75 family was seen to be the key residue. Computer simulations revealed a preferred molecular conformation of the 5-carboxamide group of carbamazepine and the sidechain of Arg62 on the B pocket of HLA-B*15:02. Furthermore, prefer-ential T-cell receptor (TCR)-Vβ and TCR-Vα usages and oligo-clonal expansion of TCR were identified in the ex vivo drug-expanded cultures of peripheral blood mononuclear cells (PBMC) of patients with carbamazepine-induced SJS/TEN (Ko et al. Citation2011). That study showed that the expression of TCR was drug-specific and phenotype-specific, and that TCR transfectants showed an immune response to the drug in a HLA-restricted manner.
Binding of the allopurinol metabolite, oxypurinol, to HLA-B*58:01 molecule was also identified by T-cell proliferation assays and in silico analysis (Lin et al. Citation2015). A high concentration of oxypurinol stimulated the T-cells of PBMC of patients with allopurinol-SCAR to produce granulysin and granzyme B (Chung et al. Citation2015). The Arg97 located between the E and C pocket of HLA-B*58:01 was identified as a key residue for oxypurinol binding (Lin et al. Citation2015). Furthermore, the technology of next-generation sequencing was applied to study TCR clonotypes of allopurinol-induced SCAR (Chung et al. Citation2015). Preferential TCR-V-β usage and clonal expansion of specific CDR3 (third complementarity-determining region) were identified in blister cells from skin lesions and oxypurinol-activated T-cells from allopurinol-SCAR patients (Chung et al. Citation2015).
In summary, these molecular and functional studies on carbamazepine and allopurinol revealed that in susceptible subjects, the plasma levels of drug antigens increased due to the defects of drug metabolism, and the genetically associated HLA-B molecules have the ability to present drug antigens to clonotype-specific T-cells, supporting the p–i model. On recognizing the drug antigen-HLA protein complex, T-cells express cytotoxic proteins (e.g. granulysin) and cytokines (e.g. IL-15), resulting in extensive skin lesions in SCAR. As viral reactivation was found in the acute stage of DRESS patients and several viral infections share the clinical presentation of SJS/TEN (Chiou et al. Citation2008; Chung et al. Citation2013), previous immune history or microbial infections may contribute to the diversity of the TCR repertoire (White et al. Citation2015). Further studies are needed to investigate the immunological basis of different types of drug hypersensitivity.
The innate immune response: inflammasome activation in the skin
An important question in understanding the etiology of drug-induced skin reactions is how APC are activated in the skin. Contact hypersensitivity is similar to drug-induced skin rashes in that in both cases many, if not most, appear to involve chemically reactive species. It was found that mice that lack components of the inflammasome are resistant to contact hypersensitivity (Watanabe et al. Citation2007). Inflammasomes are multimeric protein complexes that are involved in producing an inflammatory response to pathogen-associated molecular pattern (PAMP) and DAMP molecules. They convert procaspase-1 to active caspase-1 that, in turn, converts pro-IL-1β, and pro-IL-18 into their active forms (Man and Kanneganti Citation2015). The most studied inflammasome is NLRP3. Inflammasome activation has been implicated in many chronic diseases including multiple sclerosis, Alzheimer’s disease, atherosclerosis, and Type 2 diabetes (Guo et al. Citation2015).
The ability of a drug to activate inflammasomes in macrophages appears to predict whether it can cause skin rashes or not (Weston and Uetrecht Citation2014). However, the drugs tested had intrinsic chemical reactivity and could directly activate macrophages. Many drugs presumably require metabolic activation, and most drug-metabolizing enzymes in the skin appear to reside in keratinocytes. Therefore, in most cases, it may be that various DAMP produced by keratinocytes lead to the activation of inflammasomes in APC and other macrophages. This, in turn, could lead to an immune-mediated skin rash in susceptible patients.
Reactive metabolite formation in the skin
Circumstantial evidence suggests that many IDR are due to reactive metabolites or chemically reactive drugs, such as the β-lactams. Reactive metabolites can be formed by almost any metabolic pathway including oxidation, reduction, acetylation, sulfation, and glucuronida-tion. The metabolic enzymes with the greatest activity in the skin lead to sulfation and acetylation (Baron and Merk Citation2001). Although it is possible that reactive metabolites formed in the liver or elsewhere could cause an immune response in the skin, given the short half-life of most reactive metabolites, it seems most likely that for a reactive metabolite to cause a skin rash it must be formed in the skin. For example, the reactive benzylic sulfate of nevirapine is also formed in the liver and could be detected in the serum of treated rats. However, inhibition of sulfation in the liver did not prevent the skin rash while a topical sulfotransferase inhibitor did prevent it (Sharma et al. Citation2013).
Most reactive metabolites are formed by oxidation, and the major enzymes involved in the oxidation of drugs to reactive metabolites are the cytochromes P450 (CYPs). CYP1A1, 1B1, and 2E1 have been identified in Langerhans cells differentiated from CD34+ cells, adult normal keratinocytes, fibroblasts and melanocytes, and CYP2C, 2D6, and 3A5 were identified in fibro-blasts (Saeki et al. Citation2002). These results were replicated by a microarray analysis, in which the CYP1A1, 1B1, and 2E1 were expressed at comparable levels with the liver (Baron et al. Citation2008). Additionally, several other drug-metabolizing enzymes including sulfotransferases, N-acetyl-transferases, UDP-glucuronosyltransferases, flavin mono-oxygenase (FMO), and carboxyl-esterases are reported to be expressed in the skin (Hu et al. Citation2010). For further extensive discussion on this subject, please see the review by Sharma et al. (Citation2013).
Although mRNA coding for various P450 are found in the skin, the expression of these proteins in the skin is low, and there are very few examples in which drugs have been shown to be oxidized to reactive metabolites in the skin. Furthermore, most drugs that form P450-mediated reactive metabolites and cause idiosyncratic liver injury are not associated with a significant risk of skin rash though it is much easier to induce an immune response in the skin than in the liver.
Many drugs associated with IDRs have the potential to form several reactive metabolites. For example, the skin rash due to nevirapine is clearly caused by a reactive benzylic sulfate formed in the skin while most of the covalent binding in the liver involves the formation of a reactive quinone methide mediated by P450. Carbamazepine forms a 10,11-epoxide, which is weakly reactive (Yip et al. Citation2017). Its phenolic metabolites suggest that it can also form a 2,3 epoxide. 2-hydroxycarbamazepine can be further oxidized to an iminoquinone (Ju and Uetrecht Citation1999), 3-hydoxycarbamazepine can be oxidized to a relatively stable free radical (Lu and Uetrecht Citation2008), and the glycol metabolite formed by hydrolysis of the 10,11-epoxide has the potential to form a benzylic sulfate analogos to that of nevirapine. These pathways are shown in . At present, it is unknown which, if any, of these potential reactive metabolites is responsible for the IDRs associated with carbamazepine, which includes TEN. The observation that polymorphisms in epoxide hydrolase are not associated with an altered risk in developing carbamazepine-induced hypersensitivity reactions (including TEN) suggests, but does not prove, that an epoxide is not responsible for the IDRs associated with this drug (Green et al. Citation1995). In addition, although the 10,11-epoxide has a long half-life and can reach the skin, oxcarbazepine also causes TEN. The same HLA is a risk factor for carbamazepine- and oxcarbazepine-induced TEN, but oxcarbazepine cannot form the 10,11-epoxide (Chen et al. Citation2017). It is possible that the iminoquinone could be formed in the liver, be reduced to an aminophenol, and then be re-oxidized in the skin, as aminophenols are very easily oxidized even by molecular oxygen.
Figure 1. Possible reactive metabolites of carbamazepine (shown in red), which illustrates how difficult it can be to determine which chemical species is responsible for a given idiosyncratic drug reaction. The major metabolite of carbamazepine is the 10,11-epoxide. It has a relatively long half-life, and it is easily detected in the serum of patients taking carbamazepine. However, it is chemically reactive and forms protein adducts. Therefore, it could reach the skin and be responsible for skin rashes. However, as noted in the text, oxcarbazepine also causes serious skin rashes such as toxic epidermal necrolysis, and the same HLA gene is a risk factor; however, oxcarbazepine cannot form a 10,11-epoxide. The 10,11-epoxide is hydrolyzed to a diol and this could form a sulfate conjugate, which is a benzylic sulfate analogous to the sulfate of 12-hydroxynevirapine and predicted to be reactive. Carbamazepine is believed to form a 2,3-epoxide based on the observed 2-, and 3-hydroxycarbamazepine metabolites. However, this epoxide has not been detected in serum and is unlikely to reach the skin in significant concentrations. The 2,3-epoxide could be formed in the skin, but the cytochrome P450 levels in skin are low. The 2-hydroxycarbamazepine can be further oxidized to a reactive iminoquinone. This is likely to occur in the liver, but it can redox cycle back to 2-hydroxyiminostibine, which could reach the skin and be easily re-oxidized to the iminoquinone. There are many more resonance structures for the free radical of 3-hydroxycarbamazepine than for 2-hydroxycarba-mazepine, and such a free radical could easily be formed and cause oxidative stress. Either the 2- or 3-hydroxycarbamazepine could be oxidized to a catechol, and this could be further oxidized to a reactive o-quinone. Carbamazepine can also cause agranulocytosis and aplastic anemia. It is oxidized to an acridine derivative by myeloperoxidase, and this involves a ring contraction that presumably goes through a reactive carbocation intermediate. Thus there are many candidates for the chemical species that is responsible for serious carbamazepine-induced skin rashes, and it may be a combination of metabolites or it is also possible that carbamazepine itself could be responsible for causing skin rashes. (Adapted from Ju and Uetrecht [Citation1999], Lu et al. [Citation2008], and Yip and Pirmohamed [Citation2017]).
![Figure 1. Possible reactive metabolites of carbamazepine (shown in red), which illustrates how difficult it can be to determine which chemical species is responsible for a given idiosyncratic drug reaction. The major metabolite of carbamazepine is the 10,11-epoxide. It has a relatively long half-life, and it is easily detected in the serum of patients taking carbamazepine. However, it is chemically reactive and forms protein adducts. Therefore, it could reach the skin and be responsible for skin rashes. However, as noted in the text, oxcarbazepine also causes serious skin rashes such as toxic epidermal necrolysis, and the same HLA gene is a risk factor; however, oxcarbazepine cannot form a 10,11-epoxide. The 10,11-epoxide is hydrolyzed to a diol and this could form a sulfate conjugate, which is a benzylic sulfate analogous to the sulfate of 12-hydroxynevirapine and predicted to be reactive. Carbamazepine is believed to form a 2,3-epoxide based on the observed 2-, and 3-hydroxycarbamazepine metabolites. However, this epoxide has not been detected in serum and is unlikely to reach the skin in significant concentrations. The 2,3-epoxide could be formed in the skin, but the cytochrome P450 levels in skin are low. The 2-hydroxycarbamazepine can be further oxidized to a reactive iminoquinone. This is likely to occur in the liver, but it can redox cycle back to 2-hydroxyiminostibine, which could reach the skin and be easily re-oxidized to the iminoquinone. There are many more resonance structures for the free radical of 3-hydroxycarbamazepine than for 2-hydroxycarba-mazepine, and such a free radical could easily be formed and cause oxidative stress. Either the 2- or 3-hydroxycarbamazepine could be oxidized to a catechol, and this could be further oxidized to a reactive o-quinone. Carbamazepine can also cause agranulocytosis and aplastic anemia. It is oxidized to an acridine derivative by myeloperoxidase, and this involves a ring contraction that presumably goes through a reactive carbocation intermediate. Thus there are many candidates for the chemical species that is responsible for serious carbamazepine-induced skin rashes, and it may be a combination of metabolites or it is also possible that carbamazepine itself could be responsible for causing skin rashes. (Adapted from Ju and Uetrecht [Citation1999], Lu et al. [Citation2008], and Yip and Pirmohamed [Citation2017]).](/cms/asset/0c0aa83d-6f4e-42c5-83bb-cbb5fd48a99b/iimt_a_1514444_f0001_c.jpg)
Other drugs that can redox cycle and are very easily oxidized are aromatic amines such as sulfamethoxazole, which are associated with skin rashes. Likewise, 3-hydroxycarbamazepine may be oxidized by a peroxidase or some other enzyme in the skin. Finally, the benzylic sulfate of the diol metabolite could be formed analogous to the bioactivation of nevirapine. The fact that drug, such as carbamazepine, often causes systemic hypersensitivity reactions suggesting that they also form reactive metabolites outside the skin. Without a valid animal model, it is very difficult to determine which chemical entity is responsible for carbamazepine-induced hyper-sensitivity reactions, and different species could be responsible for different IDRs, as appears to be the case for nevirapine.
Sulfation is an attractive metabolic pathway for the formation of reactive metabolites in the skin because, unlike with the various P450, sulfotransferase activity is high in the skin. One of the few drugs that causes skin rashes and has been shown to be oxidized by these P450 in the skin is lamotrigine (Chen et al. Citation2010). It forms a glutathione conjugate when incubated with keratino-cytes, and the glutathione conjugate is presumably formed from an epoxide. However, the reaction constant Km is very high, and the glutathione conjugate was not detectable unless very high concentrations of lamotrigine were used. Another metabolite of lamotrigine is the N-oxide. N-Oxides, such as minoxidil, are substrates for sulfotransferase. The sulfate of lamotrigine N-oxide is reactive (Lu and Uetrecht Citation2007), and preliminary evidence suggests that it is formed by human sulfotransferases (unpublished observation). Presumably IDRs caused by lamotrigine could be due to the parent drug through the p–i mechanism. Without a valid animal model, it will be very difficult to test these various hypotheses. A similar problem may exist for other drugs that have the potential to form reactive sulfate conjugates.
If, as discussed below, the dominant immune response to drugs that can cause IDRs is immune tolerance, blocking immune tolerance may unmask the ability of a drug to cause skin rashes. A major molecule involved in immune tolerance is the checkpoint molecule PD-1. Therefore, it may be possible to develop an animal model using the PD-1−/− mouse; however, the benzylic alcohol of nevirapine is not a substrate for murine sulfotransferases, and nevirapine does not induce a skin rash in mice.
Animal models of hypersensitivity reactions
Nevirapine-induced skin rash model
Nevirapine causes skin rash in about 10% of patients treated with drug; however, in early clinical trials almost everyone developed a rash when the dose was escalated (Uetrecht: personal communication). In most patients, the rash is mild; unfortunately, severe rashes such as TEN can occur. Nevirapine also caused a skin rash in rats; an accidental finding while trying to identify potential reactive metabolites of nevirapine (Uetrecht, personal communication). The rash was idiosyncratic in that it did not occur in male rats, the incidence was about 20% in female Sprague Dawley rats, but it was 100% in female Brown Norway rats (Shenton et al. Citation2003). The rash was clearly immune mediated as it occurred more rapidly in rats that had been previously sensitized by treatment with nevirapine, and this sensitivity could be transferred to naïve rats with lymphocytes from sensitized rats. In addition, depletion of CD4 T-cells prevented rash (Shenton et al. Citation2005). The characteristics of rash in rats were similar to the rash in humans, especially in patients with low CD4 T-cell count and are at decreased risk, and incubation of PBMC from patients with a history of nevirapine-induced rash with nevirapine also resulted in the release of interferon (IFN)-γ (Chen et al. Citation2009). Therefore, this appears to be a very good animal model of an IDR, and possibly the only animal model of an IDR with characteristics similar to that of the IDR in humans that occurs without manipulation of the immune system.
The nevirapine model also allowed for testing the basis for the p–i hypothesis. Peripheral mononuclear leukocytes from animals that have had a nevirapine-induced skin rash were activated by the parent drug, despite the fact that it is the reactive benzylic sulfate that induces the rash (Chen et al. Citation2009). Therefore, what the lymphocytes from a patient who has had an IDR respond to in vitro is not necessarily what induced the IDR in the first place, which suggests that the basis for the p–i hypothesis is false. Thus, although the in vitro results suggest that nevirapine-induced skin rash involves the p–i hypothesis, the rash is due to reactive metabolite. That calls into question other in vitro studies that have been used to support the p–i hypothesis; therefore, the p–i hypothesis remains a hypothesis without definitive supportive evidence. These are two examples of the power of a valid animal model that made it possible to test hypotheses that would be virtually impossible to test in humans.
An important factor for developing animal models of IDRs is immune tolerance
Nevirapine is an exception; other attempts to reproduce adverse reactions in animals similar to IDRs in humans have failed. In addition, even though nevirapine can also cause idiosyncratic liver injury in humans, and Brown Norway rats develop a skin rash similar to the rash that it causes in humans, the rats did not develop liver injury. Extensive efforts to develop animal models based on the current hapten and danger hypotheses have been unsuccessful. Based on the danger hypothesis, and in addition to treating animals with drugs that cause IDRs in humans and covalently bind to hepatic proteins, the Uetrecht labaratory tried to activate APC through toll-like receptors, for example, lipopolysaccharide (LPS) and polyinosinic:polycytidylic acid (poly-IC). This was done with several drugs, but without success; most of this work was never published. This is consistent with the clinical observation that patients with inflammatory conditions such as ulcerative colitis do not appear to have a higher risk of IDR. In most cases, the immune system seems to be able to determine whether the danger signal came from the drug or some other factor, and only respond to the drug if it is the drug that caused the injury. Immunizing animals with isoniazid- or amodiaquine-modified hepatic proteins followed by treatment with the drug, only led to immune tolerance (Mak and Uetrecht Citation2015b). This is consistent with the fact that patients with mild IDRs often have resolution of the IDR despite continued treatment with the drug. If the injury is immune mediated, then the resolution of the mild injury must involve immune tolerance. Therefore, if it is immune tolerance that keeps most animals and humans from having an IDR to a drug that commonly causes IDR, it might be possible to develop an animal model by inhibiting immune tolerance. In fact, this strategy works. Specifically, immune checkpoint inhibition using a combination of PD-1−/− mice and anti-CTLA-4 antibodies unmasks the potential of drugs to cause idiosyncratic drug-induced liver injury (Mak and Uetrecht Citation2015a; Metushi et al. Citation2015), and therefore has the potential to test hypotheses that have not been possible to test in the past.
In vitro strategies to study immunological drug reactions
Susceptibility to drug hypersensitivity is a function of the chemistry of the drug, the genetics of the patient, and environmental factors the patient was exposed to at the time of reaction. It is incredibly difficult to apply each of these factors to an in vitro cell system; as such there are no currently no assays available for predicting drug immunogenicity. The purpose of this section of the article is to provide a brief summary of what is known about drug-specific T-cell responses and how this knowledge is being used to develop novel T-cell priming assays that may in the future be used to screen for drug immunogenicity and skin sensitization.
Whenever possible T-cells isolated from blood and tissues of hypersensitive patients should be used to investigate the cellular pathophysiology of the adverse event and the way in which drugs interact with immune receptors (major histocompatibility complex [MHC] and the T-cell receptor [TCR]), as with these cells one can reimagine in vitro the relevant drug immune cell interactions. As discussed earlier, the discovery of strong associations between expression of single HLA alleles and susceptibility to drug hypersensitivity has changed the way in which we view hypersensitivity reactions to drugs; they are no longer totally unpredictable. As such, it is important that all mechanistic studies use cells from HLA-typed patients alongside a battery of in vitro assays to characterize the effects of drug treatment on DC signaling and the activation of cells of the adaptive immune system. Moreover, state-of-the-art mass spectrometers should be available to study the metabolism of drugs in in vitro culture systems, characterize and quantify the binding of drug metabolites to protein, and relate the chemistry of adduct formation to the biological response (i.e. activation of T-cells). illustrates a research strategy that could be applied to study drug hypersensitivity using patient cells.
Figure 2. Drug hypersensitivity research strategy. Drug hypersensitivity research strategy using PBMC from hypersensitive patient and healthy volunteers. Patient PBMC are used directly in diagnostic tests and for mechanistic studies to assess antigen specificity and mechanisms of HLA allele-restricted T-cell activation. PBMC from volunteers are used to investigate the priming of naïve T-cells to drugs and drug metabolites. It is important that samples are collected from all immunological investigations to explore the cellular distribution of drugs and the irreversible binding of drugs to protein.
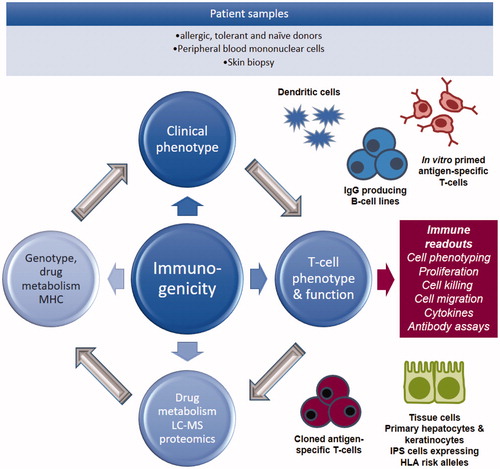
Activation of hypersensitive patient T-cells with drugs
Several drugs are directly protein-reactive, while others gain protein reactivity through drug metabolism. Reactive drugs bind covalently to nucleophilic amino acid residues on proteins to form adducts that, following protein processing, yields MHC binding peptides that activate T-cells (Meng et al. Citation2017). APC express low levels of drug metabolizing enzymes, when compared with hepatocytes; however, the levels of adducts formed from some drugs have been shown to be sufficient to bind to protein and form adducts that activate T-cells (Elsheikh et al. Citation2011). Reactive drug metabolites in in vitro culture also bind directly to MHC bound peptides to activate T-cells (Schnyder et al. Citation2000; Castrejon et al. Citation2010). Whether this represents an in vitro artifact mimicking a naturally processed protein adduct is currently not known. Drugs can also interact with MHC and T-cell receptors directly.
Classical studies demonstrated that T-cell receptors can be triggered with drugs rapidly (within minutes) through a pathway that does not require protein processing (Schnyder et al. Citation1997; Zanni et al. Citation1998). Abacavir has been shown to bind deep within the MHC peptide binding cleft, altering the space available for peptides to bind (Illing et al. Citation2012; Norcross et al. Citation2012; Ostrov et al. Citation2012). As such, a novel repertoire of peptides interact with the abacavir-modified MHC molecules, and it is believed that T-cells responsible for abacavir hypersensitivity are triggered by these peptide sequences. For all other drugs shown to activate T-cells directly (e.g. sulfa-methoxazole [Schnyder et al. Citation1997], oxypurinol [Yun et al. Citation2013], lamotrigine [Naisbitt et al. Citation2003]), the nature of the MHC binding interaction has not been defined.
Development of assays to study the priming of naïve T-cells
Studies utilizing hypersensitive patient T-cells are important, but they tell nothing about the origin of the drug-specific T-cell response. For this reason, researchers at the Medical Research Council (MRC)-funded Liverpool Centre for Drug Safety Science have spent 5 year developing cellular platforms using PBMC from healthy drug-naïve volunteers to study T-cell priming. The following section details the evolution of such assays, their current application domain and how further development might result in in vitro test systems to predict the intrinsic immunogenicity of new compounds. The availability of such assays is important as hyper-sensitivity often fails to be detected until post-marketing. As such, hypersensitivity reactions are a leading cause for drug withdrawal, resulting in patients needing to wait until an alternative is brought to the market (Negrini and Becquemont Citation2017).
Newman et al. (Citation1977) were the first to report that human lymphocytes could be primed in vitro against chemical sensitizers. Subsequently, Seldin and Rich (Citation1978) devised an in vitro system to study both primary and secondary responses of lymphocytes to hapten-conjugated proteins. Co-culture of lymphocytes with hapten-modified PBMC resulted in antigen-specific proliferative responses. The method has been modified in several subsequent studies; specifically, different forms of DC have been used as APC with naïve T-cells as responders. This allowed for discrimination of strong sensitizers from irritants. However, weak sensitizers failed to activate T-cells. In recent years, improved protocols have been used to generate DC, and T-regulatory (Treg) cells have been removed from responders to enhance sensitivity. Furthermore, multiple readouts are now available to detect the antigen-specific T-cell responses (Dietz et al. Citation2010; Martin et al. Citation2010).
Researchers at Liverpool developed methods to study the origin of drug- and drug metabolite-specific T-cell responses in healthy donors (Faulkner et al. Citation2012, Citation2016; Monshi et al. Citation2013). The assay relies on isolation and culture of autologos monocyte-derived DC and naive T-cells as APC and responder cells, respectively. After a 7–10 d culture period, T-cells are re-exposed to the drug antigen and DC before antigen specificity is measured shortly after. In addition to classical readouts for proliferation, cytokine secretion and cytotoxicity, a change in phenotype from naive T-cell to memory T-cell is quantified by flow cytometry. Strong responses are detected to haptenic drug metabolite(s), such as piperacillin and nitroso sulfamethoxazole (Faulkner et al. Citation2016). T-Cell priming is also detected against direct MHC binding drug metabolite(s), including sulfamethoxazole, carbamazepine, and oxypurinol. The platform is flexible in that it can be used to study (1) the phenotype of activated T-cells, (2) HLA-allele-restricted forms of hypersensitivity, (3) immune regulation, and (4) tissue-specific signaling, as briefly discussed below.
Immune cells activated by drugs
Chemically distinct drugs and metabolites have been shown to prime naïve T-cells, produce polarized cytokine secretion profiles following re-stimulation and display selective chemokine migration properties (Faulkner et al. Citation2012, Citation2016). In contrast, abacavir preferentially activates pre-existing memory T-cells from healthy donors that must have been primed by earlier exposure to another foreign antigen (Lucas et al. Citation2015). Most recently, focusing on sulfamethox-azole, we have shown that naïve and memory T-cells from healthy donors can be stimulated by parent drug and drug metabolites (Gibson et al. Citation2017). These data highlight the need to study naïve and memory T-cell populations.
HLA alleles
Several strong associations between susceptibility to specific forms of immunological drug reaction and expression of HLA alleles have been identified. For certain drugs, it has been possible to relate the genetic association to the disease by showing that T-cells are selectively activated in individuals expressing the risk allele. To study the propensity of drugs to initiate T-cell responses, it is critical to work with genetically characterized cells. For this to be feasible, the Naisbitt laboratory has established a unique HLA genotyped cell bank containing ≈100 million PBMC from 1000 healthy donors.
Immune regulation
Most HLA associations with drug hypersensitivity have a low prognostic value (e.g. HLA-B*57:01 is associated with flucloxacillin-induced liver injury, but only 1/1000 HLA-B*57:01-expressing individuals develop hypersensitivity). Therefore, additional immunological parameters are involved in regulating the activation of drug-specific T-cells. Co-stimulatory and co-inhibitory pathways signal simultaneously to regulate whether antigen exposure will lead to T-cell activation. Thus, it is conceivable that dysregulation of these pathways may manipulate the threshold of T-cell activation to such an extent that they enhance a patient’s susceptibility to drug hypersensitivity. Indeed, an inhibitory role for the PD-1 and CTLA-4 pathways during the priming of naïve T-cells to drugs has been reported (Gibson et al. Citation2014, Citation2017). As research moves forward, it will be crucial to elucidate the effect of modulation of multiple cosignaling pathways together to effectively analyze the role of inhibitory signaling during T-cell activation.
Tissue microenvironment
A limitation of existing T-cell priming assays is that immune cells are exposed to drugs directly or conjugated to a carrier protein. Assay development is complicated by the fact that many drugs are not directly protein-reactive, and as such, antigenic determinants are not always generated in the assay. Furthermore, the cross-talk between tissue and immune cells is important in determining the outcome of compound exposure. For example, hepatocytes treated with model hepatotoxins release signals that promote the secretion of polarizing cytokines from DC (Ogese et al. Citation2017). Thus, new strategies need to be developed to enhance exposure of immune cells to relevant tissue-specific stress signaling and tissue-derived drug-protein adducts. Development of a tissue/immune cell culture system has been a challenge for the investigation of drug immuno-genicity in humans as autologous cells are required. One way to overcome this shortcoming will be the generation of induced pluripotent stem cells (iPS)-derived keratinocyte lines and antigen-specific T-cells from the same donors expressing an important HLA risk allele.
Development of screening assays
Existing T-cell priming assays are still far from perfect. A major drawback is the experimental design, which is time-consuming and is more suited for laboratory-based investigation of a few variables. As such, they cannot currently be used to screen new compound for the potential to cause immune responses when administered to humans. A reason for this is the absence of medium/high throughput assays. Efforts to transform existing assays into suitable platforms for the screening of drugs are focused on miniaturization to assess (1) multiple HLA-typed individuals simultaneously and (2) multiple experiments with cells from one individual. The eventual transfer of such assays to industrial partners provides a pathway for assessment of immunogenicity of novel drugs and for initial application of the newly established methods.
Conclusions
Skin rashes continue to represent a significant clinical problem, and our mechanistic understanding is superficial. A simplified scheme of a reasonable hypothesis for the mechanism of serious skin rashes is shown in . The components of this hypothesis should be tested, and the only practical way to do this is by developing valid animal models that replicate the metabolic pathways in humans to accurately predict which drug candidates are likely to pose a significant risk of serious skin rashes.
Figure 3. Hypothesis by which a drug can cause a serious drug rash. Keratinocytes can metabolize some drugs to reactive metabolites, especially through the formation of reactive sulfate conjugates. The reactive metabolites can both covalently bind to proteins and cause cell damage leading to the release of danger associated molecular pattern (DAMP) molecules. These DAMP can activate dermal DC through toll-like receptors (TLR) that, in turn, can activate inflammasomes. In addition, the DC also process drug-modified proteins leading to drug-modified peptides. When presented in the context of major histocompatibility complex-1 (MHC-1), these peptides can be recognized by T-cells with the appropriate T-cell receptor. This can lead to the production of CTL that kill keratinocytes that express the drug modified peptides. This is grossly oversimplified as many other cells, such as TH cells, are also involved, many other molecules such as chemokines and cytokines are involved, and the initial activation of T-cells occurs in lymph nodes, not in the skin. There are also mechanisms that can lead to immune tolerance, such as activation of Treg cells that prevent serious skin rashes in most individuals.
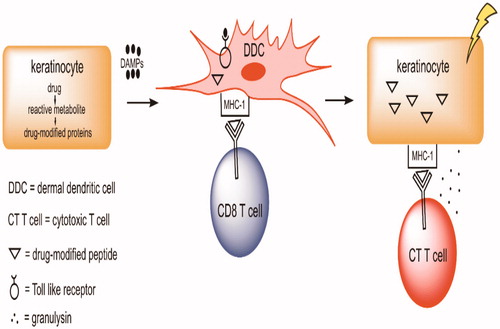
Even though the skin is less tolerogenic than most organs, the dominant immune response to drugs that can cause skin rashes in humans appears to be immune tolerance. Therefore, it may be possible to develop animal models of skin rashes by impairing immune tolerance. However, it is necessary that the animal has the same metabolic pathways as humans; this is not the case for the formation of a reactive sulfate metabolite of nevirapine in mice. With such models, it might be possible to predict as to which drug candidates are likely to pose a significant risk of serious skin rashes.
Disclosure statement
The authors alone are responsible for the content of this review.
References
- Adam J, Eriksson K, Schnyder B, Fontana S, Pichler W, Yerly D. 2012. Avidity determines T-cell reactivity in abacavir hypersensitivity. Eur J Immunol. 42:1706–1716.
- Baron J, Merk H. 2001. Drug metabolism in the skin. Curr Opin Allergy Clin Immunol. 1:287–291.
- Baron J, Wiederholt T, Heise R, Merk H, Bickers D. 2008. Expression and function of cytochrome p450-dependent enzymes in human skin cells. Curr Med Chem. 15:2258–2264.
- Calne R, Sells R, Pena J, Davis D, Millard P, Herbertson B, Binns R, Davies D. 1969. Induction of immunological tolerance by porcine liver allografts. Nature 223:472–476.
- Castrejon J, Berry N, El-Ghaiesh S, Gerber B, Pichler W, Park B, Naisbitt D. 2010. Stimulation of human T-cells with sulfonamides and sulfonamide metabolites . J Allergy Clin Immunol. 125:411–418.
- Chen C, Hsiao Y, Wu T, Hsih M, Tassaneeyakul W, Jorns T, Sukasem C, Hsu C, Su S, Chang W, et al. 2017. Risk and association of HLA with oxcarbazepine-induced cutaneous adverse reactions in Asians. Neurology 88:78–86.
- Chen H, Grover S, Yu L, Walker G, Mutlib A. 2010. Bioactivation of lamotrigine in vivo in rat and in vitro in human liver microsomes, hepatocytes, and epidermal keratinocytes: Characterization of thioether conjugates by liquid chromatography/mass spectrometry and high field nuclear magnetic resonance spectroscopy. Chem Res Toxicol. 23:159–170.
- Chen P, Lin J, Lu C, Ong C, Hsieh P, Yang C, Tai C, Wu S, Lu C, Hsu Y, et al. 2011. Carbamazepine-induced toxic effects and HLA-B*1502 screening in Taiwan. N Engl J Med. 364:1126–1133.
- Chen X, Tharmanathan T, Mannargudi B, Gou H, Uetrecht J. 2009. A study of the specificity of lymphocytes in nevirapine-induced skin rash. J Pharmacol Exp Ther. 331:836–841.
- Chiou C, Yang L, Hung S, Chang Y, Kuo T, Ho H, Hu S, Hong H, Chung W. 2008. Clinicopathological features and prognosis of drug rash with eosinophilia and systemic symptoms: A study of 30 cases in Taiwan. J Eur Acad Dermatol Venereol. 22:1044–1049.
- Cho T, Uetrecht J. 2017. How reactive metabolites induce an immune response that sometimes leads to an idiosyncratic drug reaction. Chem Res Toxicol. 30:295–314.
- Cho Y, Lin J, Chen Y, Chang C, Hsiao C, Chung W, Chu CY. 2014. Generalized bullous fixed drug eruption is distinct from Stevens-Johnson syndrome/toxic epidermal necrolysis by immunohistopathological features. J Amer Acad Dermatol. 70:539–548.
- Chuang W, Strobel P, Gold R, Nix W, Schalke B, Kiefer R, Opitz A, Klinker E, Muller-Hermelink H, Marx A. 2005. A CTLA4high genotype is associated with myasthenia gravis in thymoma patients. Ann Neurol. 58:644–648.
- Chung W, Chang W, Stocker S, Juo C, Graham G, Lee M, Williams K, Tian Y, Juan K, Jan Y, et al. 2015. Insights into the poor prognosis of allopurinol-induced severe cutaneous adverse reactions: Impact of renal insufficiency, high plasma levels of oxypurinol and granulysin. Ann Rheum Dis. 74:2157–2164.
- Chung W, Hung S. 2014. Genetic factors associated with severe cutaneous adverse reactions-reply. JAMA 312:2166.
- Chung W, Hung S, Hong H, Hsih M, Yang L, Ho H, Wu J, Chen Y. 2004. Medical genetics: A marker for Stevens-Johnson syndrome. Nature 428:486.
- Chung W, Hung S, Yang J, Su S, Huang S, Wei C, Chin S, Chiou C, Chu S, Ho H, et al. 2008. Granulysin is a key mediator for disseminated keratinocyte death in Stevens-Johnson syndrome and toxic epidermal necrolysis. Nat Med. 14:1343–1350.
- Chung W, Pan R, Chu M, Chin S, Huang Y, Wang W, Chang J, Hung S. 2015. Oxypurinol-specific T-cells possess preferential TCR clonotypes and express granulysin in allopurinol-induced severe cutaneous adverse reactions. J Invest Dermatol. 135:2237–2248.
- Chung W, Shih S, Chang C, Lin T, Huang Y, Chang S, Liu M, Ko Y, Deng M, Liau Y, et al. 2013. Clinico-pathologic analysis of Coxsackievirus a6 new variant induced widespread mucocutaneous bullous reactions mimicking severe cutaneous adverse reactions. J Infect Dis. 208:1968–1978.
- Chung W, Wang C, Dao R. 2016. Severe cutaneous adverse drug reactions. J Dermatol. 43:758–766.
- Clausen B, Stoitzner P. 2015. Functional specialization of skin dendritic cell subsets in regulating T-cell responses. Front Immunol. 6:534.
- Di Meglio P, Perera G, Nestle F. 2011. The multitasking organ: recent insights into skin immune function. Immunity 35:857–869.
- Dietz L, Esser P, Schmucker S, Goette I, Richter A, Schnolzer M, Martin S, Thierse H. 2010. Tracking human contact allergens: From mass spectrometric identification of peptide-bound reactive small chemicals to chemical-specific naive human T-cell priming. Toxicol Sci. 117:336–347.
- Duong T, Valeyrie-Allanore L, Wolkenstein P, Chosidow O. 2017. Severe cutaneous adverse reactions to drugs. Lancet 390:1996–2011.
- Elsheikh A, Castrejon L, Lavergne S, Whitaker P, Monshi M, Callan H, El-Ghaiesh S, Farrell J, Pichler W, Peckham D, et al. 2011. Enhanced antigenicity leads to altered immunogenicity in sulfamethoxazole-hypersensitive patients with cystic fibrosis. J Allergy Clin Immunol. 127:1543–1551.
- Faulkner L, Gibson A, Sullivan A, Tailor A, Usui T, Alfirevic A, Pirmohamed M, Naisbitt D, Park B. 2016. Detection of primary T-cell responses to drugs and chemicals in HLA-typed volunteers: Implications for prediction of drug immunogenicity. Toxicol Sci. 154:416–429.
- Faulkner L, Martinsson K, Santoyo-Castelazo A, Cederbrant K, Schuppe-Koistinen I, Powell H, Tugwood J, Naisbitt D, Park B. 2012. The development of in vitro culture methods to characterize primary T-cell responses to drugs. Toxicol Sci. 127:150–158.
- Fernandez T, Canto G, Blanca M. 2009. Molecular mechanisms of maculopapular exanthema. Curr Opin Infect Dis. 22:272–278.
- Gallucci S, Matzinger P. 2001. Danger signals: SOS to the immune system. Curr Opin Immunol. 13:114–119.
- Genin E, Chen D, Hung S, Sekula P, Schumacher M, Chang P, Tsai S, Wu T, Bellon T, Tamouza R, et al. 2014. HLA-A*31:01 and different types of carbamazepine-induced severe cutaneous adverse reactions: An international study and meta-analysis. Pharmacogen J. 14:281–288.
- Gibson A, Faulkner L, Lichtenfels M, Ogese M, Al-Attar Z, Alfirevic A, Esser PR, Martin SF, Pirmohamed M, Park B, et al. 2017. The effect of inhibitory signals on the priming of drug hapten-specific T-cells that express distinct Vβ receptors. J Immunol. 199:1223–1237.
- Gibson A, Faulkner L, Wood S, Park B, Naisbitt D. 2017. Identification of drug- and drug-metabolite immune responses originating from both naive and memory T-cells. J Allergy Clin Immunol. 140:578–581.
- Gibson A, Ogese M, Sullivan A, Wang E, Saide K, Whitaker P, Peckham D, Faulkner L, Park B, Naisbitt D. 2014. Negative regulation by PD-L1 during drug-specific priming of IL-22-secreting T-cells and the influence of PD-1 on effector T-cell function . J Immunol. 192:2611–2621.
- Green V, Pirmohamed M, Kitteringham N, Gaedigk A, Grant D, Boxer M, Burchell B, Park B. 1995. Genetic analysis of microsomal epoxide hydrolase in patients with carbamazepine hypersensitivity. Biochem Pharmacol. 50:1353–1359.
- Guo H, Callaway J, Ting J. 2015. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med. 21:677–687.
- Hodi F, O'Day S, McDermott D, Weber R, Sosman J, Haanen J, Gonzalez R, Robert C, Schadendorf D, Hassel J, et al. 2010. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 363:711–723.
- Hu T, Khambatta Z, Hayden P, Bolmarcich J, Binder R, Robinson M, Carr G, Tiesman J, Jarrold B, Osborne R, et al. 2010. Xenobiotic metabolism gene expression in the EpiDermin vitro 3D human epidermis model compared to human skin. Toxicol In Vitro 24:1450–1463.
- Hung S, Chung W, Liou L, Chu C, Lin M, Huang H, Lin Y, Lan J, Yang L, Hong H, et al. 2005. HLA-B*5801 allele as a genetic marker for severe cutaneous adverse reactions caused by allopurinol. Proc Natl Acad Sci. USA 102:4134–4139.
- Illing P, Vivian J, Dudek N, Kostenko L, Chen Z, Bharadwaj M, Miles J, Kjer-Nielsen L, Gras S, Williamson N, et al. 2012. Immune self-reactivity triggered by drug-modified HLA-peptide repertoire. Nature 486:554–558.
- Ju C, Uetrecht J. 1999. Detection of 2-hydroxyiminostilbene in the urine of patients taking carbamazepine and its oxidation to a reactive iminoquinone intermediate. J Pharmacol Exp Ther. 288:51–56.
- Kaniwa N, Saito Y. 2013. Pharmacogenomics of severe cutaneous adverse reactions and drug-induced liver injury. J. Human Genetics 58:317–326.
- Ko T, Chung W, Wei C, Shih H, Chen J, Lin C, Chen Y, Hung S. 2011. Shared and restricted T-cell receptor use is crucial for carbamazepine-induced Stevens-Johnson syndrome. J Allergy Clin Immunol. 128:1266–1276.
- Kongpan T, Mahasirimongkol S, Konyoung P, Kanjanawart S, Chumworathayi P, Wichukchinda N, Kidkeukarun R, Preechakul S, Khunarkornsiri U, Bamrungram W, et al. 2015. Candidate HLA genes for prediction of co-trimoxazole-induced severe cutaneous reactions. Pharmacogen Genomics 25:402–411.
- Kouki T, Sawai Y, Gardine C, Fisfalen M, Alegre M, DeGroot L. 2000. CTLA-4 gene polymorphism at position 49 in exon 1 reduces the inhibitory function of CTLA-4 and contributes to the pathogenesis of Graves' disease. J Immunol. 165:6606–6611.
- Lally A, Turner R. 2007. A survey of approach to wound healing by secondary intention among British Society for Dermatological Surgery members. Br J Dermatol. 156:575–576.
- Lamoreux M, Sternbach M, Hsu W. 2006. Erythema multiforme. Am Fam Physician 74:1883–1888.
- Landsteiner K, Jacobs J. 1935. Studies on the sensitization of animals with simple chemical compounds. J Exp Med. 61:643–656.
- Lin C, Chen J, Ko T, Wei C, Wu J, Chung W, Chen S, Liao Y, Hung S, Chen Y. 2015. Immuno-logic basis for allopurinol-induced severe cutaneous adverse reactions: HLA-B*58:01-restricted activation of drug-specific T-cells and molecular interaction. J Allergy Clin Immunol. 135:1063–1065 e1065.
- Lonjou C, Borot N, Sekula P, Ledger N, Thomas L, Halevy S, Naldi L, Bouwes-Bavinck J, Sidoroff A, de Toma C, et al. 2008. European study of HLA-B in Stevens-Johnson syndrome and toxic epidermal necrolysis related to five high-risk drugs. Pharmacogen Genomics 18:99–107.
- Lu W, Li X, Uetrecht J. 2008. Changes in gene expression induced by carbamazepine and phenytoin: Testing the danger hypothesis. J Immunotoxicol. 5:107–113.
- Lu W, Uetrecht J. 2007. Possible bioactivation pathways of lamotrigine. Drug Metab Dispos. 35:1050–1056.
- Lu W, Uetrecht J. 2008. Peroxidase-mediated bioactivation of hydroxylated metabolites of carbamazepine and phenytoin. Drug Metabol Disposit 36:1624–1636.
- Lucas A, Lucas M, Strhyn A, Keane N, McKinnon E, Pavlos R, Moran E, Meyer-Pannwitt V, Gaudieri S, D'Orsogna L, et al. 2015. Abacavir-reactive memory T-cells are present in drug naïve individuals. PLoS One 10:e0117160
- Mak A, Uetrecht J. 2015a. The combination of anti-CTLA-4 and PD1−/− mice unmasks the potential of isoniazid and nevirapine to cause liver injury. Chem Res Toxicol. 28:2287–2291.
- Mak A, Uetrecht J. 2015b. Immunization with amodiaquine-modified hepatic proteins prevents amodiaquine-induced liver injury. J Immunotoxicol. 12:361–367.
- Mallal S, Nolan D, Witt C, Masel G, Martin A, Moore C, Sayer D, Castley A, Mamotte C, Maxwell D, et al. 2002. Association between presence of HLA-B*5701, HLA-DR7, and HLA-DQ3 and hypersensitivity to HIV-1 reverse-transcriptase inhibitor abacavir. Lancet 359:727–732.
- Mallal S, Phillips E, Carosi G, Molina J, Workman C, Tomazic J, Jagel-Guedes E, Rugina S, Kozyrev O, Cid J, et al. 2008. HLA-B*5701 screening for hypersensitivity to abacavir. N Engl J. Med 358:568–579.
- Man S, Kanneganti T. 2015. Regulation of inflammasome activation. Immunol Rev. 265:6–21.
- Martin S, Esser P, Schmucker S, Dietz L, Naisbitt D, Park B, Vocanson M, Nicolas J, Keller M, Pichler W, et al. 2010. T-Cell recognition of chemicals, protein allergens and drugs: towards the development of in vitro assays. Cell Mol Life Sci. 67:4171–4184.
- Matzinger P. 1998. An innate sense of danger. Semin Immunol. 10:399–415.
- McCormack M, Alfirevic A, Bourgeois S, Farrell JJ, Kasperavičiūtė D, Carrington M, Sills GJ, Marson T, Jia X, de Bakker PIW, et al. 2011. HLA-A*3101 and carbamazepine-induced hypersensitivity reactions in Europeans . N Engl J Med 364:1134–1143.
- Meng X, Al-Attar Z, Yaseen F, Jenkins R, Earnshaw C, Whitaker P, Peckham D, French N, Naisbitt D, Park B. 2017. Definition of the nature and hapten threshold of the β-lactic antigen required for t-cell activation in vitro and in patients. J Immunol. 198:4217–4227.
- Metushi I, Hayes M, Uetrecht J. 2015. Treatment of PD-1(−/−) mice with amodiaquine and anti-CTLA4 leads to liver injury similar to idiosyncratic liver injury in patients. Hepatology. 61:1332–1342.
- Monshi M, Faulkner L, Gibson A, Jenkins R, Farrell J, Earnshaw C, Alfirevic A, Cederbrant K, Daly A, French N, et al. 2013. Human leukocyte antigen (HLA)-B*57:01-restricted activa-tion of drug-specific T-cells provides the immunological basis for flucloxacillin-induced liver injury. Hepatology 57:727–739.
- Naisbitt D, Farrell J, Wong G, Depta J, Dodd C, Hopkins J, Gibney C, Chadwick D, Pichler W, Pirmohamed M, et al. 2003. Characterization of drug-specific T-cells in lamotrigine hypersensitivity. J Allergy Clin Immunol. 111:1393–1403.
- Nassif A, Bensussan A, Boumsell L, Deniaud A, Moslehi H, Wolkenstein P, Bagot M, Roujeau J. 2004. Toxic epidermal necrolysis: Effector cells are drug-specific cytotoxic T-cells. J Allergy Clin Immunol. 114:1209–1215.
- Negrini S, Becquemont L. 2017. HLA-associated drug hypersensitivity and the prediction of adverse drug reactions. Pharmacogenomics 18:1441–1457.
- Newman W, Stoner G, Bloom B. 1977. Primary in vitro sensitisation of human T-cells. Nature 269:151–153.
- Ng C, Yeh Y, Wang C, Hung S, Yang C, Chang Y, Chang W, Lin Y, Chang C, Su S, et al. 2016. Impact of the HLA-B(*)58:01 allele and renal impairment on allopurinol-induced cutaneous adverse reactions. J Invest Dermatol. 136:1373–1381.
- Norcross M, Luo S, Lu L, Boyne M, Gomarteli M, Rennels A, Woodcock J, Margulies D, McMurtrey C, Vernon S, et al. 2012. Abacavir induces loading of novel self-peptides into HLA-B*57: 01: An autoimmune model for HLA-associated drug hypersensitivity. AIDS 26:F21–F29.
- Ogese M, Ahmed S, Alferivic A, Betts C, Dickinson A, Faulkner L, French N, Gibson A, Hirschfield G, Kammuller M, et al. 2017. New approaches to investigate drug-induced hypersensitivity. Chem Res Toxicol. 30:239–259.
- Ostrov D, Grant B, Pompeu Y, Sidney J, Harndahl M, Southwood S, Oseroff C, Lu S, Jakoncic J, de Oliveira C, et al. 2012. Drug hypersensitivity caused by alteration of the MHC-presented self-peptide repertoire. Proc Natl Acad Sci USA. 109:9959–9964.
- Pan R, Dao R, Hung S, Chung W. 2017. Pharmacogenomic advances in the prediction and prevention of cutaneous idiosyncratic drug reactions. Clin Pharmacol Ther. 102:86–97.
- Pichler W. 2002. Pharmacological interaction of drugs with antigen-specific immune receptors: The p-i concept. Curr Opin Allergy Clin Immunol. 2:301–305.
- Pichler W. 2003. Delayed drug hypersensitivity reactions. Ann Intern Med. 139:683–693.
- Pichler W, Tilch J. 2004. The lymphocyte transformation test in the diagnosis of drug hypersensitivity. Allergy 59:809–820.
- Saeki M, Saito Y, Nagano M, Teshima R, Ozawa S, Sawada J. 2002. mRNA expression of multiple cytochrome P450 isozymes in four types of cultured skin cells. Int Arch Allergy Immunol. 127:333–336.
- Schnyder B, Burkhart C, Schnyder-Frutig K, von Greyerz S, Naisbitt D, Pirmohamed M, Park B, Pichler W. 2000. Recognition of sulfamethoxazole and its reactive metabolites by drug-specific CD4+ T-cells from allergic individuals. J Immunol. 164:6647–6654.
- Schnyder B, Mauri-Hellweg D, Zanni M, Bettens F, Pichler W. 1997. Direct, MHC-dependent presentation of the drug sulfamethoxazole to human alphabeta T-cell clones. J Clin Invest. 100:136–141.
- Seldin M, Rich R. 1978. Human immune responses to hapten-conjugated cells. I. Primary and secondary proliferative responses in vitro. J Exp Med. 147:1671–1683.
- Sharma A, Novalen M, Tanino T, Uetrecht J. 2013. 12-OH-nevirapine sulfate, formed in the skin, is responsible for nevirapine-induced skin rash. Chem Res Toxicol. 26:817–827.
- Shenton J, Popovic M, Chen J, Masson M, Uetrecht J. 2005. Evidence of an immune-mediated mechanism for an idiosyncratic nevirapine-induced reaction in the female Brown Norway rat. Chem Res Toxicol. 18:1799–1813.
- Shenton J, Teranishi M, Abu-Asab M, Yager J, Uetrecht J. 2003. Characterization of a potential animal model of an idiosyncratic drug reaction: Nevirapine-induced skin rash in the rat. Chem Res Toxicol. 16:1078–1089.
- Shiohara T, Inaoka M, Kano Y. 2006. Drug-induced hypersensitivity syndrome (DIHS): A reaction induced by a complex interplay among herpesviruses and anti-viral and antidrug immune responses. Allergol Intl. 55:1–8.
- Su S, Mockenhaupt M, Wolkenstein P, Dunant A, le Gouvello S, Chen C, Chosidow O, Valeyrie-Allanore L, Bellon T, Sekula P, et al. 2017. IL-15 is associated with severity and mortality in Stevens-Johnson syndrome/toxic epidermal necrosis. J Invest Dermatol. 137:1065–1073.
- Sullivan A, Wang E, Farrell J, Whitaker P, Faulkner L, Peckham D, Park B, Naisbitt D. 2018. β-lactam hypersensitivity involves expansion of circulating and skin-resident TH22 cells. J Allergy Clin Immunol. 141:235–249.
- Tassaneeyakul W, Jantararoungtong T, Chen P, Lin P, Tiamkao S, Khunarkornsiri U, Chucherd P, Konyoung P, Vannaprasaht S, Choonhakarn C, et al. 2009. Strong association between HLA-B*5801 and allopurinol-induced Stevens-Johnson syndrome and toxic epidermal necrolysis in a Thai population. Pharmacogen Genomics. 19:704–709.
- Tate S, Singh R, Hung C, Tai J, Depondt C, Cavalleri G, Sisodiya S, Goldstein D, Liou H. 2006. A common polymorphism in the SCN1A gene associates with phenytoin serum levels at maintenance dose. Pharmacogenet Genomics. 16:721–726.
- Ueta M, Kannabiran C, Wakamatsu T, Kim M, Yoon K, Seo K, Joo C, Sangwan V, Rathi V, Basu S, et al. 2014. Trans-ethnic study confirmed independent associations of HLA-A*02:06 and HLA-B*44:03 with cold medicine-related Stevens-Johnson syndrome with severe ocular surface complications. Sci Rep. 4:5981
- Watanabe H, Gaide O, Petrilli V, Martinon F, Contassot E, Roques S, Kummer J, Tschopp J, French L. 2007. Activation of the IL-1β-processing inflammasome is involved in contact hypersensitivity. J Invest Dermatol. 127:1956–1963.
- Wei C, Chung W, Huang H, Chen Y, Hung SI. 2012. Direct interaction between HLA-B and carbamazepine activates T-cells in patients with Stevens-Johnson syndrome. J Allergy Clin Immunol. 129:1562–1569. e1565.
- Weston J, Uetrecht J. 2014. Activation of inflammasomes by agents causing idiosyncratic skin reactions: a possible biomarker. Chem Res Toxicol. 27:949–951.
- Whitaker P, Meng X, Lavergne S, El-Ghaiesh S, Monshi M, Earnshaw C, Peckham D, Gooi J, Conway S, Pirmohamed M, et al. 2011. Mass spectrometric characterization of circulating and functional antigens derived from piperacillin in patients with cystic fibrosis. J Immunol. 187:200–211.
- White K, Chung W, Hung S, Mallal S, Phillips E. 2015. Evolving models of the immunopathogenesis of T-cell-mediated drug allergy: the role of host, pathogens, and drug response. J Allergy Clin Immunol. 136:219–234.
- Yang C, Hung S, Juo C, Lin Y, Fang W, Lu I, Chen S, Chen Y. 2007. HLA-B*1502-bound peptides: Implications for the pathogenesis of carbamazepine-induced Stevens-Johnson syndrome. J Allergy Clin Immunol. 120:870–877.
- Yang C, Chen C, Deng S, Huang C, Lin Y, Chen Y, Wu C, Hung S, Chung W. 2015. Allopurinol use and risk of fatal hypersensitivity reactions: a nationwide population-based study in Taiwan. JAMA Intern Med. 175:1550–1557.
- Yip V, Pirmohamed M. 2017. The HLA-A*31:01 allele: Influence on carbamazepine treatment. Pharmgenomics Pers Med. 10:29–38.
- Yip V, Meng X, Maggs J, Jenkins R, Marlot P, Marson A, Park B, Pirmohamed M. 2017. Mass spectrometric characterization of circulating covalent protein adducts derived from epoxide metabolites of carbamazepine in patients. Chem Res Toxicol. 30:1419–1435.
- Yun J, Mattsson J, Schnyder K, Fontana S, Largiader C, Pichler W, Yerly D. 2013. Allopurinol hypersensitivity is primarily mediated by dose-dependent oxypurinol-specific T-cell response. Clin Exp Allergy. 43:1246–1255.
- Zanni M, von Greyerz S, Schnyder B, Brander K, Frutig K, Hari Y, Valitutti S, Pichler W. 1998. HLA-restricted, processing- and metabolism-independent pathway of drug recognition by human α β T lymphocytes. J Clin Invest. 102:1591–1598.
- Zhang F, Liu H, Irwanto A, Fu X, Li Y, Yu G, Yu Y, Chen M, Low H, Li J, et al. 2013. HLA-B*13:01 and the dapsone hypersensitivity syndrome. N Engl J Med. 369:1620–1628.