
Abstract
In preclinical toxicity studies, species-foreign proteins administered to animals frequently leads to formation of anti-drug antibodies (ADA). Such antibodies may form circulating immune complexes (CIC) with the administered protein. These CIC can activate the classical complement pathway, thereby forming complement-bound CIC (cCIC); if large of amounts of CIC or cCIC is formed, the clearance mechanism may become saturated which potentially leads to vascular immune complex (IC) deposition and inflammation. Limited information is available on the effect of different treatment related procedures as well as biomarkers of IC-related vascular disease. In order to explore the effect of different dose regimens on IC formation and deposition, and identification of possible biomarkers of IC deposition and IC-related pathological changes, C57BL/6J and BALB/c mice were dosed subcutaneously twice weekly with bovine serum albumin (BSA) for 13 weeks without adjuvant. After 6 and 13 weeks, CIC and cCIC were detected in plasma; after 13 weeks, IC deposition was detected in kidney glomeruli. In particular immunohistochemistry double-staining was shown to be useful for detection of IC deposition. Increasing dosing frequency or changing BSA dose level on top of an already established CIC and cCIC response did not cause changes in IC deposition, but CIC and cCIC concentrations tended to decrease with increased dose level, and increased cCIC formation was observed after more frequent dosing. The presence of CIC in plasma was associated with glomerular IC deposits in the dose regimen study; however, the use of CIC or cCIC as potential biomarkers for IC deposition and IC-related pathological changes, needs to be explored further.
Introduction
Accurate assessment of safety and efficacy of therapeutic proteins is an essential part of the costly drug development process. Even though the predictive value of preclinical studies for evaluation of immunogenicity of therapeutic proteins in humans is low (US FDA Citation1997; Bugelski and Treacy Citation2004; Brinks et al. Citation2011; van Meer et al. Citation2013), regulatory agencies expect that assessment of immune responses caused by drug treatment are performed during preclinical development (EMA Citation2006). Anti-drug antibodies (ADA) are often expected to be analyzed to aid in interpretation of the toxicology studies (ICH S6 [R1] Citation2011).
When human therapeutic proteins are administered to animals during preclinical development, ADA are commonly formed and may - together with the administered protein – form circulating immune complexes (CIC) (Leach et al. Citation2014; Rojko et al. Citation2014; Krishna and Nadler Citation2016). The CIC differ in size, depending on a variety of factors including the ADA concentration and affinity as well as the ratio between ADA and administered protein (Schifferli and Taylor Citation1989; Rojas et al. Citation2005; Ponce et al. Citation2009; Rojko et al. Citation2014; Krishna and Nadler Citation2016). In particular, CIC formed under condition of ADA:protein molar equivalence tend to be large and insoluble (Mannik Citation1980; Lucisano Valim and Lachmann Citation1991; Tsokos et al. Citation2007; Rojko et al. Citation2014). Such CIC of large lattice size efficiently activate the classical complement cascade and form complement-bound circulating CIC (cCIC) (Rojko et al. Citation2014). If large amounts of insoluble cCIC are formed, the intrinsic clearance mechanisms may potentially be overwhelmed leading to immune complex (IC) deposition and complement mediated inflammation in the vasculature in various tissues (Mannik Citation1980; Rojko et al. Citation2014), in particular at sites of ultra-filtration or where there is turbulent or loss of laminar blood flow (Ponce et al. Citation2009; Leach et al. Citation2014; Rojko et al. Citation2014; Frazier et al. Citation2015; Frazier and Obert Citation2018).
There has been an increased reporting of IC-related findings in toxicity studies in the recent years (Rojko et al. Citation2014). However, the underlying treatment-related factors, including dose level and regimen that may affect risk of immunogenicity (Shankar et al. Citation2007; Kijanka et al. Citation2013; Leach et al. Citation2014; Jiskoot et al. Citation2016) needs to be explored further. Moreover, identification of biomarkers of IC deposition and IC-related pathology will be useful to ease identification of IC-related findings.
The present studies sought to investigate effects of different bovine serum albumin (BSA) dose regimens on CIC and cCIC formation, and on IC deposition in two different mouse strains, in order to better understand and facilitate interpretation of IC-related findings. BSA was selected as a model molecule for a species foreign protein, and was expected to induce ADA and CIC formation, which possibly could lead to IC deposition and pathological changes (Noble et al. Citation1981; Devey et al. Citation1982; Chen et al. Citation2004), as seen in toxicity studies.
The first study, a dose-finding study, was performed to identify the dose levels that could initiate a CIC response in the mice. Different dose levels of BSA without adjuvant were administered subcutaneously (SC) twice weekly (2QW) to C57BL/6J and BALB/c mice for 13 weeks, thereby mimicking a sub-chronic toxicity study. The level of BSA giving the most robust CIC response was then chosen for further investigation in the second study. In this dose regimen study, dosing frequency or BSA dose level was changed on top of the already-established CIC response in order to reach the best molar equivalence between ADA and BSA that potentially could lead to further increase of CIC, cCIC formation, and IC deposition in C57BL/6J mice. Moreover, to identify potential biomarkers for IC deposition and IC-related pathological changes, the possible association between the presence of CIC or cCIC in plasma and the presence of glomerular IC deposits and/or IC-related tissue injury was examined.
Materials and methods
Animals
Female C57BL/6J and BALB/c mice were obtained from Charles River Laboratories (Sulzfeld, Germany) at 10–12-weeks-of-age. The mice were housed under standardized conditions (12/12 hr light/dark cycle, temperature at 21 °C, 60% relative humidity, water and chow ad libitum) and group housed 12 mice/cage. After a 7-d acclimatization period, the mice were randomized into groups for use in the studies. Throughout the studies, all mice were observed daily for clinical findings by cage-side observation. All animal experiments were approved by the Danish Animal Experiments Council under the Danish Ministry of Environment and Food of Denmark, as well as the Novo Nordisk Ethical Review Council.
Experimental design
Dose-finding study
Groups of C57BL/6J and BALB/c mice (n = 12/group) received vehicle, 10 mg/kg BSA (C57BL/6J mice only), or 50 mg/kg BSA (Sigma, St. Louis, MO) SC 2QW for 13 weeks. The BSA dose levels were selected based on the literature (see for a study design overview). The concentrations of BSA in the dose formulations were 3 and 14 mg/ml for the two dose levels. Blood (ethylenediaminetetraacetic acid [EDTA]-stabilized) was sampled at Week 6 (1 d after injection #12) and Week 13 (1 d after injection #26) from sublingual or periorbital plexus blood, respectively. Plasma was separated by centrifugation (4000 × g, 5 min, 5 °C), aliquoted, and stored at −80 °C for later analysis. After the final blood sampling (performed under isoflurane anesthesia), mice were euthanized by abdominal aorta puncture, and the kidneys were harvested and processed for immunohistochemistry (IHC) and histopathological analysis.
Table 1. Number of mice with granular staining, IC deposits, and glomerulonephritis.
Dose regimen study
Forty-eight C57BL/6J mice were administered 50 mg/kg BSA SC 2QW for six weeks. Thereafter, the animals received 10, 50, or 250 mg/kg 2QW or 50 mg/kg four times weekly (4QW) for another seven weeks (n = 12/group). The concentrations of BSA in the dose formulations were 3, 15 and 75 mg/ml for the three dose levels, respectively. A control group received vehicle 2QW for 13 weeks (n = 12). Similar to the dose-finding study, plasma was obtained at Weeks 6 and 13, and kidneys were collected at Week 13 for IHC and histopathology.
Immunoassays
BSA exposure
Plasma concentrations of BSA were analyzed using a competitive ELISA kit (Biomatik, Ontario, Canada), according to manufacturer protocols. The BSA in study samples and biotin-labelled BSA competed with pre-coated anti-BSA, which provided a reverse proportional relationship between BSA concentration and assay signal. Study samples were diluted 1:10 or 1:20; the assay lower limit of quantification was 2.41 nM. All samples were assayed in duplicate.
CIC Assay
Any CIC formed by ADA in a complex with BSA were analyzed in a sandwich ELISA, as described in Boysen et al. (Citation2019a), with modifications. In brief, rabbit anti-BSA (Thermo Fisher, Waltham, MA) diluted 1:10.000 in carbonate buffer (0.1 M; pH 9.6) was used as the coating antibody; peroxidase-conjugated goat anti-mouse IgG (mIgG) (κ light chain-specific) (Jackson Immunoresearch, Newmarket Suffolk, UK) was used as detecting antibody. Calibration samples and quality controls (QC) were prepared by chemical conjugation of BSA and mouse IgG (mIgG) (both Sigma). Study samples were diluted 1:40 in dilution buffer (Sigma: 0.01 M phosphate, 0.65 M sodium chloride, 0.05% Tween 20, 0.00025% phenol red and 0.2% fish gelatin [pH 7.2]) before analysis. The signal was expressed as signal-to-noise (S/N; optical density/QCnegative). Samples above a study specific cut-point which allowed for 5% false positive were considered CIC positive (CIC+).
cCIC assay
Complement C3 bound to CIC was detected by ELISA as described in Boysen et al. (Citation2019b). Here, solid phase binding rabbit anti-C3 (Bioss, Boston, MA) and fluid phase detecting peroxidase-conjugated goat anti-mIgG (κ light chain-specific) (Jackson Immunoresearch) were used to capture cCIC. Study samples were diluted 1:80 (dose-finding study) and 1:2000 (dose regimen study) and cCIC level was expressed as S/N. The assay allowed a 5% false positive as determined by a cut-point that distinguished between cCIC positive (cCIC+) and negative (cCIC−) at Week 6 or 13. Samples from C57BL/6J mice dosed with 50 mg/kg BSA were partly used for qualification of the cCIC assay (Boysen et al. Citation2019b).
Histopathology
Kidney tissues were preserved in neutral buffered 4% paraformaldehyde for 48 h and then embedded in paraffin. Slides of ≈ 3-μm thickness were prepared and then stained with hematoxylin and eosin (H&E) dye or periodic acid-Schiff (PAS) stain. Thereafter, the slides were evaluated using light microscopy.
Immunohistochemistry
IHC staining using antibodies against test-item (BSA), mouse complement C3d, mouse IgM (mIgM) and mIgG (ADA) were performed to identify granular staining in kidney tissue, indicating IC deposition (Rojko et al. Citation2014). Sections (3-µm thick) were de-paraffinized, rehydrated, treated with TEG [pH 9] buffer (Ampliqon, Odense, Denmark) for antigen retrieval, and then treated with Peroxidase Block (Agilent, Santa Clara, CA) to block endogenous peroxidase activities.
For C3d, mIgG, and mIgM staining, this was followed by an Avidin-biotin Block (Invitrogen, Carlsbad, CA) and 3% BSA block before incubation with primary antibody. Biotinylated primary antibodies were: donkey anti-mIgG (Jackson Immunoresearch) diluted 1:10.000 in Tris-Buffered Saline (TBS) (Apliqon) containing 3% BSA (Sigma), donkey anti-mIgM (Jackson Immunoresearch) diluted 1:20.000 in TBS containing 3% BSA, and goat anti-mouse C3d (R&D Systems, Minneapolis, MN) diluted 1:500 in TBS containing 3% BSA. Thereafter, the slides were incubated with ABC-HRP (horseradish peroxidase; Vectastain®, Burlingame, CA) and 3,3'-diaminobenzidine (DAB) + substrate chromogen system (Agilent).
For anti-BSA staining, slides were incubated with rabbit anti-BSA (Life technologies, Carlsbad, CA) diluted 1:40.000 in TBS without BSA. Thereafter, slides were incubated with Bright Vision goat anti-rabbit HRP (Immunologic, Amsterdam, the Netherlands) and DAB + substrate chromogen system. In all four stainings, slides were washed in TBST (TBS containing 0.05% Tween 20) between incubations.
The specificity of the C3d antibody was successfully evaluated by pre-incubation of the antibody with 10 C3-peptides (Caslo, Kongens Lyngby, Denmark) targeting the His1002-Arg1303 sequence in mouse C3 protein, which was equal to the sequence used for immunization. C3d-antibody was incubated 1 hr with up to 5-time higher concentrations of C3 peptides, prior to a 30 min incubation with primary antibody. The pre-incubation with peptides resulted in a total block of the C3d signal.
IHC score
IHC slides from both kidneys were assessed for granular deposits in kidney glomeruli in a blinded manner. Frequency of granular staining within the glomerular tufts was evaluated for each staining (BSA, mIgG, mIgM, and C3d) and assigned a score of 0-3 if at least three glomeruli were affected (0, no granular staining, 1, mild; 2, moderate; 3, severe) (, ). Likewise, frequency of affected glomeruli within one slide was evaluated with scores from 0 to 3 (0, none; 1, up to 20%; 2, 20–50%; 3, 50–100%) (). From each kidney and each staining, a total IHC score was determined by: Total IHC score = Frequency of granular deposits within glomerular tufts plus frequency of affected glomeruli within the slide. Each animal was assigned the score from the kidney with highest total IHC score. An IHC score cut-point was determined based on the background levels in the vehicle animals. If the IHC score was above the cut-point in at least two stainings, including the criteria that either IHC score for BSA or C3d was above cut-point, the animal was regarded as positive for IC deposits (IHC+).
Figure 1. IHC score of granular deposits within glomerular tufts. For each of the four staining targets (BSA, mIgG, mIgM, and C3d), frequency of granular staining within the glomerular tuft was evaluated from mice dosed with (A) vehicle or (B–D) bovine serum albumin (BSA). A score 0 (A) was assigned if no granular was observed; score 1 (B) if granular staining was shown as mild; score 2 if shown as moderate (C); or score 3 if shown as severe (D) in at least three glomeruli within the IHC cross section. Original objective 60×.
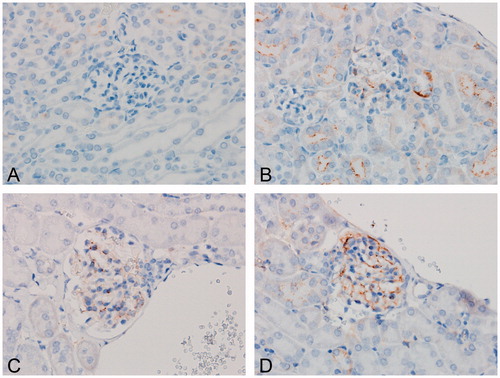
Table 2. Immunohistochemistry (IHC) scores.
Double IHC staining
To confirm co-localization of granular staining containing BSA and C3d or mIgM and C3d in kidney tissues, samples from selected mice (from dose regimen study) that were administered with vehicle or BSA were double-stained with fluorescent antibodies. Except for minor changes, the IHC staining procedure and the primary antibodies were those as already described above.
The primary goat anti-mouse C3d (R&D Systems; diluted 1:100 in TBS) was co-incubated with rabbit anti-BSA (Life Technologies; 1:4.000 in TBS). In the secondary incubation, donkey anti-goat Alexa Fluor (AF)-594 (1:2.000 in TBS) (Abcam) and anti-rabbit AF488 conjugate (1:500 in TBS) (Abcam) were used to detect anti-C3d or anti-BSA, respectively. In another staining, primary goat anti-mouse C3d (R&D Systems; 1:100 in TBS-3% BSA) was co-incubated with biotinylated donkey anti-mIgM (Jackson Immunoresearch; 1:2.000 in TBS-3% BSA. In the secondary incubations, donkey anti-goat AF594 (Abcam; 1:2.000 in TBS) and streptavidin AF488 conjugate (Invitrogen; 1:500 in TBS) were used to detect anti-C3d or anti-mIgM, respectively. The signal from each sample was assessed by immunofluorescent microscopy using an Olympus BX53 scope (fitted with a DP74 Camera) (Olympus, Tokyo, Japan), containing Olympus AF488 and AF594 filters. Olympus cell Sens Entry (v.2.1) software was used to combine the images obtained with each filter.
Statistical analysis
For statistical analysis of exposure, CIC, and cCIC levels, a Student’s unpaired t-test with or without a Welch’s correction was used to compare dose groups in the dose-finding study. A one-way analysis of variance (ANOVA) and a Bonferroni’s multiple comparison or Kruskal Wallis and Dunn’s multiple comparison were used to compare dose groups with vehicle in both studies and to compare between dose groups in dose regimen study, dependent on parametric or non-parametric conditions. Data was expressed as mean ± SD unless otherwise stated. A p-value < 0.05 was considered significant. A possible association between the presence of CIC and IC deposits was evaluated by Fischer’s exact test, by comparing the proportion of mice being IHC+ and IHC− with mice being CIC+ and CIC− across all dose levels. Data was analyzed using Prism Software (v.7, GraphPad, San Diego, CA) and SAS JMP® v.13.0.0 (SAS, Marlow, UK).
Results
Dose-finding study
Clinical observations
No treatment-related clinical signs were observed.
BSA exposure
BSA concentration increased dose-dependently, reaching 415 [±114] nM in C57BL/6J mice administered 2QW with 50 mg/kg at Week 13. Even higher concentrations were noted in the BALB/c mice (748 [± 428] nM) at Week 13 (p < 0.05) ().
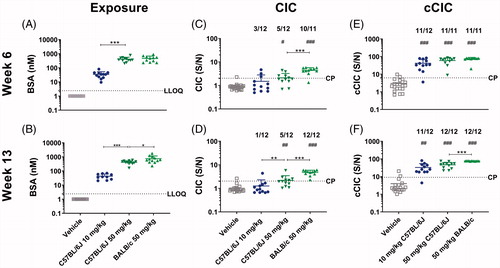
Figure 2. Exposure, CIC and cCIC formation in mice in BSA dose-finding study. C57BL/6J mice or BALB/c mice were administered with vehicle, 10 mg/kg BSA (C57BL/6J only), or 50 mg/kg BSA SC twice weekly for 13 weeks. Exposure (A,B), circulating immune complexes (CIC) (C,D), and complement-bound CIC (cCIC) (E-F) concentrations in plasma were measured at Weeks 6 and 13. Data were expressed as means ± SD. A Student’s unpaired t-test/t-test with Welch’s correction or Mann-Whitney, 1-way ANOVA/Bonferroni or Kruskal Wallis/Dunns, #p < 0.05, ##p < 0.01, ###p < 0.001 compared to vehicle. *p < 0.05, **p < 0.01, ***p < 0.001 compared to indicated mouse group. Number above graph: Number of mice in a dose group above cut-point (CP). LLOQ: Lower limit of quantification; S/N: Signal to noise.
Circulating immune complexes (CIC)
The CIC concentration increased dose-dependently, with only small changes from Week 6 to Week 13 in C57BL/6J mice. This reached statistical significance (compared to the vehicle-administered animals) at 50 mg/kg (p < 0.05-0.01), where 5 of 12 animals were CIC+ (). In BALB/c mice administered 50 mg/kg, the CIC concentration was significantly higher compared to vehicle and to C57BL/6J mice at same dose level (p < 0.001) at both Weeks 6 and 13, with all mice being CIC+ at Week 13.
Complement-bound circulating immune complexes (cCIC)
The cCIC concentration increased dose-dependently with higher cCIC concentration in all dose groups of the C57BL/6J and BALB/c mice as compared to vehicle (p < 0.001–0.01) at both Weeks 6 and 13. Except for one C57BL/6J mouse administered 10 mg/kg, all BSA-administered C57BL/6J and BALB/c mice were cCIC positive (cCIC+) at Weeks 6 and 13 ().
Histopathology
Minimal glomerulonephritis (GN) – characterized by thickening of the glomerular base-ment membrane and infiltration of mononuclear cells with few affected glomeruli in a cross-section – was detected in two C57BL/6J mice administered with 10 mg/kg and one C57BL/6J mice administered 50 mg/kg BSA () ().
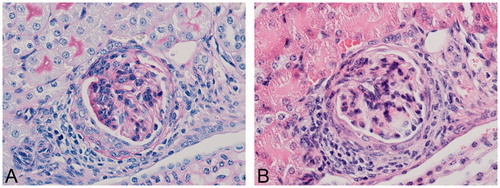
Figure 3. Glomerulonephritis in BSA-dosed mouse. (A) Glomerular basement membrane thickening (PAS) and (B) increased cellularity in a C57BL/6J mouse dosed with 10 mg/kg BSA SC for 13 weeks in dose-finding study. Original objective 60×.
Immunohistochemistry
Granular staining containing C3d was present above the IHC cut-point in kidney glomeruli in eight, four, and two out of 12 C57BL/6J mice administered, respectively, 10 mg/kg BSA, 50 mg/kg BSA, or vehicle. Only a few mice had granular deposits containing BSA and/or endogenous mIgG; no mice had granular deposits containing mIgM. Only one C57BL/6J mouse administered 50 mg/kg BSA had an IHC score above the cut-point in at least two stainings and was therefore considered IHC+. For the three mice with GN, granular staining above IHC cut-point was only present in one of the stainings, and the mice were therefore considered IHC− (). The granular staining in glomeruli was observed along the glomerular basement membrane of kidney glomeruli, especially in glomeruli located areas in the inner cortex close to the pelvis. In addition, granular staining containing C3d () was observed along tubular basement membranes. Occasionally, granular staining containing C3d, BSA, and mIgM were observed in smaller vasculature of the kidney (data not shown).
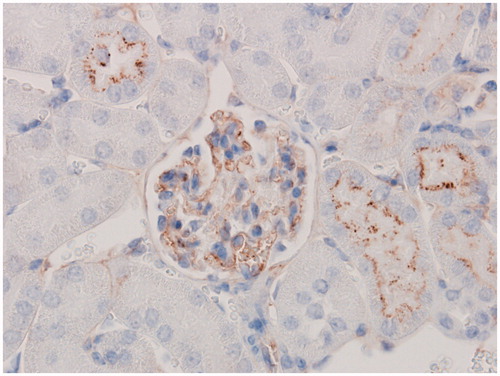
Figure 4. IC-associated granular staining containing C3d. Immunohistochemical staining with anti-mouse C3d showed IC associated granular deposits containing mouse C3d in glomeruli from a C57BL/6J mouse administered 10 mg/kg BSA SC for 13 weeks in dose-finding study. Original objective 60×.
Association between CIC formation and IC deposition
A possible association between the presence of CIC in plasma at any timepoint and the presence of glomerular IC deposits and GN at Week 13 was investigated. However, since two out of the four mice which were considered IHC+ or had GN were detected as CIC+, and 22 of 36 BSA-treated C57BL/6J and BALB/c mice were considered CIC+, no association was shown between these two variables.
Dose regimen study
Clinical signs
No treatment-related clinical signs were observed.
BSA exposure
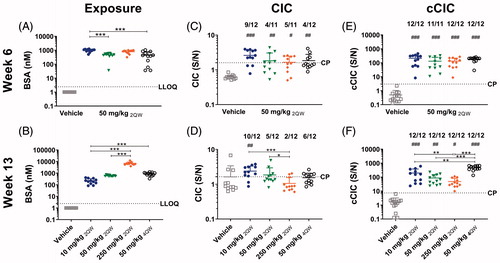
At Week 6, small differences in average BSA concentrations were noted between three dose groups. At Week 13, BSA concentration increased dose-dependently, reaching 6967 [± 2111] nM in the 250 mg/kg dose group. At this same point, the BSA concentrations in the 50 mg/kg 2QW and the 4QW groups were comparable ().
Figure 5. Exposure, and CIC and cCIC formation in mice in BSA dose regimen study. C57BL/6J mice were dosed with vehicle or 50 mg/kg BSA SC twice weekly (2QW) for 6 weeks. Thereafter, dose levels were changed to 10, 50 (unchanged), 250 mg/kg 2QW, or the dosing frequency was changed to 50 mg/kg four times weekly (4QW) for another 7 weeks. At Weeks 6 and 13, (A-B) exposure, (C-D) circulating immune complexes (CIC), and (E-F) complement-bound CIC (cCIC) concentrations in plasma were measured. Data were expressed as means ± SD. A Student’s unpaired t-test/t-test with Welch’s correction or Mann-Whitney, 1-way ANOVA/Bon-ferroni or Kruskal Wallis/Dunns, #p < 0.05, ##p < 0.01, ###p < 0.001 compared to vehicle. *p < 0.05, **p < 0.01, ***p < 0.001 compared to indicated mouse group. Number above graph: Number of mice in dose group above cut-point (CP). LLOQ: Lower limit of quantification, S/N: Signal to noise.
Circulating immune complexes (CIC)
In all dose groups, CIC concentrations were significantly higher compared to vehicle (p < 0.001–0.05), with no significant differences between the dose groups at Week 6. However, the incidence of mice being CIC+ at Week 6 varied between four and nine out of 12 mice. At Week 13, CIC concentration decreased with increasing BSA exposure level, with the highest CIC concentrations seen in the 10 mg/kg dose group (p < 0.05). Increased dosing frequency had no significant effect on CIC concentration. Similar to in the dose-finding study, the incidence of CIC+ mice was comparable at Weeks 6 and 13, with ten, five, two, and six out of 12 mice given 10, 50, 250 mg/kg BSA 2QW, or 50 mg/kg 4QW, respectively, being CIC+ at Week 13.
Complement-bound circulating immune complexes (cCIC)
In all BSA-administered mice, cCIC was detected, with higher cCIC concentrations in all dose groups compared to in vehicle at both Weeks 6 and 13 (p < 0.001–0.05). No significant differences in cCIC concentration were seen between dose groups at Week 6; however, at Week 13, the cCIC concentration in 2QW dose groups tended to decrease with increasing BSA exposure, in contrast to in the dose-finding study, with statistically significance obtained between 10 and 250 mg/kg. The highest cCIC concentration was seen in the 50 mg/kg 4QW dose group (p < 0.001–0.01 compared to the other three dose groups) ().
Histopathology
In contrast to in the dose-finding study, no GN was observed.
Immunohistochemistry
Granular staining primarily contained C3d, BSA, mIgM, and/or to a lesser extent mIgG, and was observed at higher incidence than in dose-finding study. In total, 19 mice were considered IHC+. Of these, seven, six, five and one out of 12 mice administered with 10, 50, or 250 mg/kg 2QW and 50 mg/kg 4QW, respectively, were considered IHC+ (). Similar to in the dose-finding study, granular staining was observed along the glomerular basement membrane of kidney glomeruli, particularly in glomeruli located in the inner cortex close to the pelvis. Occasionally, granular staining containing C3d, BSA, and mIgM were observed in smaller vasculature.
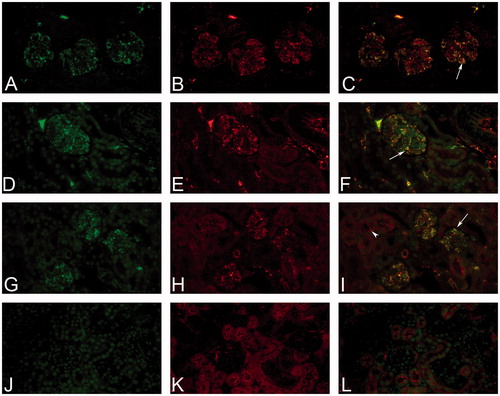
Double IHC staining with fluorescence showed co-localization of granular containing C3d and BSA and/or C3d and mIgM in kidney glomeruli and vasculature of animals administered with BSA (). Granular staining containing C3d was observed along the tubular epithelial membrane, but did not co-localize with mIgM () or BSA in the double IHC staining.
Figure 6. Immune complex deposition shown by double-staining with fluorescent antibodies. Immune complex (IC) deposition in a C57BL/6J mouse administered with 50 mg/kg BSA (A-I) SC for 13 weeks. IHC staining with fluorescence showed granular staining containing BSA (green, AF488) (A), or mIgM (green, AF488) (D, G); mouse granular staining containing C3d (red, AF594) (B, E, H) within glomeruli and kidney vasculature. Co-localization of granular deposits was shown by a yellow/orange fluorescent signal (arrow) (C, F, I). Granular staining containing C3d (H) was observed along the tubular basement membrane, but did not co-localize with mIgM (arrowhead) (I). In a C57BL/6J mouse administered with vehicle no granular deposits within the glomeruli was observed in IHC staining of mouse IgM (green, AF488) (J) and mouse C3d (red, AF594) (K) or in co-localization (L). Original objective 60× (Color Figure available online).
Association between CIC formation and IC deposition
The presence of CIC in plasma at Week 6 or Week 13 was associated with a presence of glomerular IC deposits at Week 13. Sixteen out of 19 mice considered IHC+ were also seen to be CIC+, and 17 out of 29 mice considered IHC− were assessed as CIC−. However, three mice considered IHC+ did not prove positive for CIC and 12 mice were assessed as CIC+ but considered IHC− (). Thus, an association between plasma IC and IHC status was supported by a Fischer’s exact test (p < 0.01).
Table 3. Number of mice with plasma CIC and IC deposits.
Discussion
The aim of the present studies was to investigate the effects of different BSA dose regimens on CIC and cCIC formation, and IC deposition and, if possible, to identify potential biomarkers of IC deposition and IC-related pathological changes in mice. In a dose-finding study, dosing of BSA SC for 13 weeks caused CIC and cCIC formation in C57BL/6J and BALB/c mice. In a few C57BL/6J mice given BSA without use of any adjuvant, IC deposition in kidney glomeruli and GN was observed. The formation of CIC and cCIC did not increase from Week 6 to 13 at any dose level in either strain, suggesting the CIC and cCIC responses were established before 6 weeks of dosing. A higher CIC concentration was found with 50 mg/kg BSA in C57BL/6J mice compared to with a 10 mg/kg dose; this was in line with other studies where immunogenicity to foreign proteins was intensified with increased dose level (Braun et al. Citation1997; Kijanka et al. Citation2013).
While IC deposition and GN was found in a few C57BL/6J mice, no IC deposition or GN was found in BALB/c mice, in spite of their higher CIC and cCIC concentrations. Genetic differences between BALB/c and C57BL/6 strains (Noble et al.Citation1981; Morokata et al. Citation1999; Chen et al. Citation2004; Watanabe et al. Citation2004; Fukushima et al. Citation2006) may lead to differences in formation of ADA with different IgG subtypes and different affinities to test substances (Alpers et al. Citation1972; Petty et al. Citation1972; Devey and Steward Citation1980; Steward et al. Citation1981; Devey et al. Citation1982; Sarvas et al. Citation1983). As an example, some inbred mouse strains show relatively good elimination of human serum albumin due to production of high affinity ADAs, which is on contrast to poor elimination in mice producing low affinity ADAs (Alpers et al. Citation1972; Petty et al. Citation1972). This could possibly explain a faster elimination of CIC and cCIC in BALB/c mice compared to by C57BL/6 in the present study. However, neither the ADA affinity nor IgG subtypes were characterized in the present study.
Based on these results, a dose regimen study was conducted to study if changes in dose regimens after 6 weeks of dosing with 50 mg/kg BSA 2QW, the dose level that caused high level of CIC and cCIC in the dose-finding study, would lead to further increased CIC, cCIC formation and IC deposition in C57BL/6J mice, as this has been shown to be dependent on molar equivalence between ADA and the dosed protein (Rojko et al. Citation2014). Apart from a tendency to decreased CIC and cCIC concentrations with increasing exposure level in mice administered 2QW, no substantial changes in CIC or cCIC formation, or IC deposition, was found after changing of the BSA dose level. This could possibly have been due to development of an early immune tolerance in the high dose group (Hebert et al. Citation1991), or CIC formation in large excess of test-substance, leading to smaller CIC that are cleared or dissociated faster and cause less complement activation (Hebert et al. Citation1994). Moreover, increased dosing frequency caused increased cCIC formation – as reflected by increased cCIC concentration in the 50 mg/kg 4QW compared to 50 mg/kg 2QW mice in the dose regimen study. However, BSA exposure or CIC level was not significantly affected by an increased dosing frequency. In a few mice, BSA exposure declined already at Week 6 (), most likely due to ADA formation, as indicated by the presence of CIC and cCIC and correspondingly lower BSA concentrations in most of these mice.
Another aim of the present studies was to identify possible associations between presence of CIC or cCIC in plasma and glomerular IC deposits and IC-related pathological changes. In the dose regimen study, a significant association between a presence of plasma CIC and glomerular IC deposits was shown. However, in the dose-finding study, no association was seen. Thus, the present study could not support use of CIC as a biomarker. Since CIC can be present without pathological changes (Bessa et al. Citation2015; Kronenberg et al. Citation2017), the presence of cCIC or both CIC and cCIC in plasma could be hypothesized to serve as a better biomarker for IC deposition and/or IC-related tissue injury. However, since almost all mice were positive for cCIC in both studies, no conclusion on cCIC as a potential biomarker for IC deposition and IC-related pathological changes could be reached.
Whereas little difference in the incidence of GN was found between the two studies, IC deposition occurred at a higher incidence in the dose regimen study compared to in the dose-finding study. The studies were conducted using the same mouse strain and the same methodologies for detection of IC-related variables, however, different batches of BSA were used. This may explain the differences between the two studies, but this may also reflect the general variation in immune response to a given antigen. Thus, further optimization and test of reproducibility are needed.
The IC-related pathological findings shown in the dose-finding study were small compared to rodent models of IC mediated GN, where high doses of species foreign serum albumin in combination with Freund’s adjuvant were used (Peress et al. Citation1977b; Arisz et al. Citation1979; Noble et al. Citation1980, Citation1981, Citation1987; Koffler et al. Citation1983; Hogendoorn et al. Citation1990; Saglam et al. Citation2010). For example, in a mouse study, daily (for up to six weeks with BSA) intraperitoneal immunization of C3H.NB mice pre-immunized with Freund’s adjuvant caused severe GN and granular staining (containing C3, mIgG, and/or BSA) within the glomeruli (Noble et al. Citation1980). Granular staining containing IgM was not examined. The more intense dosing schedule and pre-immunization with adjuvant in some of these studies were probably the reason of the increased immunogenicity and severe pathological findings which in some mice and rats led to clinical signs and lethality (Peress et al. Citation1977b; Arisz et al. Citation1979). On the contrary, no immunogenicity of BSA without adjuvant was found in a previous pilot study (Arisz et al. Citation1979). The clinical signs associated with use of adjuvant (Soothill and Steward Citation1971; Petty et al. Citation1972; Broderson Citation1989; Pilegaard and Madsen Citation2004) precluded its use in the present studies. Other studies have used cationic BSA that targets anionic sites in the glomeruli to induce IC-mediated vascular disease (Border et al. Citation1981; Furness and Turner Citation1988; Urizar et al. Citation1989; Chen et al. Citation2004; Wu et al. Citation2008). In contrast to the present studies where IC deposition was caused by in vivo CIC formation, dosing with cationic BSA mainly led to direct in situ IC deposition.
Detection of IC formation and deposition was performed by supplemental measures of BSA exposure, CIC and cCIC concentrations by ELISA, pathological changes by H&E and PAS stainings, and IC deposits by IHC. The CIC assay measures ADA bound to BSA, which in similar assay set-ups have been used to detect ADA bound to human IgG in mice and non-human primates (Stubenrauch et al. Citation2010, Citation2012; Pierog et al. Citation2015; Boysen et al. Citation2019a). This assay set-up was suggested to have improved drug-tolerance compared to a standard bridging ELISA for detection of monomeric ADA (Pierog et al. Citation2015; Boysen et al. Citation2019a). Assay drug interference could explain the low CIC concentrations measured in C57BL/6J mice administered 250 mg/kg BSA in the dose regiment study; however, this was not expected to be entirely caused by drug interference, since cCIC levels and IC deposition also were low.
The high cCIC concentration in almost all mice administered BSA suggested that the dosing caused CIC formation, leading to activation of the complement cascade and binding of C3 to various CIC. In majority of the animals, cCIC were detected even in the absence of CIC. Drug interference is a typical factor for false negative results in these kinds of assays, and whereas BSA in the CIC assay set-up could be interfering, the presence of BSA in the cCIC assay set-up was not expected to interfere with cCIC measurements (Boysen et al. Citation2019b). In samples containing high amounts of BSA, the sensitivity of the cCIC assay would therefore be expected to be higher than the CIC assay. The cCIC assay is not drug-specific; this allows for simultaneous screening of different drugs within one assay. However, other immunological reactions which involve complement activity (Pangburn and Muller-Eberhard Citation1983; Murphy and Weaver Citation2016) may contribute to a positive signal and may explain a presence of cCIC in a few of the vehicle-treated mice in the present study.
As expected, the kidney was seen as a target organ for IC deposition after BSA dosing, due in main part to its filtering capacity such that CIC and cCIC are easily caught in the glomeruli (Leach et al. Citation2014; Rojko et al. Citation2014). Consequently, the present studies examined kidneys for IC-related changes using H&E, PAS, and IHC staining. In the current studies, GN was only seen with a low incidence (3 three out of 72 C57BL/6J mice dosed with BSA) and low severity in spite of CIC and cCIC formation; this was most likely due to rapid clearance of CIC and cCIC from the glomeruli. In spite of the negative IHC status, granular staining containing C3d was detected in all three mice with levels above the IHC cut-point. Therefore, this GN was most likely caused by IC deposition. However, it cannot be excluded that the GN developed spontaneously (Poskitt and Poskitt Citation1982).
In the present studies, the granular deposits detected by IHC contained C3d, BSA, mIgG, and mIgM. Such outcomes suggested that BSA clearance, in part, was facilitated by C3d and mIgG/mIgM type ADA. All IC components were not always contained within the same glomeruli or even within the same mice, potentially due to unstable IC components, steric hindrance caused by some IC components, granular deposits below the IHC detection limit, or possible clearance of IC by the mononuclear phagocytic system (Rojko et al. Citation2014). In addition to the above-mentioned IHC limitation, the high exposure level complicated evaluation of the BSA IHC staining and may have caused a false higher frequency of granular deposits within the glomerular tufts for this staining.
In several toxicity studies in non-human primates, IC deposits containing IgM were most frequently detected (Leach et al. Citation2014; Rojko et al. Citation2014; Husar et al. Citation2017), which is in line with the present dose regimen study where IgM were detected more frequently than IgG. Importantly, while the IHC approach differentiated between ADA of either the IgM or IgG subtype, the CIC and cCIC assays did not (Boysen et al. Citation2019a, Citation2019b). Double-staining with fluorescent antibodies in the present dose regimen study provided information about co-localization and presence of two IC components. The co-localization of C3d and IgM or BSA, observed in glomeruli and occasionally in small blood vessels outside the glomeruli of BSA-treated mice, points towards IC-mediated vascular deposition. In contrast, the C3d signal seen in the kidney tubules (by IHC) did not co-localize with BSA and mIgM, indicating that this presence of C3d not was IC-related. A normal physiological deposition of C3 fragments and IgM in the kidneys (Peress et al. Citation1977a; Devey and Steward Citation1980; Koffler et al. Citation1983; Thurman et al. Citation2003; Rojko et al. Citation2014) may explain why some vehicle animals were found to be positive for IgM or C3. However, in those cases, the double-staining was useful to show that any IgM and C3 deposits were not co-localized. Lastly, it should be noted that CIC may deposit in other organs. Thus, histological evaluation of the kidneys exclusively does not preclude the potential detection of IC deposits in other organs (Leach et al. Citation2014; Rojko et al. Citation2014).
Conclusions
In the present studies, SC administration of BSA without adjuvant for 13 weeks caused CIC and cCIC formation in C57BL/6J mice, which led to IC deposition in kidney glomeruli in 20, and GN in 3, of 72 C57LB/6J mice. CIC and cCIC concentrations tended to decrease with increasing exposure level and cCIC concentration increased with more frequent dosing when BSA dose level or dosing frequency was changed after 6 weeks of dosing to C57BL/6J mice. The potential use of CIC as a possibly biomarker for IC deposition needs to be explored further. In future studies, IHC double staining instead of single staining is proposed.
Acknowledgements
The authors wish to thank Karsten Marckstrøm, Helle Wagner, and Louise Nielsen for all of their excellent technical assistance.
Disclosure statement
The authors declare no conflicts of interest. The authors alone are responsible for the content of this manuscript.
Additional information
Funding
References
- Alpers J, Steward M, Soothill J. 1972. Differences in immune elimination in inbred mice. The role of low affinity antibody. Clin Exp Immunol. 12(1):121–132.
- Arisz L, Noble B, Milgrom M, Brentjens J, Andres G. 1979. Experimental chronic serum sickness in rats. A model of immune complex glomerulonephritis and systemic immune complex deposition. Int Arch Allergy Immunol. 60(1):80–88.
- Bessa J, Boeckle S, Beck H, Buckel T, Schlicht S, Ebeling M, Kiialainen A, Koulov A, Boll B, Weiser T. 2015. The immunogenicity of antibody aggregates in a novel transgenic mouse model. Pharm Res. 32(7):2344–2359.
- Border W, Kamil E, Ward H, Cohen A. 1981. Antigenic changes as a determinant of immune complex localization in the rat glomerulus. Lab Invest. 45(5):442–449.
- Boysen L, Lauritzen B, Viuff BM, Lykkesfeldt J, Landsy LH. 2019a. An ELISA for detection of complement-bound circulating immune complexes in mice. J Immunotoxicol. 16(1):82–86.
- Boysen L, Sprinkel AME, Lauritzen B, Breinholt J, Lykkesfeldt J, Viuff BM, Landsy LH. 2019b. Generic immune complex assay for detection of murine anti-drug antibodies in complex with human IgG. Biologicals. 60:42–48.
- Braun A, Kwee L, Labow M, Alsenz J. 1997. Protein aggregates seem to play a key role among the parameters influencing the antigenicity of interferon (IFN)-γ in normal and transgenic mice. Pharm Res. 14(10):1472–1478.
- Brinks V, Jiskoot W, Schellekens H. 2011. Immunogenicity of therapeutic proteins: The use of animal models. Pharm Res. 28(10):2379–2385.
- Broderson J. 1989. A retrospective review of lesions associated with the use of Freund's adjuvant. Lab Animal Sci. 39(5):400–405.
- Bugelski P, Treacy G. 2004. Predictive power of preclinical studies in animals for the immunogenicity of recombinant therapeutic proteins in humans. Curr Opin Mol Ther. 6(1):10–16.
- Chen J, Chen A, Chang L, Chang W, Lee H, Lin S, Lin Y. 2004. Mouse model of membranous nephropathy induced by cationic bovine serum albumin: Antigen dose-response relations and strain differences. Nephrol Dial Transplant. 19(11):2721–2728.
- Devey M, Steward M. 1980. The induction of chronic antigen-antibody complex disease in selectively-bred mice producing either high or low affinity antibody to protein antigens. Immunology. 41(2):303–311.
- Devey ME, Bleasdale K, Collins M, Steward MW. 1982. Experimental antigen-antibody complex disease in mice. The role of antibody levels, antibody affinity and circulating antigen-antibody complexes. Int Arch Allergy Immunol. 68(1):47–53.
- European Medicines Agency (EMA). 2006. Guideline on immunogenicity assessment of therapeutic proteins. Available from: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-immunogenicity-assessment-therapeutic-proteins-revision-1_en.pdf
- Frazier K, Obert L. 2018. Drug-induced glomerulonephritis: The spectre of biotherapeutic and antisense oligonucleotide immune activation in the kidney. Toxicol Pathol. 46(8):904–917.
- Frazier K, Engelhardt J, Fant P, Guionaud S, Henry S, Leach M, Louden C, Scicchitano M, Weaver J, Zabka T. 2015. Drug-induced vascular injury associated with non-small molecule therapeutics in preclinical development: part I. Toxicol Pathol. 43(7):915–934.
- Fukushima A, Yamaguchi T, Ishida W, Fukata K, Taniguchi T, Liu FT, Ueno H. 2006. Genetic background determines susceptibility to experimental immune-mediated blepharoconjunctivitis: Comparison of Balb/c and C57BL/6 mice. Exp Eye Res. 82(2):210–218.
- Furness P, Turner D. 1988. Chronic serum sickness glomerulonephritis: removal of glomerular antigen and electron-dense deposits is largely dependent on plasma complement. Clin Exp Immunol. 74(1):126–130.
- Hebert L, Birmingham D, Mahan J, Shen X, McAllister C, Cosio F, Dillon J. 1994. Effect of chronically-increased erythrocyte complement receptors on immune complex nephritis. Kidney Intl. 45(2):493–499.
- Hebert L, Cosio F, Birmingham D, Mahan J, Sharma H, Smead W, Goel R. 1991. Experimental immune complex-mediated glomerulonephritis in the non-human primate. Kidney Intl. 39(1):44–56.
- Hogendoorn P, Bruijn J, Gelok E, van den Broek L, Fleuren G. 1990. Development of progressive glomerulosclerosis in experimental chronic serum sickness. Nephrol Dial Transplant. 5(2):100–109.
- Husar E, Solonets M, Kuhlmann O, Schick E, Piper-Lepoutre H, Singer T, Tyagi G. 2017. Hypersensitivity reactions to obinutuzumab in cynomolgus monkeys and relevance to humans. Toxicol Pathol. 45(5):676–686.
- ICH S6 (R1) 2011. ICH guideline S6 (R1) - Preclinical safety evaluation of biotechnology-derived pharmaceuticals. Available from: https://database.ich.org/sites/default/files/S6_R1_Guideline_0.pdf
- Jiskoot W, Kijanka G, Randolph TW, Carpenter JF, Koulov AV, Mahler H-C, Joubert MK, Jawa V, Narhi LO. 2016. Mouse models for assessing protein immunogenicity: lessons and challenges. J Pharm Sci. 105(5):1567–1575.
- Kijanka G, Jiskoot W, Schellekens H, Brinks V. 2013. Effect of treatment regimen on the immunogenicity of human IFN-γ in immune-tolerant mice. Pharm Res. 30(6):1553–1560.
- Koffler D, Biesecker G, Noble B, Andres G, Martinez-Hernandez A. 1983. Localization of the membrane attack complex (MAC) in experimental immune complex glomerulonephritis. J Exp Med. 157(6):1885–1905.
- Krishna M, Nadler S. 2016. Immunogenicity to biotherapeutics: The role of anti-drug immune complexes. Front Immunol. 7:1–13.
- Kronenberg S, Husar E, Schubert C, Freichel C, Emrich T, Lechmann M, Giusti AM, Regenass F. 2017. Comparative assessment of immune complex-mediated hypersensitivity reactions with biotherapeutics in the non-human primate: critical parameters, safety and lessons for future studies. Regul Toxicol Pharmacol. 88:125–137.
- Leach M, Rottman J, Hock M, Finco D, Rojko J, Beyer J. 2014. Immunogenicity/hypersensitivity of biologics. Toxicol Pathol. 42(1):293–300.
- Lucisano Valim Y, Lachmann P. 1991. The effect of antibody isotype and antigenic epitope density on the complement-fixing activity of immune complexes: A systematic study using chimaeric anti-NIP antibodies with human Fc regions. Clin Exp Immunol. 84(1):1–8.
- Mannik M. 1980. Physicochemical and functional relationships of immune complexes. J Invest Dermatol. 74(5):333–338.
- Morokata T, Ishikawa J, Ida K, Yamada T. 1999. C57BL/6 mice are more susceptible to antigen-induced pulmonary eosinophilia than BALB/c mice, irrespective of systemic TH1/TH2 responses. Immunology. 98(3):345–351.
- Murphy K, and Weaver C. (Eds.). 2016. Janeway's Immunobiology. New York: Garland Science.
- Noble B, Milgrom M, van Liew J, Brentjens J. 1981. Chronic serum sickness in the rat: Influence of antigen dose, route of antigen administration and strain of rat on the development of disease. Clin Exp Immunol. 46(3):499–507.
- Noble B, Olson K, Milgrom M, Albini B. 1980. Tissue deposition of immune complexes in mice receiving daily injections of bovine serum albumin. Clin Exp Immunol. 42(2):255–262.
- Noble B, Steward M, Vladutiu A, Brentjens J. 1987. Relationship of the quality and quantity of circulating anti-BSA antibodies to the severity of glomerulonephritis in rats with chronic serum sickness. Clin Exp Immunol. 67(2):277–282.
- Pangburn M, Muller-Eberhard H. 1983. Initiation of the alternative complement pathway due to spontaneous hydrolysis of the thioester of C3. Ann NY Acad Sci. 421(1):291–298.
- Peress N, Miller F, Palu W. 1977a. The choroid plexus in passive serum sickness. J Neuropathol Exp Neurol. 36(3):561–566.
- Peress N, Miller F, Palu W. 1977b. The immunopathophysiological effects of chronic serum sickness on rat choroid plexus, ciliary process and renal glomeruli. J Neuropathol Exp Neurol. 36(4):726–733.
- Petty R, Steward M, Soothill J. 1972. The heterogeneity of antibody affinity in inbred mice and its possible immuno-pathologic significance. Clin Exp Immunol. 12:231–241.
- Pierog P, Krishna M, Yamniuk A, Chauhan A, DeSilva B. 2015. Detection of drug-specific circulating immune complexes from in vivo cynomolgus monkey serum samples. J Immunol Meth. 416:124–136.
- Pilegaard K, Madsen C. 2004. An oral Brown Norway rat model for food allergy: Comparison of age, sex, dosing volume, and allergen preparation. Toxicology. 196(3):247–257.
- Ponce R, Abad L, Amaravadi L, Gelzleichter T, Gore E, Green J, Gupta S, Herzyk D, Hurst C, Ivens IA, et al. 2009. Immunogenicity of biologically-derived therapeutics: Assessment and interpretation of non-clinical safety studies. Regul Toxicol Pharmacol. 54(2):164–182.
- Poskitt P, Poskitt T. 1982. Absence of circulating IgG immune complexes in C57Bl/6 mice with age-associated renal IgG deposits. Immunol Commun. 11(2):97–104.
- Rojas J, Taylor R, Cunningham M, Rutkoski T, Vennarini J, Jang H, Graham M, Geboes K, Rousselle S, Wagner C. 2005. Formation, distribution, and elimination of infliximab and anti-infliximab immune complexes in cynomolgus monkeys. J Pharmacol Exp Ther. 313(2):578–585.
- Rojko JL, Evans MG, Price SA, Han B, Waine G, DeWitte M, Haynes J, Freimark B, Martin P, Raymond JT, et al. 2014. Formation, clearance, deposition, pathogenicity, and identification of biopharmaceutical-related immune complexes: Review and case studies. Toxicol Pathol. 42(4):725–764.
- Saglam F, Celik A, Tayfur D, Cavdar Z, Yilmaz O, Sarioglu S, Kolatan E, Oktay G, Camsari T. 2010. Decrease in cell proliferation by a matrix metalloproteinase inhibitor, doxycycline, in a model of immune-complex nephritis. Nephrology. 15(5):560–567.
- Sarvas H, Seppala I, Tahtinen T, Peterfy F, Makela O. 1983. Mouse IgG antibodies have subclass-associated affinity differences. Mol Immunol. 20(3):239–246.
- Schifferli J, Taylor R. 1989. Physiological and pathological aspects of circulating immune complexes. Kidney Intl. 35(4):993–1003.
- Shankar G, Charles C, Stein K. 2007. A risk-based bioanalytical strategy for the assessment of antibody immune responses against biological drugs. Nat Biotechnol. 25(5):555–561.
- Soothill J, Steward M. 1971. The immuno-pathological significance of the heterogeneity of antibody affinity. Clin Exp Immunol. 9:193–199.
- Steward M, Collins M, Stanley C, Devey M. 1981. Chronic antigen-antibody-complex glomerulonephritis in mice. Br J Exp Pathol. 62(6):614–622.
- Stubenrauch K, Mackeben K, Vogel R, Heinrich J. 2012. Generic anti-drug antibody assay with drug tolerance in serum samples from mice exposed to human antibodies. Anal Biochem. 430(2):193–199.
- Stubenrauch K, Wessels U, Essig U, Vogel R, Schleypen J. 2010. Evaluation of a generic immunoassay with drug tolerance to detect immune complexes in serum samples from cynomolgus monkeys after administration of human antibodies. J Pharm Biomed Anal. 52(2):249–254.
- Thurman J, Ljubanovic D, Edelstein C, Gilkeson G, Holers V. 2003. Lack of a functional alternative complement pathway ameliorates ischemic acute renal failure in mice. J Immunol. 170(3):1517–1523.
- Tsokos G, Gordon C, Smolen J. (Eds.) 2007. Systemic Lupus Erythematosus: A Companion to Rheumatology. Philadelphia: Mosby Elsevier.
- Urizar R, Cerda J, Reilly A. 1989. Glomerulitis induced by cationized bovine serum albumin in the rat. Pediatr Nephrol. 3(2):149–155.
- US FDA (U.S. Food and Drug Administration). 1997. International Conference on Harmonization: Guidance on Preclinical Safety Evaluation of Biotechnology-derived Pharmaceuticals. Vol. 62. Bethesda: US FDA, pp. 515–519.
- van Meer P, Kooijman M, Brinks V, Gispen-de Wied C, Silva-Lima B, Moors E, Schellekens H. 2013. Immunogenicity of mAbs in non-human primates during non-clinical safety assessment. MAbs. 5(5):810–816.
- Watanabe H, Numata K, Ito T, Takagi K, Matsukawa A. 2004. Innate immune response in TH1- and TH2-dominant mouse strains. Shock. 22(5):460–466.
- Wu C, Chen J, Lin S, Chen A, Sytwu H, Lin Y. 2008. Experimental model of membranous nephropathy in mice: Sequence of histological and biochemical events. Lab Anim. 42(3):350–359.