
Abstract
While the detailed mechanisms for how particulate matter (PM) causes adverse health effects in the lungs remain largely unknown, endoplasmic reticulum (ER) stress has been implicated in PM-induced lung injury. The present study was undertaken to examine how/if ER stress might regulate PM-induced inflammation, and to begin to define potential underlying molecular mechanisms. Here, ER stress hallmarks were examined in human bronchial epithelial (HBE) cells exposed to PM. To confirm roles of certain pathways, siRNA targeting ER stress genes and an ER stress inhibitor were employed. Expression of select inflammatory cytokines and related signaling pathway components by the cells were assessed as well. The results showed that PM exposure induced elevations in two ER stress hallmarks, i.e. GRP78 and IRE1α, in time-and/or dose-related manners in the HBE cells. Inhibition of ER stress by siRNA for GRP78 or IRE1α significantly alleviated the PM-induced effects. Further, ER stress appeared to regulate PM-induced inflammation – likely through downstream autophagy and NF-κB pathways – as implied by studies showing that inhibition of ER stress by siRNA of GRP78 or IRE1α caused significant amelioration of PM-induced autophagy and subsequent activation of NF-κB pathways. Moreover, the ER stress inhibitor 4-PBA were used to confirm the protective effects against PM-induced outcomes. Together, the results suggest ER stress plays a deleterious role in PM-induced airway inflammation, possibly through activation of autophagy and NF-κB signaling. Accordingly, protocols/treatments that could lead to inhibited ER stress could potentially be effective for treatment of PM-related airway disorders.
Introduction
It is well known that airborne pollutants, specifically particulate matter (PM), pose a risk to human health (Dockery et al. Citation1993). Because the lung is the initial target for effects from inhaled pollutants, it follows that epidemiological data has shown that PM exposures are associated with the increases in morbidity/mortality from cardiopulmonary diseases (Gehring et al. Citation2010; Guan et al. Citation2016; Traboulsi et al. Citation2017). Among the arrays of pathologies induced in the lungs, there is evidence to show that exposures to environmental PM are also linked to increases in impaired lung function, airway hyperresponsiveness, and granulocyte inflammation (Gehring et al. Citation2010; Karakatsani et al. Citation2012; Guan et al. Citation2016). At the more macro level, both short- and long-term PM exposures have been found to be involved in the pathogenesis and exacerbation of several respiratory diseases, including chronic obstructive pulmonary disease and asthma (Ebelt et al. Citation2000; Gehring et al. Citation2010; Jacquemin et al. Citation2015). As such, developing novel therapeutics that could alleviate PM-induced adverse effects remain urgent.
Airway epithelial cells serve as the first line of airway defense and also act as effector cells that can respond to endogenous and exogenous stimuli. Inhaled particles that deposit in the respiratory tract can interact with pulmonary epithelial cells and potentially cause disruption of the airway epithelial barrier, as well as increases in local oxidative stress and inflammation (Duarte et al. Citation2012; Byrd et al. Citation2016; Guan et al. Citation2016). To date, precise mechanisms regarding how PM mediates this damage to the airway epithelium remain poorly defined.
The endoplasmic reticulum (ER), which consist of membrane-bound flattened sacs, is an important organelle that serves multiple functions in cells, especially in the biosynthesis, folding, and post-translational modifications of both secretory and membrane-bound proteins. The pathology of ER stress has been found to occur in several respiratory diseases, including chronic obstructive pulmonary disease (COPD), asthma, and acute lung injury (Kim et al. Citation2013; Zeng et al. Citation2017; Wang et al. Citation2018). In the context of pollutant exposures, accumulated evidence has demonstrated that exposures to airborne PM can cause ER stress in the lungs (Wang and Tang Citation2020; Ding et al. Citation2021). At the cellular level, ER stress occurs in states wherein unfolded/mis-folded proteins accumulate in the ER lumen, and subsequently induce unfolded protein responses mediated by three ER signaling proteins, i.e. inositol requiring protein 1 (IRE1), activating transcription factor-6 (ATF6), and protein kinase RNA (PKR)-like ER kinase (Davenport et al. Citation2008). Glucose-regulated protein 78 (GRP78), a prominent ER chaperone, is generally also activated and its levels elevated in these processes. At this point, it is not clear if any of these signaling proteins are impacted by PM exposure, much less so if these changes occur at the level of the airway epithelium.
The current study was undertaken to explore the potential roles of ER stress in the regulation of PM-induced inflammatory responses in airway epithelium. It was also hoped that some underlying mechanisms for any PM-induced effects could be elucidated.
Materials and methods
PM preparation
PM (Standard Reference Material [SRM] 1649b-Urban Dust) was purchased from the National Institute of Standards and Technology (NIST, Gaithersburg, MD). The particle size distribution is shown in Supplemental Figure 1, and the estimated volume median diameter of the particle was ≈ 2.5–10 µm based on the distribution plot. For use in all the experiments here, fresh solutions of PM were generated by suspending the particles in sterile saline (to 2 mg/mL final concentration) and heavily sonicating the material prior to its use in the assays below.
Chemicals and reagents
4-phenylbutyric acid (4-PBA, inhibitor of ER stress) was purchased from Selleck Corp. (Houston, TX) and freshly-suspended in phosphate-buffered saline (PBS, pH 7.4) to create a stock solution for each set of experiments. Primary antibodies against human GRP78, IRE1α, and β-actin were obtained from Cell Signaling Technology (Danvers, MA), while those against human LC3B and p-p65 were purchased from AB Clonal Technology (Wuhan, China). Secondary horseradish peroxidase (HRP)-conjugated anti-rabbit/anti-mouse IgG antibodies were also purchased from AB Clonal Technology.
HBE cells and treatments
The human bronchial epithelial cells (HBE, CRL-2741) used here were purchased from American Type Culture Collection (ATCC, Manassas, VA). Throughout, the HBE cells were cultured in RPMI 1640 containing 10% heat-inactivated fetal bovine serum (FBS) and both antibiotic/antimycotic supplements (all from Gibco, Grand Island, NY). Cells were maintained in T-25 flasks at 37°C in a humidified 5% CO2 incubator. Cells were passaged when they reached a confluence of ≈ 80–90% and grown to near-confluence prior to harvest for use in the experiments.
To provide baseline information about effects of the PM on the cells, cells were seeded at 1.5 × 105 cells/well (1 ml/well) in 6-well plates and then exposed to various concentrations of PM (0, 25, 50, 75, 100, 150 µg/mL) for fixed times (0, 1, 2, 4, 6, 8 hr) or exposed to a fixed level of PM (i.e. 100 µg PM/ml) for various times (0, 2, 4, 8, 12 hr). From measures of p-p65 and LC3B protein expression or of interleukin IL-6 and IL-8 protein and mRNA expressions at each timepoint (and at each time in the context of specific level of PM), the optimal PM dosing regimen for each endpoint to be measured was obtained. Similarly, to ascertain what would be the optimal potential concentration of 4-PBA to be used in these in vitro studies, dosing regimens of 100 µg PM/ml (optimal level based on aforementioned study outcomes) + fixed concentrations of 4-PBA (0, 0.5, 1, 1.5, 2 mM) were tested for fixed periods (times specific to each measured outcome).
Transfection experiments for blocking PM-induced effects in HBE cells
The HBE cells were cultured in 6-well plates at 37°C. At ≈ 35–45% confluence, the cells were transfected with 5 nmol of siRNA duplexes for GRP78, IRE1α, LC3B, or p65, as well as control nonspecific siRNA; each siRNA duplex was designed and synthesized by Tsingke Biotechnology (Nanjing, China). Each siRNA was transfected into cells using GenMuteTM siRNA Transfection Reagent (SignaGen Labs, Frederick, MD), following manufacturer protocols. After transfection for 24 hr, the cells were treated with 100 µg PM/ml (1 ml/well) for 2 hr to permit measures of p-p65 and LC3B protein expression or for 8 hr to permit measures of IL-6 and IL-8 protein and mRNA expressions.
Testing of 4-PBA blocking action against PM-induced effects in HBE cells
Normal (i.e. non-transfected) HBE cells were seeded as above into 6-well plates and treated with 100 µg PM/mL (or vehicle) for 6 hr only to permit inducibility of GRP78 and IRE1α expression in the stressed cells. Concurrently, other sets of cells were treated with 100 µg PM/ml, 4-PBA (2 mM), PM + 4-PBA (2 mM), or vehicle for 8 hr and then evaluated for IL-6 and IL-8 mRNA expressions. The choice to use only 2 mM 4-PBA in the mRNA studies was based on the outcomes from the above-outlined range-finding studies. In all cases, at the end of a given exposure, the culture supernatant from each well was isolated for use in ELISA measures of IL-6 and IL-8 release by the cells using commercial kits (R&D Systems, Minneapolis, MN). The level of sensitivity of the kits was 0.7 pg IL-6/ml and 7.5 pg IL-8/ml.
The remaining cells in each well were washed with PBS, collected by gentle scraping, and then prepared for use in RNA analytical studies of the expression of IL-6 and IL-8 mRNA (see below) and for use in Western blot assays of expressions of GRP78, IRE1α, LC3B, and p-p65 (see below).
RNA isolation and quantitative real-time polymerase chain reaction (qRT-PCR)
After recovery from the test wells, cells were counted and then total RNA from 106 HBE cells in each regimen was isolated using Trizol (Takara Bio, Beijing, China), following manufac-turer instructions. The concentration and purity of the total RNA isolated each time from was confirmed using an Eppendorf Bio Photometer (Hamburg, Germany). From each sample, 0.5 µg RNA was then reverse-transcribed using HiFair® III 1st Strand cDNA Synthesis Super Mix for qPCR (Yeasen Biotechnology, Shanghai, China). Subsequently, qRT-PCR was carried out using a SYBR Green system (Yeasen Biotechnology) to quantify cell expression of IL-6 and IL-8 mRNA. The primers used for this study, as well as to measure β-actin (ACTB) gene expression, are indicated in . In the end, IL-6 and IL-8 mRNA expression was normalized to that of ACTB and all outcomes were expressed in terms of relative units using the 2−ΔΔCt method of Livak and Schmittgen (Citation2001).
Table 1. Primer sequences used for the qRT-PCR assays here.
Western blotting
Total protein from 106 HBE cells/regimen was extracted by suspending the cells in RIPA lysis buffer (containing PMSF protease inhibitor-phosphatase inhibitor cocktail; all Beyotime, Shanghai) and placing them on ice for 20 min. After centrifugation to pellet cell debris, the resulting supernatant was collected and total protein concentration evaluated with a BCA assay kit (Beyotime). Thereafter, an aliquot containing 20 µg protein was removed and placed into the well of a 10% SDS-PAGE gel, and all proteins present resolved. The materials were then electro-transferred to a PVDF membrane that, in turn, was blocked for 1 hr at room temperature in a solution of TBST (Tris-buffered saline [pH 7.4] containing 0.05% [v/v] Tween-20) supple-mented with 5% nonfat milk powder. Dedicated membranes were generated for each primary antibody to avoid issues of antigen loss that are associated with repeated stripping-probing of a single membrane.
Each membrane was placed in TBST containing a primary rabbit antibody against GRP78 or IRE1α, and primary mouse antibodies against LC3B or p-p65 (all at 1:1000 manufacturer-recommended dilution) and incubated overnight at 4 °C with gentle rocking. Each solution also contained antibody against β-actin (1:10000) to monitor for membrane loading. After gentle rinsing with TBST, each membrane was placed in a solution of TBST containing horse-radish peroxidase (HRP)-conjugated anti-rabbit/anti-mouse IgG antibody and incubated at room temperature for 2 hr. After a final TBST rinsing, enhanced chemiluminescence reagent (ECL, Shanghai Epizyme Biomedical Technology, Shanghai) was applied, and band detection/intensity measurements were performed in a Tanon 5200 Chemiluminescent Imaging System (Tanon, Shanghai). All data were reported in terms of band density after normalization to level of β-actin.
Statistical analysis
All data are presented as means ± SD. Differences between experimental and control groups were evaluated using an unpaired two-tailed Student’s t-test. When multiple comparisons were required, a one-way analysis of variance (ANOVA) was employed. A p-value < 0.05 was considered significant. All statistical analyses were performed using Prism software (v.7.0; GraphPad, San Diego, CA).
Results
PM exposure induces ER stress and inflammation-related responses in HBE cells
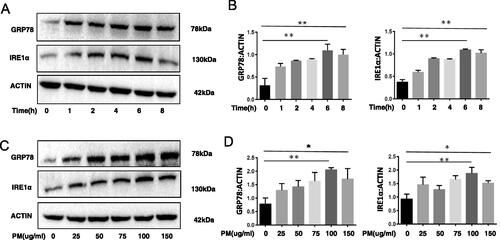
To determine whether ER stress and inflammation-related responses was activated in HBE cells due to PM exposure, HBE cells were treated with PM, and levels of ER stress-related molecules GRP78 and IRE1α and IL-6 and IL-8 cytokines then measured. It was found that PM exposure significantly increased cell expression of GRP78 and IRE1α proteins in time- () and dose- () related manners, moreover, the IL-6 and IL-8 cytokines level were increases after exposure to PM (Supplemental Figure 2(A,B)). The data clearly suggests in vitro PM exposure induces ER stress and inflammation-related responses in HBE cells.
Figure 1. PM exposure and ER stress in HBE cells. HBE cells were treated with 100 µg PM/ml for various lengths of time or with various concentrations of PM for 6 hr. Expression of GRP78 and IRE1α were then evaluated using Western blot analysis (A–D). Relative expression of GRP78 and IRE1α after normalization to lysate β-actin levels. *p < 0.05; **p < 0.01.
Inhibition of ER stress alleviates PM-induced inflammation-related responses in HBE cells
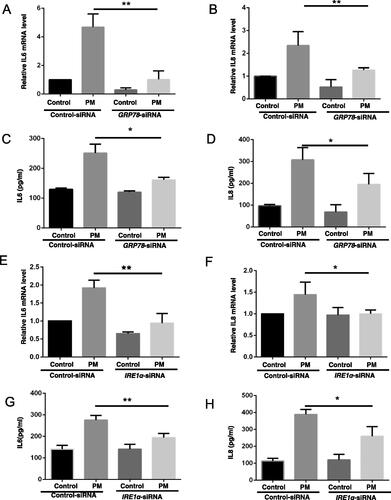
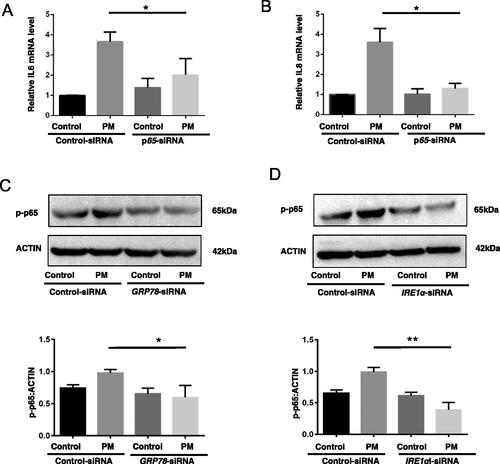
As IL-6 and IL-8 cytokines play an important part in the pathogenesis of PM-induced airway injury (see Tao and Kobzik [Citation2002]), the impact of PM on expression of these two proteins by the HBE cells was investigated. The data showed PM caused increases in the levels of both IL-6 and IL-8 mRNA transcripts and protein in the cells, and that these effects from the PM were significantly alleviated by use of the GRP78-siRNA (). Similar outcomes were observed in cells treated with IRE1α-siRNA (). Control siRNA had no ameliorating effect against PM-induced outcomes.
Figure 2. Inhibition of ER stress impact on PM-induced inflammation-related responses in HBE cells. HBE cells were transfected with indicated siRNA for 24 hr, and then treated with 100 µg PM/ml for additional 8 hr. Relative levels of IL-6 (A and E) and IL-8 (B and F) mRNA were then evaluated (qRT-PCR). IL-6 (C and G) and IL-8 (D and H) protein levels were measured in culture supernatants (ELISA). *p < 0.05; **p < 0.01.
4-PBA decreases PM-induced HBE cell inflammation-related responses
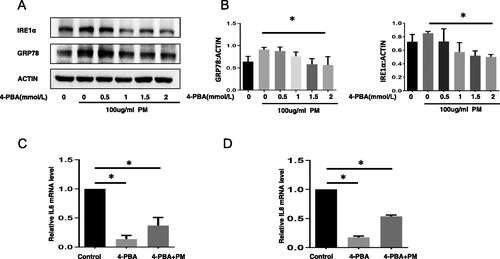
We found other study shows that using 2 mM 4-PBA can significantly alleviate the harmful effect by ER stress (such as Wang et al. [Citation2021]), so 2 mM 4-PBA dose was used in the subsequent in vitro protocols here. The results of studies to assess potential protective effects of 4-PBA in vitro indicated that while PM exposure alone caused increases in HBE cell levels of GRP78 and IRE1α protein, this change could be blocked/mitigated by use of 4-PBA (). Further, use of 4-PBA also abrogated most of the expected effects from PM on cell mRNA levels of IL-6 and IL-8 ().
Figure 3. ER stress inhibitor 4-PBA impact on PM-induced airway inflammation-related outcomes in HBE cells. After treatment with various concentrations of 4-PBA for 1 hr, HBE cells were exposed to 100 µg PM/ml for an additional 6 hr, expression of GRP78 and IRE1α were then evaluated by Western blot analysis (A and B). After the HBE cells were treated with 4-PBA (2 mM), PM + 4-PBA (2 mM), or vehicle for 8 hr, IL-6 (C) and IL-8 (D) mRNA expressions were measured (qRT-PCR).
ER stress impacts PM-induced inflammation-related responses in HBE cells via autophagy
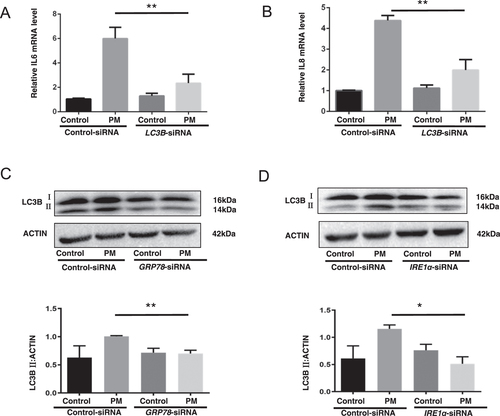
Since PM exposure induces autophagy in lung epithelial cells and autophagy is essential to PM-induced inflammatory responses in lung epithelium (Deng et al. Citation2014; Chen et al. Citation2016; Xu et al. Citation2017), levels of autophagy (and corresponding cytokine formation) were evaluated in the treated HBE cells. Consistent with earlier data, it was seen that inhibition of autophagy by LC3B-siRNA significantly reduced the mRNA levels of IL-6 and IL-8 that could be induced by PM (). In ascertaining the function, if any, of ER stress in the regulation of autophagy in these cells, it was found that inhibition of ER stress by use of GRP78 or IRE1α-siRNA led to marked decreases in levels of the LC3B autophagy hallmark that could be induced by the PM (). The totality of these outcomes suggested that ER stress may up-regulate PM-induced inflammatory responses in the lungs through autophagy.
Figure 4. ER stress regulation of PM-induced inflammation-related responses in HBE cells via autophagy. Control-siRNA or LC3B-siRNA-transfected HBE cells were treated with or without 100 µg PM/mL for 8 hr, and then IL-6 (A) and IL-8 (B) mRNA levels were evaluated (qRT-PCR). Control-siRNA or GRP78-siRNA or IRE1α-siRNA-transfected HBE cells were treated with or without 100 µg PM/ml for 2 hr, and then protein levels of LC3B in the cells were evaluated using Western blot analysis (C and D). *p < 0.05; **p < 0.01.
ER stress regulates PM-induced HBE inflammation-related responses via NF-κB activation
As earlier studies showed that inhibition of autophagy suppressed PM-induced IL-6 and IL-8 expression partly by dysregulation of NF-κB pathways (see Chen et al. [Citation2016] and Xu et al. [Citation2017]), HBE cells were evaluated for IL-6 and IL-8 expression in the context of co-expressed p65 (actually p-p65). It was observed that PM-induced IL-6 and IL-8 mRNA expressions were increased by PM exposure and this was significantly reduced by use of p65-siRNA (). Control siRNA had no impact. With regard to whether ER stress might be regulating PM-induced inflammatory responses in the cells through NF-κB pathways, post-exposure p65/p-p65 levels were evaluated. As expected, an inhibition of ER stress by use of GRP78- or IRE1α-siRNA significantly (almost completely) suppressed any PM-induced NF-κB pathway activation as reflected by a lack of increase in p-p65 levels in the cells (). As such, it may seem that ER stress along with downstream molecules LC3B up-regulate PM-induced inflamma-tory events in lung epithelial cells in part via activation of NF-κB pathways.
Figure 5. ER stress as regulator of PM-induced inflammation-related events in HBE cells through NF-κB pathway activation. Control-siRNA or p65-siRNA-transfected HBE cells were treated with or without 100 µg PM/ml for 8 hr and then IL-6 (A) and IL-8 (B) mRNA expression was measured (qRT-PCR). Control-siRNA or GRP78-siRNA or IRE1α-siRNA-transfected HBE cells were treated with or without 100 µg PM/mL for 2 hr and then expression of p-p65 was evaluated by Western blotting (C and D). Blots representative of three independent experiments. *p < 0.05; **p < 0.01.
Discussion
In the study here, it was seen that PM exposure could induce ER stress in HBE airway epithelial cells, and that this one step might be essential for the development of inflammation in situ. This view was supported by the findings that inhibition of ER stress dramatically alleviated PM-induced ‘inflammatory’ responses in vitro. Examination of potential mechanisms for how ER stress regulated PM-induced inflammatory events indicated that this was, in part, via effects on downstream autophagy and NF-κB pathways.
ER stress seems to be involved in several different pathologies, including colitis, obesity-associated chronic systemic inflammation, and nephrotic syndrome (Brooks-Worrell and Palmer Citation2019; Park et al. Citation2019). Studies have also shown that ER stress is induced during episodes of bronchial asthma, acute lung injury, and chronic obstructive pulmonary disease (Kim et al. Citation2013). Other studies have yielded data that indicated that inhibition of ER stress by 4-PBA (an inhibitor of ER stress) could significantly attenuate inflammatory responses in asthma or acute lung injury mice model (Kim et al. Citation2013; Zeng et al. Citation2017). In agreement with these results, it was observed here that PM exposure elicited ER stress in airway epithelial cells and that inhibition of such stress markedly alleviated any PM-induced inflammation-related outcomes in both the HBE cells and a mouse model. The study here also found that PM initially triggered ER stress in the HBE cells and this then led to dysregulation of inflammation in a mechanism involving modulation of autophagy and cell NF-κB pathways. This work thus provides new insights into the potential use of agents to block ER stress in airway epithelia to reduce/block PM-induced inflammatory responses.
Inflammation is a cardinal feature for lung injury and causes the release of inflammatory mediators and substantial proteases, thus ultimately contributing to tissue damage. It is well-established that lung is an important target for PM exposure and PM-induced inflammation in the lung is partly associated with early mortality (Dagher et al. Citation2005; Seagrave et al. Citation2005). ER stress has been implicated in inflammatory processes, however, the eventual roles of ER stress-induced inflammation are either deleterious or protective in disease progression (Garg et al. Citation2012). For example, XBP1 abnormalities in intestinal epithelial cells elicits ER stress and subse-quently causes spontaneous enteritis (Kaser et al. Citation2008), whereas ER stress-induced inflammation impedes tumorigenesis through anti-tumor immunity (Verfaillie et al. Citation2013). The outcomes in the study here are consistent with those noted in previous ones in that it was clear that the inhibiting of ER stress relieved PM-induced inflammation-related events in lung epithelial cells and that ER stress appeared to play a key deleterious role in PM-induced lung inflammation.
Autophagy is a dynamic process responsible for the degradation and recycling of cytosolic proteins and organelles (Kim and Guan Citation2015) and the conversion of the autophagy hallmark LC3B from LC3B-I to LC3B-II represents autophagosome formation. Emerging evidence shows that inhibition of autophagy can significantly alleviate PM-induced airway inflammation (Chen et al. Citation2016; Xu et al. Citation2017). The study here confirmed that autophagy was essential for PM-induced inflammation marker expression in HBE cells. The present study further showed that ER stress modulated autophagy protein LC3B expression in the context of this ‘induced’ inflamma-tion, suggesting such stress was an important regulator of autophagy activation.
During inflammatory events, NF-κB signaling plays a large role. In general, in NF-κB p50/p65 heterodimers, p65 often controls NF-κB transcriptional activation. Prior studies have shown that inhibiting autophagy decreased PM-induced inflammation-related events via NF-κB pathway activation (Xu et al. Citation2017). Along those lines, it was seen here that NF-κB pathway was essential to PM-induced inflammatory events in the HBE cells and that ER stress modulated NF-κB activation. This suggested to us that ER stress – along with activation of downstream autophagy – positively-regulated PM-induced inflammation via NF-κB activation.
In summary, the results from the present study show that PM exposure may induce ER stress in airway epithelial cells that, in turn, promotes autophagy and modulation of NF-κB pathway activity. Together, these steps might prove to be essential for how inflammation within the airways is induced following host exposure(s) to PM.
Author contributions
Authors YW and AX designed the study; LP, FY, and YW performed the study and collected/analyzed the data using reagents and materials from YT. All authors participated in drafting the manuscript, and approving its submission. Authors Pu and Yi contributed equally to this article.
Supplemental Material
Download MS Power Point (92.1 KB)Data availability statement
Access to the data generated during this study can be obtained by reasonable request to the corresponding author.
Additional information
Funding
References
- Brooks-Worrell B, Palmer J. 2019. Setting the stage for islet autoimmunity in Type 2 diabetes: obesity-associated chronic systemic inflammation and endoplasmic reticulum (ER) stress. Diabetes Care. 42(12):2338–2346. doi: 10.2337/dc19-0475.
- Byrd J, Morishita M, Bard R, Das R, Wang L, Sun Z, Spino C, Harkema J, Dvonch J, Rajagopalan S, et al. 2016. Acute increase in blood pressure during inhalation of coarse particulate matter air pollution from an urban location. J Am Soc Hypertens. 10(2):133–139.e4. doi: 10.1016/j.jash.2015.11.015.
- Chen Z, Wu Y, Wang P, Wu Y, Li Z, Zhao Y, Zhou J, Zhu C, Cao C, Mao Y, et al. 2016. Autophagy is essential for ultrafine particle-induced inflammation and mucus hyper-production in airway epithelium. Autophagy. 12(2):297–311. doi: 10.1080/15548627.2015.1124224.
- Dagher Z, Garçon G, Gosset P, Ledoux F, Surpateanu G, Courcot D, Aboukais A, Puskaric E, Shirali P. 2005. Pro-inflammatory effects of Dunkirk city air pollution PM2.5 in human epithelial lung cells (L132) in culture. J Appl Toxicol. 25(2):166–175. doi: 10.1002/jat.1050.
- Davenport E, Morgan G, Davies F. 2008. Untangling the unfolded protein response. Cell Cycle. 7(7):865–869. doi: 10.4161/cc.7.7.5615.
- Deng X, Zhang F, Wang L, Rui W, Long F, Zhao Y, Chen D, Ding W. 2014. Airborne fine particulate matter induces multiple cell death pathways in human lung epithelial cells. Apoptosis. 19(7):1099–1112. doi: 10.1007/s10495-014-0980-5.
- Ding H, Jiang M, Li D, Zhao Y, Yu D, Zhang R, Chen W, Pi J, Chen R, Cui L, et al. 2021. Effects of real-ambient PM2.5 exposure on lung damage modulated by Nrf2. Front Pharmacol. 12:662664. doi: 10.3389/fphar.2021.662664.
- Dockery D, Pope C, Xu X, Spengler J, Ware J, Fay M, Ferris B Jr, Speizer F. 1993. An association between air pollution and mortality in six U.S. cities. N Engl J Med. 329(24):1753–1759. doi: 10.1056/NEJM199312093292401.
- Duarte F, Teodoro J, Rolo A, Palmeira C. 2012. Exposure to dibenzofuran triggers autophagy in lung cells. Toxicol Lett. 209(1):35–42. doi: 10.1016/j.toxlet.2011.11.029.
- Ebelt S, Petkau A, Vedal S, Fisher T, Brauer M. 2000. Exposure of chronic obstructive pulmonary disease patients to particulate matter: relationships between personal and ambient air concentrations. J Air Waste Manag Assoc. 50(7):1081–1094. doi: 10.1080/10473289.2000.10464166.
- Garg A, Kaczmarek A, Krysko O, Vandenabeele P, Krysko D, Agostinis P. 2012. ER stress-induced inflammation: does it aid or impede disease progression? Trends Mol Med. 18(10):589–598. doi: 10.1016/j.molmed.2012.06.010.
- Gehring U, Wijga A, Brauer M, Fischer P, De Jongste J, Kerkhof M, Oldenwening M, Smit H, Brunekreef B. 2010. Traffic-related air pollution and the development of asthma and allergies during the first 8 years of life. Am J Respir Crit Care Med. 181(6):596–603. doi: 10.1164/rccm.200906-0858OC.
- Guan W, Zheng X, Chung K, Zhong N. 2016. Impact of air pollution on the burden of chronic respiratory diseases in China: time for urgent action. Lancet. 388(10054):1939–1951. doi: 10.1016/S0140-6736(16)31597-5.
- Jacquemin B, Siroux V, Sanchez M, Carsin A, Schikowski T, Adam M, Bellisario V, Buschka A, Bono R, Brunekreef B, et al. 2015. Ambient air pollution and adult asthma incidence in six European cohorts (ESCAPE). Environ Health Perspect. 123(6):613–621. doi: 10.1289/ehp.1408206.
- Karakatsani A, Analitis A, Perifanou D, Ayres J, Harrison R, Kotronarou A, Kavouras I, Pekkanen J, Hämeri K, Kos G, et al. 2012. Particulate matter air pollution and respiratory symptoms in individuals having either asthma or chronic obstructive pulmonary disease: a European multicentre panel study. Environ Health. 11:75. doi: 10.1186/1476-069X-11-75.
- Kaser A, Lee A, Franke A, Glickman J, Zeissig S, Tilg H, Nieuwenhuis E, Higgins D, Schreiber S, Glimcher L, et al. 2008. XBP1 links ER stress to intestinal inflammation and confers genetic risk for human inflammatory bowel disease. Cell. 134(5):743–756. doi: 10.1016/j.cell.2008.07.021.
- Kim S, Kim D, Kang M, Lee K, Park S, Jeong J, Lee Y. 2013. Endoplasmic reticulum stress influences bronchial asthma pathogenesis by modulating NF-κB activation. J Allergy Clin Immunol. 132(6):1397–1408. doi: 10.1016/j.jaci.2013.08.041.
- Kim Y, Guan K. 2015. mTOR: a pharmacologic target for autophagy regulation. J Clin Invest. 125(1):25–32. doi: 10.1172/JCI73939.
- Livak K, Schmittgen T. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCt Method. Methods. 25(4):402–408. doi: 10.1006/meth.2001.1262.
- Park S, Kim Y, Yang S, Henderson M, Yang W, Lindahl M, Urano F, Chen Y. 2019. Discovery of endoplasmic reticulum calcium stabilizers to rescue ER-stressed podocytes in nephrotic syndrome. Proc Natl Acad Sci U S A. 116(28):14154–14163. doi: 10.1073/pnas.1813580116.
- Seagrave J, Gigliotti A, Mcdonald J, Seilkop S, Whitney K, Zielinska B, Mauderly J. 2005. Composition, toxicity, and mutagenicity of particulate and semivolatile emissions from heavy-duty compressed natural gas-powered vehicles. Toxicol Sci. 87(1):232–241. doi: 10.1093/toxsci/kfi230.
- Tao F, Kobzik L. 2002. Lung macrophage-epithelial cell interactions amplify particle-mediated cytokine release. Am J Respir Cell Mol Biol. 26(4):499–505. doi: 10.1165/ajrcmb.26.4.4749.
- Traboulsi H, Guerrina N, Iu M, Maysinger D, Ariya P, Baglole C. 2017. Inhaled pollutants: the molecular scene behind respiratory and systemic diseases associated with ultrafine particulate matter. IJMS. 18(2):243. doi: 10.3390/ijms18020243.
- Verfaillie T, Garg A, Agostinis P. 2013. Targeting ER stress induced apoptosis and inflammation in cancer. Cancer Lett. 332(2):249–264. doi: 10.1016/j.canlet.2010.07.016.
- Wang J, Zhang J, Ma Y, Zeng Y, Lu C, Yang F, Jiang N, Zhang X, Wang Y, Xu Y, et al. 2021. WTAP promotes myocardial ischemia/re-perfusion injury by increasing endoplasmic reticulum stress via regulating m(6)A modification of ATF4 mRNA. Aging (Albany NY). 13(8):11135–11149. doi: 10.18632/aging.202770.
- Wang Y, Tang M. 2020. PM2.5 induces autophagy and apoptosis through endoplasmic reticulum stress in human endothelial cells. Sci Total Environ. 710:136397. doi: 10.1016/j.scitotenv.2019.136397.
- Wang Y, Zhou J, Xu X, Li Z, Chen H, Ying S, Li W, Shen H, Chen Z. 2018. Endoplasmic reticulum chaperone GRP78 mediates cigarette smoke-induced necroptosis and injury in bronchial epithelium. Int J Chron Obstruct Pulmon Dis. 13:571–581. doi: 10.2147/COPD.S150633.
- Xu X, Wu Y, Zhou J, Chen H, Wang Y, Li Z, Zhao Y, Shen H, Chen Z. 2017. Autophagy inhibitors suppress environmental particulate matter-induced airway inflammation. Toxicol Lett. 280:206–212. doi: 10.1016/j.toxlet.2017.08.081.
- Zeng M, Sang W, Chen S, Chen R, Zhang H, Xue F, Li Z, Liu Y, Gong Y, Zhang H, et al. 2017. 4-PBA inhibits LPS-induced inflammation through regulating ER stress and autophagy in acute lung injury models. Toxicol Lett. 271:26–37. doi: 10.1016/j.toxlet.2017.02.023.