
Abstract
The importance of alveolar macrophages has been reported in many toxicology/immunology studies. Alveolar macrophages release interleukin (IL)-1α as a damage-associated molecular pattern (DAMP) when stimulated by fine particles. However, it is unclear whether cell isolation procedures affect ex vivo particle-induced responses in primary mouse alveolar macrophages (mAM). In this study, effects of injection buffer volume used to perform bronchoalveolar lavage fluid (BALF) collection to isolate mAM for use in ex vivo particle-induced responses were assessed. Among the mAM obtained from BALF collected using a 0.55 or 0.75 ml, but not a 1.0 ml buffer injection volume, decreased cell viability and IL-1α release were observed when cells were stimulated ex vivo with silica crystal or aluminum salt. Injected buffer composition did not affect the IL-1α release. On the other hand, IL-6 secretion induced by lipopolysaccharide (LPS) did not differ among mAM obtained from BALF collected using the different volumes. Expression levels of cell surface markers like CD11c, SiglecF, and CD64 did not differ among mAM obtained from BALF collected using the different injection buffer volumes. IL-1α release (and also necroptosis) induced by ex vivoparticle stimulation was suppressed by RIPK3 inhibitor or cytochalasin D co-treatment. Decreases in RIPK3 phosphorylation were noted in mAM obtained in BALF collected using the 1.0 ml injection volume compared with mAM obtained in BALF using 0.55 or 0.75 ml buffer. These observations illustrate that larger volumes of buffer used to collect BALF from mice can affect sensitivity of the isolated mAM to ex vivo particle-induced responses by inhibiting their functions.
Introduction
Alveolar macrophages (AM) have critical roles in immune regulation, surfactant homeostasis, and play a crucial role in the initiation of inflammation and innate immunity (Kopf et al. Citation2016; Fan and Fan Citation2018). As AM are located in alveoli, they are directly exposed to inhaled air, pathogens, and aerosolized particles. These cells play an important role in host defense against inhaled pathogens and particulates (Gordon and Read Citation2002) as they respond to pathogens and particulates to regulate the immune response via secretion of cytokines, chemokines, and growth factors (Murray and Wynn Citation2011). As a result of their importance in host health, many studies have examined mechanisms of particle-induced pulmonary injury, pulmonary allergy reactions, and pulmonary infectious diseases (i.e. Hodge et al. Citation2007; Sun and Metzger Citation2008; Happle et al. Citation2014; Schneider, Nobs, Heer, et al. Citation2014; Schneider, Nobs, Kurrer, et al. Citation2014; Fan and Fan Citation2018).
Among the different cytokines released from macrophages, interleukin (IL)-1α has been reported to be a unique cytokine that is involved local inflammatory responses (Chen et al. Citation2007), but ultimately systemically as well. In the lungs, IL-1α is secreted by AM in response to intranasal or intratracheal fine particle instillation in mice (Kuroda et al. Citation2016). The IL-1 family of ligands and receptors are associated with both acute and chronic inflammation induced by bacteria and bacterial products (Ulich, Yin, Guo, del Castillo, et al. 1991; Ulich, Yin, Guo, Yi, et al. 1991; Dinarello Citation2011). There are two related but distinct IL-1 genes (Il1a and Il1b) that encode IL-1α and IL-1β, respectively (Dinarello Citation2011). Both IL-1α and IL-1β bind to the same cell surface IL-1R1 receptor that is present on nearly all cells. While IL-1α, unlike IL-1β, can be passively released during necrotic cell death to induce inflammation, it has also been to act as a damage-associated molecular pattern (DAMP) protein under these circumstances (Chen and Nunez Citation2010).
Release of IL-1α from AM is known to also occur as a result of influenza virus infection (Momota et al. Citation2020), aluminum salt (alum), or silica crystal stimulation (Kuroda et al. Citation2016), and lipopolysaccharide (LPS) stimulation (Dagvadorj et al. Citation2015) through a process thought to require AM necroptosis. Release of IL-1α by AM has been reported to be more sensitive to particle stimulation than that by other tissue-resident macrophages, i.e. intestinal and peritoneal macrophages, in mice (Kuroda et al. Citation2016). Thus, it would seem that AM have unique properties, and analyses of primary AM rather than other tissue macrophages or lines like the RAW264.7 or J774.1 cells (Ralph and Nakoinz Citation1975; Raschke et al. Citation1978) or oncogene-transformed cells like MH-S cells (Mbawuike and Herscowitz Citation1989), would be more important to better and more accurately elucidate pulmonary immune responses to particles.
The AM can be isolated from mice with minimal tissue disturbance through the collection of bronchoalveolar lavage fluid (BALF). Many researchers have adopted this method as it allows for collection of a large number of cells and is convenient (Steele et al. Citation2003; Zhang et al. Citation2008). BALF can be obtained by injecting phosphate-buffered saline (PBS, often containing EDTA and fetal bovine serum) into the lung via a cannula, inflating the lung, and then aspirating the buffer. A previous report showed the temperature of the buffer is crucial for increasing the efficiency of mAM harvesting (Busch et al. Citation2019). provides a review of commonly used buffer compositions, injected BALF volumes, buffer temperatures, numbers of flushes employed, and the number of collected mAM from a representative series of studies.
Table 1. Comparison of different mAM isolation protocols.
Improving the alveolar lavage fluid parameters would appear to increase the efficiency of AM collection. However, it is still necessary to evaluate effects of changes in different harvesting parameters on AM collection and the AM themselves. To date, an optimal collection method to assess particle-induced responses, as reflected by IL-1α release or particle uptake activity by harvested cells has not yet been verified. Many reports have shown the recovered cell number can be increased by the adding EDTA and that cell viability can be increased by adding FBS. The flushing required to recover AM from the lung can also cause mild cell damage due to osmotic or mechanical stress (Denkert et al. Citation1998; Aikawa et al. Citation2002; Shiratsuchi and Basson Citation2005). This latter phenomenon suggests to us that the volume of injection buffer used to perform BALF collection may impact on collected mAM functions.
In general, ex vivo cultures of AM provide a useful tool to study lung tissue-resident macrophage behavior and function both under normal and toxicant (particle)-exposure conditions (Hetland et al. Citation2005; Hamilton et al. Citation2006; Chavez-Santoscoy et al. Citation2012; Kuroda et al. Citation2016). Because tissue macrophages in situ differentiate under the control of local environmental factors (Davies and Taylor Citation2015), isolated cultured AM have an advantage over those cells (as well as most macrophage cell lines that have been transformed to immortalize) from the perspective of reflecting realistic “in vivo”functions of normal AM in experiments (Zhang et al. Citation2008). Nevertheless, there are experimental factors that can impact on the yield, viability, and functionality – including in response to particles – of such isolated AM. The latter is an important issue in that AM have an important role in particle-induced acute pulmonary toxicity (i.e.Hodge et al. Citation2007; Sun and Metzger Citation2008; Happle et al. Citation2014; Schneider, Nobs, Heer, et al. Citation2014; Schneider, Nobs, Kurrer, et al. Citation2014; Fan and Fan Citation2018).
In the study reported here, the impact of one particular experimental factor was the focus of interest. Specifically, effects on ex vivo mAM functions from the volume of the injection buffer volume employed to perform BALF collection during cell isolation procedure was evaluated.
Materials and methods
Animals
BALB/c mice (female, 6–7-week-of-age, 16–22 g) were obtained from Japan SLC, Inc. (Tokyo, Japan) and housed in pathogen-free facilities maintained at 23 [± 1]°C with a 50 [± 10]% relative humidity and a 12-h light/dark cycle. All mice had ad libitum access to standard rodent chow and filtered tap water. Mice were acclimated for at least 3–7 d before experiments began. All animal experiments were performed according to the guidelines of the Institutional Animal Care and Use Committee (IACUC) of the National Institute of Infectious Diseases (Tokyo) and reviewed and approved by this committee (approvals #117085 and #120101).
Alveolar macrophage isolation
Mouse alveolar macrophages were obtained using the method in Busch et al. (2019). In brief, each mouse was euthanized by intraperitoneal injection of 100 mg/kg ketamine (Ketalar; Daiichi-Sankyo Co., Tokyo, Japan) and 10 mg/kg xylazine (Selactar; Bayer, Tokyo, Japan). At necropsy, the lung was cannulated and then flushed 10 times with the fixed volume (i.e. 0.55, 0.75, or 1.0 ml) of lavaging solution warmed to 37 °C to obtain BALF. For the experiments, PBS, 2 mM EDTA PBS, or 2% FBS/2 mM EDTA/PBS were used as the injection buffer. Experiments were also repeated using ice-cold lavaging solution in place of the 37 °C buffers. In all cases, aliquots of the isolated BALF were cytocentrifuged (Cytospin centrifuge, Thermo Fisher Scientific, Waltham, MA, USA) and the cells on the slides subsequently stained with May–Grünwald–Giemsa dye solution (Muto Pure Chemicals Co., Tokyo, Japan) to permit examinations of both cell morphologies and cell populations present in each BALF.
This isolation approach is distinct from another widelyused format (especially with larger rodent models) wherein the first instilled fluid is refluxed 1–3 times, collected, and saved on ice apart from any further (1–5 total) instillates that are performed. This approach is thought to maximize collection of soluble agents from the lungs (including cytokines, etc.) and minimize risk of damage from excessive insufflation on those particular endpoints. Ultimately, all subsequent lavages are pelleted and their cells harvested/combined with those of the first tube to maximize total cell yield.
In all cases here, collected BALF was immediately placed on ice until centrifugation (300 × g, 5 min, 4 °C) after which the supernatant was removed. Each cell pellet was re-suspended in RPMI 1640 medium containing 10% FBS, 100 U penicillin/ml, and 100 μg streptomycin/ml (Sigma, St. Louis, MO, USA), counted/checked for viability, and then the isolated cells were seeded into 96-well plates at 5 × 105 live cells/ml (200 µl/well). The plates were cultured at 37 °C under 5% CO2 for 2 h, after which the medium was removed from each well and the cells were gently washed with warmed PBS. All cells then received fresh 10% FBS-RPMI 1640-antibiotic solution and were incubated again for 30 min. The final set of adherent cells were considered “freshly-isolated alveolar macrophages (AM).”
To prepare cell lysates, after stimulation (see below), the AM were washed three times with PBS. Thereafter, to each final pellet, 100 µl of 1% Nonidet P-40 (NP-40, Sigma, St. Louis, MO, USA) in PBS was added, and the cells were placed on ice for 30 min. The plates were then centrifuged (12,000 × g, 15 min, 4 °C), and well supernatants collected. Total protein concentration in each lysate was measured using a Pierce BCA Protein Assay Kit (Thermo Scientific, Waltham, MA, USA).
Macrophage stimulation
The above-isolated macrophages (105 live cells/well) were stimulated with 200 µl fresh medium containing 100 µg/ml alum, crystalline silica particles (MIN-U-SIL5, average particle size 1.5–2.0 µm; U.S. Silica Co., Frederick, MD, USA), fluorescent-conjugated silica particles (FL-silica; Sicaster-greenF [excit485 nm; emiss510 nm]; 1-µm particle size) (Micromod Partikeltechnologie, Rostock/Warnemünde, Germany), or 1 µg/ml LPS (from Escherichia coli Type O55:B5) (Sigma), and then incubated at 37 °C. In some cases using dedicated sets of cultured cells, after 6 h of stimulation, cell-free culture supernatant in each well was collected to analyze lactate dehydrogenase (LDH) and cytokine levels. LDH levels were assessed to measure cell viability using a Cytotoxicity LDH Assay Kit-WST (Dojindo, Kumamoto, Japan) following manufacturer protocols. IL-1α, -1β, and -6 levels were measured using ELISA kits (BioLegend, San Diego, CA, USA). The level of sensitivity of the kits was0.5 pg IL-1α/ml, 16 pg IL-1β/ml, and 2.0 pg IL-6/ml.
In some experiments, 2 µM cytochalasin D (Sigma, St. Louis, MO, USA; actin polymerization inhibitor) or 5 µM GSK’872 (Abcam, Cambridge, UK; RIPK3 inhibitor) was added to the wells containing medium + 100 µg/ml alum. As controls for the inhibitors, additional wells receiving medium + 100 µg/ml alum received dimethyl sulfoxide (DMSO; Sigma, St. Louis, MO, USA). After collection of the medium, cell lysates were prepared to assess cell cytokine levels. After determining total protein concentration in each lysate, IL-1α, IL-1β, and IL-6 levels were measured using ELISA kits as above.
Cell surface marker analyses
Immediately after BALF collection, cells in BALF collected using different injection buffer volumes were counted on a hemocytometer. Thereafter, separate aliquots each containing 2 × 105 cells were incubated for 30 min at 4 °C with a solution of PBS containing a manufacturer-recommended dilution of a conjugated monoclonal antibody (mAb) against specific mouse antigen, i.e. fluorescein isothiocyanate (FITC)-conjugated rat anti-SiglecF [clone 1A8], allophycocyanin (APC)-hamster anti-CD11c [clone N418], phycoerythin (PE)-rat anti I-A/I-E [clone M5/114.15.2] (all Invitrogen, San Diego, CA, USA), Brilliant Violet 421-mouse anti-CD64 [clone X54-5/7.1] or AlexaFluor (AF)-700-rat anti-CD11b [clone M1/70] (the latter two from BioLegend, San Diego, CA, USA). After the incubation, each cell population was washed twice with ice-cold FACS buffer (PBS containing 5% FBS) buffer. To exclude dead cells during the flow cytometric analyses, each final cell pellet was re-suspended in FACS buffer bearing 50 µg propidium iodide/ml (Invitrogen, Carlsbad, CA, USA) immediately before analyses. All samples were then evaluated in a CytoFLEX Flow Cytometer (Beckman Coulter, La Brea, CA, USA) and resulting data was analyzed using FlowJo Software (v.10.0.8, TreeStar, San Carlos, CA, USA). A minimum of 100,000 events/sample was acquired.
Measurement of fluorescent-silica particle uptake
For measures of the impact of injection buffer volume on potential particle uptake activity by the mAM obtained from each resulting BALF, dedicated sets of untreated cultured cells (1 × 105/well) were incubated for 4 h at 37 °C in the presence of 100 µg/ml FL-silica (200 µl/well). The treated mAM were then gently washed with warmed PBS three times and then harvested on ice using 2 mM EDTA-PBS and gentle scraping. Isolated cells were centrifuged (300 × g, 10 min, 4 °C), gently washed twice with PBS, and then the pelleted cells were re-suspended in FACS buffer (5% FBS-PBS). The cells were then immediately analyzed in the CytoFLEX flow cytometer. All acquired data were analyzed for fluorescence intensity (excit485 nm; emiss510 nm); a minimum of 20,000 events per sample was acquired. Based on fluorescence of known levels of the particles in solution (evaluated in parallel), levels of the silica particles in the cells (reflecting silica uptake) could then be calculated.
Immunoblotting
Immunoblotting was performed according to a previous report with slight modifications (Higham et al. Citation2014). In brief, using other dedicated sets of cultured cells, the treated cells were lysed in their wells using RIPA buffer (10 mM Tris-HCl [pH 7.4], 150 mM NaCl, 1 mM EDTA, 1% NP-40, 0.25% sodium deoxycholate; Invitrogen, Carlsbad, CA, USA) supplemented with phosphatase- protease inhibitor cocktails (Merck Millipore, Darmstadt, Germany). The resulting lysates were collected, their protein concentration measured, and then an aliquot was diluted in sample buffer (62.5 mM Tris, 10% glycerol, 1% SDS, 1% β-mercaptoethanol, and 0.01% bromophenol blue [pH 6.8]). A total of 25 µg protein/sample was then loaded into a 10% polyacrylamide gel and the proteins electrophoresed. After resolution of the proteins, the gel contents were electrotransferred to a polyvinylidene fluoride (PVDF) membrane (Merck Millipore, Billerica, MA, USA).
For protein immunoblotting, each membrane was incubated with blocking buffer (2% BSA [bovine serum albumin] in Tris-buffered saline containing 0.1% Tween 20 [TBS-T]) for 1 h at room temperature. Afterwards, each membrane was coated with a solution of blocking buffer containing a manufacturer-recommended 1:1000 dilution of rabbit anti-mouse RIPK3 [clone D4G2A] or rabbit anti-mouse p-RIPK3 [Thr231/Ser232, clone E7S1R] (both Cell Signaling, Hertfordshire, UK) and then incubated at 4 °C overnight. Dedicated membranes were generated for each protein of interest to avoid the issue of stripping/re-probing affecting data outcomes. A third membrane was probed only with horseradish peroxidase (HRP)-labeled goat anti-mouse glyceraldehyde-3-phosphate dehydrogenase (GAPDH; Santa Cruz Biotechnology, Santa Cruz, CA, USA) to confirm sample loading. After washing in TBS-T, the membranes (excluding those for GAPDH) were incubated for 1 h at room temperature in a solution of blocking buffer containing a 1:10,000 dilution of HRP-conjugated anti-rabbit secondary antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA). After a final gentle washing with TBS, labeled proteins were visualized using an enhanced chemiluminescence kit (Thermo Scientific, Waltham, MA, USA). Digital images of each spot were acquired with a LAS-3000 LCD camera system and the spots were analyzed using MultiGuage software (version 2.1, Fuji Film, Tokyo, Japan). All data were normalized to GAPDH levels and outcomes expressed as relative expression levels.
Data analysis
All data are shown as means ± SEM of untransformed data. An unpaired Student’s t-test was used to compare means between two groups. For a single variable, the means of three or more groups were compared using one-way analysis of variance (ANOVA). In cases where significant differences were observed, a Dunnett’s multiple-comparison test was used to identify differences between the control and another group with an unequal number of samples. A p-value <0.05 was considered statistically significant. All statistical analyses were performed using Prism software (v.6.0, GraphPad, La Jolla, CA, USA).
Results
Fine particle-/LPS-induced mAM IL-1α secretion associated with different injection buffer volumes
To assess if a presence of fine particles for 6 h in the lungs can induce IL-1α secretion in BALF, in a pilot study, lung IL-1α concentrations were measured at 6 h after intranasal fine particle administration into mice. The results showed clearly that alum or crystalline silica particles induced IL-1α secretion in situ (data not shown). Knowing the test particles did induce mAM to form/release IL-1α, this study then assessed IL-1α secretion inducible by the fine particles using primary mAM isolated under varying lavage conditions. The concentrations of the test particles and the LPS were set according to a previous study (Kuroda et al. Citation2016). The variables included cells isolated using various lavaging fluid compositions (PBS, 2 mM EDTA/PBS, 2 mM EDTA/2% FBS/PBS) at 37 °C or with ice-cold 2 mM EDTA/2% FBS/PBS. In each case, the injected buffer volume to perform the BALF collection was set to 0.75 or 1.0 ml.
For actual analyses of ex vivo particle-stimulated cell functions, mAM isolated in each scenario were seeded at 5 × 105 cells/ml (200 µl/well) in 96-well plates. The mAM were then treated with medium containing the alum and incubated at 37 °C for 6 h. The results showed that alum stimulation-induced IL-1α secretions were not affected by lavaging fluid composition or temperature (). However, differences in volume of injected buffer to perform BALF collection did affect mAM alum-induced IL-1α secretion.
Figure 1. Effects of injected buffer volume and composition for BALF collection on particle-induced IL-1α release from isolated mAM. (A) mAM obtained from BALF collected using each injection buffer volume (1.0 or 0.75 ml) or compositions were stimulated ex vivo with 100 µg/ml alum. After 6 h, cell-free supernatants were collected, and IL-1α and IL-6 concentrations assessed by ELISA. (B) mAM obtained from BALF collected using each injection buffer volume (0.55, 0.75, or 1.0 ml) were stimulated ex vivo with 100 µg/ml alum, crystalline silica particles, or LPS for 6 hr. Cell-free culture supernatants were collected, and IL-1α and IL-6 concentrations assessed by ELISA. (C) Cells in each well were isolated, pelleted, and then lysed with RIPA buffer. IL-1α and total protein concentrations were measured by ELISA and BCA assay, respectively. (D) Cell viability was assessed by measuring LDH activity in cell-free supernatant. All data shown are means (± SEM) of three independent experiments. Statistical significance assessed by one-way ANOVA and Dunnett’s multiple-comparison test to determine differences between PBS only and other groups. *p < 0.05, **p < 0.01, ***p < 0.001 versus the saline or PBS group.
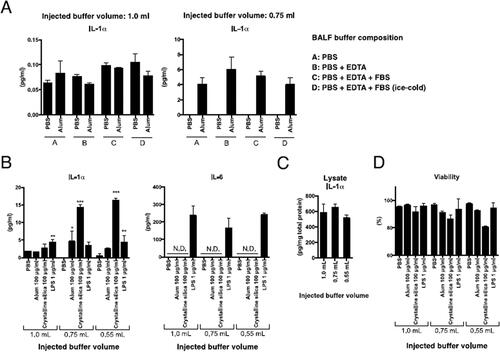
In the next set of studies, mAM from BALF collected using a 0.55, 0.75, or 1.0 ml buffer volume of 0.5 mM EDTA/2% FBS/PBS solution were stimulated with alum, crystalline silica particles, or LPS and incubated at 37 °C for 6 h. Thereafter, culture supernatant IL-1α, IL-6, and IL-1β levels were measured (). Elevation of IL-1α was observed with alum or crystalline silica particles in cells isolated using 0.55 or 0.75 ml injection buffer, but not with mAM collected using 1.0 ml buffer. In contrast, LPS-induced IL-1α responses were not affected by the buffer volume. Similarly, IL-6 levels were uniformly elevated by LPS regardless of the injected buffer volume used to perform the BALF collection. IL-1β was not detected after any stimulation (data not shown).
To assess IL-1α protein expression levels in the mAM, IL-1α levels in cell lysates were assessed. The results showed there were no notable differences in IL-1α levels between mAM obtained from BALF collected using the varying injected buffer volumes (). As IL-1α is secreted by necroptotic mAM (Dagvadorj et al. Citation2015; Kuroda et al. Citation2016), viabilities of mAM obtained using 0.55 or 0.75 ml injected buffer were evaluated. The results showed that mAM viabilities in these populations were reduced (versus mAM obtained using 1.0 ml injected buffer) after particle stimulation (). This suggested that the volume of buffer used to perform BALF collection could ultimately affect mAM IL-1α secretion, in part, by affecting cell viabilities in response to fine particles.
Morphology of cells isolated using different buffer volumes
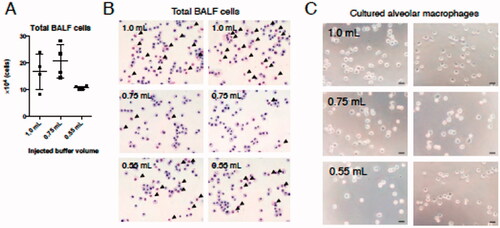
While the results of the effects of buffer volume on total cell recovery showed that the number of cells obtained in BALF collected using 0.75 or 1.0 ml buffer tended to be higher than that isolated using 0.55 ml fluid (), any effect on cell morphology required a deeper analysis. The data showed that mAM obtained using 1.0 ml buffer were more likely to also be accompanied by alveolar epithelial cells more often than when mAM were obtained from BALF collected using the smaller injection volumes (). On the other hand, after washing away epithelial cells that did not attach to dishes that lacked any extracellular matrix coating (Corti et al. Citation1996), the morphology of cells remaining after the attachment period did not appear to differ based on buffer volume used to perform BALF collection (). These results indicated that recovery efficiency and mAM morphology were not markedly impacted by lavaging fluid volume employed.
Figure 2. Number and morphology of cells obtained from BALF collected using each injection buffer volume. (A) Total cell numbers in harvested BALF were determined. (B) Aliquots were then placed on slides and stained with May–Grünwald–Giemsa. (C) Thirty minutes after initiating mAM culture, cell images were acquired by phase contrast microscopy. Black arrows in (B) indicate alveolar epithelial cells. Data shown in (A) are means (± SEM) of four independent experiments.
Characterization of surface antigen levels on mAM isolated using different buffer volumes
Flow cytometry was used to assess potential changes in cell surface antigen expression levels on cells obtained from BALF collected using different injected buffer volumes. The data showed that the main cell population in all isolated BALF was CD11c+SiglecF+CD64+ cells, which corresponded to mAM (). There was a larger CD11b-CD11c-SiglecF- cell population among the cells in BALF collected with 1.0 ml injected buffer relative to levels among all cells in BALF collected using 0.75 ml or 0.55 ml buffer. The percentage of mAM among total BALF cells was also significantly higher in fluid collected using 0.55 and 0.75 ml buffer compared to with 1.0 ml buffer. These results –along with that presented in –suggest to us that these CD11b-CD11c−SiglecF− cells were alveolar epithelial cells.
Figure 3. Characterization of mAM collected using the differing buffer volumes. (A) Cell surface antigen expression on cells analyzed by FACS. Percentage of mAM among total cells is shown as a representative dot-plot. (B) Viability assessed by LDH assay. (C) HMC Class II expression analyzed by FACS. In graphs, each dot indicates result for an individual animal; mean of three independent experiments is indicated by horizontal line. Data shown are means (± SEM) of four independent experiments. Significance of differences between 1.0 ml and other groups assessed by one-way ANOVA and Dunnett’s multiple-comparison test. ***p < 0.001 and *p < 0.05 versus the 1.0 ml group.
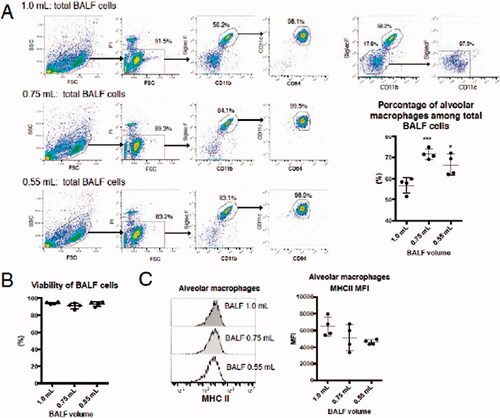
Viability of total BALF cells immediately before seeding was not significantly affected by the volume of buffer fluid used (). Cell surface expression levels of MHC Class II (mouse I-A/I-E) were not significantly affected either (). Though there were cells other than mAM in BALF isolated using 1.0 ml buffer, the viability of mAM and their expression level of cell surface antigens did not differ from those of mAM collected using 0.55 or 0.75 ml buffer. From this it is concluded that the observed decrease IL-1α release in response to particle interactions by mAM isolated using 1.0 ml injected buffer was not caused by a reduction in cell viability at isolation or differences in macrophage subsets.
Viability of cells isolated in different lavaging fluid volumes
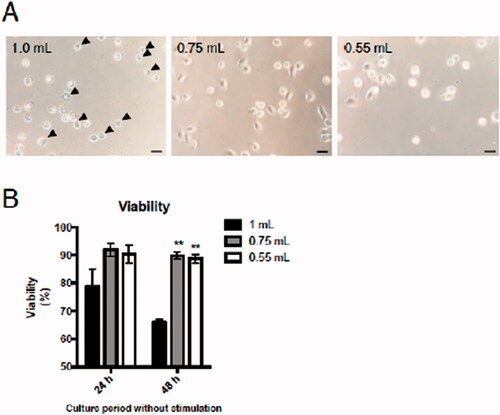
Though there was no change in cell viability immediately after BALF collection (), it was speculated changes in viability during culturing might affect mAM functions in response to ex vivo fine particle stimulation. Thus, background mAM viability and any changes in cell morphology in the absence of stimulation/particle treatment were assessed. The data showed that 24 h after seeding, mAM obtained from BALF collected using 1.0 ml buffer seemed to undergo some shrinkage (). Cell shrinkage is a ubiquitous characteristic of programmed cell death observed in all examples of apoptosis, independent of the type of cell death stimulus (Kerr et al. Citation1972). The data here thus suggest that apoptosis occurred among the mAM obtained using a 1.0 ml buffer volume. Of note, at both 24 and 48 h of culture, the viability of these mAM was reduced (significantly so at 48 h) versus that of cells from BALF collected using the smaller buffer volumes (). Though measures of LDH release cannot distinguish directly between apoptotic and necrotic cell death, the results here may be “partially”reflecting the cell apoptosis we surmise occurred among the mAM obtained using the 1.0 ml buffer volume (). Future studies will need to evaluate markers of necrosis (i.e. by staining with annexin V and 7-amino-actinomycin D (7-AAD), annexin V+/7-AAD+ cell is considered as necrosis (Dagvadorj et al. Citation2015) among the isolated cells to better delineate the type(s) of cell death that appear to have occurred.
Figure 4. Effects of injected buffer volume on mAM viability during culture. Isolated mAM were seeded in uncoated dishes and cell viability assessed. (A) Viability assessed via cell morphology 24 h after seeding and by (B) LDH assay 24 and 48 h after seeding. Data shown in (B) are means (± SEM) of three independent experiments. Significance between 1.0 ml and other groups assessed by one-way ANOVA and Dunnett’s multiple-comparison test. **p < 0.01 versus the 1.0 ml group.
Involvement of particle uptake activity and RIPK3-dependent necroptosis in sensitivity of mAM to particle-induced IL-1α secretion
To investigate potential mechanisms by which sensitivity to fine particle stimulation of mAM obtained from BALF collected using 1.0 ml buffer was decreased, particle uptake activity and necroptosis signals were analyzed. mAM IL-1α release induced by alum was inhibited by cytochalasin D (potent inhibitor of actin polymerization and a general inhibitor of phagocytosis) co-treatment, suggesting particle uptake was crucial (; cell viability and changes in IL-1α release were recovered by cytochalasin D co-treatment). Particles can activate RIPK3 pathways (via lysosome disruption) that then trigger necroptosis. Co-treatment with the RIPK3 selective inhibitor GSK’872 inhibited the IL-1α release/cell death induced by alum (), suggesting mAM IL-1α release was dependent on RIPK3-dependent necroptosis. Taken together, these findings suggested both particle uptake and RIPK3-dependent cell death were critical for ex vivo particle-inducedIL-1α release by the isolated mAM.
Figure 5. Effects of injected buffer volume on ex vivo IL-1α secretion by mAM in the context of changes in particle uptake activity and/or RIPK3-dependent necroptosis. (A) mAM isolated using 0.75 ml buffer stimulated 6 h with 100 µg/ml alum with or without 2 µM cytochalasin D. (B) mAM isolated using 0.75 ml buffer stimulated 6 h with 100 mg/ml alum with or without 5 µM GSK’872. In each case, DMSO was used as a vehicle control for inhibitor. Cell-free supernatants were then measured for IL-1α and cell viability. (C) mAM isolated from BALF collected using 0.75 or 1.0 ml buffer were stimulated 4 h with 100 µg/ml FL-silica, after which the cells were analyzed by FACS to assess silica uptake/cell by fluorescence intensity. As a control, PBS-treated mAM obtained from BALF collected using 1.0 ml buffer and those isolated using 0.75 ml buffer were mixed 1:1 prior to FACS analyses. (D) mAM from BALF collected using 0.75 or 1.0 ml buffer were stimulated 4 h with 100 µg/ml alum. Cells were then lysed and p-RIPK3, total RIPK3, and GAPDH protein levels evaluated by immunoblot. Data shown in (A) and (B) are means (± SEM) of three independent experiments. ***p < 0.001.
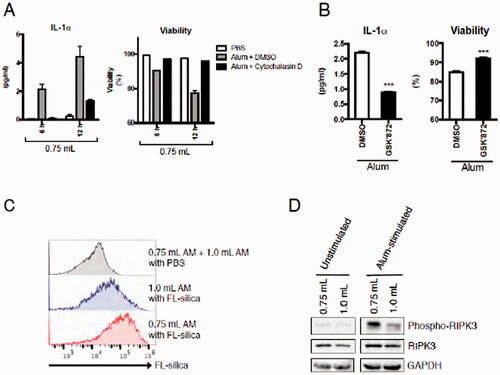
To assess particle uptake activity of mAM from BALF isolated using different buffer volumes, isolated mAM were stimulated 4 h with fluorescent-silica particles and their uptake was assessed by flow cytometry. The results showed that mAM isolated from BALF using 1.0 ml buffer had a lower capacity for silica uptake than mAM from BALF isolated using 0.75 ml fluid (). Immunoblotting performed to verify phosphorylation levels of RIPK3 in unstimulated and alum-stimulated mAM revealed no differences in mAM expression of total or p-RIPK3 in an absence of stimulation (). On the other hand, after alum stimulation, there was a marked increase in p-RIPK3 levels in mAM collected from BALF isolated using 0.75 ml buffer, indicating that there were cells that could release IL-1α in response to alum (). p-RIPK3 levels were markedly lower in mAM obtained from BALF collected using 1.0 ml buffer than mAM from BALF collected using 0.75 ml buffer (). This indicated that though total RIPK3 expression in mAM was not affected by the volume of buffer used to perform BALF collection, levels of p-RIPK3 induced by alum were affected. This latter effect might have impacted particle-inducing necroptosis and IL-1α release by these mAM.
Discussion
While buffer composition used for lavaging the lungs did not affect fine particle-induced IL-1α release in collected mAM, the volume used to harvest BALF did so. Specifically, fine particle-induced IL-1α release levels by mAM obtained from BALF collected using 1.0 ml injected buffer was decreased compared to that by mAM obtained from BALF collected using 0.55 ml or 0.75 ml buffer. Moreover, a decrease in cell viability induced by fine particles was suppressed in mAM obtained from BALF collected using the 1.0 ml volume. However, there was no change in protein expression levels of intracellular IL-1α. Because release of IL-1α induced by fine particles requires their uptake by phagocytosis followed by an RIPK3-dependent necroptosis, each of those endpoints were also examined. It was seen that in mAM obtained from BALF collected using the 1.0 ml buffer volume, both fine particle uptake and particle-induced RIPK3 phosphorylation were lower than in mAM obtained from BALF collected using 0.75 ml buffer. Such results indicates that ex vivo functions of obtained mAM could likely differ depending on the buffer volume used to perform BALF from mice.
There have been numerous reports on efficient collection methods for primary mAM isolation (). These reports mainly focused on increasing the number of obtained cells and cell viability. A few of the reports evaluated cytokine secretion by and viability/purity of the collected mAM; however, mAM functions in response to fine particle stimulation were not assessed. Necroptosis is required for the release of IL-1α (Chen et al. Citation2007), which involves a different mechanism than the release of other cytokines, such as IL-6 and tumor necrosis factor (TNF)-α. Thus, it was important for this study to verify mechanisms of IL-1α secretion distinct from those for other cytokines. Here, it was first clarified that the injected buffer volume used to perform BALF collection during the mAM isolation procedure affected those cells ex vivo IL-1α release induced by fine particles. Mechanistically, particle uptake capacity and RIPK3 pathways were affected by the volume of buffer used to perform the BALF collection. It was revealed that mAM obtained from BALF collected in >1.0 ml buffer did not release IL-1α in response to fine particle stimulation; this has methodological implications. This outcome suggested that mAM obtained from BALF collected in ≥1.0 ml buffer could not be used to correctly assess in vivo functions of mAM, especially in response to fine particles. Many studies have assessed effects of different lavage fluid compositions (presence or absence of FBS, EDTA, etc.), fluid temperatures, and number of flushes on mAM isolation; however, these studies did not examine effects of the volume of injected buffer to perform BALF collection. It can also be inferred from the reported data that IL-6 and TNFα formation by mAM is maintained regardless of buffer volume used to perform BALF collection. This study is the first to reveal that the volume of buffer used to perform BALF collection in mAM isolation procedures could adversely impact on the ex vivo functionality of these cells, particularly in response to fine particles.
Among the causes for differences in IL-1α release by isolated mAM are mechanical or osmotic stresses (Denkert et al. Citation1998; Aikawa et al. Citation2002; Shiratsuchi and Basson Citation2005) that occur during the lung-flushing process. However, the results of the present study showed that there were no differences in cell viability immediately after BALF isolation regardless of the volume of buffer injected for the collection. In addition, the purity of mAM obtained from BALF collected using the 1.0 ml buffer volume tended to be low, an outcome thought to be due to significant contamination in the wash by alveolar epithelial cells. However, as these cells cannot attach to uncoated dishes and are removed during washing steps prior to cell stimulation, this contamination effect likely had little impact on any changes in IL-1α release induced by the fine particles.
To better understand if use of the 1.0 ml buffer to isolate BALF caused significant physical stress to the mAM, cultures of the cells were evaluated after 24 and 48 incubations at 37 °C. Interestingly, only mAM collected using 1.0 ml buffer showed a decrease in cell viability under unstimulated conditions. On the other hand, analyses of cell surface antigen levels showed no differences among the mAM collected using the differing buffer volumes. Taken together, these results suggested that use of the different buffer volumes to isolate mAM did not cause any shift in macrophage subsets that could be isolated, but instead that is might be a loss of some mAM functions/viability after culturing that ultimately contribute to differences in the ability of the isolated cells to respond to the fine particles and secrete IL-1α.
A previous study using J774.1 cells and bone marrow-derived macrophages (BMDM) showed that cyclic strength force reduces IL-1β release via the caspase-1 cascade (Maruyama et al. Citation2019). IL-1α can be cleaved by caspase-1; however, mechanisms of IL-1β release differ from that of IL-1α as necroptosis is the rate-determining process for IL-1α release (Chen et al. Citation2007). The present study showed RIPK3 phosphorylation induced by fine particle stimulation was decreased in mAM from BALF collected using 1.0 ml buffer. It was reported that RIPK3 cascade activation by fine particles is triggered by lysosomal disruption induced by the incorporated fine particles (Nakayama Citation2018). Lysosomes are formed by fusion of phagosomes that develop when extracellular substances are taken up by phagocytosis. However, fine particles cannot be digested in lysosomes and so accumulate there, eventually causing lysosomal disruption (Hornung et al. Citation2008) and then RIPK3 cascade activation (Nakayama Citation2018). The present study showed that a more marked decrease in silica uptake levels was seen with mAM from BALF isolated using the 1.0 ml buffer volume than in mAM collected in a lower volume. In addition, this study showed that mAM co-treatment with cytochalasin D suppressed alum-induced IL-1α release Based on these results, mAM isolated from BALF harvested using 1.0 ml buffer had reduced amounts of fine particles taken up, thus making it difficult for p-RIPK3 expression to reach a level sufficient to trigger necroptosis, i.e. and so no induced IL-1α release. That the same results for these mAM were seen even if the cells were incubated for 18 or 24 h (data not shown) suggested that stimulation time did not affect IL-1α release in the case of cells obtained from BALF collected using the 1.0 ml buffer conditions.
It was also seen here that though buffer volume did not affect collected cell number, mAM purity among all isolated cells was higher in BALF isolated using smaller buffer volumes. Thus, if one were conducting fine particle stimulation assays, it would be desirable to collect lung cells using buffer volumes of <0.75 ml. Further, as total lung capacity and functional residual capacity in mice are 1.05 [± 0.04] and 0.25 [± 0.01] ml, respectively (Lai and Chou Citation2000), infusion of 1.0 ml buffer during BALF harvest would likely place a physical stress on alveoli. Since these stresses deleteriously affect macrophages (Denkert et al. Citation1998; Aikawa et al. Citation2002; Shiratsuchi and Basson Citation2005; Maruyama et al. Citation2019), the total volume of fluid used for lung lavage should be considered a key factor in subsequent evaluations of ex vivo mAM functions.
The present study does have some important limitations to acknowledge. Primarily, it only focused on the impact of buffer volume during AM isolation on subsequent ex vivo fine particle-induced IL-1α release and particle uptake by the isolated cells. Examination of effects on a broader range of ex vivo mAM functions are clearly necessary, i.e. responses to pathogens, cytokine secretion, a wider array of surface marker expression, etc. to better clarify how this single experimental factor might be impacting on data interpretation in ex vivo particle exposure studies. Furthermore, analyzing effects of lavage buffer volume used during isolation of AM from rats, another important animal model used in particle-induced toxicity studies, is also important. In the end, the rat and mouse data that would be obtained would allow investigators to better design protocols to harvest lung AM for use in ex vivo studies of particle toxicity going forward.
Conclusions
The study here is the first to show that the volume of buffer used to lavage mouse lungs in mAM isolation procedures could subsequently impact on ex vivo particle uptake, as well as normal and particle-induced activity by the cells. Data here suggest that the latter may be due, in part, to effects on the process of necroptosis (secondary to changes in phosphorylation of RIPK3). On the other hand, the ex vivo ability of the isolated mAM to secrete select cytokines that do not require necroptosis (like IL-6) did not appear to have been affected by buffer volume. Thus, effects of buffer volume used in mAM isolation procedures, though important, would seem to be limited. In the end, the results of this study reinforce the import of optimizing the buffer volume used during mAM isolation as it clearly could impact on select mAM functions and thus the data obtained in the ex vivo cell response to particles.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The authors confirm the data supporting the findings here are available within the article.
Additional information
Funding
References
- Aikawa R, Nagai T, Kudoh S, Zou Y, Tanaka M, Tamura M, Akazawa H, Takano H, Nagai R, Komuro I. 2002. Integrins play a critical role in mechanical stress-induced p38 MAPK activation. Hypertension. 39:233.
- Busch C, Favret J, Geirsdóttir L, Molawi K, Sieweke M. 2019. Isolation and long-term cultivation of mouse alveolar macrophages. Bio Protoc. 9:e3302.
- Chavez-Santoscoy A, Huntimer L, Ramer-Tait A, Wannemuehler M, Narasimhan B. 2012. Harvesting murine alveolar macrophages and evaluating cellular activation induced by polyanhydride nanoparticles. J Vis Exp. 64:e3883.
- Chen C, Kono H, Golenbock D, Reed G, Akira S, Rock K. 2007. Identification of a key pathway required for the sterile inflammatory response triggered by dying cells. Nat Med. 13:851–856.
- Chen G, Nunez G. 2010. Sterile inflammation: sensing and reacting to damage. Nat Rev Immunol. 10:826–837.
- Corti M, Brody A, Harrison J. 1996. Isolation and primary culture of murine alveolar Type II cells. Am J Respir Cell Mol Biol. 14:309–315.
- Dagvadorj J, Shimada K, Chen S, Jones H, Tumurkhuu G, Zhang W, Wawrowsky K, Crother T, Arditi M. 2015. Lipopolysaccharide induces alveolar macrophage necrosis via CD14 and the P2X7 receptor leading to IL-1α release. Immunity. 42:640–653.
- Davies L, Taylor P. 2015. Tissue-resident macrophages: Then and now. Immunology. 144:541–548.
- Denkert C, Warskulat U, Hensel F, Haussinger D. 1998. Osmolyte strategy in human monocytes and macrophages: Involvement of p38MAPK in hyperosmotic induction of betaine and myoinositol transporters. Arch Biochem Biophys. 354:172.
- Dinarello C. 2011. Interleukin-1 in the pathogenesis and treatment of inflammatory diseases. Blood. 117:3720–3732.
- Dong Y, Poon G, Arif A, Lee-Sayer S, Dosanjh M, Johnson P. 2018. The survival of fetal and bone marrow monocyte-derived alveolar macrophages is promoted by CD44 and its interaction with hyaluronan. Mucosal Immunol. 11:601–614.
- Fan E, Fan J. 2018. Regulation of alveolar macrophage death in acute lung inflammation. Respir Res. 19:50.
- Gordon S, Read R. 2002. Macrophage defenses against respiratory tract infections. Br Med Bull. 61:45–61.
- Hamilton R, Thakur S, Mayfair J, Holian A. 2006. MARCO mediates silica uptake and toxicity in alveolar macrophages from C57BL/6 mice. J Biol Chem. 281:34218–34226.
- Happle C, Lachmann N, Skuljec J, Wetzke M, Ackermann M, Brennig S, Mucci A, Jirmo A, Groos S, Mirenska A, et al. 2014. Pulmonary transplantation of macrophage progenitors as effective and long-lasting therapy for hereditary pulmonary alveolar proteinosis. Sci Transl Med. 6:250.
- Hetland R, Cassee F, Låg M, Refsnes M, Dybing E, Schwarze P. 2005. Cytokine release from alveolar macrophages exposed to ambient particulate matter: Heterogeneity in relation to size, city and season. Part Fibre Toxicol. 2:4.
- Higham A, Lea S, Ray D, Singh D. 2014. Corticosteroid effects on COPD alveolar macrophages: Dependency on cell culture methodology. J Immunol Methods. 405:144–153.
- Hodge S, Hodge G, Ahern J, Jersmann H, Holmes M, Reynolds P. 2007. Smoking alters alveolar macrophage recognition and phagocytic ability: Implications in chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol. 37:748–755.
- Hornby S, Kellington J. 1990. DNA synthesis in alveolar macrophages and other changes in lavaged cells following exposure of CBA/H mice to cigarette smoke. Environ Health Perspect. 85:107–112.
- Hornung V, Bauernfeind F, Halle A, Samstad E, Kono H, Rock K, Fitzgerald K, Latz E. 2008. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol. 9:847–856.
- Imperatore F, Maurizio J, Vargas Aguilar S, Busch C, Favret J, Kowenz-Leutz E, Cathou W, Gentek R, Perrin P, Leutz A, et al. 2017. SIRT1 regulates macrophage self-renewal. Embo J. 36:2353–2372.
- Kerr J, Wyllie A, Currie A. 1972. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 26:239–257.
- Kopf M, Schneider C, Nobs S. 2016. Development and function of lung-resident macrophages and dendritic cells. Nat Immunol. 16:36–44.
- Kuroda E, Ozasa K, Temizoz B, Ohata K, Koo C, Kanuma T, Kusakabe T, Kobari S, Horie M, Morimoto Y, et al. 2016. Inhaled fine particles induce alveolar macrophage death and IL-1α release to promote inducible bronchus-associated lymphoid tissue formation. Immunity. 45:1299–1310.
- Lai Y, Chou H. 2000. Respiratory mechanics and maximal expiratory flow in anesthetized mouse. J Appl Physiol. 88:939–943.
- Machiels B, Dourcy M, Xiao X, Javaux J, Mesnil C, Sabatel C, Desmecht D, Lallemand F, Martinive P, Hammad H, et al. 2017. A gamma-herpesvirus provides protection against allergic asthma by inducing the replacement of resident alveolar macrophages with regulatory monocytes. Nat Immunol. 18:1310–1320.
- Maruyama K, Nemoto E, Yamada S. 2019. Mechanical regulation of macrophage function-cyclic tensile force inhibits NLRP3 inflammasome-dependent IL-1β secretion in murine macrophages. Inflamm Regen. 39:3.
- Mbawuike I, Herscowitz H. 1989. MH-S, a murine alveolar macrophage cell line: Morphological, cytochemical, and functional characteristics. J Leukoc Biol. 46:119–127.
- Momota M, Lelliott P, Kubo A, Kusakabe T, Kobiyama K, Kuroda E, Imai Y, Akira S, Coban C, Ishii K. 2020. ZBP1 governs inflammasome-independent IL-1α and neutrophil inflammation that play a dual role in anti-influenza virus immunity. Intl Immunol. 32:203–212.
- Murray P, Wynn T. 2011. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol. 11:723.
- Nakayama M. 2018. Macrophage recognition of crystals and nanoparticles. Front Immunol. 9:103.
- Qian G, Jiang W, Zou B, Feng J, Cheng X, Gu J, Chu T, Niu C, He R, Chu Y, et al. 2018. LPS inactivation by a host lipase allows lung epithelial cell sensitization for allergic asthma. J Exp Med. 215:2397–2412.
- Ralph P, Nakoinz I. 1975. Phagocytosis and cytolysis by a macrophage tumor and its cloned cell line. Nature. 257:393–394.
- Raschke W, Baird S, Ralph P, Nakoinz I. 1978. Functional macrophage cell lines transformed by Abelson leukemia virus. Cell. 15:261–267.
- Schneider C, Nobs S, Heer A, Kurrer M, Klinke G, van Rooijen N, Vogel J, Kopf M. 2014. Alveolar macrophages are essential for protection from respiratory failure and associated morbidity following influenza virus infection. PLoS Pathog. 10:e1004053.
- Schneider C, Nobs S, Kurrer M, Rehrauer H, ThieleC, Kopf M. 2014. Induction of nuclear receptor PPARγ by cytokine GM-CSF is critical for the differentiation of fetal monocytes into alveolar macrophages. Nat Immunol. 15:1026–1037.
- Shiratsuchi H, Basson M. 2005. Activation of p38 MAPKα by extracellular pressure mediates stimulation of macrophage phagocytosis by pressure. Am J Physiol. 288:C1083–C1093.
- Soucie E, Weng Z, Geirsdóttir L, Molawi K, Maurizio J, Fenouil R, Mossadegh-Keller N, Gimenez G, van Hille L, Beniazza M, et al. 2016. Lineage-specific enhancers activate self-renewal genes in macrophages and embryonic stem cells. Science. 351:aad5510.
- Steele C, Marrero L, Swain S, Harmsen A, Zheng M, Brown G, Gordon S, Shellito J, Kolls J. 2003. Alveolar macrophage-mediated killing of Pneumocystis carinii f. sp. muris involves molecular recognition by the dectin-1 beta-glucan receptor. J Exp Med. 198:1677–1688.
- Sun K, Metzger D. 2008. Inhibition of pulmonary antibacterial defense by IFNγ during recovery from influenza infection. Nat Med. 14:558–564.
- Suzuki T, Arumugam P, Sakagami T, Lachmann N, Chalk C, Sallese A, Abe S, Trapnell C, Carey B, Moritz T, et al. 2014. Pulmonary macrophage transplantation therapy. Nature. 514:450–454.
- Ueno M, Maeno T, Nishimura S, Ogata F, Masubuchi H, Hara K, Yamaguchi K, Aoki F, Suga T, Nagai R, et al. 2015. Alendronate inhalation ameliorates elastase-induced pulmonary emphysema in mice by induction of apoptosis of alveolar macrophages. Nat Commun. 6:6332.
- Ulich T, Yin S, Guo K, del Castillo J, Eisenberg S, Thompson R. 1991a. The intratracheal administration of endotoxin and cytokines. III. The interleukin-1 (IL-1) receptor antagonist inhibits endotoxin- and IL-1-induced acute inflammation. Am J Pathol. 138:521–524.
- Ulich T, Yin S, Guo K, Yi E, Remick D, del Castillo J. 1991b. Intratracheal injection of endotoxin and cytokines. II. Interleukin-6 and transforming growth factor-β inhibit acute inflammation. Am J Pathol. 138:1097–1101.
- van de Laar L, Saelens W, de Prijck S, Martens L, Scott C, van Isterdael G, Hoffmann E, Beyaert R, Saeys Y, Lambrecht B, et al. 2016. Yolk sac macrophages, fetal liver, and adult monocytes can colonize an empty niche and develop into functional tissue-resident macrophages. Immunity. 44(4):755–768.
- Yu X, Buttgereit A, Lelios I, Utz S, Cansever D, Becher B, Greter M. 2017. The cytokine TGFβ promotes development and homeostasis of alveolar macrophages. Immunity. 47:903–912.
- Yuan X, Shan M, You R, Frazier M, Hong M, Wetsel R, Drouin S, Seryshev A, Song L, Cornwell L, et al. 2015. Activation of C3a receptor is required in cigarette smoke-mediated emphysema. Mucosal Immunol. 8:874–885.
- Zhang X, Goncalves R, Mosser D. 2008. Chapter 14. The isolation and characterization of murine macrophages. Curr Protocols Immunol. 83:1–14.