
Abstract
A variety of anticancer agents employed in standard chemotherapy or novel targeted therapy induce autophagy. A cytoprotective autophagic response often counteracts apoptosis triggered by such agents, potentially contributing to acquired drug-resistance. It is recognized that autophagy and apoptosis share molecular regulatory mechanisms primarily governed by multidomain anti-apoptotic members (e.g., BCL2/Bcl-2 and BCL2L1/Bcl-xL) of the BCL2 family. However, the role of pro-apoptotic BH3-only proteins (e.g.,, BCL2L11/Bim), another class of BCL2 family proteins that critically determine therapeutic responses, in autophagy regulation remains largely unexplored, particularly with respect to mechanisms of acquired drug resistance.
In response to various anticancer therapies, 2 major forms of programmed cell death i.e., apoptosis (“self-killing”) and autophagy (“self-eating” or “self-cleaning”), often occur simultaneously. While apoptosis represents a well-established effector mechanism for both standard chemotherapeutic and novel targeted agents, autophagy generally plays a cytoprotective role through which cancer cells escape the lethal effects of various noxious stimuli. Consequently, the autophagic response may counteract apoptosis triggered by these agents, potentially contributing to acquired drug resistance. Mechanistically, autophagy and apoptosis share molecular regulatory mechanisms and signaling pathways primarily governed by multidomain anti-apoptotic BCL2 family proteins. Specifically, BCL2 or BCL2L1 prevent both apoptosis and autophagy by sequestering different BH3-only proteins (e.g., pro-apoptotic BCL2L11, BID, and pro-autophagic BECN1/Beclin 1). Thus, BH3-only proteins may play critical roles in maintaining the balance between autophagy and apoptosis, thereby determining tumor cell responses to drug treatment by promoting cell death (via apoptosis) or survival (through autophagy).
Unlike their multidomain anti-apoptotic counterparts, the role of pro-apoptotic BH3-only proteins in autophagy regulation has been minimally explored. The BH3-only protein BIK/NBK, an endoplasmic reticulum-associated pro-apoptotic protein, cooperates with NAF1 to promote autophagy by facilitating BECN1 release from BCL2 at the endoplasmic reticulum. As BIK is a ubiquitination substrate, it accumulates following failure of autophagic degradation (e.g.,, in the case of “inefficient” autophagy), thereby triggering apoptosis. Therefore, such a BH3-only protein could act as a switch converting autophagy to apoptosis. BCL2L11 is another BH3-only protein, which governs the activity of diverse agents targeting oncogene-driven pathways. Often, in new therapies targeting specific driver oncoproteins to which tumor cells are addicted (e.g., mutant BRAF, KRAS, EGFR, FLT3, and BCR-ABL, etc.), BCL2L11 is upregulated due to inhibition of signaling pathways (e.g., MAPK/ERK [RAS/RAF/MEK/ERK] and PI3K/AKT) that repress its expression through transcriptional regulation and/or post-translational modifications e.g. phosphorylation. BCL2L11 is also upregulated by impaired degradation secondary to inhibition of ubiquitination or proteasomal degradation e.g., by proteasome inhibitors such as bortezomib. In this context, BCL2L11 upregulation represents an important mechanism of action for such agents. Conversely, BCL2L11 deletion/downregulation could contribute to acquired drug resistance to both targeted and standard agents.
In contrast to its well-established role in apoptosis, a recent study demonstrated that BCL2L11 sequesters BECN1 at microtubules by promoting the BECN1-DYNLL/LC8 interaction and thereby inhibits autophagy by mislocalizing BECN1 to the dynein motor complex. Specifically, whereas BCL2L11 directly binds to BECN1, cells with BCL2L11 siRNA knockdown or gene knockout display higher LC3-II levels in vitro and in vivo, indicating autophagy induction. Similarly, BCL2L11 shRNA knockdown in human multiple myeloma (MM) cells triggers a marked increase in LC3-II expression associated with pronounced bortezomib resistance. Notably, BCL2L11 is highly expressed in both human MM cell lines (BCL2L11/Bimhi), with rare exceptions (e.g., H929, BCL2L11/Bimlow), and primary CD138+ MM cells, suggesting that such cells are primed by BCL2L11 for death. However, while basal BCL2L11 levels do not correlate with intrinsic bortezomib-resistance, several lines of evidence suggest that loss of BCL2L11 contributes to acquired bortezomib-resistance in MM cells, in part through enhanced autophagy. First, gene expression profiling targeting apoptosis-pathways identified BCL2L11 as a downregulated gene in bortezomib-resistant cells generated by continuous exposure to increasing bortezomib concentrations. Moreover, marked BCL2L11 downregulation was observed in resistant cell lines and in primary CD138+ MM cells from patients who initially responded to bortezomib and then relapsed. Notably, BCL2L11 loss closely correlates with increased LC3 puncta in bortezomib-resistant cells, consistent with increased autophagy observed in BCL2L11 shRNA MM cells. However, no changes in protein levels of key factors involved in autophagy initiation (e.g.,, ULK1, BECN1, and ATG5) were noted. Collectively, these observations argue that BCL2L11 downregulation contributes to acquired bortezomib-resistance by raising the death threshold through 2 mechanisms: direct inactivation of the apoptotic signaling cascade and induction of cytoprotective autophagy.
A corollary of these findings is that anticancer agents that upregulate BCL2L11 may reprime bortezomib-resistant MM cells to cell death by disabling autophagy. Indeed, histone deacetylase (HDAC) inhibitors restore BCL2L11 levels in bortezomib-resistant cells, while inhibiting autophagy (e.g., diminished LC3-II levels and GFP-LC3 puncta formation) induced by BH3-mimetics (e.g., ABT-737) that unleash BECN1 from multidomain anti-apoptotic proteins (e.g., BCL2 or BCL2L1). Consequently, HDAC inhibitors significantly potentiate BH3-mimetic lethality in bortezomib-resistant MM cells. Specifically, HDAC inhibitors increase BCL2L11-BCL2 binding, presumably due to BCL2L11 upregulation, and ABT-737 releases BCL2L11 from binding to BCL2. As a result, HDAC inhibitor/ABT-737 co-administration sharply increases the association of BECN1 with BCL2, which prevents BECN1-induced autophagy. This raises the possibility that BCL2 release from BCL2L11 contributes to increased BCL2-BECN1 binding as a consequence of increased BCL2 availability. Significantly, HDAC inhibitors fail to prevent ABT-737-induced autophagy in BCL2L11 gene knockout cells, arguing strongly for a role for BCL2L11 in autophagy regulation. Thus, a rational BCL2L11-targeting strategy combining a class of agents (e.g., BH3-mimetics) that release BCL2L11 (apoptotic response) and BECN1 (autophagic response) from BCL2 or BCL2L1 with agents (e.g., HDAC inhibitors) that upregulate BCL2L11 and inhibit autophagy, may be particularly effective in circumventing certain forms of drug resistance (e.g., acquired bortezomib-resistance) by turning autophagy off and apoptosis on ().
Figure 1. A mechanism for the role of autophagy in BCL2L11-targeting strategies. Targeted therapeutics like BH3-mimetics (e.g., ABT-737) simultaneously activate apoptosis and autophagy by releasing BCL2L11 and BECN1 from BCL2, respectively. However, apoptosis is opposed by autophagy (autophagy ON, left), conferring drug resistance. HDAC inhibitors restore BCL2L11 levels in drug-resistant cells exhibiting loss of BCL2L11, thereby repriming them to apoptosis by autophagy inhibition (autophagy OFF, right) through release of BCL2 from BCL2L11 and sequestration of BECN1 by BCL2. In intrinsically BCL2L11/Bimlow cells, autophagy disruption (e.g., by chloroquine) is required for this BCL2L11-targeting approach. Thus, while BCL2L11 downregulation contributes to acquired drug resistance, a BCL2L11-targeting strategy combining agents (e.g., HDAC inhibitors), which upregulate BCL2L11 and inhibit autophagy, with those (e.g., BH3-mimetics) that release BCL2L11 from anti-apoptotic BCL2 family proteins but induce autophagy may represent an effective approach to the problem of drug resistance which currently represents a major challenge for both standard and novel targeted therapies.
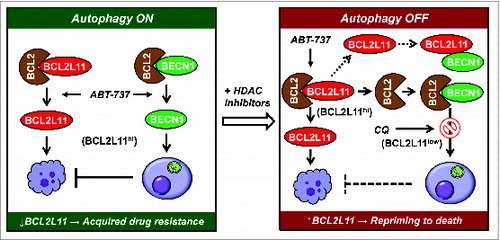
Finally, HDAC inhibitors minimally upregulate BCL2L11 and fail to increase ABT-737 lethality in BCL2L11/Bimlow MM cells. However, in this setting, disruption of autophagy by chloroquine strikingly increases apoptosis in association with pronounced LC3-II expression and BCL2L11 upregulation in such cells. These findings argue that BCL2L11, a ubiquitination substrate, may be subjected to degradation through autophagy, in addition to proteasomal degradation. In contrast, this phenomenon was not observed in BCL2L11/Bimhi MM cells. Therefore, disruption of autophagy (particularly at later stages, e.g., maturation) might be required for restoration of BCL2L11 expression and sensitivity of BCL2L11/Bimlow cells to regimens combining HDAC inhibitors with BH3-mimetics. In this regard, basal BCL2L11 levels could potentially serve as a biomarker for optimizing BCL2L11-targeting strategies in future personalized medicine approaches.