
Abstract
WIPI proteins, phosphatidylinositol 3-phosphate (PtdIns3P) binding proteins with β-propeller folds, are recruited to the omegasome following PtdIns3P production. The functions of the WIPI proteins in autophagosome formation are poorly understood. In a recent study, we reported that WIPI2B directly binds ATG16L1 and functions by recruiting the ATG12–ATG5-ATG16L1 complex to forming autophagosomes during starvation- or pathogen-induced autophagy. Our model of WIPI2 function provides an explanation for the PtdIns3P-dependent recruitment of the ATG12–ATG5-ATG16L1 complex during initiation of autophagy.
Abbreviations:
Autophagy initiation results in activation of the BECN1-PIK3C3/VPS34 PtdIns 3-kinase complex at the omegasome, the ER-derived autophagosome formation site. This localized PtdIns3P production recruits PtdIns3P-binding proteins such as the WIPI family members and ZFYVE1/DFCP1. The functions of these proteins are poorly understood. Downstream of PtdIns3P production, the ATG12–ATG5-ATG16L1 complex is recruited to the omegasome. This complex acts as an E3-like protein to recruit the E2-like protein ATG3, coupled to LC3, to the forming autophagosome. Subsequently, LC3 is conjugated to the lipid phosphatidylethanolamine. Recruitment of the ATG12–ATG5-ATG16L1 complex is an essential step in autophagosome formation, and previous studies have demonstrated that the ATG12–ATG5-ATG16L1 complex is recruited through a direct interaction between ATG16L1 and RB1CC1/FIP200 (a member of the ULK1 kinase complex) and ubiquitin. However, neither of these interactors provide an explanation for the PtdIns3P dependence of ATG12–ATG5-ATG16L1 recruitment and subsequent LC3 lipidation.
To establish the molecular function of WIPI1 and WIPI2 in autophagy we used mass spectrometry to identify binding partners of GFP-WIPI1A and GFP-WIPI2B. We found that the ATG12–ATG5-ATG16L1 complex bound GFP-WIPI2B, but not GFP-WIPI1A. Binding between WIPI2 and the ATG12–ATG5-ATG16L1 complex is detected at the endogenous level when the interaction is stabilized, suggesting that the interaction is weak and/or transient. Further analysis showed that ATG16L1 is the member of the ATG12–ATG5-ATG16L1 complex mediating the interaction with WIPI2, and that this interaction is within the middle, coiled-coil-containing, domain of ATG16L1. This domain is important for ATG16L1 binding to a number of proteins, including RAB33B and RB1CC1/FIP200. Single-residue point mutations of ATG16L1 identified acidic resides E226 and E230 as the WIPI2B binding site. Surprisingly, our mutational analysis revealed that WIPI2B and RB1CC1/FIP200 occupy adjacent binding sites (RB1CC1/FIP200 binds amino acids 235–241). Further experiments showed that WIPI2, ATG16L1, and RB1CC1/FIP200 can form a trimeric complex when overexpressed proteins are used, suggesting that RB1CC1/FIP200 and WIPI2 binding is not mutually exclusive, and preliminary experiments suggest that binding is not cooperative.
ATG16L1 binding by WIPI2 is required for WIPI2 function in autophagy: in cells depleted for WIPI2, WIPI2 mutants unable to bind ATG16L1 inhibit LC3 lipidation and LC3 puncta formation. Mutations that render WIPI2B unable to bind PtdIns3P relieve this inhibition. This implies that WIPI2B mutants unable to bind ATG16L1 inhibit autophagosome formation by sequestering PtdIns3P away from residual WIPI2B or other PtdIns3P-binding proteins, which may be required for LC3 lipidation.
Reciprocal mutational analysis of the ATG16L1 binding site on WIPI2 showed that the acidic region on ATG16L1 binds a basic cleft between blades 2 and 3 of the β-propeller of WIPI2B, suggesting WIPI2B and ATG16L1 bind directly through a charge-charge interaction between acidic (ATG16L1) and basic (WIPI2B) residues. This is supported by a rescue of WIPI2B-ATG16L1 binding seen when mutants that reverse the charge on both ATG16L1 (E230R) and WIPI2B (R108E) are used. However, this charge-charge interaction may not be the only mechanism for WIPI2B-ATG16L1 binding: the basic residues in WIPI2B required for ATG16L1 binding are well conserved in WIPI1A and so we cannot currently explain why WIPI1A binds ATG16L1 to a lesser degree than WIPI2B.
To prove that WIPI2B can directly recruit ATG12–ATG5-ATG16L1, we used a KRAS CAAX motif to direct WIPI2B to the plasma membrane. WIPI2B on the plasma membrane is able to recruit the ATG12–ATG5-ATG16L1 complex and enable LC3 lipidation. The plasma membrane-localized WIPI2B supports LC3 recruitment and lipidation independently of upstream autophagy initiation factors, including MTORC1 inhibition and PtdIns3P production, but is dependent on ATG16L1 binding. This strongly supports our model in which WIPI2B is the PtdIns3P binding protein responsible for PtdIns3P-dependent ATG12–ATG5-ATG16L1 recruitment to the omegasome. This is further supported by live-cell imaging of tagged-WIPI2B and ATG16L1 that demonstrates that WIPI2B puncta form immediately before ATG16L1 puncta. Furthermore, to test whether WIPI2 binding is required for ATG16L1 function we studied ATG16L1Δ/Δ MEFs that contain a deletion of the middle domain. ATG16L1Δ/Δ protein is not functional for autophagy and the MEFs are autophagy null. By restoring wild-type ATG16L1 or ATG16L1 mutants unable to bind WIPI2b we showed that WIPI2B binding is essential for autophagy in these rescued ATG16L1Δ/Δ MEFs. In addition, our results suggest that WIPI2B, rather than RB1CC1/FIP200, is responsible for ATG16L1 recruitment: we found that autophagy is rescued to a lesser extent by ATG16L1 defective in WIPI2 binding than ATG16L1 defective in RB1CC1/FIP200 binding.
Finally by studying xenophagy, a type of autophagy that targets intracellular bacteria, we demonstrated the requirement for WIPI2B in LC3 recruitment and functional xenophagy. ATG16L1 recruitment during xenophagy involves both RB1CC1/FIP200 and ubiquitin binding by ATG16L1, but the requirement for PtdIns3P binding proteins was poorly understood. WIPI2 is required for Salmonella clearance and LC3 targeting to Salmonella in HeLa cells, and the WIPI2B-ATG16L1 interaction is required for LC3 targeting to Salmonella in rescued ATG6L1Δ/Δ MEFs, demonstrating that WIPI2B functions as an ATG12–ATG5-ATG16L1 recruitment mechanism during xenophagy as well as starvation-induced autophagy.
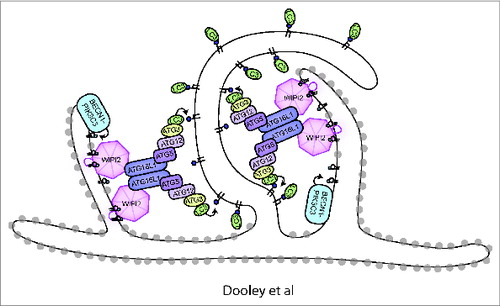
Our data have given new insight into the molecular requirement for PtdIns3P in autophagy initiation through connecting recruitment of the ATG12–ATG5-ATG16L1 complex to PtdIns3P production via WIPI2B binding. We speculate that the recruitment of the ATG12–ATG5-ATG16L1 complex positioned by WIPI2 may provide a spatial positioning of the phagophore on the omegasome (). However, there are still many questions regarding regulation of the WIPI2B-ATG16L1 interaction, including whether there is starvation-based regulation of the binding. Furthermore, our findings that RB1CC1/FIP200 and WIPI2B can both bind ATG16L1 either independently or concurrently and the close proximity of the binding sites for these 2 proteins on ATG16L1 make it tempting to speculate that a functional or regulatory interplay exists between these 2 proteins.
Figure 1. Model of WIPI2 function in autophagy. PtdIns3P is produced by the BECN1-PIK3C3/VPS34 PtdIns 3-kinase complex on the omegasome (omegasome is shown as ER cisternae with gray ribosomes). WIPI2 is recruited by binding PtdIns3P. WIPI2 subsequently binds and recruits the ATG12–ATG5-ATG16L1 complex, resulting in ATG3 recruitment, LC3 lipidation, and phagophore expansion. The expanding phagophore membrane is shown with no ribosomes.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
HCD and SAT are funded by Cancer Research UK, MIW is funded by BBSRC.