
Abstract
In the present study we have established a vital role of autophagy in retinoic acid (RA)-induced differentiation of toll-like receptor (TLR)-stimulated human B cells into Ig-secreting cells. Thus, RA enhanced autophagy in TLR9- and CD180-stimulated peripheral blood B cells, as revealed by increased levels of the autophagosomal marker LC3B-II, enhanced colocalization between LC3B and the lysosomal marker Lyso-ID, by a larger percentage of cells with more than 5 characteristic LC3B puncta, and by the concomitant reduction in the level of SQSTM1/p62. Furthermore, RA induced expression of the autophagy-inducing protein ULK1 at the transcriptional level, in a process that required the retinoic acid receptor RAR. By inhibiting autophagy with specific inhibitors or by knocking down ULK1 by siRNA, the RA-stimulated IgG production in TLR9- and CD180-mediated cells was markedly reduced. We propose that the identified prominent role of autophagy in RA-mediated IgG-production in normal human B cells provides a novel mechanism whereby vitamin A exerts its important functions in the immune system.
Abbreviations:
- ATG, autophagy-related
- BDS, bright detail similarity
- CD180, CD180 molecule
- CVID, common variable immune deficiency
- ELISA, enzyme-linked immunosorbent assay
- Ig, immunoglobulin
- IL, interleukin
- MAP1LC3B/LC3B, microtubule-associated protein 1 light chain 3 β
- MTOR, mechanistic target of rapamycin (serine/threonine kinase)
- PAMP, pathogen-associated molecular pattern, PML/RARA, promyelocytic leukemia/ retinoic acid receptor α
- RA, all-trans retinoic acid
- RAR, retinoic acid receptor
- SQSTM1, sequestosome 1
- TLR, toll-like receptor
- ULK1, unc-51 like autophagy activating kinase 1
Introduction
Autophagy is a highly conserved self-digestive process aimed at degradation and recycling of cytoplasmic components.Citation1 Macroautophagy, hereafter referred to as autophagy, involves bulk degradation of cytoplasmic constituents like damaged organelles, microbes, and long-lived proteins through their sequestration into an autophagosome, a double-membraned vesicle. The autophagosome forms as it sequesters the substances destined for degradation and later in the process, the autophagosome fuses with the lysosome where hydrolytic enzymes degrade its contents.Citation1 The function of autophagy in the immune system is an emerging research field, involving immune responses such as antigen presentation, microbe removal, and lymphocyte activation.Citation2-7 Recently, an essential role for autophagy in humoral immunity has been revealed, linked to generation and maintenance of plasma cells.Citation8-10
Nutrient supply is one of the key regulators of autophagy, and while the general suppression of autophagy by macronutrients is well established,Citation11-13 the role of micronutrients such as vitamin A remains largely unknown. Vitamin A is metabolized in target cells to retinoic acid derivatives such as all-trans retinoic acid (hereafter denoted as RA). RA binds to the nuclear retinoic acid receptors (RARs) and thereby activates transcription of numerous genesCitation14-16 involved in a variety of processes, including hematopoiesis and the immune system.Citation17-20
A functional immune system not only depends on an optimal vitamin A status, but also relies on proper activation of the innate immune system involving toll-like receptors (TLRs). TLRs recognize pathogen-associated molecular patterns/PAMPs and activate B lymphocytes to proliferate and differentiate into immunoglobulin (Ig)-secreting plasma cells.Citation21 TLR9, located in the membrane of endosomes, recognizes CpG motifs of bacterial DNA and is activated in vitro by synthetic unmethylated CpG oligomers.Citation22 WeCitation23,24 and othersCitation25 have revealed that TLR9-mediated activation of B cells can be significantly enhanced by costimulation with antibodies against the TLR-related receptor CD180/RP105. CD180 was originally identified as a radio-protective protein of 150 kDaCitation26,27 and the in vivo ligand for CD180 remains unknown.
Being well established as an important factor in protection against infections,Citation28 it is evident that vitamin A has pleiotropic effects on both the innate and adaptive parts of the immune system.Citation17,29,30 We have previously shown that the vitamin A metabolite RA is able to potentiate the interplay between innate and adaptive B cell responses by enhancing TLR9-mediated and TLR9- and CD180-mediated activation of human B cellsCitation23,31 with a particularly strong impact on TLR9- and CD180-mediated differentiation of B cells into Ig-secreting plasma cells.Citation23 Given the recent awareness of the importance of autophagy for the generation and maintenance of plasma cells,Citation8,9 we here explored the involvement of autophagy in RA-mediated differentiation of B cells into Ig-secreting cells. We revealed that RA noticeably enhances autophagy in TLR9- and CD180-stimulated human peripheral blood B cells, and we suggest that RA exerts this effect at least partly by increasing the transcription of the gene encoding the autophagy-inducing protein ULK1 (unc-51 like autophagy activating kinase 1). Blocking autophagy with siRNA against ULK1 or by specific inhibitors of autophagy selectively inhibited the RA-induced IgG production in TLR-stimulated B cells. Hence, by highlighting the importance of autophagy in RA-mediated IgG production in normal human B cells, we have identified a novel mechanism whereby RA may exert its strong immune stimulatory effect.
Results
Retinoic acid enhances the formation of autophagosomes in TLR9- and CD180-stimulated human B cells
It has recently been established that autophagy is involved in regulating the differentiation of mouse B lymphocytes into IgG- and IgM-secreting cells.Citation8,9 Having previously demonstrated the stimulatory effect of RA on IgG-production in normal human B cells activated via TLR9 alone or in combination with CD180,Citation23,24,31 we here investigated the possible involvement of autophagy in this process. The MAP1LC3/LC3 (microtubule-associated protein 1 light chain 3) protein is a widely used and accepted marker of phagophores and autophagosomes,Citation32 and is commonly used to monitor the levels of autophagy. Upon induction of autophagy, cytosolic LC3-I will become conjugated to phosphatidylethanolamine (PE), bound to the phagophore membrane and termed LC3-II. Hence, the amount of LC3-II present in a sample reflects the formation of autophagosomes.
Freshly isolated CD19+ human B cells from healthy blood donors were stimulated with RA in combination with CpG and/or anti-CD180 for 24 h before the lysosomal protease inhibitors E64d and pepstatin ACitation33 were added to the cultures. The B cells were further incubated for 72 h prior to western blot analysis of LC3B-levels and quantification of the ratio between LC3B-II and LC3B-I (). RA alone had only a minor effect on LC3B-II formation, but it enhanced the levels of LC3B-II in B cells cotreated with either CpG, anti-CD180, or with the combination of CpG and anti-CD180. Concomitantly with the enhanced levels of LC3B-II, the LC3B-I levels were reduced. (The shifts in LC3B-II/LC3B-I ratio are presented in the histograms in the lower panel of .) Interestingly, anti-CD180 alone had no effect on the level of LC3B-II, and combined with CpG there was only a marginal increased effect on LC3B-II accumulation. However, when RA was combined with CpG and anti-CD180 the most striking synergy on the LC3B-II/LC3B-I ratio was obtained.
Figure 1. Retinoic acid enhances the level of LC3B-II and the LC3B-II/LC3B-I ratio in TLR-stimulated B cells. (A) CD19+ B cells (0.5 × 106 /ml) were stimulated with CpG (1 μg/ml), anti-CD180 (1 μg/ml) and RA (100 nM) for 24 h before the lysosomal inhibitors pepstatin A (10 μg/ml) and E64d (10 μg/ml) were added. The cells were incubated for another 72 h, before the cells were lysed and subjected to western blot analysis of LC3B-I/II and TUBA1A as described in Materials and Methods. To visualize the weaker protein bands in the first 2 lanes (representing unstimulated cells and cells stimulated with RA alone) the exposure time during development of the blot was substantially increased compared to the exposure time required for visualizing the bands in the other lanes of the blot. The 2 different exposure times are separated by a vertical line in the blot. (B) B cells were stimulated as in panel (A) in the presence or absence of the lysosomal inhibitors added after 24 h of stimulation. Again the cells were incubated for another 72 h, before subjected to western blot analysis as described in panel (A). The protein bands in both panel (A) and (B) were quantified, and the LC3B-II/LC3B-I ratios were calculated and displayed in the lower diagram. The data represent the mean ± SEM of 5 independent experiments. *P < 0.05 (Wilcoxon signed-rank test, n=5). (C) B cells were stimulated in the same manner as in panel (A) in the presence and absence of the lysosomal inhibitors pepstatin A and E64d the last 3 h of a 96-h incubation. The cells were then subjected to western blot analysis and the LC3B-II/LC3B-I ratios were quantitated and displayed in the diagram below, *P < 0.05 (paired sample Student t test, n=3). (D) B cells were stimulated as in panel (A) without the presence of lysosomal inhibitors. The protein levels of SQSTM1 were detected by western blotting analysis after 96 h of stimulation. The SQSTM1/GAPDH ratios were quantitated and displayed in the lower diagram. *P < 0.05 (paired sample Student t test, n=3) All the blots were developed and quantified as described in Materials and Methods.
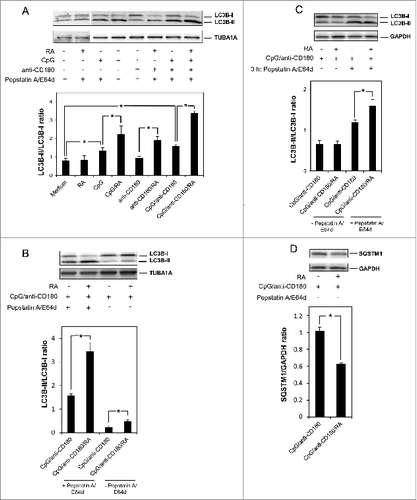
The accumulation of autophagosomes induced by RA could be due to one of 2 mechanisms; either enhanced autophagosome formation or blocked autophagosome degradation. Hence, in order to distinguish between these 2 possibilities, the same experiments as in were performed in the presence or absence of the lysosomal inhibitors E64d and pepstatin A. The results presented in reveal that the strong induction of LC3B-II induced by RA in combination with CpG and anti-CD180 was significantly diminished in the absence of the lysosomal inhibitors, supporting the notion that RA enhances the rate of autophagosome formation. That a slight RA-mediated induction of LC3B-II levels was notable even in the absence of the lysosomal inhibitors, further emphasizes the strong impact of RA on autophagosome formation in TLR9- and CD180-stimulated B cells. We also assessed the formation of LC3B-II in the presence of lysosomal inhibitors for only the last 3 h of the 96 h incubation, and also, here, we noted an RA-mediated increase in the LC3B-II/LC3B-I ratio (). However, as the effects were more pronounced when the inhibitors were added after 24 h of treatment, these conditions were used in the remaining experiments.
To further confirm that RA induces autophagy in CpG and anti-CD180 treated cells, we assessed the levels of SQSTM1/p62. The protein SQSTM1 is known to serve as an autophagy receptor for protein aggregates and damaged organelles, and is itself degraded by autophagy.Citation34 Hence, a reduction in SQSTM1 levels is consistent with induced autophagy. In order to allow SQSTM1 degradation, the analysis of SQSTM1 expression was performed in the absence of lysosomal inhibitors. As shown in , RA significantly reduced the SQSTM1 levels in cells costimulated via TLR9 and CD180.
Colocalization of autophagosomes and lysosomes is enhanced by RA
In order to further establish a possible connection between RA-mediated formation of autophagosomes and lysosomal degradation, we examined the colocalization between autophagosomes and lysosomes. To this end, B cells were stimulated with or without RA in the presence of the same combinations of CpG and anti-CD180 as used in , and the cells were costained with anti-LC3B and Lyso-ID. The extent of colocalization was determined by Image Stream analysisCitation35 where the cells with a high degree of colocalization had a bright detail similarity (BDS) value of more than 1.5 (). Although RA alone had no impact on the colocalization of autophagosomes and lysosomes, RA significantly enhanced the colocalization in both CpG- and anti-CD180-treated cells (). The highest level of colocalization between autophagosomes and lysosomes was obtained when RA was combined with both CpG and anti-CD180. Taken together, these results suggest that the RA-mediated formation of autophagosomes is part of the lysosomal degradation process.
Figure 2. RA enhances the colocalization of LC3B-II and lysosomes. B cells were stimulated as described in , and 72 h after addition of the lysosomal inhibitors the cells were fixed, permeabilized, and stained with the lysosomal marker Lyso-ID and anti-LC3B antibody. (A) The cells were subjected to Image Stream analysis as described in Materials and Methods. The degree of colocalization between LC3B puncta and lysosomes was calculated, and the percentages of cells with high degree of colocalization (bright detail similarity [BDS] > 1.5) are presented in the diagram. The data represent the mean ± SEM of 5 independent experiments *P < 0.05 (Wilcoxon signed-rank test, n=5). (B) The upper panel presents representative images of cells with BDS < 1.5 and the lower panel presents representative images of cells with BDS>1.5 from the Image Stream analysis. (C) The cells were subjected to Image Stream analysis, and the percentages of cells with more than 5 LC3B-II puncta were estimated. The data represent the mean ± SEM of 5 independent experiments. *P < 0.05 (Wilcoxon signed-rank test, n=5) (D) In addition to staining the cells with anti-LC3B antibody and Lyso-ID, the nuclei were stained with DAPI. The degree of colocalization between LC3B and the lysosomal marker was visualized by confocal microscopy, using the 60 x objective.
![Figure 2. RA enhances the colocalization of LC3B-II and lysosomes. B cells were stimulated as described in Figure 1A, and 72 h after addition of the lysosomal inhibitors the cells were fixed, permeabilized, and stained with the lysosomal marker Lyso-ID and anti-LC3B antibody. (A) The cells were subjected to Image Stream analysis as described in Materials and Methods. The degree of colocalization between LC3B puncta and lysosomes was calculated, and the percentages of cells with high degree of colocalization (bright detail similarity [BDS] > 1.5) are presented in the diagram. The data represent the mean ± SEM of 5 independent experiments *P < 0.05 (Wilcoxon signed-rank test, n=5). (B) The upper panel presents representative images of cells with BDS < 1.5 and the lower panel presents representative images of cells with BDS>1.5 from the Image Stream analysis. (C) The cells were subjected to Image Stream analysis, and the percentages of cells with more than 5 LC3B-II puncta were estimated. The data represent the mean ± SEM of 5 independent experiments. *P < 0.05 (Wilcoxon signed-rank test, n=5) (D) In addition to staining the cells with anti-LC3B antibody and Lyso-ID, the nuclei were stained with DAPI. The degree of colocalization between LC3B and the lysosomal marker was visualized by confocal microscopy, using the 60 x objective.](/cms/asset/77d624d1-4edf-4185-8af8-998e1de64074/kaup_a_1009797_f0002_c.jpg)
RA not only enhanced the colocalization of autophagosomes and lysosomes in TLR9- and CD180-stimulated cells, but the number of autophagosomes per cell was also enhanced. Hence, by Image Stream analysis we were able to show that RA significantly enhanced the percentage of TLR9- and CD180-stimulated cells with more than 5 LC3 puncta (). Finally, we used confocal microscopy to confirm the clear impact of RA on both the colocalization between autophagosomes and lysosomes and the enhanced number of LC3 puncta per cell ().
RA-mediated autophagy is associated with enhanced expression of ULK1
We investigated the time course of LC3B-II formation induced by RA in combination with CpG and anti-CD180. As shown in , a slight effect of RA was noticeable already 24 h after addition of the lysosomal inhibitors, but the effect gradually increased over the next 48 h. Concomitant with the formation of LC3B-II, RA also enhanced the TLR9- and CD180-mediated expression of one of the enzymes critical for the formation of autophagosomes, ULK1 (). Despite the very reproducible strong effect of RA on LC3B-II formation noted at 72 h, the maximal induction of ULK1 varied somewhat between 24 and 72 h in the different experiments we performed. Hence, we chose to do statistical analysis of the ULK1 expression after 72 h. As shown in , RA more than doubled the expression of ULK1 in TLR9- and CD180-stimulated cells in a statistically significant manner. The selective effect of RA on ULK1-expression was underlined by the inability of RA to enhance the expression of either ATG7 (), ATG5 (), or ATG3 (data not shown)—important autophagy-related proteins.Citation36 With the ability of RA to induce TLR9- and CD180-mediated expression of ULK1, the impact of RA on ULK1-expression alone and in combination with either TLR9 or CD180 stimulation was explored. shows that RA by itself and in combination with CpG has a moderate but significant impact on the expression of ULK1, whereas a striking effect of RA is observed when combined with TLR9 and CD180 stimulation leading to the doubling of ULK1 expression. RA has no impact on ULK1 levels in CD180-stimulated cells, even though anti-CD180 alone enhances ULK1 levels to some extent.
Figure 3. RA induces LC3B-II and ULK1-expression in a time-dependent manner. (A) CD19+ B cells (0.5 x 106 /ml) were stimulated with CpG (1 μg/ml), anti-CD180 (1 μg/ml) and RA (100 nM). The lysosomal inhibitors pepstatin A (10 μg/ml) and E64d (10 μg/ml) were added after 24 h, and the cells were harvested after 4, 24, 48, or 72 h, respectively. The cells were lysed and subjected to western blot analysis of LC3B-I/II, ULK1 or GAPDH, as described in Materials and Methods. The LC3B protein bands were quantitated, and the LC3B-II/LC3B-I ratios were calculated and displayed in the lower diagram. (B) B cells were stimulated with CpG (1 μg/ml), anti-CD180 (1 μg/ml) and RA (100 nM) and in the absence of lysosomal inhibitors. The cells were subjected to western blot analysis of ULK1, ATG5, ATG7, and GAPDH after 96 h. The lower diagram presents the quantification of the bands representing ULK1. The data represent the mean ± SEM of 5 independent experiments. *P < 0.05 (Wilcoxon signed-rank test, n=5). (C) B cells were stimulated with combinations of CpG (1 μg/ml), anti-CD180 (1 μg/ml) and RA (100 nM) for 48 h before the cells were lysed and subjected to western blot analysis. The ULK1 and CANX levels were quantitated and their ratio displayed in the lower diagram. *P < 0.05 (paired sample Student t test, n=3).
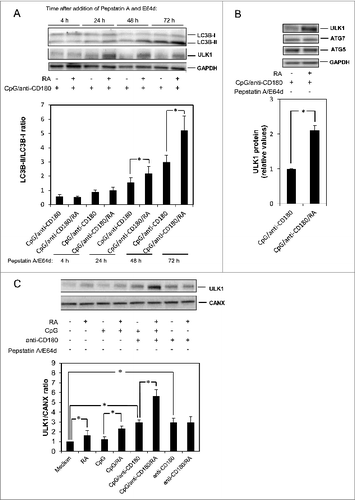
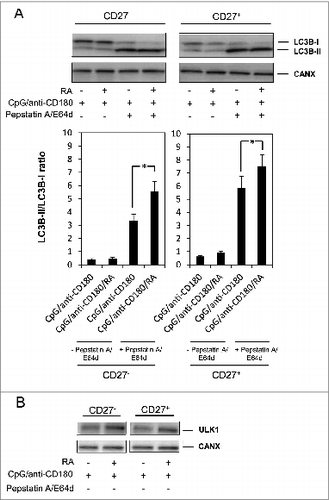
The CD19+ cells comprise a mixture of different subsets of B cells, including the distinct populations of naïve (CD19+ CD27-) and memory (CD19+ CD27+) B cells. We have previously shown that RA enhances TLR9- and CD180-mediated IgG production in both naïve and memory B cells, with the strongest effects seen in memory B cells.Citation23 In order to verify that the effects of RA on autophagy and ULK1 could be seen both in the naïve (CD19+ CD27-) and memory (CD19+ CD27-) subsets of B cells, the CD19+ B cells were fractionated into CD27+ and CD27- B cells and the 2 cell fractions subjected to analysis of the LC3B-II/LC3B-I ratio and ULK1 expression. As shown in , RA enhanced the LC3B-II/LC3B-I ratio and the ULK1 expression in both subpopulations. Finally, in order to exclude the possibility that the isolation method used for purification of the B cells (i.e., with anti-CD19-coated Dynabeads) in any way would interfere with the autophagic process, we also performed experiments on negatively isolated B cells and obtained identical results on both LC3B-II formation and ULK1 expression (data not shown).
Figure 4. RA induces ULK1 and LC3B-II formation in both memory and naïve B cells. (A) CD19+ B cells were separated into CD27+ and CD27− fractions and cells from both populations were stimulated with CpG (1 μg/ml), anti-CD180 (1 μg/ml) and RA with and without the lysosomal inhbitors pepstatin A (10 μg/ml) and E64d (10 μg/ml) for the last 72 h of a 96 h incubation. Cells were harvested and subjected to western blot analysis where the levels of LC3B and CANX were detected. The diagram below shows the quantification of the LC3B-II/LC3B-I ratios. *P < 0.05 (paired sample Student t test, n=3) (B) CD27+ and CD27− cells were stimulated for 48 h in the same manner as in A without lysosomal inhibitors. The levels of ULK1 and CANX were detected by western blot analysis. *P < 0.05 (paired sample Student t test, n=3).
RA-induced autophagosome formation involves RAR-mediated transcription of ULK1
RA is known to exert its effects primarily via the nuclear retinoic acid receptors (RARs),Citation14 but RAR-independent effects of RA have also been reported.Citation37-39 The immunoblot analysis in revealed that RA induced the accumulation of LC3B-II in a dose-dependent manner with notable effects already at 1 nM of RA. The LC3B-II-formation was mediated via RARs, as the RAR agonist TTNPB induced the expression of LC3B-II to a similar or even greater extent than 100 nM of RA, whereas a 500 times excess of the RAR antagonist Ro 41–4253 reversed the LC3B-II-formation induced by 10 nM of RA. Importantly, RA clearly also induced a dose-dependent expression of ULK1 via RARs. This was demonstrated by the TTNPB-mediated induction of ULK1 and by the finding that the RAR-antagonist Ro-41–4253 reversed the effects of RA (). No effects on ULK1 expression were noted in the presence of the RXR-agonist (SR11237) (data not shown), which is in accordance with the notion that the primary active metabolite in B cells is all-trans retinoic acid.Citation40
Figure 5. RA induces LC3B-II-formation via RAR-mediated gene expression. (A) B cells were stimulated with CpG (1 μg/ml) and anti-CD180 (1 μg/ml) in the presence or absence of increasing concentrations of RA (1, 10, or 100 nM), with the RAR-agonist TTNPB (100 nM), or with the RAR-antagonist Ro-41–4253 (5000 nM) in the presence of 10 nM of RA. Lysosomal inhibitors were added after 24 h, and the proteins were subjected to western blot analysis of LC3B-I/II, ULK1, and GAPDH after further culturing for 72 h. (B) B cells were stimulated as in A, and RNA was collected after 48 h as described in Materials and Methods. cDNA was synthesized, and the samples were subjected to quantitative real-time PCR by using primers specific for ULK1 and the controls TBP and ACTB. The diagrams represent the mean ± SEM of 5 independent experiments. *P < 0.05 (Wilcoxon signed-rank test, n=5). (C) B cells were pretreated with CpG (1 μg/ml) and anti-CD180 for 24 h before the transcriptional inhibitor actinomycin D (1 μg/ml) was added to the cell cultures. RA (100 nM) was added after another 2 h and cells were harvested at 0, 2, 4, 8, 24, and 48 h after the addition of actinomycin D. The 2 right columns are controls pretreated with CpG and anti-CD180 for 24 h and then stimulated with RA for another 48 h, all in the absence of actinomycin D. *P < 0.05 (paired sample Student t test, n=4) (D) B cells were pretreated with CpG (1 μg/ml) and anti-CD180 for 24 h before the transcriptional inhibitor actinomycin D (1 μg/ml) was added to the cell cultures. RA (100 nM) was added after another 2 h and the cells were further cultured for 48 h, before the pellets were harvested. The levels of ULK1 and CANX were detected by western blot analysis.
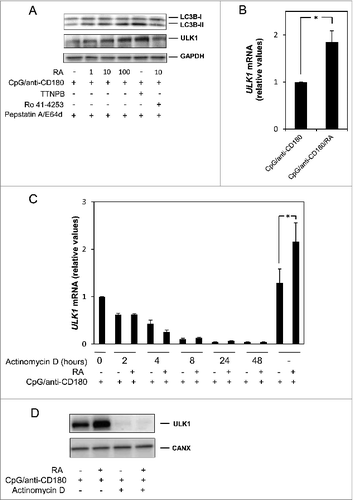
Quantitative real-time PCR (qRT-PCR) suggested that the RA-mediated upregulation of ULK1 in TLR9- and CD180-stimulated cells was at the transcriptional level (). However, in order to exclude the possibility that the enhanced levels of ULK1 mRNA detected in cells stimulated with RA in combination with CpG and anti-CD180 was due to enhanced mRNA stability, we analyzed the half-life of ULK1 mRNA after actinomycin D treatment of the cells. As shown in , the half-life of ULK1 mRNA was unaffected or even reduced (not statistically significant) by RA in the presence of actinomycin D, and the level of ULK1 protein was dramatically reduced (). Taken together, these results support our postulation that RA enhances ULK1 expression at the level of RA-mediated transcription, and this notion was strengthened by in silico analysis of the ULK1 promoter region. Hence, we identified 2 putative RAR and RXR binding sites (DR1 motifs) approximately 500 and 700 bp upstream of the transcription start site (data not shown).
RA-mediated secretion of IgG involves autophagy
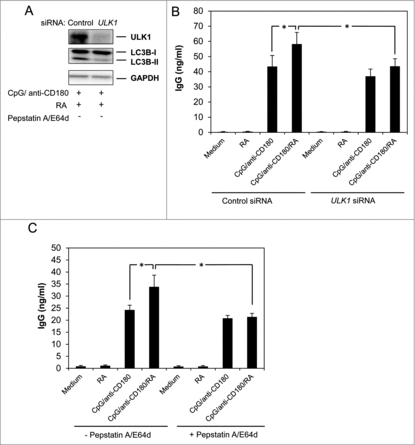
Given the recently acknowledged importance of autophagy in plasma cell formation and Ig secretion,Citation8,9 and having previously shown that RA potentiates IgG secretion in human peripheral B cells stimulated via TLRs,Citation23,24,31 we next asked whether autophagy was involved in RA-mediated induction of IgG. To this end, B cells were transfected with siRNA against ULK1 or with a control siRNA, and as shown in , knocking down ULK1 reversed the RA-induced formation of LC3B-II. Depletion of ULK1 also significantly reduced the level of IgG induced by RA in CpG and anti-CD180-stimulated cells (). To further confirm the involvement of autophagy in RA-mediated IgG secretion, the B cells were stimulated in the presence of the lysosomal inhibitors E64d and pepstatin A. As shown in , blocking autophagic degradation significantly reduced the secretion of IgG from cells stimulated with RA in the presence of CpG and anti-CD180. Of note is that blocking autophagy by either siRNA against ULK1 or by E64d and pepstatin A appeared to selectively inhibit the effect of RA on IgG-secretion in TLR9- and CD180-stimulated cells and not IgG produced by CpG and CD180-treatment alone ().
Figure 6. Knockdown of ULK1 inhibits RA-mediated IgG-secretion. B cells were transfected with siRNA (1.6 mM) against ULK1 or control siRNA (1.6 mM) and stimulated with CpG (1 μg/ml), anti-CD180 (1 μg/ml) and RA (100 nM). (A) After 48 h of stimulation, the cells were subjected to western blot analysis as described in Materials and Methods. (B) After 72 h of stimulation supernatant fractions were collected and subjected to analysis of IgG by ELISA assays. The data represent the mean ± SEM of 10 individual experiments. *P < 0.05 (Wilcoxon signed-rank test, n=10). (C) B cells were stimulated with combinations of CpG (1 μg/ml), anti-CD180 (1 μg/ml) and RA (100 nM) in the presence or absence of the lysosomal inhibitors pepstatin A (10 μg/ml) and E64d (10 μg/ml) added after 24 h of stimulation. The cells were cultured for another 48 h before supernatant fractions were collected and subjected to analysis of IgG by ELISA assays. The data represent the mean ± SEM of 5 independent experiments. *P < 0.05 (Wilcoxon signed-rank test, n=5).
Discussion
It is firmly established that a well-functioning immune system requires an optimal vitamin A status, and it is increasingly evident that vitamin A exerts its immune stimulatory effects by regulating critical processes in both the innate and adaptive parts of the immune system.Citation17,18 We have previously shown that RA enhances proliferation and Ig-production in B cells stimulated via TLRs.Citation23,24,31 In the present study we provide a novel mechanism whereby vitamin A may stimulate the immune system, by demonstrating that RA-induced secretion of IgG from TLR9- and CD180-stimulated human B cells involves ULK1-mediated autophagy. Hence, RA promoted both the formation of LC3B-II and the expression of ULK1, whereas blocking autophagy by knockdown of ULK1 reduced the RA-mediated production of IgG.
Autophagy was originally identified as a process aimed at turning over cellular components and providing building blocks for stressed cells.Citation11,13,41,42 However, during the past few years autophagy has been linked to a number of crucial functions in the immune system.Citation3,7 Of particular relevance for the present study are recent reports on the involvement of autophagy in development and functions of murine plasma cells; Conway and coworkers conclude that autophagy-deficient murine B cells were unable to mount antibody-specific responses to TNP-CGG or H.polygyrus larvae,Citation9 whereas Pengo and coworkers have demonstrated that T cell-dependent and independent antibody responses are reduced in Atg5fl/fl CD19-Cre mice.Citation8 Interestingly, the immunoglobulin synthesis is enhanced in the autophagy-deficient cells in vitro, and this is associated with more ER stress, less ATP, and more death of the mutant plasma cells.Citation8 The authors concluded that autophagy is required for plasma cell homeostasis and humoral immunity by providing a cytoprotective trade-off between Ig synthesis and plasma cell viability.
It is worth noting that the blocking of autophagy by ULK1 siRNA or by lysosomal inhibitors in the present study primarily diminished the effect of RA on IgG-secretion in TLR9- and CD180-stimulated cells and not IgG produced upon CpG and anti-CD180-treatment alone. Thus, our results suggest that although CpG alone or in combination with CD180 increases autophagosome formation in B cells, the short-term IgG response induced by stimulation of human B cells via TLR9 and CD180 may be only marginally dependent on autophagy. This is in keeping with the finding by Pengo et al.Citation8 that plasma cell differentiation is maintained in the absence of autophagy.
While the previously recognized positive control exerted in vivo by autophagy applies to both IgG and IgM responses,Citation8 the stimulatory role of RA-induced autophagy identified herein appears to apply specifically to IgG-secreting cells. Hence, it is possible that the requirement for autophagy is particularly high in B cells stimulated by RA in the presence of CpG and anti-CD180 in order to sustain the increased ER-stress associated with the higher level of IgG produced in these cells. We have not yet assessed whether RA provides a more long-term humoral immune response, but it is possible that the combination of RA, CpG, and anti-CD180 somehow reflects the in vivo environment that B cells normally are exposed to. It is worth noting that autophagy has been implicated in bridging the innate and adaptive immune receptor functions by promoting the fusion between TLR9-containing endosomes and B cell receptor (BcR)-signaling in autophagosomes.Citation43-46 Hence, one may also speculate that enhanced autophagy-mediated bridging of TLR9 and BcR signaling in the autophagosomes might be part of the costimulatory effects of RA on TLR-activated responses in B cells.
One of the key findings in the present study was the ability of RA to induce autophagy in TLR9- and CD180-stimulated B cells via transcriptional induction of ULK1. ULK1 has an important role in regulating the initiation of autophagy,Citation47,48 and it was therefore interesting to find that RA selectively induced the levels of ULK1 and not the other core autophagy-related proteins ATG5 and ATG7. Autophagy induced by TLR9 and CD180 stimulation alone was not associated with enhanced expression of ULK1, further supporting the unique role of RA in this process. RA has previously been linked to autophagy in other cell types and under other conditions.Citation38,49,50 In acute promyelocytic leukemia (APL) cells we found RA to induce autophagy-mediated degradation of the PML-RARA (promyelocytic leukemia/ retinoic acid receptor α) fusion oncoprotein.Citation49 The effect of RA in the APL cells was mediated via inhibition of the MTOR (mechanistic target of rapamycin [serine/threonine kinase]) pathway, and interestingly, siRNA against ULK1 diminished the autophagy-induced degradation of the PML-RARA fusion oncoprotein. By using antibodies specific for MTOR-mediated phosphorylation of ULK1, we could exclude the involvement of MTOR in RA-induced autophagy in TLR-activated B cells (data not shown). Instead we found clear evidence for the involvement of RARs in this process. Hence, the RAR analog TTNPB mimicked the effect of RA both on the expression of ULK1 and LC3B-II levels. Likewise, the RAR antagonist prevented the RA-induced ULK1 expression and LC3B-II formation. As we previously also have shown that RA-induced secretion of IgG in TLR9- and CD180-activated B cells is mediated via RARs,Citation24 it appears that autophagy is a link connecting RAR-mediated gene expression and RA-induced antibody production. This notion was further strengthened by the fact that 1) blocking RAR-mediated gene expression, inhibited the RA-mediated autophagy, and 2) blocking autophagy by ULK1 siRNA nearly reduced the level of RA-induced IgG to the background level observed in CpG and anti-CD180-stimulated cells. The mechanism whereby RA induces ULK1 expression appears to be at the transcriptional level, as demonstrated by the enhanced level of ULK1 mRNA induced by RA. The RAR-mediated transcriptional induction of ULK1 was also supported by in silico analysis of the ULK1 promoter region for identification of putative RAR and RXR response elements. By this method we identified 2 DR1 elements approximately 500 and 700 bp upstream of the transcription start site. Such DR1 elements are known to bind ligand-activated heterodimers (i.e RA-activated RAR-RXR),Citation51 strongly supporting a direct effect of RA on ULK1-transcription. Whether RA-activated RAR-RXR heterodimers indeed bind to the ULK1 promoter warrants future investigation.
We cannot exclude the possibility that RA-induced autophagy in B cells also involves RAR-independent mechanisms. In a recent paper it was reported that RA enhances the acidification of autophagosomes in HeLa cells by promoting fusion with endosomes or lysosomes, and that RA in this manner contributes to increase autophagosomal maturation.Citation38 Our results demonstrating that RA promotes the colocalization between the autophagosomal marker LC3B and the Lyso-ID staining of lysosomes in TLR9- and CD180-stimulated cells, suggest that RA enhances autophagosomal degradation. Whether or not RA-mediated acidification and maturation of autophagosomes is involved in the RA-induced autophagosomal degradation and IgG production is, however, not yet established.
The finding that autophagy is required for RA-mediated IgG-secretion in B cells stimulated via TLR9 and CD180, may have clinical implications. We recently reported that RA is able to partly restore several of the immune defects linked to common variable immune deficiency (CVID).Citation24 CVID is characterized by severely reduced production of Igs, particularly of the IgG type.Citation52 It will be interesting to link the diminished IgG production in CVID patients to possible defects in the autophagic machinery, and it will be particularly important to reveal the positive impact of RA on autophagy-related events in these patients—including IgG production.
Materials and Methods
Reagents and antibodies
Modified CpG oligodeoxynucleotide phosphorothionate (ODN2006) was purchased from Enzo Life Science (ALX-746–056-M001), and purified anti-human CD180/RP105 antibody (clone MHR73–11) was obtained from BioLegend (312902). All-trans retinoic acid (RA) (R2625) from Sigma was kindly provided by the Department of Nutrition (University of Oslo, Oslo, Norway) and stored in darkness at −20°C. In all experiments anti-CD180 and CpG were used at 1 μg/ml, and RA was used at 100 nM if not otherwise stated. TTNPB was purchased from Sigma (T3757), and Ro 41–5253 (BML-GR-110) was obtained from Enzo Life Science. Prolong Gold Antifade reagent (P36935) was from Life Technologies. E64d (BML-PI107–0005) was obtained from Enzo Life Science and pepstatin A (P5318) from Sigma. Monoclonal antibodies against LC3B (clone 5F10; 0231–100) were purchased from Nanotools, whereas the Lyso-ID Red Detection kit (ENZ-51005–500) was from Enzo Life Science. The Alexa Flour 488 goat anti-mouse IgG (H+L) antibody (A-11001) was obtained from Life Technologies. Antibodies against LC3B (2775), ULK1 (4773), ATG5 (2630) and CANX (2433) used in western blot analysis were purchased from Cell Signaling Technology. The GAPDH antibody (G9545) was from Sigma and the antibody against TUBA1A (NB100–690) was obtained from Novus Biologicals. The ATG7 antibody (ab89775) was from Abcam, and the SQSTM1 antibody (MABC32) was obtained from BD Biosciences PharMingen. siRNA specific for human ULK1 (UCACUGACCUGCUCCUUAA) (J-005049–08–0020) and nontargeting control siRNA (D-001810–01–20) were purchased from Dharmacon. Actinomycin D (474790) was purchased from Merck Millipore.
B cell isolation and culturing
CD19 positive human B cells were isolated from buffy coats obtained from the Blood Bank (Oslo University Hospital, Oslo, Norway) by using CD19 antibody-coated Dynabeads (11143D) purchased from Life Technologies as previously described.Citation53 The purity of the isolated B cells was > 99%. The CD27+ and CD27- B cells were isolated using anti-CD27-coated microbeads (130–051–601) from and MS columns (130–042–201), both from Miltenyi Biotech according to the manufacturer's description. The B cells were cultured in RPMI 1640 (Cell Signaling Technology, 11875–093) containing 10% fetal bovine serum, 2 mM glutamine, 125 U/ml penicillin, and 125 μg/ml streptomycin at 37°C in a humidified atmosphere with 5% CO2. The project has been approved by the Regional Ethics Committee of Norway region Sør-Øst A.
Autophagy assays
To examine autophagic flux in B cells, the cells (0.5 × 106/ml) were treated with CpG, anti-CD180 and RA for 24 h before lysosomal protease inhibitors E64d (10 μg/ml) and pepstatin A (10 μg/ml) were added to the cell cultures. The protease inhibitors block lysosomal degradation, resulting in accumulation of autophagosomes.Citation33 LC3-II is bound to the autophagosomal membrane, and is therefore a direct reflection of the numbers of autophagosomes. The level of LC3-II was detected by 3 different methods; by western blot analysis, Image Stream analysis, and by confocal microscopy of immunostained cells (see below for details). By western blot analysis we determined the level of LC3B-II (14 kDa) and the ratio between LC3B-I (16 kDa) and LC3B-II. By Image Stream analysis we determined the extent of colocalization of autophagosomes and lysosomes by staining the B cells with anti-LC3B and Lyso-ID. The extent of colocalization reflects the amount of autophagosomes destined for lysosomal degradation and is considered as a measure of autophagic flux.Citation35 The colocalization of the characteristic LC3B puncta and the lysosomal staining was also visualized by immunocytochemistry and confocal microscopy.
Western blot analysis
Western blot analysis was performed as previously described.Citation54 The signals were detected using the SyngeneChemiGenious camera, and visualized by the GeneSnap software tool (Syngene, Cambridge, UK). Quantification by densitometric analysis of the blots was performed on the primary SGD files using the GeneTools 4.01 program.
Image stream analysis
Stimulated B cells (0.5 × 106/ml) were harvested and fixed with IC Fixation buffer (eBioscience, 00–8222–49), permeabilized with permeabilization buffer (eBioscience, 00–8333–56), and stained with anti-LC3B (Nanotools, 0231–100) as well as with Lyso-ID and Hoechst from the Lyso-ID Red Detection Kit (Enzo Life Science, ENZ-51005–500). Image-based flow was performed by acquiring data of 30000–40000 cells per sample on a 12-channel ISX Imaging Flow Cytometer (Amnis Corporation, Seattle, WA) with 40x objective and EDF filter. Autophagy was quantified as previously described.Citation35 Briefly, colocalization of LC3B and lysosomes was compared between different samples by gating for high bright detail similarity. The BDS threshold was set above 1.5 to define cells with a high degree of colocalization. Punctate LC3B staining was quantified using the spot counting feature in IDEAS. The data was analyzed using the IDEAS 5.0 software (Amnis).
Immunocytochemistry
B cells were resuspended in Krebs/Ringer solution containing 1 μl/ml Lyso-ID, and stained as previously describedCitation55 with anti-LC3B (Nanotools, 0231–100) and Alexa Flour 488 goat anti-mouse (Life Technologies, A-11001). Images were obtained using an Olympus Fluoview 1000 laser scanning confocal microscope (Olympus America Inc., Center Valley, PA) using a 60x objective.
Analysis of ULK1 by real-time quantitative PCR
B cells (0.6 × 106 cells/ml) were stimulated with CpG, anti-CD180, and RA for 48 h before the pellet fractions were collected. RNA was isolated by using the RNeasy Plus Mini Kit from QIAGEN (74134) and subjected to reverse transcription using the iScript cDNA Synthesis Kit (Bio-Rad, 170–88–90). Real-time quantitative PCR (RT-qPCR) was performed using the CFX96 Real-Time PCR Detection system (Bio-Rad Laboratories, Hercules, CA), and SsoFast EvaGreen Supermix (Bio-Rad, 172–5200). Primers toward ULK1 (PPH09320A) and TBP (PPH01091G) were purchased from QIAGEN. Primers against ACTB (actin, β) (forward, 5′-CTGAACCCCAAGGCCAACAG-3′; reverse, 5′-CCAGAGAAGAGGAGGATGCG-3′) were from Sigma-Aldrich.
In silico analysis of the ULK1 promoter
A 5-prime 2 kb sequence of the ULK1 transcription start site (pos. 94657481 to 94659480 of the HSCHR12_CTG2_1 primary assembly) was subjected to in silico analysis for putative transcription factor binding sites using MatInspector v. Eight.1 (Genomatix, Munich, Germany,Citation56and the Matrix Family Librarary v. Nine.1, with a Matrix similarity cut off at 0.75.
Transfection of small-interfering RNA oligonucleotides
B cells (2 × 106/ml) were transfected with 1.6 μM small interfering RNA (siRNA) in a nucleofector device (Amaxa Biosciences, Basel, Switzerland) using the human B cell Nucleofector kit (Amaxa Biosciences, VPA-1001) according to the manufacturer's instructions and the U-15 program.
ELISA assays
B cells (0.5 × 106/ml) were cultured for 3 d in combinations of CpG, anti-CD180 and RA before the supernatant fractions were collected. The levels of IgG were measured in duplicates using an ELISA assay specific for IgG (Bethyl Laboratories, E80–104 and E101).
Statistical analysis
The Wilcoxon signed-rank test and paired sample Student t test were performed using PASW Statistics 18 (IBM, NY). The asterisk (*) represents P < 0.05 in all figures.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Nasser Bastani, Department of Nutrition, Institute of Basic Medical Science, University of Oslo, Oslo, Norway, for providing us with all-trans retinoic acid. We also thank Jannicke Holmseth Bukve and Camilla Solberg for excellent technical support.
Funding
This work has been supported by the University of Oslo, the Freia Research Foundation, the Jahre Research Foundation, the Research Council of Norway and the Norwegian Cancer Society.
References
- Klionsky DJ. Autophagy: from phenomenology to molecular understanding in less than a decade. Nat Rev Mol Cell Biol 2007; 8:931-7; PMID:17712358; http://dx.doi.org/10.1038/nrm2245
- Deretic V. Autophagy in immunity and cell-autonomous defense against intracellular microbes. Immunol Rev 2011; 240:92-104; PMID:21349088; http://dx.doi.org/10.1111/j.1600-065X.2010.00995.x
- Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature 2011; 469:323-35; PMID:21248839; http://dx.doi.org/10.1038/nature09782
- Chaturvedi A, Pierce SK. Autophagy in immune cell regulation and dysregulation. Curr Allergy Asthma Rep 2009; 9:341-6; PMID:19671376; http://dx.doi.org/10.1007/s11882-009-0050-1
- McLeod IX, He Y. Roles of autophagy in lymphocytes: reflections and directions. Cell Mol Immunol 2010; 7:104-7; PMID:20118969; http://dx.doi.org/10.1038/cmi.2009.115
- Liu G, Bi Y, Wang R, Wang X. Self-eating and self-defense: autophagy controls innate immunity and adaptive immunity. J Leukoc Biol 2013; 93:511-9; PMID:23271703; http://dx.doi.org/10.1189/jlb.0812389
- Deretic V, Saitoh T, Akira S. Autophagy in infection, inflammation and immunity. Nat Rev Immunol 2013; 13:722-37; PMID:24064518; http://dx.doi.org/10.1038/nri3532
- Pengo N, Scolari M, Oliva L, Milan E, Mainoldi F, Raimondi A, Fagioli C, Merlini A, Mariani E, Pasqualetto E, et al. Plasma cells require autophagy for sustainable immunoglobulin production. Nat Immunol 2013; 14:298-305; PMID:23354484; http://dx.doi.org/10.1038/ni.2524
- Conway KL, Kuballa P, Khor B, Zhang M, Shi HN, Virgin HW, Xavier RJ. ATG5 regulates plasma cell differentiation. Autophagy 2013; 9:528-37; PMID:23327930; http://dx.doi.org/10.4161/auto.23484
- Pengo N, Cenci S. The role of autophagy in plasma cell ontogenesis. Autophagy 2013; 9:942-4; PMID:23528926; http://dx.doi.org/10.4161/auto.24399
- Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, Ohsumi Y, Tokuhisa T, Mizushima N. The role of autophagy during the early neonatal starvation period. Nature 2004; 432:1032-6; PMID:15525940; http://dx.doi.org/10.1038/nature03029
- Schworer CM, Mortimore GE. Glucagon-induced autophagy and proteolysis in rat liver: mediation by selective deprivation of intracellular amino acids. Proc Natl Acad Sci USA 1979; 76:3169-73; PMID:290994; http://dx.doi.org/10.1073/pnas.76.7.3169
- Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell 2004; 15:1101-11; PMID:14699058; http://dx.doi.org/10.1091/mbc.E03-09-0704
- Balmer JE, Blomhoff R. Gene expression regulation by retinoic acid. J Lipid Res 2002; 43:1773-808; PMID:12401878; http://dx.doi.org/10.1194/jlr.R100015-JLR200
- Chambon P. A decade of molecular biology of retinoic acid receptors. FASEB J 1996; 10:940-54; PMID:8801176
- Napoli JL. Retinoic acid biosynthesis and metabolism. FASEB J 1996; 10:993-1001; PMID:8801182
- Hall JA, Grainger JR, Spencer SP, Belkaid Y. The role of retinoic acid in tolerance and immunity. Immunity 2011; 35:13-22; PMID:21777796; http://dx.doi.org/10.1016/j.immuni.2011.07.002
- Blomhoff R, Blomhoff HK. Overview of retinoid metabolism and function. J Neurobiol 2006; 66:606-30; PMID:16688755; http://dx.doi.org/10.1002/neu.20242
- Ertesvag A, Naderi S, Blomhoff HK. Regulation of B cell proliferation and differentiation by retinoic acid. Semin Immunol 2009; 21:36-41; PMID:18703353; http://dx.doi.org/10.1016/j.smim.2008.06.005
- Ross AC, Chen Q, Ma Y. Vitamin A and retinoic acid in the regulation of B-cell development and antibody production. Vitam Horm 2011; 86:103-26; PMID:21419269; http://dx.doi.org/10.1016/B978-0-12-386960-9.00005-8
- Ochsenbein AF, Zinkernagel RM. Natural antibodies and complement link innate and acquired immunity. Immunol Today 2000; 21:624-30; PMID:11114423; http://dx.doi.org/10.1016/S0167-5699(00)01754-0
- Krieg AM, Yi AK, Matson S et al. CpG motifs in bacterial DNA trigger direct B-cell activation. Nature 1995; 374:546-9; PMID:7700380; http://dx.doi.org/10.1038/374546a0
- Eriksen AB, Indrevaer RL, Holm KL, Landskron J, Blomhoff HK. TLR9-signaling is required for turning retinoic acid into a potent stimulator of RP105 (CD180)-mediated proliferation and IgG synthesis in human memory B cells. Cell Immunol 2012; 279:87-95; PMID:23103284; http://dx.doi.org/10.1016/j.cellimm.2012.09.003
- Indrevaer RL, Holm KL, Aukrust P, Osnes LT, Naderi EH, Fevang B, Blomhoff HK. Retinoic acid improves defective TLR9/RP105-induced immune responses in common variable immunodeficiency-derived B cells. J Immunol 2013; 191:3624-33; PMID:24006462; http://dx.doi.org/10.4049/jimmunol.1300213
- Yamazaki K, Yamazaki T, Taki S, Miyake K, Hayashi T, Ochs HD, Agematsu K. Potentiation of TLR9 responses for human naive B-cell growth through RP105 signaling. Clin Immunol 2010; 135:125-36; PMID:20133206; http://dx.doi.org/10.1016/j.clim.2009.12.013
- Miyake K, Yamashita Y, Ogata M, Sudo T, Kimoto M. RP105, a novel B cell surface molecule implicated in B cell activation, is a member of the leucine-rich repeat protein family. J Immunol 1995; 154:3333-40; PMID:7897216
- Miyake K, Yamashita Y, Hitoshi Y, Takatsu K, Kimoto M. Murine B cell proliferation and protection from apoptosis with an antibody against a 105-kD molecule: unresponsiveness of X-linked immunodeficient B cells. J Exp Med 1994; 180:1217-24; PMID:7523567; http://dx.doi.org/10.1084/jem.180.4.1217
- Stephensen CB. Vitamin A, infection, and immune function. Annu Rev Nutr 2001; 21:167-92; PMID:11375434; http://dx.doi.org/10.1146/annurev.nutr.21.1.167
- Ross AC. Vitamin A supplementation and retinoic acid treatment in the regulation of antibody responses in vivo. Vitam Horm 2007; 75:197-222; PMID:17368317; http://dx.doi.org/10.1016/S0083-6729(06)75008-7
- Ross AC, Chen Q, Ma Y. Augmentation of antibody responses by retinoic acid and costimulatory molecules. Semin Immunol 2009; 21:42-50; PMID:18819820; http://dx.doi.org/10.1016/j.smim.2008.08.004
- Ertesvag A, Aasheim HC, Naderi S, Blomhoff HK. Vitamin A potentiates CpG-mediated memory B-cell proliferation and differentiation: involvement of early activation of p38MAPK. Blood 2007; 109:3865-72; PMID:17209053; http://dx.doi.org/10.1182/blood-2006-09-046748
- Tanida I, Minematsu-Ikeguchi N, Ueno T, Kominami E. Lysosomal turnover, but not a cellular level, of endogenous LC3 is a marker for autophagy. Autophagy 2005; 1:84-91; PMID:16874052; http://dx.doi.org/10.4161/auto.1.2.1697
- Ahlberg J, Berkenstam A, Henell F, Glaumann H. Degradation of short and long lived proteins in isolated rat liver lysosomes. Effects of pH, temperature, and proteolytic inhibitors. J Biol Chem 1985; 260:5847-54; PMID:3988775
- Bjorkoy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, Stenmark H, Johansen T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol 2005; 171:603-14; PMID:16286508; http://dx.doi.org/10.1083/jcb.200507002
- Phadwal K, Alegre-Abarrategui J, Watson AS, Pike L, Anbalagan S, Hammond EM, Wade-Martins R, McMichael A, Klenerman P, Simon AK. A novel method for autophagy detection in primary cells: impaired levels of macroautophagy in immunosenescent T cells. Autophagy 2012; 8:677-89; PMID:22302009; http://dx.doi.org/10.4161/auto.18935
- Mizushima N, Yoshimori T, Ohsumi Y. The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol 2011; 27:107-32; PMID:21801009; http://dx.doi.org/10.1146/annurev-cellbio-092910-154005
- Radominska-Pandya A, Chen G, Czernik PJ, Little JM, Samokyszyn VM, Carter CA, Nowak Gd. Direct interaction of all-trans-retinoic acid with protein kinase C (PKC). Implications for PKC signaling and cancer therapy. J Biol Chem 2000; 275:22324-30; PMID:10748087; http://dx.doi.org/10.1074/jbc.M907722199
- Rajawat Y, Hilioti Z, Bossis I. Retinoic acid induces autophagosome maturation through redistribution of the cation-independent mannose-6-phosphate receptor. Antioxid Redox Signal 2011; 14:2165-77; PMID:20812861; http://dx.doi.org/10.1089/ars.2010.3491
- Ochoa WF, Torrecillas A, Fita I, Verdaguer N, Corbalán-García S, XGomez-Fernandez JC. Retinoic acid binds to the C2-domain of protein kinase C(α). Biochemistry 2003; 42:8774-79; PMID:12873138; http://dx.doi.org/10.1021/bi034713g
- Semba RD. The role of vitamin A and related retinoids in immune function. Nutr Rev 1998; 56:S38-48; PMID:9481123; http://dx.doi.org/10.1111/j.1753-4887.1998.tb01643.x
- Kroemer G, Marino G, Levine B. Autophagy and the integrated stress response. Mol Cell 2010; 40:280-93; PMID:20965422; http://dx.doi.org/10.1016/j.molcel.2010.09.023
- Rabinowitz JD, White E. Autophagy and metabolism. Science 2010; 330:1344-8; PMID:21127245; http://dx.doi.org/10.1126/science.1193497
- Chaturvedi A, Dorward D, Pierce SK. The B cell receptor governs the subcellular location of Toll-like receptor 9 leading to hyperresponses to DNA-containing antigens. Immunity 2008; 28:799-809; PMID:18513998; http://dx.doi.org/10.1016/j.immuni.2008.03.019
- Busconi L, Bauer JW, Tumang JR, Laws A, Perkins-Mesires K, Tabor AS, Lau C, Corley RB, Rothstein TL, Lund FE., et al. Functional outcome of B cell activation by chromatin immune complex engagement of the B cell receptor and TLR9. J Immunol 2007; 179:7397-405; PMID:18025183; http://dx.doi.org/10.4049/jimmunol.179.11.7397
- Kenny EF, Quinn SR, Doyle SL, Vink PM, van Eenennaam H, O'Neill LA. Bruton's tyrosine kinase mediates the synergistic signalling between TLR9 and the B cell receptor by regulating calcium and calmodulin. PLoS One 2013; 8:e74103; PMID:23967355; http://dx.doi.org/10.1371/journal.pone.0074103
- Monroe JG, Keir ME. Bridging Toll-like- and B cell-receptor signaling: meet me at the autophagosome. Immunity 2008; 28:729-31; PMID:18549794; http://dx.doi.org/10.1016/j.immuni.2008.05.006
- Wong PM, Puente C, Ganley IG, Jiang X. The ULK1 complex: sensing nutrient signals for autophagy activation. Autophagy 2013; 9:124-37; PMID:23295650; http://dx.doi.org/10.4161/auto.23323
- Mizushima N. The role of the Atg1/ULK1 complex in autophagy regulation. Curr Opin Cell Biol 2010; 22:132-9; PMID:20056399; http://dx.doi.org/10.1016/j.ceb.2009.12.004
- Isakson P, Bjoras M, Boe SO, Simonsen A. Autophagy contributes to therapy-induced degradation of the PML/RARA oncoprotein. Blood 2010; 116:2324-31; PMID:20574048; http://dx.doi.org/10.1182/blood-2010-01-261040
- Anguiano J, Garner TP, Mahalingam M, Das BC, Gavathiotis E, Cuervo AM. Chemical modulation of chaperone-mediated autophagy by retinoic acid derivatives. Nat Chem Biol 2013; 9:746; PMID:23584676; http://dx.doi.org/10.1038/nchembio1013-746a
- Durand B, Saunders M, Leroy P, Leid M, Chambon P. All-trans and 9-cis retinoic acid induction of CRABPII transcription is mediated by RAR-RXR heterodimers bound to DR1 and DR2 repeated motifs. Cell 1992; 71:73-85; PMID:1327537; http://dx.doi.org/10.1016/0092-8674(92)90267-G
- Cunningham-Rundles C. How I treat common variable immune deficiency. Blood 2010; 116:7-15; PMID:20332369; http://dx.doi.org/10.1182/blood-2010-01-254417
- Naderi S, Blomhoff HK. Retinoic acid prevents phosphorylation of pRB in normal human B lymphocytes: regulation of cyclin E, cyclin A, and p21(Cip1). Blood 1999; 94:1348-58; PMID:10438723
- Follin-Arbelet V, Torgersen ML, Naderi EH, Misund K, Sundan A, Blomhoff HK. Death of multiple myeloma cells induced by cAMP-signaling involves downregulation of Mcl-1 via the JAK/STAT pathway. Cancer Lett 2013; 335:323-31; PMID:23454584; http://dx.doi.org/10.1016/j.canlet.2013.02.042
- Torgersen ML, Engedal N, Boe SO, Hokland P, Simonsen A. Targeting autophagy potentiates the apoptotic effect of histone deacetylase inhibitors in t(8;21) AML cells. Blood 2013; 122:2467-76; PMID:23970379; http://dx.doi.org/10.1182/blood-2013-05-500629
- Cartharius K, Frech K, Grote K, Klocke B, Haltmeier M, Klingenhoff A, Frisch M, Bayerlein M, Werner T. MatInspector and beyond: promoter analysis based on transcription factor binding sites. Bioinformatics 2005; 21:2933-42; PMID:15860560; http://dx.doi.org/10.1093/bioinformatics/bti473