
Abstract
Chemical allergy can manifest into allergic contact dermatitis and asthma and the importance of skin sensitization in both of these diseases is increasingly being recognized. Given the unique characteristics of chemical allergy, coupled with the distinct immunological microenvironment of the skin research is still unraveling the mechanisms through which sensitization and elicitation occur. This review first describes the features of chemical sensitization and the known steps that must occur to develop a chemical allergy. Next, the unique immunological properties of the skin - which may influence chemical sensitization - are highlighted. Additionally, mediators involved with the development of allergy are reviewed, starting with early ones - including the properties of haptens, skin integrity, the microbiome, the inflammasome, and toll-like receptors (TLR). Novel cellular mediators of chemical sensitization are highlighted, including innate lymphoid cells, mast cells, T-helper (TH) cell subsets, and skin intrinsic populations including γδ T-cells and resident memory T-cells. Finally, this review discusses two epigenetic mechanisms that can influence chemical sensitization, microRNAs and DNA methylation. Overall, this review highlights recent research investigating novel mediators of chemical allergy that are present in the skin. It also emphasizes the need to further explore these mediators to gain a better understanding of what makes a chemical an allergen, and how best to prevent the development of chemical-induced allergic diseases.
Keywords:
Introduction
Chemical allergy
The incidence of allergic disease has increased worldwide over the past several decades and thus remains a burden on human health (Pawankar Citation2014). While proteins are known to be causative antigens for many types of allergy, low molecular weight (LMW) chemicals also contribute to allergic disease (Sasseville Citation2008). Typically, LMW chemical allergens cause dermal or respiratory allergic manifestations including allergic contact dermatitis (ACD) and asthma, and it is estimated that the prevalence of allergic disease to at least one chemical in North America and Western Europe is ≈20% (Ainscough et al. Citation2013). While the most common cause of occupational allergy is exposure to metal salts, exposure to various chemicals found in rubber additives, plastics and resins, biocides, and cosmetics are also responsible. Traditionally, LMW chemical sensitizers have been classified as either respiratory or dermal sensitizers, based on their ability to sensitize via the respiratory tract or skin, and their ability to induce T-helper (TH)-2 or TH1 cell-dominated immune responses, respectively (Dearman et al. Citation1996). Thousands of chemicals have been identified as causative agents of skin sensitization resulting in ACD, while substantially fewer chemical allergens (< 100) have been identified as causative agents of asthma (Jarvis et al. Citation2005). In general, the majority of chemical sensitizers are capable of causing dermal sensitization but do not cause respiratory sensitization (Kimber and Dearman Citation2005); however, several respiratory sensitizers can also cause skin sensitization, leading to both ACD and/or asthma following sensitization (Padoan et al. Citation2003; Vanoirbeek at al. 2006; Bello et al. Citation2007). However, for many LMW sensitizers, the immunologic mechanisms driving sensitization are not completely understood (Palm and Medzhitov Citation2009).
LMW chemical allergens are diverse in structure, reactivity, and application; however, there are several common attributes that are associated with immunogenicity, including haptenation potential (protein reactivity), ability to penetrate the skin, and irritancy potential (Kimber and Dearman Citation2005; Palm and Medzhitov Citation2009). Induction of a hypersensitivity response requires three crucial steps: (1) presentation of antigen to an antigen-specific immune cell, (2) co-stimulation, provided by an antigen-presenting cell (APC), and (3) inflammatory cytokine signaling (Curtsinger and Mescher Citation2010). For the development of chemical allergy, an additional crucial step must occur. Since the majority of chemicals are small (<1000 daltons) and cannot be processed and presented by APC, they must bind with a carrier protein to be presented on the major histocompatibility complex (MHC) molecule (on surface of cell) and subsequently be recognized by the adaptive immune system (Chipinda et al. Citation2011). These chemicals are known as haptens.
This haptenization process adds a layer of complexity in understanding chemical allergy, as a chemical may bind a range of host proteins. The type of host protein that the hapten binds to as well as its physical location within the host’s body can influence the developing immune response, resulting in a range of outcomes, from tolerance to a spectrum of clinical manifestation falling under the umbrella of “allergy/hypersensitivity”.
Classification of chemical allergy
In an early attempt to more clearly define allergic reactions, Gell and Coombs developed a clinical classification system which defined four basic types of hypersensitivity reactions (Type I–IV) based on effector molecules and cells involved in the immune response (Coombs Citation1963). Although it was proposed > 50 years ago and several revision attempts have been made to update the classification scheme, it is still widely accepted as the most valid scheme for phenotypically categorizing hypersensitivity reactions. Type I and Type IV hypersensitivity reactions are the most common with respect to chemical allergy.
A Type IV [delayed-type] hypersensitivity response is T-cell-mediated and characterized by excessive inflammation. The most distinctive feature of a Type IV-mediated hypersensitivity response is the delay observed between allergen exposure and clinical manifestation. Following sensitization, subsequent exposures result in elicitation of a non-IgE-mediated response, characterized by the secretion of TH1 and pro-inflammatory cytokines (GM-CSF, interferon [IFN]-γ, interleukin [IL]-3, IL-12, and tumor necrosis factor [TNF]-α) that activate and recruit macrophages and CD8+ cytotoxic T-cells. Due to the requirement for these cytokines to attract and activate macrophages at sites of exposure, the effector phase typically occurs 24 hr following exposure and it generally peaks 48–72 hr after exposure (Budinger and Hertl Citation2000). Typically, dermal or contact sensitizers are considered Type IV inducers. ACD is an example of a Type IV-mediated hypersensitivity reaction.
Although not as common as Type IV responses, exposure to LMW chemicals can also result in Type I or IgE-mediated responses. A Type I, or an IgE-mediated allergic reaction, is orchestrated by IgE antibody and mast cells and is sometimes called immediate-type hypersensitivity. Sensitization involves the initiation of TH2 cytokines, such as IL-4 and IL-13, leading to isotype switching and IgE production by B-cells. Once IgE is produced and secreted, it binds to mast cells. Upon subsequent re-exposure, antigen binds to mast cell-associated IgE, crosslinks two IgE molecules, and causes cell activation. With mast cells, activation results in degranulation and release of soluble allergic mediators such as histamine and leukotrienes which act on smooth muscles, sensory nerves, mucous glands, epithelial cells, arteries, and eosinophils, promoting inflammation.
Common clinical outcomes of an IgE-mediated reaction are increased vascular permeability, smooth muscle cell contraction, and vasodilation. The elicitation of IgE-mediated reactions occur within minutes to hours after exposure and can occur in the skin and lung. Depending on the site(s) and frequency of allergen exposure, these reactions may occur in one or more organs or tissues resulting in diseases such asthma, allergic rhinitis, urticaria, and/or anaphylaxis.
The classification of allergens, particularly LMW allergens, has often proven to be difficult since studies have identified that exposure to certain chemicals can result in multiple hypersensitivity pathways (i.e., both ACD and asthma). For example, toluene diisocyanate (TDI) and persulfate salts are generally classified as IgE-mediated sensitizers but may also induce non-IgE-mediated responses resulting in a variety of disease including: ACD, urticaria, rhinitis, and asthma (Kanerva et al. Citation1999; Matheson et al. Citation2005; de Vooght et al. Citation2010; Aalto-Korte et al. Citation2012; Hougaard et al. Citation2012; Nguyen and Lee, Citation2012). However, the complete immunological mechanisms of sensitization for these chemicals and other LMW sensitizers are not fully understood.
Although the Gell and Coombs classification is seminal work that has withstood decades of evolution in immunologic knowledge, its current validity has been questioned, specifically in the context of chemical allergy. The landscape of immunology has been dramatically altered since the 1960s when the classification was proposed; one would expect the need for mechanistic updates to the classification, which have been lacking. The idea that more than one defined hypersensitivity mechanism may be involved in a given reaction has been proposed as well. This concept does not invalidate the hypersensitivity mechanisms presented by Gell and Coombs; rather, it emphasizes the flexibility of the classification system, which is not generally highlighted. The broad clinical use of this classification presents several concerns, including the dismissal of underlying immune mechanisms involved in certain conditions based on their simple classification. Our current knowledge of immunology has revealed the dynamic nature of the immune system potentially influenced by the cellular microenvironment and interplay of mediators that could impact the mechanism of sensitization and ultimately hypersensitivity classification.
Skin as a distinct immunological environment
In addition to providing protection from the outside environment, as the largest organ in the body, the skin is an extremely important player in allergic disease. The skin is a highly integrated and immunologically active organ that must maintain a delicate balance between pro-inflammatory and anti-inflammatory immune responses in order to protect against pathogens and mitigate tissue damage. In order to achieve this balance, the skin is endowed with a complex immune cell repertoire that is unique to this tissue.
While development of allergic immune responses is often thought to be orchestrated in the lymph nodes, and are thus largely studied at these sites, researchers are beginning to understand that many of these crucial steps begin at the site of antigen contact, often a mucosal or barrier site such as the skin. In general, when an allergen is present in the skin (chemical or otherwise) it is picked up by skin-resident dendritic cells (DC), located either in the epidermis (Langerhans cells) or dermis (dermal DC). The antigen is transferred to the draining lymph nodes by the DC, where it will activate antigen-specific T-cells, initiating an adaptive immune response. However, it is becoming increasingly clear that this process can be influenced by the local microenvironment, which can influence the speed at which the DC migrate, the co-stimulatory molecules expressed, as well as the cytokines produced. In addition, the presence of multiple innate immune factors present in the skin including leukocytes, complement factors, antimicrobial peptides, and pattern recognition receptors (Bangert et al. Citation2011) allow the skin to be a site of immune surveillance and tolerance, although the role of these factors in the development of allergic disease has yet to be thoroughly investigated. Not only the skin involved in the development of dermal sensitization, it has also been implicated in the development of systemic sensitization leading to elicitation at various sites in the body, including the respiratory tract (Bello et al. Citation2007; Tordesillas et al. Citation2014). Additionally, factors such as skin integrity have been shown to influence sensitization and the development of the allergic response (Kasemsarn et al. Citation2016). However, additional research is needed to fully understand the role of the skin in allergic disease.
The identification of novel mechanisms of allergic disease is paramount to the objective of answering the questions of what makes a chemical an allergen and what are the immunologic events that drive the response. Furthermore, the investigation of innate events preceding full sensitization such as danger signals, cytokine and chemokine signaling, and APC phenotype and function is also necessary to answer this question. As exemplified above, the discovery and thorough understanding of mechanisms of allergic disease, specifically relating to chemical sensitization, is necessary to further the development of preventative and therapeutic strategies to combat allergic disease. Having a more complete view of the immunologic events taking place during both sensitization and elicitation will be achieved by investigating novel cellular subsets and players potentially involved in these events along with emphasizing the need for continued research on previously identified key players. Many of the potential mediators have been identified in other forms of allergic diseases, such as atopic dermatitis (AD). While likely operating through immunologically distinct mechanisms, the study of other skin inflammatory diseases can provide us with numerous “suspects” in this regard as mentioned above; however, there is a need to investigate these potential mediators in chemical allergy. While this review is not exhaustive, it is intended to describe current literature on potential novel mediators of allergic disease that are present in the skin and to identify research needs related to understanding chemical sensitization.
Early mediators of chemical allergy in the skin (danger signals)
It has long been recognized that the presence of foreign antigens alone is insufficient to generate immune responses: activation of the innate immune system is also required. Research is continuing to bring to light the importance of such “danger signals” in allergic sensitization. During an infection, one of the first forms of defense employed by the innate immune response is the recognition of molecular patterns expressed by invading pathogens using pattern recognition receptors (PRR). PRR may either be membrane-associated (cell surface or endosomal), e.g., Toll-like receptors (TLR) and C-type lectin receptors (CLR), or cytoplasmic, e.g., Nod-like receptors (NLR) and RIG-I-like receptors (RLR). There is an analogy between innate anti-infection immune responses and the innate immune response to chemical allergens as several PRR including TLR and NLR have been implicated in chemical sensitization (Santoni et al. Citation2015). These receptors specifically recognize pathogen-associated microbial patterns (PAMP) - typically expressed by microbial pathogens, or danger-associated molecular patterns (DAMP) - endogenous molecules released from necrotic/dying cells potentially induced by chemical allergens.
Early signaling events in the skin including PRR signaling are thought to provide a bridge between the innate and adaptive immune systems and are of pivotal importance for the initiation of immune responses, including those to chemical allergens resulting in skin sensitization (Ainscough et al. Citation2013). In general, these danger signals are required as a supplementary signal, together with an antigen, for the initiation of an immune response. Some chemical allergens have the ability to deliver both antigenic and danger signals, representing complete allergens capable of triggering the allergic cascade. In such instances, the danger signals are related to the irritant type properties of the inducing chemical allergens, resulting in cell and tissue trauma, which initiates subsequent inflammation. While one recognizes the importance of danger signals in chemical sensitization, there is still much to be learned with respect to how and why these signals occur in response to chemical exposure.
Haptens
Hapten formation is a critical step in forming an adaptive immune response to LMW chemicals, and its influence on the activation of innate immunity is highlighted by the clinical observation that chemical-induced irritancy correlates with the induction of ACD. In addition, in order for a LMW chemical to cause sensitization, skin penetration should occur at a rate that is quick enough to ensure that an ample amount of chemical may react with self-proteins to produce sufficiently immunogenic conjugates following conformational changes of the LMW chemical and the self-protein. Chemical allergens are naturally electrophilic or can be converted to an electrophilic species in the skin (pro-hapten) or via auto-oxidation (pre-hapten) so as to bind to nucleophilic amino acids (Martin et al. Citation2011). A chemical can become immunogenic through several complex mechanisms including: direct binding to the MHC on the APC, modification of host-proteins, allergen-induced alterations of protein processing, and allergen-mediated binding between the T-cell receptor and MHC. Uncertainty regarding the exact structure and identity of chemical hapten complexes that are relevant in vivo have posed challenges for diagnosis and understanding of the mechanisms driving allergic disease. The well-studied chemical sensitizer, toluene diisocyanate (TDI) provides a good example of these complexities. The isocyanate groups in TDI can react with amine groups to form ureas, hydroxyl groups to form urethanes, and/or thiol groups to from thioureas (Ruwona et al. Citation2010). However, the antigenic portion of TDI-hapten complexes are poorly understood along with the overall process in vivo. This is a challenging subject as TDI and reactive diisocyanate can self-polymerize and haptenate proteins at single or multiple locations, creating a complex chemical environment (Ruwona et al. Citation2010). A greater understanding of this process will likely aid in the understanding of the immunologic basis for sensitization of LMW chemicals in general.
While there is a lot to be learned with respect to haptenization, the question of “what makes a hapten immunogenic” is also a topic of great interest. For the initiation of an immune response, chemical haptens must deliver two independent signals. One is an antigenic signal that is based upon the chemical itself and its ability to induce T-cell activation. The second is the triggering of an innate immune response usually resulting from traumatic changes to cells and tissues local to the site of application. The presence or absence of the danger signal determines whether a cutaneous immune response and skin sensitization develops, or whether instead there is a failure to initiate an immune response (Martin et al. Citation2011). Historically, research into the immunology of chemical sensitization has overlooked hapten-mediated activation of the innate immune system to focus on the roles of cutaneous APC, such as Langerhans cells and dermal DC, and of conventional CD8+ and CD4+ T-cells. However, complete haptens can directly damage keratinocytes, causing them or neighboring keratinocytes to release their cellular contents generating DAMP including the release of reactive oxygen species (ROS), DNA, ATP, and/or other extracellular matrix proteins through mechanisms that are largely unknown (Martin et al. Citation2011).
While some haptens are not intrinsically sensitizing, pretreatment with an irritant such as sodium lauryl sulfate (SLS) has been shown in both mice and humans to enhance the immune response to the hapten, likely through a combination of increased allergen penetration and low-grade skin inflammation (Kligman Citation1966; Cumberbatch et al. Citation1993). This presents the question of how a mixture or co-exposure might influence chemical sensitization. Alterations in the chemical properties of single allergens within the mixture could result in changes in the inflammatory response, or could ultimately affect T-cell activation, proliferation, and differentiation. (Bonefeld et al. Citation2017). In addition, research also suggests that exposure parameters, such as dose and frequency, are critical in the sensitization response.
Toll-like receptor signaling
Toll-like receptors (TLR) are a family of receptors (TLR1-TLR13) expressed on both immune and non-immune cells (including epithelial cells) that recognize various PAMP and DAMP molecules. Stimulation of TLR by a corresponding PAMP or DAMP initiates signaling cascades leading to increases in gene transcription, and ultimately the production of pro-inflammatory cytokines such as IL-1β, -6, -12, -18, and -23, as well as TNFα (McFadden et al. Citation2013). Activation of certain TLR can result in the production of high levels of the TH2-skewing cytokine thymic stromal lymphopoietin (TSLP) that, in turn, can also promote sensitization (Cianferoni and Spergel Citation2014). During the last decade, it has become clear that TLR plays an important role in the initiation of the inflammatory response to chemical allergens (Martin et al. Citation2008; Jin et al. Citation2009; Schmidt et al. Citation2010; Nakamura et al. Citation2015). The importance of TLR in the development of skin sensitization was first highlighted in a study reported by Schmidt et al. (Citation2010) describing the role of TLR4 in nickel allergy. While nickel was shown to stimulate TLR4 by direct binding, chemical haptens can also stimulate TLR indirectly. For example, the chemical hapten TNCB has been shown to induce the production of ROS that can generate DAMP such as degradation products of hyaluronic acid (HA) that can function as TLR2 and TLR4 ligands (Esser et al. Citation2012) in a murine model. In addition, it has been shown that treatment of human keratinocytes with chemical haptens induces the production of high-mobility group protein B1, which can function as an endogenous TLR4 ligand (Galbiati et al. Citation2014). Similarly, it has been suggested that RNA released from apoptotic/necrotic keratinocytes induced by exposure to the chemical allergen TNCB can serve as ligands for TLR3 in a murine model (Nakamura et al. Citation2015).
Given similar roles and signaling pathways induced by TLR stimulation, redundancy has been demonstrated in multiple experimental models of chemical allergy. A crucial role for TLR2 and TLR4 was described using an experimental model of chemical-induced ACD (Martin et al. Citation2008). Although mice deficient in either TLR2 or TLR4 developed normal contact hypersensitivity responses, mice lacking both of these TLR were not sensitized and failed to develop contact hypersensitivity in response to trinitro-chlorobenzene (TNCB), oxazolone, or fluorescein isothiocyanate (FITC) haptens. Interestingly, it appears that triggering of TLR by non-allergens can also modify the allergic responses. Pre-treatment with the non-sensitizing TLR7 agonist imiquimod-induced ear-swelling in mice challenged with dinitrofluorobenzene (DNFB) or dinitrochlorobenzene (DNCB) as compared with mice pretreated with vehicle control (Suzuki et al. Citation2000; Thatcher et al. Citation2006).
Additionally, studies have revealed that, although not sensitizing itself, topical exposure to the antimicrobial chemical triclosan augmented the allergic response to ovalbumin (OVA) through a TSLP-mediated pathway in a mouse model of asthma (Anderson et al. Citation2013; Marshall et al. Citation2015). Further investigation demonstrated that triclosan induced abundant expression of S100A8/A9 in the skin, which acts as an endogenous ligand for TLR4. Expression of Tlr4 along with Tlr1, Tlr2 and Tlr6 were increased in skin tissues over time with triclosan exposure and high levels of TLR4 were expressed on skin-infiltrating leukocytes (Marshall et al. Citation2017). These findings suggested that the simultaneous exposure of non-allergens or irritants, environmentally, occupationally, or in the use of consumer products, with the potential to stimulate TLR may increase the risk of users developing an allergic response to other allergens. The requirement for TLR in sensitization is a relatively new discovery and there is still much to learn about this family of receptors and their specific roles in this chemical sensitization.
Inflammasome signaling
Nucleotide-binding and oligomerization domain (NOD)-like receptors (NLR) are highly conserved cytosolic PRR that survey the intracellular environment for danger signal molecules including PAMP and DAMP through an incompletely understood mechanism. The inflammasome complex is a multiprotein oligomer that contains proteins of the NLR family that can activate an inflammatory cascade upon stimulation. By sensing danger and consequently activating caspase-1, certain NLR facilitate the processing of pro-interleukins IL-1β and other pro-inflammatory cytokines including IL-18, and thus support activation of DC (Evavold and Kagan Citation2018).
NLRP3 is the most comprehensively studied NLR and evidence suggests that the inflammation resulting from NLRP3 activation is critical for the development of sensitization. Although many different substances have been shown to activate NLRP3 including ATP, silica, asbestos, and uric acid, a direct ligand for this inflammasome has yet to be identified (Pedra et al. Citation2009). Additionally, chemical haptens (TNCB, DNFB and dinitro-1-chlorobenzene) have been shown to lead to the activation of the NLRP3 inflammasome in primary human keratinocytes and murine models (Watanabe et al. Citation2007) by an indirect pathway involving ROS and ATP. This indicates that in the context of chemical allergy, inflammasome activation may occur indirectly, as a result of cell damage and the release of ATP. The importance of the inflammasome complex during hapten-induced immune responses is demonstrated by reduced sensitization to TNCB and DNFB that occurs in Asc−/−, Nlrp3−/− and Casp1−/− mice (Watanabe et al. Citation2007). In a murine model of TNCB sensitization, inflammasome activation was shown to depend on the ATP receptor P2X7 (Weber et al. Citation2010). The decisive role of the inflammasome in sensitization was further underlined by use of the weak sensitizer DNTB. Co-application of IL-1β in addition to DNTB resulted in a strong sensitization response (Watanabe et al. Citation2008).
Although not as well studied as NLRP3, NLRP1, NLRP2, and NLRP4 are also inflammasome subsets of the NLR family with suggested roles in the activation of pro-inflammatory cytokines and the development of skin sensitization (Faustin et al. Citation2007; Shaw et al. Citation2010; Zhao et al. Citation2011); these subsets have not yet been investigated in the development of chemical allergy. However, although a role for NLR is recognized in the process of skin sensitization, evidence suggests that the stimulation of the NLR alone is insufficient to induce the activation of DC. Most likely, other PRR signaling pathways such as TLR pathways must act in concert with NLR for successful activation of DC during skin sensitization. As this has been a largely unexplored area in the realm of LMW chemicals, additional research is needed to further define the role of NLR in chemical allergy.
Skin integrity
It is generally recognized that environmental influences such as solvent and soap exposure, excessive hand washing, existing skin conditions, humidity, temperature, physical trauma, and glove use are all factors that can influence skin barrier function and the development of allergic disease (Redlich Citation2010). However, the impact of sensitizing chemicals on skin integrity has not been thoroughly investigated. While a role for inflammation in skin sensitization has been demonstrated, in certain circumstances this may result in a compromised epithelium, increasing the potential for allergen exposure and subsequent allergic disease. Additionally, loss of barrier function has been identified as an important factor in the development of various allergic diseases including asthma and AD (Redlich Citation2010). This concept has been demonstrated with animal and human-based studies involving filaggrin, a protein involved in the formation of the epidermis (Palmer et al. Citation2006, Citation2007; Oyoshi et al. Citation2009). It has been suggested that barrier disruption and dysfunction followed by chemical allergen exposure may allow the perturbing agent to activate the epithelium, leading to the production of allergic cytokines (Islam and Luster Citation2012; Marshall et al. Citation2015).
Skin microbiome
While the skin utilizes its unique immune environment primarily for host protection, increasing data supports a very important role for skin commensal bacteria in human health. The evidence is accumulating to suggest that indigenous microbial communities may have a role in modulating immune disorders of the skin and there are studies that link commensal skin micro-organisms with modulation of the innate immune system through cutaneous tolerance and maintenance of the skin barrier. However, the mechanisms that underlie this protection remain an active area of investigation. In healthy skin, commensals form a physical and chemical barrier against pathogenic organisms without triggering allergic responses (Zeeuwen et al. Citation2013). Conversely, in AD skin, commensal bacteria are less diverse and ineffectively prevent colonization with pathogenic organisms, most notably Staphylococcus aureus (Wisniewski et al. Citation2013). Further support for the importance of commensal bacteria in skin immunity was demonstrated by Myles et al. (2016). Those authors found that Gram-negative bacteria taken from the skin of healthy volunteers, but not from patients with AD, were associated with enhanced barrier function, select innate immunity activation, and control of S. aureus. Treatment with the bacteria from the healthy controls also improved outcomes in a mouse model of AD suggesting therapeutic potential.
Recent murine models have also shown that commensal skin bacteria play an active role in supporting skin barrier homeostasis and defense against microbial penetration. The most common species associated with protection from the development of AD are Staphylococcus epidermidis and Staphylococcus cohnni (Wollina Citation2017). The association between the lack of colonization with these species and AD development suggests a potential functional role for these organisms in regulating the skin barrier and response to environmental allergens. Enhanced colonization with S. aureus is observed in filaggrin-deficient and cathelicidin-deficient mice, mimicking the landscape of AD skin (Nakatsuji et al. Citation2016) and entry of S. aureus below the epidermis was also shown to increase pro-inflammatory cytokines, thus enhancing the potential for environmental allergen exposure through barrier disruption.
Despite their role in established AD, how commensal organisms contribute to the development of skin sensitization is not completely understood. Recently, skin commensals have also been shown to play an important role in TH1 and TH2 balance along with the regulation of anti-inflammatory responses to chemical allergens (Knaysi et al. Citation2017). Research has also shown that germ-free mice have elevated levels of TSLP which suggests a role for the microbiota in ameliorating stress signals released by keratinocytes which could potentially influence subsequent immune responses (Yockey et al. Citation2013). Additionally, properties of chemical allergens and commensal bacteria add to the complex landscape of skin immunity. Many chemical sensitizers are also anti-microbial agents (Anderson et al. Citation2010, Citation2016; Shane et al. Citation2017). While these agents may be beneficial in protecting against pathogenic bacteria, the influence of exposure on resident bacterial populations warrants further investigations. A recent study evaluated the influence of topical antibiotics on skin commensal bacteria. The authors found that mice treated with topical antibiotics exhibited altered commensal bacterial populations, with reduction in Staphylococcus spp. These mice were more susceptible to exogenous association with S. aureus, and pre-colonization with the same Staphylococcus residents that were previously disrupted by treatment reduced pathogenic S. aureus levels by over 100-fold (SanMiguel et al. Citation2017). The findings suggest that anti-microbial agents can disrupt skin bacterial residents and that these alterations can have critical implications for cutaneous immune responses.
Cellular mediators of chemical allergy in the skin
Recently, the emergence of many novel cellular subsets and molecules involved in immunological responses has occurred, shedding light on unexplored realms of the immune system and their potential involvement in a variety of disease states, including allergic disease. While keratinocytes and DC are known to have very important roles in sensitization (Christensen and Haase Citation2012), other cellular mediators have recently been identified as potential mediators. These novel mediators include innate lymphoid cells (ILC), mast cells, and additional T-cell subsets.
Innate lymphoid cells
ILC are a recently defined family of lymphocytes that lack rearranged antigen-specific receptors; they are activated by innate signals such as pro-inflammatory cytokines. Much like T-cells, there are distinct sub-types of ILC that are classified by their phenotype and effector functions. The current ILC classification scheme is also reflective of T-cell classification, where there are cytotoxic ILC (NK cells) and non-cytotoxic helper-like ILC (ILC1s, ILC2s, ILC3s) (Artis and Spits Citation2015). ILC2s, like TH2 cells, express the transcription factors GATA-3 and RORα, produce TH2-associated cytokines IL-4, -5, -9, and -13, and contribute to anti-helminth immune responses and development of allergic disease. In addition to producing Type 2 cytokines, ILC2s can act directly on T-cells to drive TH2 differentiation and Type 2 cytokine production, due to their expression of MHC-II and CD80/CD86 co-stimulatory molecules (Mirchandani et al. Citation2014; Oliphant et al. Citation2014).
The majority of research regarding the role of ILC2s in the development of allergic disease has focused on inflammation in the lung and asthma models, and this data has been thoroughly reviewed elsewhere (Drake and Kita Citation2014; Martinez-Gonzalez et al. Citation2015). In addition to their role in the lungs, ILC2s may be particularly important in development of allergy in the skin due to their relative abundance in this tissue, even at a resting state in healthy individuals (Salimi et al. Citation2013), and their preferential activation by TSLP (Kim et al. Citation2013). Although little work has been done regarding the role of ILC in chemical allergy, other models of allergic disease have implicated ILC2 involvement. In human AD patients, ILC2s were enriched in lesional skin (Kim et al. Citation2013; Salimi et al. Citation2013). Salimi et al. (Citation2013) showed that intra-epidermal delivery of house dust mite allergen induces infiltration of skin ILC2s into human and mouse skin. Through the production of IL-13, ILC2s in the skin are important for the production of CCL17 chemokine by cutaneous DC (Halim et al. Citation2016). As CCL17 is important for TH2 cell infiltration into the skin, ILC2 derived IL-13 is likely important for delayed-type hypersensitivities. A functional role for ILC2s in the promotion of cutaneous inflammation has been identified using Rag1-/- mice, which lack a functional adaptive immune system. Despite lacking TH2 cells, IL-2 treated Rag1-/-mice produced significant amounts of IL-5 and IL-13, resulting in eosinophil infiltration (Roediger et al. Citation2013).
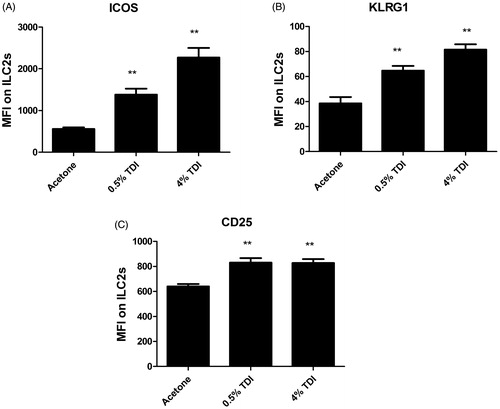
Interestingly, the dermis may be home to a phenotypically unique population of ILC2s, abundant upon resting conditions, and that constitutively produce IL-13. However, upon activation by IL-2, these cells stimulate mast cells via the production of IL-5 (Roediger et al. Citation2013). Supporting a role for ILC2 in chemical allergy, it has been previously shown in a mouse model that dermal exposure to the known chemical sensitizer, TDI, significantly increased the activation markers ICOS, KLRG1, and CD25 on ILC2s in the skin (). ILC1s and ILC3s may also have specific roles in allergic disease; however, they have not been studied extensively in this context. The scarcity of research regarding the role of ILC in chemical-induced allergy is striking and as more experimental tools are available to facilitate the study of these cells, these cells should be characterized in relevant models, as they likely play important roles in these conditions.
Figure 1. Ear ILC2 Phenotyping following TDI sensitization in mice. Flow cytometric analysis of ILC2 isolated from the ear at 2 days post dermal treatment (25 µl/ear) of female Balb/c mice with acetone, 0.5% TDI, or 4% TDI (as indicated on the X-axis). These concentrations were previously determined to be sensitizing following a single dermal application. While exposure at 4% induced irritation, 0.5% induced sensitization in the absence of irritation. Cells were isolated from the ear via enzymatic digestion (Liberase-TL) and then passed through cell strainer to obtain single cell suspensions. Cells were counted, stained, fixed, and data acquired on an LSRII flow cytometer (BD Biosciences). Following doublet-exclusion gating, ILC2s were identified as Lineage-CD45+CD90+CD25+CD127+ cells. The lineage gate was composed of antibodies detecting the following antigens: Ter119, CD19, CD3e, CD11b, Ly-6G, CD2, and CD11c. ILC2s were then further characterized by analyzing median fluorescent intensities (MFI) for activation markers (A) ICOS, (B) KLRG1, and (C) CD25. Bars represent mean MFI ± SEM. n = 5 mice/treatment group. p Values are represented by asterisks (p < .01).
Mast cells
Mast cells are important immune cells for both host defense, through activation of innate immunity (via TLR or complement receptors), and for IgE-mediated allergic inflammatory reactions. Mast cells reside long-term in barrier sites such as the gut and the skin and produce effector molecules including histamine, various cytokines, and lipid mediators. While having a well-recognized role in IgE-mediated allergy, recent research has shown that mast cells also play roles in inflammation and the development of immune responses in the skin. While limited, research has shown that mast cells can directly influence the development of adaptive immune responses and act as effectors cells that exacerbate the development of allergic or autoimmune disorders (Gilfillan and Beaven Citation2011). Additionally, it has long been hypothesized that mast cells can act as APCs due to their induced expression of MHC-II (Banovac et al. Citation1989). Using intravital two-photon microscopy, Dudeck et al. (Citation2017) showed that mast cells activated by the chemical sensitizer DNFB, were “interrogated” by dermal DC. Through this interaction, DC transferred functional MHC-II complexes to mast cells, providing a mechanism through which mast cells in the skin acquired APC functions and were capable of driving T-cell-induced skin inflammation to DNFB.
While these studies suggested a role for mast cells in generation of an adaptive immune response, their potential role in contact hypersensitivity (CHS) is controversial. Several groups have investigated this relationship using mast cell-deficient mice, with seemingly contradictory results. Mast cell-deficient mice were identified to exhibit reduced inflammation in TNCB-induced CHS in several studies (Askenase et al. Citation1983; Biedermann et al. Citation2000), while other studies showed undiminished CHS induced with TNCB or 2,4-dinitrobenzene sulfonic acid sodium salt (DNFS) (Galli and Hammel Citation1984, Mekori and Galli Citation1985 ).More recently, it was demonstrated that mast cells might play a regulatory role in hypersensitivity responses in the skin through the production of IL-10 and subsequent suppression of chronic CHS (Grimbaldeston et al. Citation2007). Reasons for these discrepancies are unknown, but it may be reflective of the model of mast cell deficiency used. Earlier studies investigating mast cells in CHS used a mouse model with a mutation in the stem cell factor or its receptor c-Kit. Although these mice lack mast cells, they also have other immunological alterations due to additional hematopoietic abnormalities making data interpretation difficult. More recent work has utilized mast cell ablation, which conditionally depletes mast cells through the administration of diphtheria toxic (DT) and does not impact other cell types. Studies conducted using the DT-depleted mast cell model suggest the importance of mast cells for both the sensitization and elicitation phases of contact hypersensitivity to FITC and oxazolone through the promotion of early immune responses and enhancement of DC migration to draining lymph nodes (Dudeck et al. Citation2011). Other studies have provided evidence mast cells initiate CHS via activation of DC (Otsuka et al. Citation2016).
Mast cells have also been shown to enhance the innate immune response through the recruitment of neutrophils during the sensitization phase of a CHS response to TNCB (Weber et al. Citation2015); this occurs via secretion of the chemokines CXCL1/CXCL2 (de Filippo et al. Citation2013). In addition to driving hypersensitivity responses, mast cells have been identified to play a regulatory role in hypersensitivity responses in the skin by enhancing skin barrier function (Sehra et al. Citation2016). However, these suppressive responses are somewhat controversial, and seem to be dependent on the severity of the inflammatory response, with a more severe hypersensitivity response (Reber et al. Citation2017) and chronic models of hypersensitivity (Gimenez-Rivera et al. Citation2016), resulting in mast-cell induced suppression of inflammation.
Another emerging role for mast cells in development of allergic skin diseases is involvement in the orchestration of ILC responses. Mast cells and ILC2s, both constitutively found in skin, have been shown to have a high level of interaction. Using in vivo multiphoton microscopy, Roediger et al. (Citation2013) determined that dermal ILC2s form stable interactions with skin-resident mast cells (lasting 20–30 min), however the full significance of this interaction is not yet known. In AD, following IgE crosslinking, mast cells release the lipid mediator prostaglandin D2 (PGD2) in addition to histamine. The receptor for PGD2, i.e., CRTH2, is expressed on ILC2s and this interaction induces the migration of ILC2s into the skin along with subsequent TH2 cytokine production (Xue et al. Citation2014). Recent studies also suggest that the skin microbiome may influence mast cells in the dermis and that a normal microbiome is necessary for correct function and maturation of mast cells (Wang et al. Citation2017) further indicating the importance of this cell population in the skin. While the studies described above support a role for mast cells in immune responses in the skin, additional research is required to further define the role of this cell type in chemical-induced allergy in the skin.
Diverse T-helper (TH) cell subsets
T-Cells have long been recognized as important mediators of chemical sensitization and allergic responses. Many factors can influence the phenotype and function of a T-cell, including the immune microenvironment, the level and duration of TCR signaling, and the presence of signaling molecules such as cytokines. The cellular phenotype and effector functions of a T-cell help determine its unique subset. Our knowledge of T-cells has increased exponentially in the past 30 years since Mosmann and Coffman proposed the seminal T-cell classification scheme, including TH1 and TH2 cells (Eyerich and Eyerich Citation2015). Currently, there are many recognized T-cell subsets, including the CD4+ TH1, TH2, TH9, TH17, TH22, T follicular helper (TFH), and regulatory T-cells (Treg), natural killer T-cells (NKT), γδ T-cells, and cytotoxic CD8+ cells. While TH1, TH2, and cytotoxic CD8+ T-cells have a relatively well-defined role in allergic disease and chemical sensitization, the potential roles of the remaining T-cell subsets are largely unexplored, particularly in the skin.
TH17 cells
TH17 cells have been implicated in the pathogenesis of autoimmune diseases and the clearance of extracellular pathogens due to their ability to recruit and activate neutrophils and macrophages, and cause acute and chronic inflammation (Cosmi et al. Citation2011). Recently, these cells have also been associated with allergic disease. TH17 cells are named after their signature cytokine, IL-17, and are regulated by their master transcription factor RORγτ (Manel et al. Citation2008). While TH17 cells are shown to selectively produce IL-17A (IL-17), they have also been recognized as producing IL-22, IL-17F, IL-21, GM-CSF, and potentially TNFα, IL-6, and IL-8 (Qu et al. Citation2013). Apart from the established role of IL-17-producing cells in psoriasis, there are indications these cells also contribute to the pathogenesis of AD (Dhingra and Guttman-Yassky Citation2014). Additionally, filaggrin-deficient mice display elevated levels of IL-17 and TH17 dominated skin inflammation suggesting a role for this subset in barrier function (Oyoshi et al. Citation2009).
IL-17 was first associated with skin allergy in a study using nickel-specific T-cells isolated from ACD patients (Albanesi et al. Citation1999). Both human-derived nickel-specific T-cells and keratinocytes expressed IL-17 and il17r, respectively. That suggested IL-17 was involved in T-cell-mediated allergic responses in the skin. Several years later, IL-17-deficient mice were utilized in several allergic disease models in an effort to define the role of TH17 cells in these conditions (Nakae et al. Citation2002). Evidence of a function for TH17 cells in CHS was provided by studies with IL-17 deficient mice that demonstrated strongly reduced ear swelling response to skin sensitizers. The role of IL-17 in human ACD was also suggested by a study showing that approximately half of nickel-specific CD4+ T-cell clones isolated from nickel-allergic patients produce IL-17 (Wong et al. Citation2009). Additionally, direct evidence for the role of IL-17 in the murine acute CHS response was shown by using IL-17-/- mice wherein a markedly suppressed ear swelling response was observed (Tsushima et al. Citation2003). CD4+ TH17 cells have been regarded as one main source of IL-1 in CHS, although some subpopulations of CD8+ T-cells have been implicated as well (Kim et al. Citation2006). The discovery of TH17 cells challenged the TH1/TH2 dualism and initiated the quest for other TH cell subsets.
TH9 cells
TH9 cells are a more recent addition to the growing family of TH cells. Data have suggested TH9 cells are a discrete skin-tropic or skin-resident memory T-cell subset that produces IL-9 (Ma et al. Citation2014). IL-9 itself acts mainly as a T-cell growth factor whose function is to drive T-cell survival, proliferation, and secretion of inflammatory mediators and has been shown to enhance the production of IFNγ, IL-9, IL-13, and IL-17 from skin-tropic T-cells, indicating an ability to amplify related immune responses (Schlapbach et al. Citation2014). IL-9 also promotes tissue accumulation, survival and activation of mast cells, eosinophils, and ILC, all key cellular contributors to allergic disorders in the skin. In particular, IL-9 seems to play an important role in early activation of ILC, in which it enhances the secretion of IL-5 and IL-13, which are intimately linked to AD pathogenesis. While there is still much to be learned about Th9 cells and allergic disease, atopic human patients were found to exhibit increased TH9 cell numbers compared to non-atopic patients, and these levels correlated with increased serum IgE levels (Jones et al. Citation2012). Additionally, the number of TH9 cells was increased in skin lesions of AD and psoriasis patients, with IL-9 levels significantly correlating with clinical severity of AD (Ciprandi et al. Citation2013). In ACD, IL-9 expression in both the skin and peripheral blood mononuclear cells seem to correlate with the strength of the allergen (Liu et al. Citation2014). While the majority of published reports/studies on the role of TH9 cells in skin disorders are related to TH2-mediated responses and allergic inflammation, there are reports suggesting a role for TH9 cells in other inflammatory and neoplastic disorders of the skin, such as psoriasis and cutaneous T-cell lymphoma, respectively.
There remain several unanswered questions regarding TH9 cells in skin disorders. Specifically, a transcription factor that serves as a master regulator of TH9 cells has not been identified which makes it challenging to unambiguously identify this subset from other TH cell subsets with the ability to secrete IL-9. The mechanisms by which IL-9 contributes to allergic disease as well as the cellular sources of IL-9 are incompletely understood. However, given the association with Th9 cells, IL-9 and skin disease, and the skin homing and skin-resident properties of these cells, further characterization of this subset and increased knowledge of its role in chemical sensitization may be crucial to our understanding of the mechanisms involved in allergic disease in the skin.
TH22 cells
Another newly-characterized T-cell subset thought to play a role in allergic disease is TH22 cells. These cells are characterized by production of IL-22 in the absence of IFNγ, IL-4, and IL-17, and are thought to contribute to host defense against microbial pathogens and promote tissue repair or remodeling (Fujita Citation2013). TH22 cells produce IL-22 in response to TNFα and IL-6 in the skin and express the skin homing chemokine receptors CCR4 and CCR10 (Wang and Xu Citation2015). In the skin, IL-22 plays a major role in homeostasis and pathogenesis of skin diseases by inducing keratinocyte proliferation and epidermal hyperplasia, inhibiting keratinocyte terminal differentiation, and promoting the production of anti-microbial proteins. The intracellular molecular mechanism involved in TH22 cell differentiation is not fully-characterized; however, it has been demonstrated that TH22 cells are dependent on the aryl hydrocarbon receptor (AHR) as the main transcription factor (Jia and Wu Citation2014). While information about the role of TH22 cells in chemical allergy in the skin is lacking, this subset has been implicated in the pathogenesis of inflammatory skin disorders such as psoriasis and AD (Mirshafiey et al. Citation2015). In addition, IL-22 levels were found to be increased in the skin of AD, ACD, and allergic asthma patients (Jia and Wu Citation2014) and have been suggested as a potential biomarker for allergic disease (Zissler et al. Citation2016). Much remains to be investigated regarding the role of TH22 cells in allergic disease, particularly those caused by chemical allergens, including the overall role of this cell type, the specific effector mechanisms involved in modulating the allergic response, and the potential interaction with additional cell types with recognized involvement in allergic disease. Considering the strong association between activation of the TH22 pathway and skin diseases, this additional research is warranted.
Gamma Delta (γδ) T-cells
CD4+ and CD8+ T-cells express a T-cell receptor composed of an α and β subunit and are thus known as αβ T-cells. These cells are responsible for the majority of general functions that one attributes to T-cells. Another population of T-cells (γδ T-cells) expresses a T-cell receptor comprised of γ and δ subunits. These cells have different requirements for activation than αβ T-cells and are enriched in peripheral sites such as the gut mucosa and skin. In the skin, γδ T-cells exist in both the epithelium and in the dermis, and the location of these cells seems to confer specific roles, an area of research which is under active investigation (O’Brien and Born Citation2015; Nielsen et al. Citation2017). There is abundant evidence that γδ T-cells participate in the development of allergic responses, through both antigen recognition and the production of allergic cytokines (Zheng and Yang Citation2014). Dermal application of the TLR7 and TLR9 agonist, imiquimod, induces γδ T-cell accumulation in the skin, and these cells are capable of rapidly promoting inflammation upon secondary challenge, via production of IL-17 (Ramirez-Valle et al. Citation2015). Additionally, γδ T-cells have been shown to be producers of IL-4 in a corticosteroid-induced ACD model (Baeck et al. Citation2013).
In hypersensitivity models, chemical sensitizers increase the total number of γδ T-cells in the lymph nodes and at the site of antigen challenge. Additionally, data suggests they assist the classical T-cell mediated response, as depletion of γδ T-cells abrogated CHS, yet both γδ T-cells and αβ T-cells must be adoptively transferred to induce CHS (Dieli et al. Citation1998). Importantly, in the context of chemical allergy, γδ T-cells are able to recognize haptens. While their location in the epidermis brings them into contact with haptens during the sensitization stage of CHS, it is unknown whether the recognition is direct or indirect. Zeng et al. (Citation2014) showed that γδ T-cells can directly recognize the synthetic fluorescent molecule cyanine 3 and 4-hydroxy-3-nitrophenyl-acetate, and are able to mount a hapten-specific response by up-regulating the activation marker CD44 and producing IL-17. Additionally, mice lacking the TCR δ gene (δ-/-) have significantly reduced levels of ear swelling in response to DNFB in a CHS model, compared to mice sufficient in dermal γδ T-cells, specifically due to the loss of IL-17-induced neutrophil infiltration (Jiang et al. Citation2017). While there is a scarcity of research on this subject currently, given the propensity of γδ T-cells in the skin, it is quite likely they influence the development of allergic disease. Determining the role they play in chemical sensitization will be important to the overall understanding of the process.
Regulatory T (Treg)-cells
True to their name, Treg cells are a subset of CD4+ T-cells that are involved in the maintenance of immune tolerance and homeostasis by acting as effectors to prevent overzealous adaptive responses to foreign antigens and allergens (Sojka et al. Citation2008; Yadav et al. Citation2012). Classical Treg cells (CD4+CD25+) were initially identified based on their suppressive capabilities in mice (Sakaguchi et al. Citation1995). Following this discovery, a transcription factor known as forkhead box P3 (FoxP3) was identified as the master transcription factor of Treg cells, allowing for their identification and functional manipulation (Hori et al. Citation2003), permitting a more comprehensive analysis of their phenotype and functional diversity. Recently published studies indicate that the supervisory functions of these cells are compromised in the pathogenesis of autoimmune and neoplastic diseases of the skin. This decrease in function is most likely a result of domination from other immune cells in the skin. The suppressive function of Treg cells is mediated by a variety of mechanisms including, but not limited to: the control of conventional T-cell proliferation through the inhibition of co-stimulation via cytotoxic T-lymphocyte associated protein 4 (CTLA4), expression and/or IL-2 consumption, immunosuppressive cytokine secretion (IL-10 and TGFβ), metabolic interference, and disruption of DC function (Sojka et al. Citation2008; Corthay Citation2009; Kimber et al. Citation2012).
Overall, Treg cells represent a heterogeneous population, with multiple subsets defined by their ontogeny, function and tissue residence (Zhang et al. Citation2014). Independent of their role in immune suppression, Tregs are endowed with tissue-specific functionality. In the skin of adult mice, 20–60% of CD4+ T-cells are Treg cells; in normal human adult skin, ≈20% of tissue-resident CD4+ T-cells are Treg cells. Treg cells in both murine and human skin appear to occupy a specialized anatomic niche and in non-inflamed healthy human skin, CD4+ Foxp3+ Treg cells preferentially reside in close association with hair follicles (Chow et al. Citation2013; Sanchez Rodriguez et al. Citation2014).
Noteworthy roles for Treg cells in allergic disease are beginning to be recognized (Zhang et al. Citation2014). A depletion of Treg cells was found to lead to significantly exacerbated skin inflammation, including increased recruitment of inflammatory cells, expression of TH2 cytokines, and serum IgE levels in a murine model of AD (Fyhrquist et al. Citation2012). A functional role for Treg cells has also been suggested in models of chemical-induced CHS (Christensen et al. Citation2015). Results from these studies suggest that circulating Treg cells infiltrate the periphery, traffic to draining lymph nodes, then recirculate back to the skin, contributing to the down-regulation of cutaneous immune responses following antigen exposure (Tomura et al. Citation2010). These migratory Treg cells were identified to have strong immunosuppressive effects and expressed high levels of mRNA for inhibitory mediators compared to lymph node-resident Treg cells, supporting an important role for these cells in the skin.
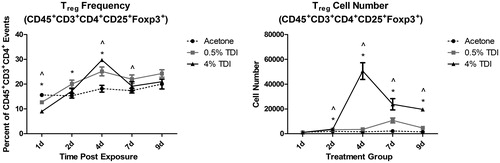
Recently, our laboratory demonstrated that epicutaneous exposure to the occupational allergen TDI resulted in expansion of Treg cells in the skin-draining lymph nodes, and that these cells were functionally more suppressive than those isolated from non-TDI exposed mice (Long et al. Citation2016). Depletion of Treg cells prior to TDI exposure led to an increased sensitization response. Subsequent studies have shown that activated Treg cells also accumulate in the skin following epidermal TDI exposure. These Treg cells expanded in number and frequency between 1 and 9 days following a single dermal application, with an identified peak around 4 days post exposure (). The collection of data regarding Treg cells and chemical allergy is growing, but remains limited. Given their critical role in suppressing inflammation, defining the cellular and molecular pathways involved in activating and maintaining Treg cells in chemical allergy is of fundamental importance.
Figure 2. Treg Phenotyping in mouse skin following TDI sensitization. Flow cytometric analysis of dermal Treg cells following treatment (25 µl/ear) of female Balb/c mice with acetone, 0.5% TDI, or 4% TDI (as indicated on X-axis). These concentrations were previously determined to be sensitizing following a single dermal application. While exposure at 4% induced irritation, 0.5% induced sensitization in the absence of irritation. Cells were isolated from the ear via enzymatic digestion (Liberase-TL) and then passed through a cell strainer to obtain single cell suspensions. Cells were counted, stained, fixed, and data was acquired on an LSRII flow cytometer (BD Biosciences). Following doublet exclusion gating, Treg cells were first gated on their expression of CD45, CD3, and CD4 then were further identified by CD25 and Foxp3 expression (defined as Treg cells) at indicated timepoints. Treg cell frequency and number were determined based on flow cytometry analysis and extrapolation of this data with total ear cellularity. Graph symbols represent mean (±SE) of 5 mice/group. p Values are presented as ^0.5% TDI and *4% TDI (p < .05). Dermal treatment groups are indicated by the following symbols: circle = acetone, square = 0.5% TDI, and triangle =4% TDI.
Resident memory T-cells
Memory T-cells, those which have been previously activated and are “primed” to promote a secondary response, have been traditionally categorized into two subsets based on their tissue preference and homing capabilities, CD62Llow effector memory cells (TEM) and CD62Lhi central memory cells (TCM). Due to interactions of CD62L with ligands expressed on lymph node high endothelial venules (HEV), cells with low CD62L expression (TEM) are more prone to circulate throughout the body and peripheral tissues than those with high expression (TCM). More recently, a specialized memory T-cell has been identified that responded to antigen upon initial encounter - without T-cell recruitment from the blood - and was maintained in tissues after antigen clearance, poised to respond with accelerated kinetics upon subsequent antigen exposure. These cells were defined as resident memory T-cells (TRM) and are retained long-term within barrier tissues that interface between the host and the environment, such as the skin and respiratory tract (Klonowski et al. Citation2004; Park and Kupper Citation2015). TRM cells in a barrier tissue are often specific for pathogens and other antigens that have been encountered previously through that barrier epithelium. TRM CD4+ and CD8+ T-cells have been defined within the skin following multiple types of immunogenic challenges (Jiang et al. Citation2012).
The role of skin specific TRM cells in immune and inflammatory diseases is just beginning to be appreciated. A recent study in mice and humans found that ACD was mediated by TRM cells that had been generated in response to topically applied allergens (Gaide et al. Citation2015). The expansion of both CD4+ and CD8+ TRM cells was observed in the skin of mice following multiple topical applications of the hapten DNFB and in a human model of contact dermatitis using the chemical allergen diphenylcyclopropenone (DPCP). Importantly, the function was attributed to TRM cells in the elicitation of cell-mediated responses observed 24 hr after challenge, as blocking lymphocyte migration and parabiosis studies neither abrogated nor augmented the hypersensitivity response. However, this study did not take into account the role of other skin-resident cellular populations such as γδ T-cells or ILC.
Interestingly, TRM cells generated as a result of localized vaccinia virus skin infection reside not only in the site of infection, but also populate the entire skin surface and remain present for many months (Jiang et al. Citation2012), demonstrating the potential for a discrete dermal sensitization event to change the overall immunological landscape of the skin. Interestingly, CD8+ TRM at mucosal sites have been shown to function as activators of innate cells through rapid production of cytokines resulting in DC maturation and natural killer cell activation, through a non-antigen specific method (Schenkel et al. Citation2014). This innate-like function of TRM results in the potential for TRM established through previous pathogen encounters to contribute to allergic sensitization. While little is known about the role of TRM in the development of dermal allergy it is likely they play a role, especially in an atopic individual where multiple, repetitive, immune responses may lead to an accumulation of these otherwise rare cell populations. Based on their potential immunomodulatory roles in the skin, additional research in chemical allergy is warranted.
Epigenetic mediators of chemical allergy in the skin
Epigenetics is currently defined as the study of heritable changes in gene expression or cellular phenotype caused by mechanisms that do not alter the nucleotide sequence. These alterations are the result of changes in DNA binding elements, which can be influenced by environmental factors, as opposed to alterations in the basic DNA sequence (Yang and Schwartz Citation2012). Epigenetic mechanisms include DNA methylation, histone modification, and non-coding RNA regulation. Both genetics and the environment are thought to influence epigenetic mechanisms that, in turn, can contribute to the development of allergic disease. While there is a clear role for genetics in the development of atopic allergic diseases, with one of the most common predictors for AD being a genetic defect in the production of filaggrin, the role of epigenetics in the development of chemical allergy in the skin has not been as well described. The general epidemiological evidence suggests that singular genetic influences do not strongly influence ACD susceptibility, but there is a tendency for general susceptibility to run in families. This implies that it is unlikely that a single gene will be the control point, and it is more likely that a combination of effector molecules will make a cooperative contribution. Several known atopy and asthma genes have been found to be susceptible to epigenetic regulation, including genes important to T-effector pathways, T-regulatory pathways, and airway inflammation (Lovinsky-Desir and Miller Citation2012). As epigenetic mechanisms are inducible and reversible, investigations into epigenetic regulation and allergic disease are critical.
microRNAs
Non-coding RNA elements such as microRNA (miRNA), transfer RNA, small interfering RNA, ribosomal RNA, and long non-coding RNA, are molecules that have exhibited great regulatory potential in a variety of disease conditions and models. Non-coding RNA exert functional effects by regulation of gene expression during chromatin remodeling, transcription, RNA splicing, editing, translation, and turn-over (Mattick and Makunin 2006). Specifically, regulatory noncoding RNA molecules function by base pairing with complementary sequences on other RNA or DNA to form complexes that are recognized by RNA-induced silencing complexes (RISC) or RNA-editing enzymes. There is increasing evidence for the importance of miRNA in normal immune cell functions and numerous studies suggest that abnormal miRNA expression identified in many inflammatory skin diseases is a result of infiltration of immune cells and disruption of normal immune cell development, homeostasis and function. Additionally, there is emerging evidence of a clinical potential for miRNA as both biomarkers and possible therapeutic targets in skin diseases.
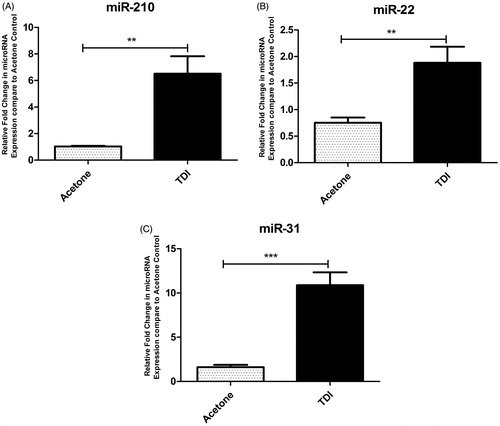
The role of miRNA in allergic disease has been demonstrated in numerous studies. For example, inactive lesions from patients with AD, miR-155 was found to be up-regulated and expressed mainly in infiltrating immune cells, including CD4+ T-cells and DC (Sonkoly et al. Citation2010). Studies also identified up-regulation of miR-146a in keratinocytes of patients with AD and during AD skin inflammation (Rebane et al. Citation2014). It was suggested miR-146a improved chronic skin inflammation in AD through suppression of innate immune responses in keratinocytes. Similarly, studies have also shown that miR-124 may play a protective role in AD (Yang et al. Citation2017). While numerous studies have investigated miRNAs in AD, limited research has investigated miRNA specifically in chemical allergy. Human skin samples were identified to have augmented levels of select miRNA, including miR-21, miR-142-3p, miR-142-5p, and miR-223 following DPCP challenge. These miRNAs were also elevated in the skin following DNFB challenge in a mouse model of ACD (Vennegaard et al. Citation2012). Gulati et al. (2015) utilized next-generation sequencing to evaluate global miRNA expression in DPCP-induced DTH responses. Their data suggested miRNA may be uniquely expressed at different time points during the immune response and that up-regulation of select miRNA was found to persist at time points up to 120 days post challenge. While those studies evaluated the elicitation phase of the allergic response, even fewer studies have described roles for miRNA in the sensitization phase of an allergic response. Anderson et al. (Citation2014) investigated global lymph node miRNA expression profiles in a murine model of dermal TDI sensitization. Increases were observed in miR-21, miR-22, miR-155, miR-126, miR-27b, miR-210, miR-31, and miR-301a expression after a single dermal sensitizing application of TDI. Peak increases were observed between 4–7 days post-exposure for the majority of miRNA examined. Additionally, increased expression of select miRNA, including miR-210, miR-22, and miR-31, were also identified in the skin following TDI exposure (). Additional work suggested that miR-210 may be impacting the allergic response through direct/indirect interactions with Treg cells (Long et al. Citation2016). While mounting evidence supports a role for miRNA in skin-related allergic disease, large research gaps still exist, including defining functional roles and identification of biomarkers. Addressing these areas of research will contribute to the understanding of the immunologic mechanisms of chemical-induced allergic diseases and may aid in the development of preventative strategies.
Figure 3. MiRNA expression in mouse skin following TDI sensitization. RT-PCR analysis of miRNA expression in the ears following a single dermal application (25 µl/ear) to female Balb/c mice of 4% TDI or acetone vehicle. This concentration was previously determined to be sensitizing following a single dermal application. Expression of (A) miR-210, (B) miR-22, and (C) miR-31 determined 7 days post-TDI exposure. Bars represent mean relative fold-change (±SE) of 4–5 mice/group. p Values are represented by asterisks (p < .05).
DNA methylation
DNA methylation refers to the covalent addition of a methyl group at a cytosine nucleotide of the CpG dinucleotide (i.e., CpG site). Methylation occurs after replication and almost exclusively affects position five of the pyrimidine ring of cytosines in the context of the dinucleotide sequence CpG. Approximately 75% of mammalian CpG dinucleotides are methylated. In contrast to heavily methylated, single CpG throughout the genome, CpG islands are generally much less methylated, potentially allowing transcription (Kabesch Citation2014). CpG islands are often located in gene regulatory elements such as promoters and enhancers. In general, active demethylation of a promoter region is necessary to allow gene transcription. Low levels of DNA methylation at these sites is typically associated with high transcriptional activity, and conversely, high DNA methylation is associated with low gene expression or gene silencing. Numerous studies have demonstrated the link between histone modification and DNA methylation and allergic asthma in animal models and in human studies (Yang and Schwartz 2012). Methylation of the intergenic region between IL4 and IL13 was seen to suppress the expression of TH2 signature cytokines in TH1 cells (Lee et al. Citation2002), while in TH2 cells, methylation of IFNγ was shown to be increased (Jones and Chen Citation2006).
Of greater potential relevance to chemical allergy are the observations that in a mouse model of asthma, and in subjects with occupational asthma to diisocyanates, there is down-regulation of IFNγ expression secondary to methylation of the ifng locus. Reduced expression of this cytokine favored the development of TH2 selective immune responses (Brand et al. Citation2012; Ouyang et al. Citation2013). Additionally, genome-wide DNA methylation changes associated with the initiation of selective immune responses in mouse whole lymph node populations following skin exposure to the reference contact allergen DNCB and reference respiratory allergen trimellitic anhydride (TMA), revealed unique patterns of DNA methylation associated with divergent T-cell polarization in vivo. Treatment with DNCB altered methylation of the prototypical TH1 genes, ifng and tbx21, with methylation occurring downstream and demethylation upstream of their transcription start site, respectively. Additionally, the key TH2 gene il13 was associated with demethylation of a region downstream of its transcription start site following treatment with TMA (Chapman et al. Citation2014). Further studies by these investigators found potential divergent methylation patterns between DNCB and TMA suggesting the potential utility as biomarkers (Chapman et al. Citation2016). These changes were identified to be tissue-specific as alterations were observed in the lymph node but not spleen indicating that tissue specificity is an important consideration when investigating changes in DNA methylation patterns. Research investigating the impact of DNA methylation in the skin is even more limited. However, differences in DNA methylation and tslp mRNA or TSLP protein levels were observed between inflamed skin lesion samples obtained from children with AD and normal skin tissue sections (Luo et al. Citation2014).
The study of epigenetics itself is complicated by many subject confounders such as diet, environmental influences, in utero exposures, aging, and co-morbidities. Questions remain regarding the time course of these epigenetic mechanisms in vivo as well as the sustainability of their effects and there remains much ground to be covered in the investigation of these components of allergic disease. However, recent advances in epigenomic profiling technologies provide new opportunities to gain mechanistic insights into the molecular basis of long-lasting cellular perturbations within functional immune cell populations that underlie altered immune responses following chemical exposures.
Conclusion
The identification of novel mediators of allergic disease is paramount to the objective of defining what makes a chemical an allergen. Furthermore, the investigation of innate events preceding full sensitization such as danger signals, cytokine and chemokine signaling, and APC phenotype and function is also necessary to answer these questions. Having a more complete view of the immunologic events taking place during both sensitization and elicitation will be achieved by investigating novel cellular subsets and molecular mediators potentially involved in these events. There are numerous “suspects” in this regard as mentioned above; however, many of these have not been thoroughly investigated in the skin. As exemplified above, the discovery of novel mechanisms of allergic disease, specifically relating to chemical sensitization, is necessary to further the development of preventative and therapeutic strategies to combat allergic disease. Optimistically, the discovery of novel mediators of dermal chemical allergy will broaden the general classification schemes and views of chemical allergy as a whole. This could represent a paradigm shift that may greatly improve allergy research and mechanistic understanding as a whole.
Disclosure statement
The findings and conclusions in this report are those of the authors and do not necessarily represent the official position of the National Institute for Occupational Safety and Health, Centers for Disease Control and Prevention.
References
- Aalto-Korte K, Suuronen K, Kuuliala O, Henriks-Eckerman M, Jolanki R. 2012. Occupational contact allergy to monomeric isocyanates. Cont Derm. 67:78–88.
- Ainscough J, Gerberick G, Dearman R, Kimber I. 2013. Danger, intracellular signaling, and the orchestration of dendritic cell function in skin sensitization. J Immunotoxicol. 10:223–234.
- Albanesi C, Cavani A, Girolomoni G. 1999. IL-17 is produced by nickel-specific T lymphocytes and regulates ICAM-1 expression and chemokine production in human keratinocytes: Synergistic or antagonist effects with IFN-γ and TNF-α. J Immunol. 162:494–502.
- Anderson S, Franko J, Kashon M, Anderson K, Hubbs A, Lukomska E, Meade B. 2013. Exposure to triclosan augments the allergic response to ovalbumin in a mouse model of asthma. Toxicol Sci. 132:96–106.
- Anderson S, Umbright C, Sellamuthu R, Fluharty K, Kashon M, Franko J, Jackson L, Johnson V, Joseph P. 2010. Irritancy and allergic responses induced by topical application of o-phthalaldehyde. Toxicol Sci. 115:435–443.
- Anderson S, Beezhold K, Lukomska E, Richardson J, Long C, Anderson K, Franko J, Meade B, D. H. Beezhold D. 2014. Expression kinetics of miRNA involved in dermal toluene 2,4-diisocyanate sensitization. J Immunotoxicol. 11:250–259.
- Anderson S, Shane H, Long C, Lukomska E, Meade B, Marshall N. 2016. Evaluation of the irritancy and hypersensitivity potential following topical application of didecyldimethylammonium chloride. J Immunotoxicol. 13:557–566.
- Artis D, Spits H. 2015. The biology of innate lymphoid cells. Nature. 517:293–301.
- Askenase P, van Loveren H, Kraeuter-Kops S, Ron Y, Meade R, Theoharides T, Nordlund J, Scovern H, Gerhson M, Ptak W. 1983. Defective elicitation of delayed-type hypersensitivity in W/Wv and SI/SId mast cell-deficient mice. J Immunol. 131:2687–2694.
- Baeck M, Soria A, Marot L, Theate I, Hendrickx E, van Belle A, Goossens A, Tennstedt D, Dachelet C, Jaeger J, et al. 2013. Characterization of the T-cell response in allergic contact dermatitis caused by corticosteroids. Cont Derm. 68:357–368.
- Bangert C, Brunner PM, Stingl G. 2011. Immune functions of the skin. Clin Dermatol. 29:360–376.
- Banovac K, Neylan D, Leone J, Ghandur-Mnaymneh L, Rabinovitch A. 1989. Are the mast cells antigen-presenting cells?. Immunol Invest. 18:901–906.
- Bello D, Herrick C, Smith T, Woskie S, Streicher R, Cullen M, Liu Y, Redlich C. 2007. Skin exposure to isocyanates: Reasons for concern. Environ. Health Perspect 115:328–335.
- Biedermann T, Kneilling M, Mailhammer R, Maier K, Sander C, Kollias G, Kunkel S, Hultner L, Rocken M. 2000. Mast cells control neutrophil recruitment during T cell-mediated delayed-type hypersensitivity reactions through tumor necrosis factor and macrophage inflammatory protein 2. J Exp Med. 192:1441–1452.
- Bonefeld C, Geisler C, Gimenez-Arnau E, Lepoittevin J, Uter W, Johansen J. 2017. Immunological, chemical and clinical aspects of exposure to mixtures of contact allergens. Cont Derm. 77:133–142.
- Brand S, Kesper D, Teich R, Kilic-Niebergall E, Pinkenburg O, Bothur E, Lohoff M, Garn H, Pfefferle P, Renz H. 2012. DNA methylation of TH1/TH2 cytokine genes affects sensitization and progress of experimental asthma. J Allergy Clin Immunol. 129:1602–1610.
- Budinger L, Hertl M. 2000. Immunologic mechanisms in hypersensitivity reactions to metal ions: An overview. Allergy. 55:108–115.
- Chapman V, Terranova R, Moggs J, Kimber I, Dearman R. 2016. Evaluation of 5-methylcytosine and 5-hydroxymethylcytosine as potential biomarkers for characterization of chemical allergens. Toxicology. 340:17–26.
- Chapman V, Zollinger T, Terranova R, Moggs J, Kimber I, Dearman R. 2014. Chemical allergen induced perturbations of the mouse lymph node DNA methylome. Toxicol Sci. 139:350–361.
- Chipinda I, Hettick J, Siegel P. 2011. Haptenation: Chemical reactivity and protein binding. J Allergy (Cairo). 2011:839682.
- Chow Z, Mueller S, Deane J, Hickey M. 2013. Dermal regulatory T cells display distinct migratory behavior that is modulated during adaptive and innate inflammation. J Immunol. 191:3049–3056.
- Christensen A, Haase C. 2012. Immunological mechanisms of contact hypersensitivity in mice. APMIS. 120:1–27.
- Christensen A, Skov S, Kvist P, Haase C. 2015. Depletion of regulatory T-cells in a hapten-induced inflammation model results in prolonged and increased inflammation driven by T-cells. Clin Exp Immunol. 179:485–499.
- Cianferoni A, Spergel J. 2014. The importance of TSLP in allergic disease and its role as a potential therapeutic target. Expert Rev Clin Immunol. 10:1463–1474.
- Ciprandi G, de Amici M, Giunta V, Marseglia A, Marseglia G. 2013. Serum interleukin-9 levels are associated with clinical severity in children with atopic dermatitis. Pediatr Dermatol. 30:222–225.
- Coombs P. 1963. The classification of allergic reactions underlying disease. In: Gell P, ed. Clinical aspects of immunology. Oxford: Blackwell. pages 317–337.
- Corthay A. 2009. How do regulatory T-cells work?. Scand J Immunol. 70:326–336.
- Cosmi L, Liotta F, Maggi E, Romagnani S, Annunziato F. 2011. TH17 cells: New players in asthma pathogenesis. Allergy. 66:989–998.
- Cumberbatch M, Scott R, Basketter D, Scholes E, Hilton J, Dearman R, Kimber I. 1993. Influence of sodium lauryl sulphate on 2,4-dinitrochlorobenzene-induced lymph node activation. Toxicology 77:181–191.
- Curtsinger J, Mescher M. 2010. Inflammatory cytokines as a third signal for T cell activation. Curr Opin Immunol. 22:333–340.
- de Filippo K, Dudeck A, Hasenberg M, Nye E, van Rooijen N, Hartmann K, Gunzer M, Roers A, Hogg N. 2013. Mast cell and macrophage chemokines CXCL1/CXCL2 control the early stage of neutrophil recruitment during tissue inflammation. Blood. 121:4930–4937.
- de Vooght V, Cruz M, Haenen S, Wijnhoven K, Munoz X, Hoet P, Morell F, Nemery N, Vanoirbeek J. 2010. Ammonium persulfate can initiate an asthmatic response in mice. Thorax. 65:252–257.
- Dearman R, Basketter D, Kimber I. 1996. Characterization of chemical allergens as a function of divergent cytokine secretion profiles induced in mice. Toxicol Appl Pharmacol. 138:308–316.
- Dhingra N, Guttman-Yassky E. 2014. A possible role for IL-17A in establishing TH2 inflammation in murine models of atopic dermatitis. J Invest Dermatol. 134:2071–2074.
- Dieli F, Ptak W, Sireci G, Romano G, Potestio M, Salerno A, Asherson G. 1998. Cross-talk between Vβ8+ and γδT-lymphocytes in contact sensitivity. Immunology. 93:469–477.
- Drake L, Kita H. 2014. Group 2 innate lymphoid cells in the lung. Adv Immunol. 124:1–16.
- Dudeck A, Dudeck J, Scholten J, Petzold A, Surianarayanan S, Kohler A, Peschke K, Vohringer D, Waskow C, Krieg T, et al. 2011. Mast cells are key promoters of contact allergy that mediate the adjuvant effects of haptens. Immunity. 34:973–984.
- Dudeck J, Medyukhina A, Frobel J, Svensson C, Kotrba J, Gerlach M, Gradtke A, Schroder B, Speier S, Figge M, et al. 2017. Mast cells acquire MHCII from dendritic cells during skin inflammation. J Exp Med. 214:3791–3811.
- Esser P, Wolfle R, Durr C, von Loewenich F, Schempp C, Freudenberg M, Jakob T, Martin S. 2012. Contact sensitizers induce skin inflammation via ROS production and hyaluronic acid degradation. PLoS One. 7:e41340.
- Evavold C, C. Kagan J. 2018. How inflammasomes inform adaptive immunity. J Mol Biol. 430:217–237.
- Eyerich K, Eyerich S. 2015. TH22 cells in allergic disease. Allergo J Int. 24:1–7.
- Faustin B, Lartigue L, Bruey J, Luciano F, Sergienko E, Bailly-Maitre B, Volkmann N, Hanein F, Rouiller I, Reed J. 2007. Reconstituted NALP1 inflammasome reveals two-step mechanism of caspase-1 activation. Mol Cell. 25:713–724.
- Fujita H. 2013. The role of IL-22 and TH22 cells in human skin diseases. J Dermatol Sci. 72:3–8.
- Fyhrquist N, Lehtimaki S, Lahl K, Savinko T, Lappetelainen A, Sparwasser T, Wolff H, Lauerma A, Alenius H. 2012. FoxP3+ cells control TH2 responses in a murine model of atopic dermatitis. J Invest Dermatol. 132:1672–1680.
- Gaide O, Emerson R, Jiang X, Gulati N, Nizza S, Desmarais C, Robins H, Krueger J, Clark R, Kupper T. 2015. Common clonal origin of central and resident memory T cells following skin immunization. Nat Med. 21:647–653.
- Galbiati V, Papale A, Galli C, Marinovich M, Corsini E. 2014. Role of ROS and HMGB1 in contact allergen-induced IL-18 production in human keratinocytes. J Invest Dermatol. 134:2719–2727.
- Galli S, Hammel I. 1984. Unequivocal delayed hypersensitivity in mast cell-deficient and beige mice. Science. 226:710–713.
- Gilfillan A, Beaven M. 2011. Regulation of mast cell responses in health and disease. Crit Rev Immunol. 31:475–529.
- Gimenez-Rivera V, Siebenhaar F, Zimmermann C, Siiskonen H, Metz M, Maurer M. 2016. Mast cells limit the exacerbation of chronic allergic contact dermatitis in response to repeated allergen exposure. J Immunol. 197:4240–4246.
- Grimbaldeston M, Nakae S, Kalesnikoff J, Tsai M, Galli S. 2007. Mast cell-derived interleukin 10 limits skin pathology in contact dermatitis and chronic irradiation with ultraviolet B. Nat Immunol. 8:1095–1104.
- Gulati N, Lovendorf M, Zibert J, Akat K, Renwick N, Tuschl T, Krueger J. 2015. Unique microRNAs appear at different times during the course of a delayed-type hypersensitivity reaction in human skin. Exp Dermatol. 24:953–957.
- Halim T, Hwang Y, Scanlon S, Zaghouani H, Garbi N, Fallon P, McKenzie A. 2016. Group 2 innate lymphoid cells license dendritic cells to potentiate memory TH2 cell responses. Nat Immunol. 17:57–64.
- Hori S, Nomura T, Sakaguchi S. 2003. Control of regulatory T-cell development by the transcription factor FoxP3. Science. 299:1057–1061.
- Hougaard M, Menné T, Søsted H. 2012. Occupational eczema and asthma in a hairdresser caused by hair-bleaching products. Dermatitis. 23:284–287.
- Islam S, Luster A. 2012. T cell homing to epithelial barriers in allergic disease. Nat Med. 18:705–715.
- Jarvis J, Seed M, Elton R, Sawyer L, Agius R. 2005. Relationship between chemical structure and the occupational asthma hazard of low molecular weight organic compounds. Occup Environ Med. 62:243–250.
- Jia L, Wu C. 2014. The biology and functions of TH22 cells. Adv Exp Med Biol. 841:209–230.
- Jiang X, Clark R, Liu L, Wagers A, Fuhlbrigge R, Kupper T. 2012. Skin infection generates non-migratory memory CD8+ T(RM) cells providing global skin immunity. Nature. 483:227–231.
- Jiang X, Park C, Geddes Sweeney J, Yoo M, Gaide O, Kupper T. 2017. Dermal γδ T cells do not freely re-circulate out of skin and produce IL-17 to promote neutrophil infiltration during primary contact hypersensitivity. PLoS One. 12:e0169397.
- Jin H, Kumar L, Mathias C, Zurakowski D, Oettgen H, Gorelik L, Geha R. 2009. Toll-like receptor 2 is important for the T(H)1 response to cutaneous sensitization. J Allergy Clin Immunol. 123:875–882.
- Jones B, Chen J. 2006. Inhibition of IFN transcription by site-specific methylation during T-helper cell development. Embo J. 25:2443–2452.
- Jones C, Gregory L, Causton B, Campbell G, Lloyd C. 2012. Activin A and TGFβ promote TH9 cell–mediated pulmonary allergic pathology. J Allergy Clin Immunol. 129:1000–1010.
- Kabesch M. 2014. Epigenetics in asthma and allergy. Curr Opin Allergy Clin Immunol. 14:62–68.
- Kanerva L, Alanko K, Jolanki R, Aalto-Korte K, Estlander T. 1999. Occupational allergic contact dermatitis from potassium persulfate. Cont Derm. 40:116–117.
- Kasemsarn P, Bosco J, Nixon R. 2016. The role of the skin barrier in occupational skin diseases. Curr. Probl. Dermatol 49:135–143.
- Kim B, Siracusa M, Saenz S, Noti M, Monticelli L, Sonnenberg G, Hepworth M, van Voorhees A, Comeau M, Artis D. 2013. TSLP elicits IL-33-independent innate lymphoid cell responses to promote skin inflammation. Sci Transl Med. 5:170ra16.
- Kimber I, Dearman R. 2005. What makes a chemical a respiratory sensitizer?. Curr Opin Allergy Clin Immunol. 5:119–124.
- Kimber I, Travis M, Martin S, Dearman R. 2012. Immunoregulation of skin sensitization and regulatory T cells. Cont Derm. 67:179–183.
- Kim H, Guan H, Zu G, Li H, Wu L, Feng X, Elmets C, Fu Y, Xu X. 2006. High-level expression of B7-H1 molecules by dendritic cells suppresses the function of activated T-cells and desensitizes allergen-primed animals. J Leukocyte Biol. 79:686–695.
- Kligman A. 1966. The identification of contact allergens by human assay. II Factors influencing the induction and measurement of allergic contact dermatitis. J Invest Dermatol. 47:375–392.
- Klonowski K, Williams K, Marzo A, Blair D, Lingenheld E, Lefrancois L. 2004. Dynamics of blood-borne CD8 memory T cell migration in vivo. Immunity. 20:551–562.
- Knaysi G, Smith A, Wilson J, Wisniewski J. 2017. The skin as a route of allergen exposure: Part II. allergens and role of the microbiome and environmental exposures. Curr Allergy Asthma Rep. 17:7
- Lee D, Agarwal S, Rao A. 2002. TH2 lineage commitment and efficient IL-4 production involves extended demethylation of the IL-4 gene. Immunity. 16:649–660.
- Liu J, Harberts E, Tammaro A, Girardi N, Filler R, Fishelevich R, Temann A, Licona-Limon P, Girardi M, Flavell R, et al. 2014. IL-9 regulates allergen-specific TH1 responses in allergic contact dermatitis. J Invest Dermatol. 134:1903–1911.
- Long C, Lukomska E, Marshall N, Nayak A, Anderson S. 2016. Potential inhibitory influence of miRNA 210 on regulatory T-cells during epicutaneous chemical sensitization. Genes. 8:1.
- Long C, Marshall N, Lukomska E, Kashon M, Meade B, Shane H, Anderson S. 2016. A role for regulatory T-cells in a murine model of epicutaneous toluene diisocyanate sensitization. Toxicol Sci. 152:85–98.
- Lovinsky-Desir S, Miller R. 2012. Epigenetics, asthma, and allergic diseases: A review of the latest advancements. Curr Allergy Asthma Rep. 12:211–220.
- Luo Y, Zhou B, Zhao M, Tang J, Lu Q. 2014. Promoter demethylation contributes to TSLP over-expression in skin lesions of patients with atopic dermatitis. Clin Exp Dermatol. 39:48–53.
- Ma L, Xue H, Guan X, Shu C, Zhang J, Yu J. 2014. Possible pathogenic role of T helper type 9 cells and interleukin (IL)-9 in atopic dermatitis. Clin Exp Immunol. 175:25–31.
- Manel N, Unutmaz D, Littman D. 2008. The differentiation of human T(H)-17 cells requires transforming growth factor-β and induction of the nuclear receptor RORγt. Nat Immunol. 9:641–649.
- Marshall N, Lukomska E, Long C, Kashon M, Sharpnack D, Nayak A, Anderson K, Meade B, Anderson S. 2015. Triclosan induces thymic stromal lymphopoietin in skin promoting TH2 allergic responses. Toxicol Sci. 147:127–139.
- Marshall N, Lukomska E, Nayak A, Long C, Hettick J, Anderson S. 2017. Topical application of the anti-microbial chemical triclosan induces immunomodulatory responses through the S100A8/A9-TLR4 pathway. J Immunotoxicol. 14:50–59.
- Martin S, Esser P, Weber F, Jakob T, Freudenberg M, Schmidt M, Goebeler M. 2011. Mechanisms of chemical-induced innate immunity in allergic contact dermatitis. Allergy. 66:1152–1163.
- Martin S, Dudda J, Bachtanian E, Lembo A, Liller S, Durr C, Heimesaat M, Bereswill S, Fejer G, Vassileva R, et al. 2008. Toll-like receptor and IL-12 signaling control susceptibility to contact hypersensitivity. J Exp Med. 205:2151–2162.
- Martinez-Gonzalez I, Steer C, Takei F. 2015. Lung ILC2s link innate and adaptive responses in allergic inflammation. Trends Immunol. 36:189–195.
- Matheson J, Johnson V, Luster M. 2005. Immune mediators in a murine model for occupational asthma: Studies with toluene diisocyanate. Toxicol Sci. 84:99–109.
- Mattick J, Makunin I. 2006. Non-coding RNA. Hum Mol Genet. 15 Spec No.1:R17-29.
- McFadden J, Puangpet P, Basketter D, Dearman R, Kimber I. 2013. Why does allergic contact dermatitis exist?. Br J Dermatol. 168:692–699.
- Mekori Y, Galli S. 1985. Undiminished immunologic tolerance to contact sensitivity in mast cell-deficient W/Wv and Sl/Sld mice. J Immunol. 135:879–885.
- Mirchandani A, Besnard A, Yip E, Scott C, Bain C, Cerovic V, Salmond R, Liew F. 2014. Type 2 innate lymphoid cells drive CD4+ TH2 cell responses. J Immunol. 192:2442–2448.
- Mirshafiey A, Simhag A, El Rouby N, Azizi G. 2015. T-helper 22 cells as a new player in chronic inflammatory skin disorders. Int J Dermatol. 54:880–888.
- Myles I, Williams K, Reckhow J, Jammeh M, Pincus N, Sastalla I, Saleem D, Stone D, Datta S. 2016. Transplantation of human skin microbiota in models of atopic dermatitis. JCI Insight. 1:10.
- Nakae S, Komiyama Y, Nambu A, Sudo K, Iwase M, Homma I, Sekikawa K, Asano M, Iwakura Y. 2002. Antigen-specific T cell sensitization is impaired in IL-17-deficient mice, causing suppression of allergic cellular and humoral responses. Immunity. 17:375–387.
- Nakamura N, Tamagawa-Mineoka R, Ueta M, Kinoshita S, Katoh N. 2015. Toll-like receptor 3 increases allergic and irritant contact dermatitis. J Invest Dermatol. 135:411–417.
- Nakatsuji T, Chen T, Two A, Chun K, Narala S, Geha R, Hata T, Gallo R. 2016. Staphylococcus aureus exploits epidermal barrier defects in atopic dermatitis to trigger cytokine expression. J Invest Dermatol. 136:2192–2200.
- Nguyen R, Lee A. 2012. Allergic contanct dermatitis caused by isocyanates in resin jewellery. Cont Derm. 67:56–57.
- Nielsen M, Witherden D, Havran W. 2017. γδ T cells in homeostasis and host defence of epithelial barrier tissues. Nat Rev Immunol. 17:733–745.
- O'Brien R, Born W. 2015. Dermal γδ T cells-what have we learned?. Cell Immunol. 296:62–69.
- Oliphant C, Hwang Y, Walker J, Salimi M, Wong S, Brewer J, Englezakis A, Barlow J, Hams E, Scanlon S, et al. 2014. MHC-II-mediated dialog between Group 2 innate lymphoid cells and CD4+ T-cells potentiates Type 2 immunity and promotes parasitic helminth expulsion. Immunity. 41:283–295.
- Otsuka A, Nonomura Y, Kabashima K. 2016. Roles of basophils and mast cells in cutaneous inflammation. Semin Immunopathol. 38:563–570.
- Ouyang B, Bernstein D, Lummus Z, Ying J, Boulet L, Cartier A, Gautrin D, Ho S. 2013. Interferon-γ promoter is hypermethylated in blood DNA from workers with confirmed diisocyanate asthma. Toxicol Sci. 133:218–224.
- Oyoshi M, Murphy G, Geha R. 2009. Filaggrin-deficient mice exhibit TH17-dominated skin inflammation and permissiveness to epicutaneous sensitization with protein antigen. J Allergy Clin Immunol. 124:485–493.
- Padoan M, Pozzato V, Simoni M, Zedda L, Milan G, Bononi I, Piola C, Maestrelli P, Boschetto P, Mapp C. 2003. Long-term follow-up of toluene diisocyanate-induced asthma. Eur Respir J. 21:637–640.
- Palm N, Medzhitov R. 2009. Immunostimulatory activity of haptenated proteins. Proc Natl Acad Sci USA. 106:4782–4787.
- Palmer C, Irvine A, Terron-Kwiatkowski A, Zhao Y, Liao H, Lee S, Goudie D, Sandilands A, Campbell L, Smith F, et al. 2006. Common loss-of-function variants of the epidermal barrier protein filaggrin are a major predisposing factor for atopic dermatitis. Nat Genet. 38:441–446.
- Palmer C, Ismail T, Lee S, Terron-Kwiatkowski A, Zhao Y, Liao H, Smith F, McLean W, Mukhopadhyay S. 2007. Filaggrin null mutations are associated with increased asthma severity in children and young adults. J Allergy Clin Immunol. 120:64–68.
- Park C, Kupper T. 2015. The emerging role of resident memory T cells in protective immunity and inflammatory disease. Nat Med. 21:688–697.
- Pawankar R. 2014. Allergic diseases and asthma: A global public health concern and a call to action. World Allergy Organ J. 7:12
- Pedra J, Cassel S, Sutterwala S. 2009. Sensing pathogens and danger signals by the inflammasome. Curr Opin Immunol. 21:10–16.
- Qu N, Xu M, Mizoguchi I, Furusawa J, Kaneko K, Watanabe K, Mizuguchi J, Itoh M, Kawakami Y, Yoshimoto T. 2013. Pivotal roles of T-helper 17-related cytokines, IL-17, IL-22, and IL-23, in inflammatory diseases. Clin Dev Immunol. 2013:968549.
- Ramirez-Valle F, Gray E, Cyster J. 2015. Inflammation induces dermal Vγ4+ γδT17 memory-like cells that travel to distant skin and accelerate secondary IL-17-driven responses. Proc Natl Acad Sci USA. 112:8046–8051.
- Rebane A, Runnel T, Aab A, Maslovskaja J, Ruckert B, Zimmermann M, Plaas M, Karner J, Treis A, Pihlap M, et al. 2014. MicroRNA-146a alleviates chronic skin inflammation in atopic dermatitis through suppression of innate immune responses in keratinocytes. J Allergy Clin Immunol. 134:836–847.
- Reber L, Sibilano R, Starkl P, Roers A, Grimbaldeston M, Tsai M, Gaudenzio N, Galli S. 2017. Imaging protective mast cells in living mice during severe contact hypersensitivity. JCI Insight. 2:9.
- Redlich C. 2010. Skin exposure and asthma: Is there a connection?. Proc Am Thorac Soc. 7:134–137.
- Roediger B, Kyle R, Yip K, Sumaria N, Guy T, Kim B, Mitchell A, Tay S, Jain R, Forbes-Blom E, et al. 2013. Cutaneous immunosurveillance and regulation of inflammation by Group 2 innate lymphoid cells. Nat Immunol. 14:564–573.
- Ruwona T, Johnson V, Schmechel D, Simoyi R, Beezhold D, Siegel P. 2010. Monoclonal anti-bodies against toluene diisocyanate haptenated proteins from vapor-exposed mice. Hybridoma. 29:221–229.
- Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. 1995. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol. 155:1151–1164.
- Salimi M, Barlow J, Saunders S, Xue L, Gutowska-Owsiak D, Wang X, Huang L, Johnson D, Scanlon S, McKenzie A, et al. 2013. A role for IL-25 and IL-33-driven Type-2 innate lymphoid cells in atopic dermatitis. J Exp Med. 210:2939–2950.
- Sanchez Rodriguez R, Pauli M, Neuhaus I, Yu S, Arron S, Harris H, Yang S, Anthony B, Sverdrup F, Krow-Lucal E, et al. 2014. Memory regulatory T cells reside in human skin. J Clin Invest. 124:1027–1036.
- SanMiguel A, Meisel J, Horwinski J, Zheng Q, Grice E. 2017. Topical anti-microbial treatments can elicit shifts to resident skin bacterial communities and reduce colonization by Staphylococcus aureus competitors. Antimicrob Agents Chemother. 61:9.
- Santoni G, Cardinali C, Morelli M, Santoni M, Nabissi M, Amantini C. 2015. Danger- and pathogen-associated molecular patterns recognition by pattern-recognition receptors and ion channels of the transient receptor potential family triggers the inflammasome activation in immune cells and sensory neurons. J Neuroinflamm. 12:21.
- Sasseville D. 2008. Occupational contact dermatitis. Allergy Asthma Clin Immunol. 4:59–65.
- Schenkel J, Fraser K, Beura L, Pauken K, Vezys V, Masopust D. 2014. T cell memory. Resident memory CD8 T cells trigger protective innate and adaptive immune responses. Science. 346:98–101.
- Schlapbach C, Gehad A, Yang C, Watanabe R, Guenova E, Teague J, Campbell L, Yawalkar N, Kupper T, Clark R. 2014. Human TH9 cells are skin-tropic and have autocrine and paracrine pro-inflammatory capacity. Sci Transl Med. 6:219ra8.
- Schmidt M, Raghavan B, Muller V, Vogl T, Fejer G, Tchaptchet S, Keck S, Kalis C, Nielsen P, Galanos C, et al. 2010. Crucial role for human Toll-like receptor 4 in the development of contact allergy to nickel. Nat Immunol. 11:814–819.
- Sehra S, Serezani A, Ocana J, Travers J, Kaplan M. 2016. Mast cells regulate epidermal barrier function and the development of allergic skin inflammation. J Invest Dermatol. 136:1429–1437.
- Shane H, Lukomska E, Stefaniak A, Anderson S. 2017. Divergent hypersensitivity responses following topical application of the quaternary ammonium compound, didecyldimethylammonium bromide. J Immunotoxicol. 14:204–214.
- Shaw P, Lamkanfi M, Kanneganti T. 2010. NOD-like receptor (NLR) signaling beyond the inflammasome. Eur J Immunol. 40:624–627.
- Sojka D, Huang Y, Fowell D. 2008. Mechanisms of regulatory T-cell suppression - a diverse arsenal for a moving target. Immunology. 124:13–22.
- Sonkoly E, Janson P, Majuri M-L, Savinko T, Fyhrquist N, Eidsmo L, Xu N, Meisgen F, Wei T, Bradley M, et al. 2010. MiR-155 is overexpressed in patients with atopic dermatitis and modulates T-cell proliferative responses by targeting cytotoxic T lymphocyte-associated antigen 4. J Allergy Clin Immunol. 126:581–589.
- Suzuki H, Wang B, Shivji G, Toto P, Amerio P, Tomai M, Miller R, Sauder D. 2000. Imiquimod, a topical immune response modifier, induces migration of Langerhans cells. J Invest Dermatol. 114:135–141.
- Thatcher T, Luzina I, Fishelevich R, Tomai M, Miller R, Gaspari A. 2006. Topical imiquimod treatment prevents UV-light induced loss of contact hypersensitivity and immune tolerance. J Invest Dermatol. 126:821–831.
- Tomura M, Honda T, Tanizaki H, Otsuka A, Egawa G, Tokura Y, Waldmann H, Hori S, Cyster J, Watanabe T, et al. 2010. Activated regulatory T-cells are the major T-cell type emigrating from the skin during a cutaneous immune response in mice. J Clin Invest. 120:883–893.
- Tordesillas L, Goswami R, Benede S, Grishina G, Dunkin D, Jarvinen K, Maleki S, Sampson H, Berin M. 2014. Skin exposure promotes a TH2-dependent sensitization to peanut allergens. J Clin Invest. 124:4965–4975.
- Tsushima F, Iwai H, Otsuki N, Abe M, Hirose S, Yamazaki T, Akiba H, Yagita H, Takahashi Y, Omura K, et al. 2003. Preferential contribution of B7-H1 to programmed death-1-mediated regulation of hapten-specific allergic inflammatory responses. Eur J Immunol. 33:2773–2782.
- Vanoirbeek J, Tarkowski M, Vanhooren H, de Vooght V, Nemery B, Hoet P. 2006. Validation of a mouse model of chemical-induced asthma using trimellitic anhydride, a respiratory sensitizer, and dinitrochlorobenzene, a dermal sensitizer. J Allergy Clin Immunol. 117:1090–1097.
- Vennegaard M, Bonefeld C, Hagedorn P, Bangsgaard N, Lovendorf M, Odum N, Woetmann A, Geisler C, Skov L. 2012. Allergic contact dermatitis induces up-regulation of identical microRNAs in humans and mice. Cont Derm. 67:298–305.
- Wang Z, Mascarenhas N, Eckmann L, Miyamoto Y, Sun X, Kawakami T, di Nardo A. 2017. Skin microbiome promotes mast cell maturation by triggering stem cell factor production in keratinocytes. J Allergy Clin Immunol. 139:1205–1216.
- Wang A, Xu N. 2015. New insights into T cells and their signature cytokines in atopic dermatitis. IUBMB Life. 67:601–610.
- Watanabe H, Gaide O, Petrilli V, Martinon F, Contassot E, Roques S, Kummer J, Tschopp J, French L. 2007. Activation of the IL-1β-processing inflammasome is involved in contact hypersensitivity. J Invest Dermatol. 127:1956–1963.
- Watanabe H, Gehrke S, Contassot E, Roques S, Tschopp J, Friedmann P, French L, Gaide O. 2008. Danger signaling through the inflammasome acts as a master switch between tolerance and sensitization. J Immunol. 180:5826–5832.
- Weber F, Esser P, Muller T, Ganesan J, Pellegatti P, Simon M, Zeiser R, Idzko M, Jakob T, Martin S. 2010. Lack of the purinergic receptor P2X(7) results in resistance to contact hypersensitivity. J Exp Med. 207:2609–2619.
- Weber F, Nemeth T, Csepregi J, Dudeck A, Roers A, Ozsvari B, Oswald E, Puskas L, Jakob T, Mocsai A, et al. 2015. Neutrophils are required for both the sensitization and elicitation phase of contact hypersensitivity. J Exp Med. 212:15–22.
- Wisniewski J, Agrawal R, Minnicozzi S, Xin W, Patrie J, Heymann P, Workman L, Platts-Mills T, Song T, Moloney M, et al. 2013. Sensitization to food and inhalant allergens in relation to age and wheeze among children with atopic dermatitis. Clin Exp Allergy. 43:1160–1170.
- Wollina U. 2017. Microbiome in atopic dermatitis. Clin Cosmet Investig Dermatol. 10:51–56.
- Wong S, Tan A, Lam K. 2009. Functional hierarchy and relative contribution of CD28/B7 and ICOS/B7-H2 co-stimulatory pathways to T-cell-mediated delayed-type hypersensitivity. Cell Immunol. 256:64–71.
- Xue L, Salimi M, Panse I, Mjosberg J, McKenzie A, Spits H, Klenerman P, Ogg G. 2014. Prostaglandin D2 activates group 2 innate lymphoid cells through chemoattractant receptor-homologous molecule expressed on TH2 cells. J Allergy Clin Immunol. 133:1184–1194.
- Yadav M, Louvet C, Davini D, Gardner J, Martinez-Llordella M, Bailey-Bucktrout S, Anthony B, Sverdrup F, Head R, Kuster D, et al. 2012. Neuropilin-1 distinguishes natural and inducible regulatory T cells among regulatory T cell subsets in vivo. J Exp Med. 209:1713–1722.
- Yang Z, Zeng N, Wang C, Wang H, Huang P, Pan Y. 2017. MicroRNA-124 alleviates chronic skin inflammation in atopic eczema via suppressing innate immune responses in keratinocytes. Cell Immunol. 319:53–60.
- Yang I, Schwartz D. 2012. Epigenetic mechanisms and the development of asthma. J Allergy Clin Immunol. 130(6):1243–55.
- Yockey L, Demehri S, Turkoz M, Turkoz A, Ahern P, Jassim O, Manivasagam S, Kearney J, Gordon J, Kopan R. 2013. The absence of a microbiota enhances TSLP expression in mice with defective skin barrier but does not affect the severity of their allergic inflammation. J Invest Dermatol. 133:2714–2721.
- Zeeuwen PLJM, Kleerebezem M, Timmerman HM, Schalkwijk J. 2013. Microbiome and skin diseases. Curr Opin Allergy Clin Immunol. 13:514–520.
- Zeng X, Meyer C, Huang J, Newell E, Kidd B, Wei Y, Chien Y. 2014. Γδ T-cells recognize haptens and mount a hapten-specific response. E-Life. 3:e03609
- Zhang H, Kong H, Zeng X, Guo L, Sun X, He S. 2014. Subsets of regulatory T cells and their roles in allergy. J Transl Med. 12:125
- Zhao Y, Yang J, Shi J, Gong Y, Lu Q, Xu H, Liu L, Shao F. 2011. The NLRC4 inflammasome receptors for bacterial flagellin and Type III secretion apparatus. Nature. 477:596–600.
- Zheng R, Yang Q. 2014. The role of the γδ T-cell in allergic diseases. J Immunol Res. 2014:963484
- Zissler U, Esser-von Bieren J, Jakwerth C, Chaker A, Schmidt-Weber C. 2016. Current and future biomarkers in allergic asthma. Allergy. 71:475–494.