
Abstract
Damaged mitochondria are selectively degraded via autophagy in a regulated pathway known as mitophagy. Parkinson disease-linked proteins PINK1 (PTEN induced putative kinase 1) and PARK2 (parkin RBR E3 ubiquitin protein ligase) are recruited to the outer mitochondrial membrane upon mitochondrial damage, leading to the PARK2-mediated ubiquitination of mitochondrial proteins. Here, we discuss our recent work demonstrating that OPTN (optineurin) is recruited to damaged mitochondria, serving as an autophagy receptor for autophagosome formation around mitochondria. Using high-resolution live-cell imaging, we find that OPTN is recruited to ubiquitinated mitochondria downstream of PARK2, and induces autophagosome assembly around mitochondria via its LC3-interacting region. Mutations in OPTN are linked to both glaucoma and ALS (amyotrophic lateral sclerosis), and an ALS-associated E478G mutation in OPTN's ubiquitin binding domain leads to defective mitophagy and accumulation of damaged mitochondria. Importantly, our results highlight a role for mitophagy defects in ALS pathogenesis, and demonstrate that defects in the same pathway for mitochondrial homeostasis are causal for both familial Parkinson disease and ALS.
Selective autophagy of ubiquitinated proteins and organelles is mediated by autophagy receptors, which bind ubiquitinated cargo and recruit the autophagosome protein MAP1LC3/LC3 (microtubule-associated protein 1 light chain 3) via their LC3-interacting region (LIR). The selective autophagy of damaged mitochondria, known as mitophagy, is crucial for degrading damaged mitochondria and thus the maintenance of mitochondrial homeostasis. During mitophagy, the kinase PINK1 is stabilized on the outer mitochondrial membrane (OMM) and recruits the E3 ubiquitin ligase PARK2, leading to the ubiquitination of OMM proteins. This is followed by autophagosome formation around ubiquitinated mitochondria, leading to their autophagic degradation. However, the receptor responsible for recruiting phagophores (the autophagosome precursor) to ubiquitinated mitochondria during PARK2-dependent mitophagy has not been previously identified.
The 6 currently known mammalian autophagy receptors are SQSTM1/p62, NBR1 (neighbor of BRCA1 gene 1), OPTN, CALCOCO2/NDP52 (calcium binding and coiled-coil domain 2), TAX1BP1/T6BP (Tax1 [human T-cell leukemia virus type I] binding protein 1), and TOLLIP (toll interacting protein). Previous reports on the role of SQSTM1 in mitophagy have been controversial, with initial reports proposing it as an autophagy receptor for damaged mitochondria. However, subsequent work has found that SQSTM1 instead aggregates neighboring mitochondria via its PB1 oligomerization domain. As mutations in the autophagy receptor OPTN are linked to glaucoma and ALS, 2 neurodegenerative diseases in which mitochondrial defects have been implicated, we investigated a possible role for OPTN as an autophagy receptor in PARK2-dependent mitophagy.
Live-cell imaging in HeLa cells indicates that in the absence of mitochondrial damage, OPTN does not stably localize to mitochondria. However, depolarization of mitochondria via CCCP (carbonyl cyanide m-chlorophenyl hydrazone) causes recruitment of OPTN to damaged mitochondria in cells overexpressing PARK2. Spatiotemporally controlled damage of a mitochondrial subpopulation via localized generation of reactive oxygen species also induces PARK2-dependent OPTN recruitment to mitochondria. Thus, upon mitochondrial damage, OPTN is robustly recruited to the outer mitochondrial membrane downstream of PARK2 recruitment.
PARK2 activity is required for the stable recruitment of OPTN, as expression of a catalytically inactive Parkinson disease-associated T240R mutation in the RING1 domain of PARK2 does not block PARK2 recruitment to damaged mitochondria, but is sufficient to block OPTN recruitment. Furthermore, an ALS-associated E478G mutation in OPTN's UBAN domain, which inhibits binding to ubiquitin, also blocks the stable recruitment of OPTN to damaged mitochondria, despite continued robust PARK2 recruitment. Thus, OPTN recruitment to damaged mitochondria is driven by the binding of its UBAN domain to PARK2-mediated ubiquitinated mitochondrial proteins.
Live-cell imaging was also used to investigate the dynamics of autophagosome formation during mitophagy. Autophagosome biogenesis begins, at least in some cases, with the omegasome, a PtdIns3P-enriched ER omega-shaped membrane that recruits ZFYVE1/DFCP1 (zinc finger, FYVE domain containing 1) and extends around autophagic cargo. During mitophagy, a ZFYVE1-positive omegasome transiently assembles on the side of a damaged mitochondria, marking the initial site of autophagosome formation. Omegasome assembly occurs subsequent to both PARK2 and OPTN recruitment, but does not require OPTN. Omegasome formation is followed by assembly of the phagophore protein LC3 around OPTN-positive damaged mitochondria. LC3 is first recruited as punctate structures, which grow into a sphere that engulfs the entire mitochondria. As expected, autophagosome biogenesis around damaged mitochondria occurs downstream of both PARK2 and OPTN recruitment.
To demonstrate that OPTN is an autophagy receptor for mitochondria, we depleted OPTN using siRNA and found that OPTN knockdown dramatically impairs LC3 recruitment and thus the formation of autophagosomes around damaged mitochondria. LC3 recruitment is also disrupted by expression of an F178A mutation in OPTN that disrupts the interaction of OPTN with LC3. In contrast, overexpression of wild-type OPTN accelerates LC3 recruitment around damaged mitochondria. To confirm that OPTN indeed regulates mitochondrial degradation via mitophagy, we examined mitochondrial levels 24 h after CCCP-induced damage. We find that both mitochondrial DNA content and the levels of mitochondrial matrix protein HSPD1/Hsp60 are increased upon OPTN siRNA knockdown, indicating that OPTN depletion results in inefficient mitochondrial degradation. Importantly, this defect is rescued by siRNA-resistant wild-type OPTN but not by expression of the ALS-associated E478G ubiquitin binding-deficient mutation in OPTN or the F178 LC3 binding-deficient OPTN mutant, demonstrating that OPTN is a critical autophagy receptor for PARK2-dependent mitophagy.
In contrast to the PARK2-dependent recruitment of OPTN to the entire surface of damaged mitochondria, SQSTM1 preferentially localizes to domains between adjacent mitochondria and induces mitochondrial aggregation as previously reported. We further examined the interplay between OPTN and SQSTM1, and found that SQSTM1 depletion does not disrupt the recruitment of either OPTN or LC3 to damaged mitochondria, but rather induces tubular mitochondria morphology. Importantly, unlike OPTN, SQSTM1 depletion does not disrupt mitochondrial degradation following CCCP-induced damage. As OPTN also does not regulate SQSTM1 recruitment or localization on mitochondria, we find that OPTN and SQSTM1 are independently recruited to different domains on damaged mitochondria and have distinct roles in mitophagy.
In summary, our recent work demonstrates a novel role for OPTN as an autophagy receptor for damaged mitochondria in PARK2-dependent mitophagy, and establishes a PARK2-OPTN-ZFYVE1-LC3 recruitment pathway using high-resolution live cell microscopy (). Interestingly, we find that in the absence of PARK2, OPTN transiently localizes to tips of damaged mitochondria. As CCCP-induced damage in the absence of PARK2 induces non-selective autophagosome formation at ER-mitochondrial junctions, this transient localization may represent OPTN recruitment to these junctions during nonselective autophagosome biogenesis.
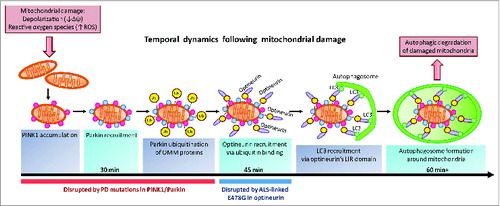
Figure 1. OPTN is an autophagy receptor for damaged mitochondria in PINK1- and PARK2-mediated mitophagy. Mitochondrial damage induced by depolarization or increased ROS production leads to the accumulation of the kinase PINK1 on the outer mitochondrial membrane (OMM). PINK1 subsequently recruits PARK2 to ubiquitinate OMM proteins. OPTN binds ubiquitinated OMM proteins via its ubiquitin-binding domain, and recruits the phagophore protein LC3 via its LC3-interacting region, inducing autophagosome formation around damaged mitochondria leading to autophagic degradation of the organelle. PARK2 is recruited within 30 min, followed by OPTN recruitment within 45 min of mitochondrial damage. Autophagosome formation begins 60 min after mitochondrial damage. Parkinson disease (PD) mutations in PINK1 and PARK2 disrupt PINK1 accumulation, PARK2 recruitment and PARK2 E3 ubiquitin ligase activity. The ALS-associated E478G mutation in OPTN's ubiquitin binding domain disrupts OPTN recruitment to mitochondria. Damaged mitochondria accumulate upon disruption of mitophagy and may contribute to neurodegeneration in both Parkinson disease and ALS.
Parkinson disease is characterized by degeneration of the substantia nigra and the formation of SNCA/α-synuclein-positive Lewy body inclusions; affected patients exhibit tremors, bradykinesia, and rigidity. In contrast, ALS is marked by motor neuron loss and formation of TARDBP/TDP-43-positive inclusions; affected patients first develop muscle weakness followed by paralysis. While these diseases preferentially affect distinct populations of neurons and are clinically distinct, our observations now demonstrate that causative mutations for each of these diseases affect a common pathway. As Parkinson disease-associated mutations in PINK1 and PARK2, and an ALS-associated mutation in OPTN lead to defective mitophagy, our study suggests that mitochondrial homeostasis is crucial for maintaining neuronal health and demonstrates that defects in the same mitochondrial degradation pathway may contribute to multiple neurodegenerative diseases.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by the National Institutes of Health Grant RO1 NS060698 (to E.L.F.H.).