
Abstract
Genetic variations in the autophagic pathway influence genetic predispositions to Crohn disease. Autophagy, the major lysosomal pathway for degrading and recycling cytoplasmic material, constitutes an important homeostatic cellular process. Of interest, single-nucleotide polymorphisms in ATG16L1 (autophagy-related 16-like 1 [S. cerevisiae]), a key component in the autophagic response to invading pathogens, have been associated with an increased risk of developing Crohn disease. The most common and well-studied genetic variant of ATG16L1 (rs2241880; leading to a T300A conversion) exhibits a strong association with risk for developing Crohn disease. The rs2241880 variant plays a crucial role in pathogen clearance, resulting in imbalanced cytokine production, and is linked to other biological processes, such as the endoplasmic reticulum stress/unfolded protein response. In this review, we focus on the importance of ATG16L1 and its genetic variant (T300A) within the elementary biological processes linked to Crohn disease.
Abbreviations:
- ATG16L1, autophagy-related 16-like 1 (S. cerevisiae)
- BCL2, B-cell CLL/lymphoma 2
- DCs, dendritic cells
- ER, endoplasmic reticulum
- IBD, inflammatory bowel disease
- GWAS, genome-wide association studies
- MDP, muramyl dipeptide
- MTOR, mechanistic target of rapamycin
- NFKB, nuclear factor of kappa light polypeptide gene enhancer in B-cells
- NOD2, nucleotide-binding oligomerization domain containing 2
- RIPK2, receptor-interacting serine-threonine kinase 2
- SNP, single-nucleotide polymorphism
- T300A, threonine-to-alanine substitution at amino acid position 300
- TNF/TNF-α, tumor necrosis factor
- UC, ulcerative colitis
- ULK1, unc-51 like autophagy-activating kinase 1
- XBP1, X-box binding protein 1
Introduction
The chronic relapsing intestinal disorders, ulcerative colitis (UC) and Crohn disease, constitute the 2 most common inflammatory bowel disease (IBD) subtypes. The inflammation in UC is continuous and restricted to the mucosal layer of the colon, whereas Crohn disease is characterized by segmental transmural lesions that can affect any part of the gastrointestinal tract.Citation1 Accumulating evidence indicates that these inflammatory conditions occur due to dysfunctional antimicrobial responses combined with genetic susceptibilities.Citation2 Thus, identifying the complex host-microbe interactions and the underlying signaling cascades is of particular importance in revealing the pathogenesis of IBD.
During the past decade, genome-wide association studies (GWAS) and subsequent meta-analyses have been used with great success to identify novel IBD-associated genetic loci. More than 160 IBD-associated loci have been identified thus far,Citation3 often including regions that regulate gene expression in immune cells and the intestinal epithelium.Citation4 The majority of loci are shared by both Crohn disease and UC, although some loci are specific for each of the 2 different disease phenotypes.Citation3
Several single-nucleotide polymorphisms (SNPs) linked to the pathogenesis of Crohn disease are located in genes involved in the pathway of autophagy,Citation5 an important homeostatic cellular process that plays various key roles in both the innate and adaptive immune systems.Citation6 SNPs in ATG16L1 (autophagy-related 16-like 1 [S. cerevisiae]), an essential component of the autophagic pathway, have been associated with an increased risk of Crohn disease. The most common disease-associated ATG16L1 SNP, rs2241880, encodes a missense variant resulting in a threonine-to-alanine substitution at amino acid position 300 (T300A).Citation7 The T300A variant is the most prevalent of all ATG16L1 variants, representing approximately 55% of the alleles in the European population and 20–40% of alleles in other populations.Citation7 Properly functioning ATG16L1 is reported to be required for bacterial clearance and the generation of antigen-specific T-cell responses.Citation8,9 Additionally, crosstalk between ATG16L1 and the cytosolic pathogen receptor NOD2 (nucleotide-binding oligomerization domain-containing 2), which initiates autophagy in response to invading pathogens, suggests the importance of these pathways in the pathogenesis of Crohn disease.Citation10,11 Consequently, there is growing interest in revealing the association between autophagy and this disorder.
The aim of this review is to present an overview of the general mechanisms of autophagy with an emphasis on ATG16L1. Furthermore, we wish to present the available evidence of the role of ATG16L1-mediated responses in the host defense against microorganisms and inflammatory responses in Crohn disease. In summary, targeting autophagy in combination with other pathways might lead to new strategies for the rational management of Crohn disease.
The Process and Regulation of Autophagy
The term autophagy, or “self-eating,” refers primarily to an intracellular lysosomal degradative process for the recycling of long-lived and damaged proteins in the maintenance of normal cellular homeostasis. Autophagy is a pivotal process for cell survival that acts by degrading proteins and membrane lipids, thus providing amino acids and free fatty acids to be used for both protein synthesis and ATP production. The first morphological description of autophagy in mammalian cells was reported in the 1950s.Citation12-14 However, a more detailed understanding of autophagy was accelerated in the 1990s after the identification of autophagy-related (ATG) genes in the yeast S. cerevisiae.Citation15 Since then, several related ATGs have been identified and are highly conserved in various species. Moreover, autophagy is involved in several human diseases (e.g., cancer, neurodegenerative diseases).Citation16,17 Although its precise role is often ambiguous, context-specific, and still under intense investigation, accumulating evidence links autophagy to various cellular processes, including cell maintenance, oxidative cell stress, bacterial handling, and cell death.Citation18
Autophagy is characterized by the formation of double-membrane sequestering compartments, phagophores, which engulf part of the cytoplasm. Upon completion, these form autophagosomes that function to allow the degradation of organelles or cytosolic proteins after fusion with lysosomes. The autophagic process, including the formation of autophagosomes, progresses through several distinct phases: initiation, phagophore elongation, closure, autophagosome maturation, and degradation (). The initiation of autophagy requires an efficient inducer. For instance, nutrient-rich conditions (e.g., amino acids and glucose) suppress autophagy following the activation of negative autophagic regulators, including MTOR (mechanistic target of rapamycin).Citation19 MTOR is in many ways a key hub for autophagy regulation, integrating the signaling of several different pathways capable of controlling autophagy, including the phosphoinositide 3-kinase (PI3K)-AKT pathway. PI3K-AKT-MTOR can be activated by various growth factors and promotes cell growth and differentiation as well as inhibition of apoptosis signaling through MTOR activation.Citation20 Inhibition of MTOR, e.g., through nutrient starvation or rapamycin treatment (an inhibitor of MTOR complex 1, also known as sirolimus), thus promotes autophagy.Citation21 The kinase MTOR is a critical regulator of a downstream protein complex that contains the autophagic protein ULK1 (unc-51 like autophagy activating kinase 1), which interacts with ATG13, ATG101, and RB1CC1/FIP200 (RB1-inducible coiled-coil 1; an Atg17 ortholog).Citation22–24 The ULK1 complex is, however, negatively regulated by MTOR.Citation23,25 In energy-depleting conditions, activation of AMP-activated protein kinase (AMPK) at high AMP levels phosphorylates ULK1 and blocks the inhibitory effect of MTOR.Citation23,26 Activation of the ULK1 complex results in the translocation of the BECN1-containing class III phosphatidylinositol 3-kinase (PtdIns3K) complex to the assembly site of a phagophoreCitation27 to promote the generation of phosphatidylinositol-3-phosphate (PtdIns3P), which is involved in recruiting a number of ATG proteins.Citation28,29 There are multiple PtdIns3K complexes that, in addition to BECN1, include combinations of the proteins ATG14, SH3GLB1/Bif-1 (SH3-domain GRB2-like endophilin B1), UVRAG (UV radiation resistance associated), and AMBRA1 (autophagy/Beclin 1 regulator 1), as well as the negative regulator KIAA0226/Rubicon; BECN1 can also be prevented from binding the complex due to its interaction with BCL2 (B-cell CLL/lymphoma 2).Citation30–34
Figure 1. Schematic stages of the autophagic pathway. The process of autophagy progresses through several stages, including initiation, elongation, maturation, and degradation. Several stimuli are implicated in activation and inhibition of autophagy where the negative autophagic regulator; MTOR, is directly involved. Inhibition of MTOR, e.g., through nutrient starvation, rapamycin, or activation of AMP-activated protein kinase (AMPK) in energy-depleting conditions promotes autophagy. Activated AMPK inhibits also the MTOR downstream protein complex; the autophagic protein ULK1, which interacts with ATG13, ATG101 and RB1CC1. Conversely, nutrient-rich conditions deactivate MTOR and thereby suppress autophagy. Additionally, induction of the PI3K-AKT pathway by growth factors inhibits autophagy by activating MTOR and suppressing the BECN1-containing class III PtdIns3K complex. Activation of the ULK1 complex results in the translocation of the BECN1 complex to the assembly site of a phagophore and generates phosphatidylinositol-3-phosphate (PtdIns3P), which is involved in recruiting a number of ATG proteins. The BECN1 complex is activated by ATG14, SH3GLB1, UVRAG, and AMBRA1, and is suppressed by KIAA0226/Rubicon or BCL2. Elongation of the phagophore requires 2 ubiquitin-like conjugation systems, the ATG12–ATG5-ATG16L1 conjugation system and the microtubule-associated protein 1 light chain 3 (LC3) conjugation system. These conjugation systems require participation a range of proteases and ligases such as ATG3, ATG4B, ATG7, and ATG10 to trigger oligomerization on the outside of the membrane of the growing autophagosome. ATG9 and/or ATG16L1-positive vesicles are involved in membrane trafficking and formation of these autophagosomes. Under conditions of autophagosomal maturation, the ATG proteins are released back into the cytosol, and the final stages of autophagy can then be initiated by autophagosome-lysosome fusion using, for example, interaction between STX17, as an autophagosomal SNARE and its partners SNAP29 and VAMP8.
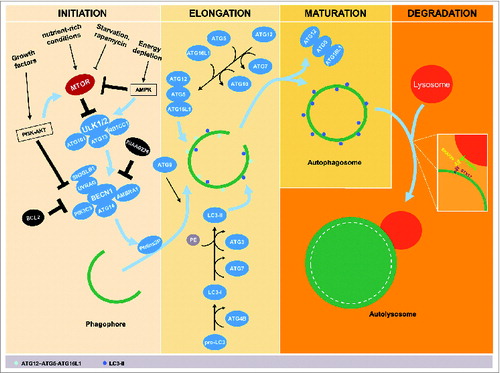
The elongation and expansion of the phagophore involves 2 ubiquitin-like conjugation systems, the ATG12–ATG5-ATG16L1 conjugation system, and the microtubule-associated protein 1 light chain 3 (LC3; Atg8 homolog) conjugation system. The ubiquitin-activating enzyme homolog ATG7, along with the ubiquitin conjugating enzyme ortholog ATG10, mediates conjugation of ATG12 to ATG5. Similarly, ATG4B (a cysteine protease), ATG7, and another conjugating enzyme, ATG3, are involved in the processing of pro-LC3 to form LC3-I followed by modification with the lipid phosphatidylethanolamine (PE) to generate the membrane-associated LC3-II; the latter protein is associated with the membrane of the growing phagophore.Citation35-37 It is important to mention that LC3 is most likely not the only substrate required for autophagy as LC3β (an isoform of LC3) knockout mice only shows minimal phenotypesCitation38 and additionally in humans at least 7 ATG8 orthologs have been identified, suggesting at least partial redundancy.Citation39 Next, the expansion of the phagophore may depend on the trafficking of ATG9- and/or ATG16L1-containing vesicles from multiple membrane sources.Citation40-42 Moreover, the ATG12–ATG5-ATG16L1 conjugate facilitates the lipidation of LC3-I.Citation43 Upon autophagosome closure, the LC3-II on the outer surface of the autophagosome is cleaved from PE and released back into the cytosol, along with the other ATG proteins; the LC3-II that is present on the concave surface remains associated with the completed autophagosome.Citation44-46 As a consequence, LC3-II has widely been used as an autophagic marker.Citation47,48 The final stage of autophagy is initiated by autophagosome-lysosome fusion, a process recently shown to involve interaction between STX17 (syntaxin 17) as an autophagosomal soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) with its targets SNAP29 and VAMP8, which drives the fusion between the outer autophagosomal membrane and the lysosomal membrane.Citation49,50 The intermediate filaments of the cytoskeleton, e.g., the adaptor proteins (YWHA/14-3-3 and VIM/vimentin) that maintain the cell shape and stabilize the cellular organelles, enable the autophagosome to fuse with lysosomes, stabilizing autolysosome formation.Citation51 Fusion with lysosomes creates an acidic environment that results in the degradation of the autophagosome's contents ().
ATG16L1 and Susceptibility to Crohn Disease
The association between SNPs in autophagy and the pathogenesis of Crohn disease has been widely studied. Numerous GWAS reports have identified several SNPs that regulate or communicate with the autophagic pathways and represent risk factors for Crohn disease.Citation7,52-58 For instance, the SNPs in NOD2 are the most prominent risk-associated SNPs and can lead to early disease onset and a more complicated clinical course of Crohn disease, including fibrostenosis and fistulization.Citation11 These genetic variations also exhibit a marked reduction in bacterial autophagy due to the fact that NOD2 recruits ATG16L1 to the site of bacterial entry,Citation10 as described in more detail below. Genetic associations with Crohn disease have also been reported in the following other autophagy-related genes: IRGM (immunity-related GTPase family, M),Citation59 PTPN2 (protein tyrosine phosphatase, non-receptor type 2),Citation60 XBP1 (X-box binding protein 1),Citation61 LRRK2 (leucine-rich repeat kinase 2),Citation60,62 ULK1Citation63 and ATG16L1.Citation7,57
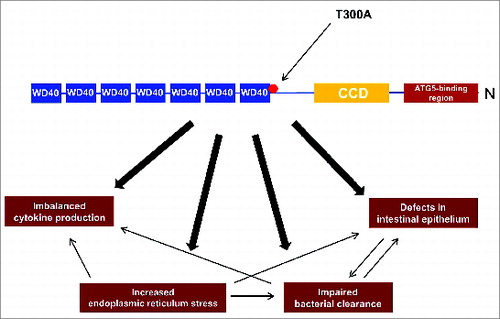
ATG16L1 exists in 3 alternative splicing isoforms (α, β and γ) and is an adaptor protein composed of an N-terminal ATG5-binding region, and an amino-terminal coiled-coil domain (CCD; involved in self-dimerization) followed by 7 tryptophan-aspartic acid (WD40)-repeat domains ().Citation36 ATG16L1 binds noncovalently to the ATG12–ATG5 conjugate and forms approximately 350-kDa tetrameric complexes.Citation35
Figure 2. The involvement of the T300A risk variant in the pathogenesis of Crohn disease. The schematic structure of ATG16L1 contains an N-terminal ATG5-binding region, and an amino-terminal coiled-coil domain (CCD; involved in self-dimerization) followed by 7 tryptophan-aspartic acid (WD40)-repeat domains. In the presence of the T300A variant, several cellular processes are affected. Among other things, defects in the morphology of the intestinal epithelium in key cells, including Paneth cells and goblet cells, are reported and the removal of pathogens is largely defective. In addition the T300A variant results in elevated endoplasmic reticulum stress, which plays a crucial role in impaired pathogen clearance and leads to imbalanced pro-inflammatory cytokines.
In particular, 9 genetic variants of ATG16L1 (rs13412102, rs12471449, rs6431660, rs1441090, rs2289472, rs2241880, rs2241879, rs3792106, and rs4663396) are associated with Crohn disease, although only rs6431660 is weakly associated with UC.Citation52 The variant rs3792106 is linked to sex differences due to a correlation between this gene variant and the fact that women with Crohn disease are more prone to surgical procedures.Citation64 The variants rs2241879 and rs2241880 are, however, more frequent and exhibit the strongest association with this disorder.Citation52 Additionally, these 2 ATG16L1 variants were recently identified at a higher frequency among patients with the chronic inflammatory skin disease palmoplantar pustulosis.Citation65 The role of the remaining ATG16L1 variants remains largely unknown in other contexts.
Although this review focuses on ATG16L1, it is worthwhile to mention that a novel isoform, ATG16L2, containing 2 alternative splicing isoforms (α and β), has recently been discovered.Citation66 SNPs in ATG16L2 are also associated with Crohn disease based on data from a new GWAS conducted in a Korean population.Citation67 Similar to ATG16L1, ATG16L2 forms tetrameric complexes with ATG5 and ATG12. However, ATG16L2 is unable to compensate for the function of ATG16L1 in autophagosome formation,Citation66 indicating nonredundant roles for ATG16L1 and L2 isoforms in the process of autophagy.
Since the first GWAS identification of the common ATG16L1 variant rs2241880 as a risk allele in Crohn disease,Citation7,58,62 this association has been replicated in several independent European disease populations.Citation57,60,68 Nevertheless, the described ATG16L1 association with Crohn disease has not been observed in any Asian meta-analyses.Citation69-71 The rs2241880 variant can be observed in all isoforms of ATG16L1Citation72 and comprises a nonsynonymous coding SNP (adenine (A) → guanine (G) polymorphism) in the coiled-coil domain encoding a threonine-to-alanine substitution. Despite intensive investigation of T300A, the precise mechanism linking this allele to the reduced autophagic state is unknown. Recently, Murthy et al. provided a compelling answer to this question.Citation72 Through multiple sequence alignments of the ATG16L1 protein sequence from several species, Murthy and colleagues predicted and then demonstrated directly that the T300A variant (or the murine equivalent, T316A) is located in the cleavage site for the enzyme CASP3/caspase 3, an endoprotease that hydrolyses peptide bonds at specific sites.Citation72 Moreover, the authors studied the role of CASP3 in the cleavage of the T300A variant in human cells and found that stress signals, such as starvation-induced metabolic stress, death receptor activation by TNF/TNF-α (tumor-necrosis factor) or infections with the pathogenic gut bacterium Yersinia enterocolitica, all result in enhanced CASP3-dependent degradation of ATG16L1T300A. Finally, the data suggest that the therapeutic inhibition of pathways that lead to CASP3 activation might restore autophagy and gut homeostasis, in part by stabilizing the mutant form of ATG16L1.Citation72 A recent paper has shown that ATG16L1T300A might also be susceptible to CASP7 degradation.Citation73,73 However, cells lacking CASP3 are completely incapable of cleaving ATG16L1 even in the presence of activated CASP7.Citation72 Thus, physiologically CASP7 is not capable of cleaving ATG16L1, possibly due to its lower abundance or potency compared to CASP3. Interestingly, ATG16L1 expression can also be affected by microRNA (miRNA) interaction via several distinct miRs, including MIR142-3p, MIR106B, and MIR93.Citation74 This opens the possibility that ATG16L1 might additionally be regulated by epigenetic factors, which implies that autophagy may be impaired in patients with normal genotypes. In fact, adherent-invasive Escherichia coli (AIEC) might suppress ATG16L1 expression and subsequently autophagy and bacterial clearance through upregulation of MIR30C and MIR130A.Citation75
ATG16L1-Dependent Signaling in Crohn Disease
Intestinal epithelium and pathogen clearance
The rapid advances in our understanding of the roles of genetic involvement in the pathogenesis of Crohn disease have demonstrated that deficiencies in ATG16L1 could play a crucial role in the homeostasis of the intestinal epithelium. In fact, in mice hypomorphic for expression of the ATG16L1 protein, the Paneth cells, which comprise highly specialized epithelial cells of the intestine that function in protecting the intestinal stem cell niche using, for instance, the granule exocytosis pathway to secrete antimicrobial peptides and lysozyme, exhibit notable abnormalities in granule exocytosis based on lysozyme staining.Citation76 This confirms the importance of ATG16L1 in maintaining the integrity of the Paneth cell granule exocytosis pathway. In another study, Saitoh et al. generated chimeric mice with a deficient CCD in ATG16L1 in the haematopoietic compartment.Citation77 These mice are highly susceptible to dextran sulfate sodium-induced acute experimental colitis.Citation77 Recently, Lassen et al.Citation73 generated a knock-in mouse model expressing ATG16L1T300A. Such mice do not develop spontaneous inflammation, although they exhibit morphological defects in both Paneth cells and goblet cells. Furthermore, the presence of the T300A mutation in Atg16l1 leads to aberrant functionality of Paneth cells. Coculturing LGR5+ stem cellsCitation78 with Paneth cells from Atg16l1 T300A mice causes a reduced organoid formation, while coculturing with Paneth cells from wild-type mice conversely enhances organoid formation,Citation73 demonstrating a crucial role of ATG16L1 not only in the control of inflammatory immune responses but also for epithelial stem cell maintenance and function in the intestine. Furthermore, elevated secretion of the pro-inflammatory cytokines IL1B (interleukin 1 β) and IL18 can be observed after stimulation with lipopolysaccharide in ATG16L1-deficient myeloid cells.Citation77 Similar observations in both Paneth cells and myeloid cells have been noted in humansCitation76,79 where disease-linked NOD2 and ATG16L1 gene variants have been shown to affect Paneth cell morphology and transcriptome, and to correlate with a more fulminant clinical course of Crohn disease.Citation76,80,81
The elevated production of IL1B in autophagy-deficient mice is associated with an increased inflammasome-mediated processing of pro-IL1B, whereas inflammasome-independent mechanisms, such as IL1B gene transcription, have been described in human cells.Citation77,82,83 Moreover, Lee et al.Citation84 found that in loss-of-functionality of ATG16L1, elevated receptor protein SQSTM1/p62 levels cause increased activation of IL1B.Citation84 This role of SQSTM1 in IL1B signaling has not been confirmed in any other cellular contexts than murine embryonic fibroblasts;Citation84 thus, additional evidence in cell types of relevance for intestinal biology might be useful. Although many of the results are based on knockdown of Atg16l1, and thus may not be representative of the conditions in T300A ATG16L1 Crohn patients, the combined findings from mice and humans indicate that IL1B signaling is increased in ATG16L1-deficient conditions through both transcriptional and post-transcriptional mechanisms resulting in a hyper-inflammatory state. However, results on expression of another cytokine, TNF, have been inconsistent with these findings concerning IL1B,Citation83 and a more complex picture of how ATG16L1 deficiency affects the cytokine microenvironment of the intestinal mucosa in Crohn disease might therefore evolve in the future.
Several studies have reported that autophagy is crucial for the degradation and elimination of intracellular pathogens.Citation85 Thus, a strong association between defective autophagy and impaired properties counteracting bacterial infections has been reported by numerous studies.Citation73,86-89 However, a recent study has revealed a protective role of Atg16l1 deficiency against intestinal disease induced by the bacterial pathogen model, Citrobacter rodentium. This immunosuppressive role of ATG16L1 deficiency is dependent on the presence of NOD2Citation90 and adds to the complexity of the role of ATG16L1 in bacterial clearance.
Of interest, the ATG16L1 deficiency abolishes the ability of cells to form autophagosomes,Citation77 which leads to the disruption of antigen uptakeCitation91 and an insufficient enteric bacterial clearance along with a hyper-inflammatory state of increased secretion of IL1B and IL6.Citation8 This deficiency may affect the composition of the microbiota favoring the growth of AIEC and Y. enterocolica during flares in patients with ileal Crohn disease.Citation76 Studies of the common risk-associated ATG16L1 variant, i.e. T300A, have reported a strong impact on bacterial handling and the generation of antigen-specific CD4+ T-cell responses due to impaired innate immune function.Citation9 For example, studies in human intestinal epithelial cells have demonstrated that the T300A variant leads to dysfunctional bacterial handling, and the efficacy of Salmonella clearance by autophagy is markedly decreased in the presence of the T300A variant relative to the wild-type ATG16L1.Citation92 Muramyl dipeptide (MDP) is a constituent of the bacterial cell wall and the minimal molecular motif capable of activating the NOD2 pathway; ATG16L1T300A blocks MDP-enhanced Salmonella killing in epithelial cells.Citation93 Furthermore, the dendritic cells (DCs) of pediatric patients with Crohn disease carrying the T300A allele reveal a marked impairment in the uptake and processing of bacterial particles from E. coli, which might lead to defects in the interaction between the DCs and intestinal epithelium.Citation91 Inhibition of ATG16L1 by siRNA has further been shown to decrease IL10 secretion and to impair the maturation of dendritic cells.Citation94 The altered function of ATG16L1-underexpressing dendritic cells causes stabilization of the interaction between dendritic cells and T cells, leading to an increased activation of T cells and T helper 17 cell responses characterizing the adaptive immune response in Crohn disease.Citation95 Loss-of-function of ATG16L1 or other autophagic proteins leads to the increased intramacrophagic replication of E. coli and to elevated secretion of pro-inflammatory cytokines, such as IL6 and TNF.Citation96 Moreover, in a recent study monocytes from patients with Crohn disease show enhanced phagocytosis associated with the presence of ATG16L1 and NOD2 variants,Citation97 which might give rise to an accumulation of bacterial products and an increased inflammatory reaction. ATG16L1-mediated autophagy impairment has also been investigated in 2 separate cohorts, which demonstrated a significant association between Crohn disease and the increased susceptibility to Helicobacter pylori infection.Citation98 In another study on T300A Crohn disease patients, a significantly increased presence of AIEC was observed in Paneth cells.Citation99 Therefore, as a result of this deficiency in bacterial clearance, the ATG16L1 risk variant has been associated with shifts in the microbial compositions of the intestineCitation100 and, additionally, has been related to the presence of bacterial DNA and an increased formation of antibacterial antibodies in patients with Crohn disease.Citation101,102 Of interest, bacterial DNA has been correlated with higher disease activity/flares and elevated levels of TNF.Citation101 Thus, this condition might require more aggressive therapeutic approaches to reduce the extent of inflammation and the risk of relapse in Crohn disease patients carrying the T300A variant.
Crosstalk between ATG16L1 and NOD2
As mentioned previously, the ATG16L1-NOD2 interaction plays an interesting role in linking autophagy with other pathogen-associated recognition patterns in response to bacterial clearance.Citation10 A recent major finding has suggested that NOD2 (as well as its homolog, NOD1) might activate autophagy by directly interacting with and recruiting ATG16L1 to the cell membrane at the site of bacterial entry.Citation10 In addition, NOD2 activation by binding MDP has been identified as an effective inducer of autophagy in DCs that is required for both bacterial degradation and the generation of major histocompatibility complex class II antigen-specific CD4+ T-cells.Citation9 The interaction between these 2 disease-associated risk factors underlines the importance of autophagy in the pathogenesis of this disease. However, an unanticipated regulatory role of ATG16L1 in NOD1- and NOD2-mediated inflammatory responses has recently been identified.Citation103 In an autophagy-independent manner, ATG16L1 downregulates NOD-driven inflammatory responses by interfering with and recruiting the central downstream NOD-kinase, RIPK2 (receptor-interacting serine-threonine- kinase 2), into large signaling complexes.Citation103 Interestingly, the disease-associated gene variant, T300A, has also been revealed to possess an altered capacity to downregulate NOD-dependent pro-inflammatory signaling.Citation103 Of note, it is important to realize that these (inflammatory) effects of genetic variation in ATG16L1 may be highly cell-specific, and consequently they should be further explored in different cell type-specific experimental models. Nevertheless, this study, together with other recent data, suggests that defective ATG16L1T300A alleles might cause intestinal inflammation in Crohn disease via autophagy-dependent and autophagy-independent immune processes. For a more detailed overview on the biology of NOD proteins and their interplay with ATG16L1, the reader is referred to published reviews.Citation104,105
Endoplasmic reticulum (ER) stress and unfolded protein response
The ER is a crucial cellular compartment with a key role in the secretory pathway, facilitating the synthesis, modification, and delivery of proteins to the cellular membranes and extracellular environment. Various stresses and pathological stimuli can alter the function of the ER and may result in the accumulation of unfolded or misfolded proteins (a condition called “ER stress”), which subsequently activates a regulatory signaling network known as the unfolded protein responseCitation106 to restore homeostasis. ER stress alters the function of specialized intestinal epithelial cells, named goblet cells, which are responsible for the production of mucin (the key component of the mucus barrier in the gut).Citation107 Moreover, a recent study reported a crucial interaction between ER stress and autophagy among the inflamed Paneth cells of the intestinal epithelium.Citation108 Impairment of the unfolded protein response by deleting its key transcription factor, Xbp1, or secondary to deficiency in the autophagic functions due to the genetic removal of Atg16l1 or Atg7 in intestinal epithelial cells, results in antagonistic compensatory engagement;Citation108 the induction of ER stress in Xbp1 knockout mice triggers the activation of a compensatory autophagic response and the formation of autophagosomes in Paneth cells. Accordingly, failure to remove ER stress-induced responses in mice with genetic deficiencies in either Atg16l1 or Atg7 causes a severe spontaneous Crohn disease-like transmural ileitis through NFKB (nuclear factor of kappa light polypeptide gene enhancer in B-cells 1) activation and TNF signaling.Citation108 It should be noted, however, that pharmacological autophagy inhibition (e.g., by 3-methyladenine) could not always confirm the above-described autophagy-mediated effects, with 3-methyladenine being reported to both induce and decrease toll-like receptor-mediated TNF secretion.Citation83,109 These conflicting data should encourage more careful investigations using both genetic and pharmacological inhibition, as discrepancies may result from off-target or at least autophagy-independent inhibitory effects linked to the chosen inhibition strategy.
Consistent with the genetic impairment of Atg16l1 and Atg7, Deuring et al.Citation99 illustrated that markers of ER stress are significantly elevated in the Paneth cells of patients suffering from Crohn disease with homozygosity or heterozygosity for the ATG16L1 risk allele, even during quiescent disease stages. Further, ileal biopsy samples from Crohn disease patients with ER-stressed Paneth cells exhibit a higher incidence of AIEC and an increased risk for ileal disease, fistulizing disease, and the need for intestinal surgery.Citation99 Thus, both ER stress responses and autophagy seem to be crucial regulatory mechanisms in intestinal homeostasis and pathogenic inflammation in Crohn disease. Recently, other genes related to ER stress were found to be associated with Crohn disease, which further highlights the importance of this pathophysiological mechanism for IBD.Citation5
Linking the T300A Variant to a Clinical Phenotype
As discussed above, genetic variations in autophagy-associated genes have been implicated in the clinical manifestations of Crohn disease. The most frequently described genetic variations are those in NOD2, which are strongly associated with early onset and ileal involvement, indicating a more complicated clinical course of the disease due to fibrostenosis and fistulization.Citation11,110 The contribution of the ATG16L1 variant T300A to the clinical phenotype is not well established, and more investigations are warranted.
Although the association of T300A with the onset of Crohn disease has been controversial,Citation111,112 a clear correlation of the T300A gene variant with the clinical course of ileal disease has been reported.Citation57,112 In a recent study, patients with this disorder homozygous for T300A exhibit a trend toward switching to a stricturing phenotype during the course of the disease, as compared with patients homozygous for the wild-type allele or heterozygous at T300A. In addition, homozygosity for T300A is associated with a major recurrence of clinical relapse and the earlier introduction of immunomodulators (thiopurines and methotrexate).Citation113 These findings are consistent with those of another study describing an association between the presence of bacterial DNA in blood samples and the disease activity of Crohn disease patients possessing the T300A gene variant.Citation101 Therefore, the present results might suggest a more aggressive therapeutic strategy to reduce the extent of inflammation and the risk of relapse among these patients.Citation101
In this respect, the targeting of autophagy has been investigated as a potential treatment for Crohn disease. The induction of autophagy might be a potential approach to increase bacterial killing and to reduce chronic inflammation in this disease. DCs isolated from T300A Crohn disease patients treated with the MTOR inhibitor sirolimus are able to effectively reverse the ability to degrade disease-associated bacteria.Citation9 Thus, a case report on severe refractory colonic and perianal Crohn disease described that the administration of sirolimus caused marked and sustained improvements in disease symptoms as well as in inflammation and endoscopic appearance.Citation114 Similar observations were also made during a recently conducted study of murine experimental colitis in which treatment with sirolimus resulted in a significant histological improvement and protection against mucosal ulcerations.Citation115 Sirolimus treatment also suppressed the mRNA expression of the pro-inflammatory cytokines IL6, TNF and IL17A in addition to enhancing the anti-inflammatory cytokines IL4 and TGFB (transforming growth factor, β).Citation115 A similar drug to sirolimus, everolimus, is also an inhibitor of MTOR and has previously been demonstrated to produce promising results in murine colitis.Citation116 However, everolimus treatment failed to produce a significant difference in a randomized, controlled clinical trial for Crohn disease that was prematurely terminated after only 96 patients were evaluated (36 of whom received everolimus), likely due to the influence of disease-related factors on pharmacokinetics and pharmacodynamics or on the dosage regimens applied.Citation117 Nevertheless, the successful use of sirolimus in a so far underpowered clinical setting suggests that the efficacy of MTOR-based pharmaceutical strategies should be evaluated in randomized clinical trials among patients with Crohn disease refractory to conventional treatment, i.e., immunomodulators and biologics. It might be important to investigate such potential autophagy-targeting agents based on the fact that ATG16L1 plays considerable roles in the regulation of IL1B production and NOD-mediated inflammatory responses. Preserving these activities would consequently require a therapy that induces autophagy and regulates bacterial-mediated responses. Moreover, the clinical efficiency of autophagy-targeting therapies among subgroups of Crohn disease patients with specific genotypes is warranted.
Conclusions
Numerous GWAS reports have confirmed that an enhanced risk for Crohn disease is associated with several genetic variations in the autophagic pathways, including the genetic variant of ATG16L1, T300A. Previous investigations, both in vitro and in vivo, have focused on the major role of ATG16L1 in sensing and degrading intracellular pathogens. Lately, novel functions of ATG16L1 have been reported, including the suppression of NOD-mediated inflammatory responses, the regulation of pro-inflammatory cytokines, and ER stress. In addition, several studies have reported the involvement of the T300A variant in disease onset and the clinical course of Crohn disease. Nonetheless, it must be kept in mind that these ATG16L1 SNPs are also common genetic variants in healthy individuals; as such, these SNPs alone are not sufficient to induce Crohn disease. Therefore, further efforts in revealing the exact triggers of the inflammatory processes of Crohn disease in combination with an improved understanding of the autophagy pathway should be advantageous. Promising results from experimental colitis models and a few published case reports do, however, highlight the potential for the pharmaceutical targeting of autophagy to optimize the clinical management of Crohn disease. Indeed, the current failure of everolimus in a randomized, controlled clinical trial stresses the need for further clinical investigations to optimize and validate autophagy targeting as a potential strategy for the management of Crohn disease, potentially in combination with the management of other pathways, such as IL1B and NOD1/2, or in subsets of patients with specific genotypes using a pharmacogenomic approach.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This paper was supported by grants from P.A. Messerschmidt, and the Hustrus Foundation, the Axel Muusfeldts Foundation, Aase og Ejnar Danielsens Foundation, and Beckett-fonden.
References
- Silverberg MS, Satsangi J, Ahmad T, Arnott ID, Bernstein CN, Brant SR, Caprilli R, Colombel JF, Gasche C, Geboes K, et al. Toward an integrated clinical, molecular and serological classification of inflammatory bowel disease: report of a Working Party of the 2005 Montreal World Congress of Gastroenterology. Can J Gastroenterol 2005; 19 Suppl A:5A-36A; PMID:16151544
- Zhang YZ, Li YY. Inflammatory bowel disease: pathogenesis. World J Gastroenterol 2014; 20:91-9; PMID:24415861; http://dx.doi.org/10.3748/wjg.v20.i1.91
- Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, Lee JC, Schumm LP, Sharma Y, Anderson CA, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 2012; 491:119-24; PMID:23128233; http://dx.doi.org/10.1038/nature11582
- Mokry M, Middendorp S, Wiegerinck CL, Witte M, Teunissen H, Meddens CA, Cuppen E, Clevers H, Nieuwenhuis EE. Many inflammatory bowel disease risk Loci include regions that regulate gene expression in immune cells and the intestinal epithelium. Gastroenterology 2014; 146:1040-7; PMID:24333384; http://dx.doi.org/10.1053/j.gastro.2013.12.003
- Hoefkens E, Nys K, John JM, Van SK, Arijs I, Van der Goten J, Van AG, Agostinis P, Rutgeerts P, Vermeire S, et al. Genetic association and functional role of Crohn disease risk alleles involved in microbial sensing, autophagy, and endoplasmic reticulum (ER) stress. Autophagy 2013; 9:2046-55; PMID:24247223; http://dx.doi.org/10.4161/auto.26337
- Nys K, Agostinis P, Vermeire S. Autophagy: a new target or an old strategy for the treatment of Crohn's disease? Nat Rev Gastroenterol Hepatol 2013; 10:395-401; PMID:23591407; http://dx.doi.org/10.1038/nrgastro.2013.66
- Hampe J, Franke A, Rosenstiel P, Till A, Teuber M, Huse K, Albrecht M, Mayr G, De La Vega FM, Briggs J, et al. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat Genet 2007; 39:207-11; PMID:17200669; http://dx.doi.org/10.1038/ng1954
- Conway KL, Kuballa P, Song JH, Patel KK, Castoreno AB, Yilmaz OH, Jijon HB, Zhang M, Aldrich LN, Villablanca EJ, et al. Atg16l1 is required for autophagy in intestinal epithelial cells and protection of mice from salmonella infection. Gastroenterology 2013; 145:1347-57; PMID:23973919; http://dx.doi.org/10.1053/j.gastro.2013.08.035
- Cooney R, Baker J, Brain O, Danis B, Pichulik T, Allan P, Ferguson DJ, Campbell BJ, Jewell D, Simmons A. NOD2 stimulation induces autophagy in dendritic cells influencing bacterial handling and antigen presentation. Nat Med 2010; 16:90-7; PMID:19966812; http://dx.doi.org/10.1038/nm.2069
- Travassos LH, Carneiro LA, Ramjeet M, Hussey S, Kim YG, Magalhaes JG, Yuan L, Soares F, Chea E, Le BL, et al. Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat Immunol 2010; 11:55-62; PMID:19898471; http://dx.doi.org/10.1038/ni.1823
- Salem M, Seidelin JB, Rogler G, Nielsen OH. Muramyl dipeptide responsive pathways in Crohn's disease: from NOD2 and beyond. Cell Mol Life Sci 2013; 70:3391-404; PMID:23275943; http://dx.doi.org/10.1007/s00018-012-1246-4
- CLARK SL Jr. Cellular differentiation in the kidneys of newborn mice studies with the electron microscope. J Biophys Biochem Cytol 1957; 3:349-62; PMID:13438920
- Klionsky DJ. Autophagy: from phenomenology to molecular understanding in less than a decade. Nat Rev Mol Cell Biol 2007; 8:931-7; PMID:17712358; http://dx.doi.org/10.1038/nrm2245
- Deter RL, Baudhuin P, De DC. Participation of lysosomes in cellular autophagy induced in rat liver by glucagon. J Cell Biol 1967; 35:C11-C16; PMID:6055998
- Ohsumi Y. Historical landmarks of autophagy research. Cell Res 2014; 24:9-23; PMID:24366340; http://dx.doi.org/10.1038/cr.2013.169
- Green DR, Levine B. To be or not to be? How selective autophagy and cell death govern cell fate. Cell 2014; 157:65-75; PMID:24679527; http://dx.doi.org/10.1016/j.cell.2014.02.049
- Giordano S, Darley-Usmar V, Zhang J. Autophagy as an essential cellular antioxidant pathway in neurodegenerative disease. Redox Biol 2014; 2:82-90; PMID:24494187; http://dx.doi.org/10.1016/j.redox.2013.12.013
- Choi AM, Ryter SW, Levine B. Autophagy in human health and disease. N Engl J Med 2013; 368:651-62; PMID:23406030; http://dx.doi.org/10.1056/NEJMra1205406
- Russell RC, Yuan HX, Guan KL. Autophagy regulation by nutrient signaling. Cell Res 2014; 24:42-57; PMID:24343578; http://dx.doi.org/10.1038/cr.2013.166
- Dunlop EA, Tee AR. mTOR and autophagy: a dynamic relationship governed by nutrients and energy. Semin Cell Dev Biol 2014; 36:121-9; PMID:25158238; http://dx.doi.org/10.1016/j.semcdb.2014.08.006
- Randall-Demllo S, Chieppa M, Eri R. Intestinal epithelium and autophagy: partners in gut homeostasis. Front Immunol 2013; 4:301; PMID:24137160; http://dx.doi.org/10.3389/fimmu.2013.00301
- Hara T, Takamura A, Kishi C, Iemura S, Natsume T, Guan JL, Mizushima N. FIP200, a ULK-interacting protein, is required for autophagosome formation in mammalian cells. J Cell Biol 2008; 181:497-510; PMID:18443221; http://dx.doi.org/10.1083/jcb.200712064
- Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 2011; 13:132-41; PMID:21258367; http://dx.doi.org/10.1038/ncb2152
- Mercer CA, Kaliappan A, Dennis PB. A novel, human Atg13 binding protein, Atg101, interacts with ULK1 and is essential for macroautophagy. Autophagy 2009; 5:649-62; PMID:19287211; http://dx.doi.org/10.4161/auto.5.5.8249
- Nazio F, Strappazzon F, Antonioli M, Bielli P, Cianfanelli V, Bordi M, Gretzmeier C, Dengjel J, Piacentini M, Fimia GM, et al. mTOR inhibits autophagy by controlling ULK1 ubiquitylation, self-association and function through AMBRA1 and TRAF6. Nat Cell Biol 2013; 15:406-16; PMID:23524951; http://dx.doi.org/10.1038/ncb2708
- Shang L, Chen S, Du F, Li S, Zhao L, Wang X. Nutrient starvation elicits an acute autophagic response mediated by Ulk1 dephosphorylation and its subsequent dissociation from AMPK. Proc Natl Acad Sci U S A 2011; 108:4788-93; PMID:21383122; http://dx.doi.org/10.1073/pnas.1100844108
- Russell RC, Tian Y, Yuan H, Park HW, Chang YY, Kim J, Kim H, Neufeld TP, Dillin A, Guan KL. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat Cell Biol 2013; 15:741-50; PMID:23685627; http://dx.doi.org/10.1038/ncb2757
- Wirth M, Joachim J, Tooze SA. Autophagosome formation-the role of ULK1 and Beclin1-PI3KC3 complexes in setting the stage. Semin Cancer Biol 2013; 23:301-9; PMID:23727157; http://dx.doi.org/10.1016/j.semcancer.2013.05.007
- Shibutani ST, Yoshimori T. A current perspective of autophagosome biogenesis. Cell Res 2014; 24:58-68; PMID:24296784; http://dx.doi.org/10.1038/cr.2013.159
- Matsunaga K, Saitoh T, Tabata K, Omori H, Satoh T, Kurotori N, Maejima I, Shirahama-Noda K, Ichimura T, Isobe T, et al. Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat Cell Biol 2009; 11:385-96; PMID:19270696; http://dx.doi.org/10.1038/ncb1846
- Takahashi Y, Coppola D, Matsushita N, Cualing HD, Sun M, Sato Y, Liang C, Jung JU, Cheng JQ, Mule JJ, et al. Bif-1 interacts with Beclin 1 through UVRAG and regulates autophagy and tumorigenesis. Nat Cell Biol 2007; 9:1142-51; PMID:17891140; http://dx.doi.org/10.1038/ncb1634
- Liang XH, Kleeman LK, Jiang HH, Gordon G, Goldman JE, Berry G, Herman B, Levine B. Protection against fatal Sindbis virus encephalitis by beclin, a novel Bcl−2-interacting protein. J Virol 1998; 72:8586-8596; PMID:9765397
- Maejima Y, Kyoi S, Zhai P, Liu T, Li H, Ivessa A, Sciarretta S, Del Re DP, Zablocki DK, Hsu CP, et al. Mst1 inhibits autophagy by promoting the interaction between Beclin1 and Bcl−2. Nat Med 2013; 19:1478-88; PMID:24141421; http://dx.doi.org/10.1038/nm.3322
- Fimia GM, Stoykova A, Romagnoli A, Giunta L, Di BS, Nardacci R, Corazzari M, Fuoco C, Ucar A, Schwartz P, et al. Ambra1 regulates autophagy and development of the nervous system. Nature 2007; 447:1121-5; PMID:17589504; http://dx.doi.org/10.1038/nature05925
- Kuma A, Mizushima N, Ishihara N, Ohsumi Y. Formation of the approximately 350-kDa Apg12-Apg5.Apg16 multimeric complex, mediated by Apg16 oligomerization, is essential for autophagy in yeast. J Biol Chem 2002; 277:18619-25; PMID:11897782; http://dx.doi.org/10.1074/jbc.M111889200
- Mizushima N, Kuma A, Kobayashi Y, Yamamoto A, Matsubae M, Takao T, Natsume T, Ohsumi Y, Yoshimori T. Mouse Apg16L, a novel WD-repeat protein, targets to the autophagic isolation membrane with the Apg12-Apg5 conjugate. J Cell Sci 2003; 116:1679-88; PMID:12665549; http://dx.doi.org/10.1242/?jcs.00381
- Fujita N, Itoh T, Omori H, Fukuda M, Noda T, Yoshimori T. The Atg16L complex specifies the site of LC3 lipidation for membrane biogenesis in autophagy. Mol Biol Cell 2008; 19:2092-100; PMID:18321988; http://dx.doi.org/10.1091/mbc.E07-12-1257
- Cann GM, Guignabert C, Ying L, Deshpande N, Bekker JM, Wang L, Zhou B, Rabinovitch M. Developmental expression of LC3alpha and beta: absence of fibronectin or autophagy phenotype in LC3beta knockout mice. Dev Dyn 2008; 237:187-95; PMID:18069693; http://dx.doi.org/10.1002/dvdy.21392
- Shpilka T, Weidberg H, Pietrokovski S, Elazar Z. Atg8: an autophagy-related ubiquitin-like protein family. Genome Biol 2011; 12:226; PMID:21867568; http://dx.doi.org/10.1186/gb-2011-12-7-226
- Puri C, Renna M, Bento CF, Moreau K, Rubinsztein DC. Diverse autophagosome membrane sources coalesce in recycling endosomes. Cell 2013; 154:1285-99; PMID:24034251; http://dx.doi.org/10.1016/j.cell.2013.08.044
- Puri C, Renna M, Bento CF, Moreau K, Rubinsztein DC. ATG16L1 meets ATG9 in recycling endosomes: additional roles for the plasma membrane and endocytosis in autophagosome biogenesis. Autophagy 2014; 10:182-4; PMID:24257061; http://dx.doi.org/10.4161/auto.27174
- Zavodszky E, Vicinanza M, Rubinsztein DC. Biology and trafficking of ATG9 and ATG16L1, two proteins that regulate autophagosome formation. FEBS Lett 2013; 587:1988-96; PMID:23669359; http://dx.doi.org/10.1016/j.febslet.2013.04.025
- Hanada T, Noda NN, Satomi Y, Ichimura Y, Fujioka Y, Takao T, Inagaki F, Ohsumi Y. The Atg12-Atg5 conjugate has a novel E3-like activity for protein lipidation in autophagy. J Biol Chem 2007; 282:37298-302; PMID:17986448; http://dx.doi.org/10.1074/jbc.C700195200
- Ichimura Y, Kirisako T, Takao T, Satomi Y, Shimonishi Y, Ishihara N, Mizushima N, Tanida I, Kominami E, Ohsumi M, et al. A ubiquitin-like system mediates protein lipidation. Nature 2000; 408:488-92; PMID:11100732; http://dx.doi.org/10.1038/35044114
- Fujita N, Hayashi-Nishino M, Fukumoto H, Omori H, Yamamoto A, Noda T, Yoshimori T. An Atg4B mutant hampers the lipidation of LC3 paralogues and causes defects in autophagosome closure. Mol Biol Cell 2008; 19:4651-9; PMID:18768752; http://dx.doi.org/10.1091/mbc.E08-03-0312
- Weidberg H, Shpilka T, Shvets E, Abada A, Shimron F, Elazar Z. LC3 and GATE-16 N termini mediate membrane fusion processes required for autophagosome biogenesis. Dev Cell 2011; 20:444-54; PMID:21497758; http://dx.doi.org/10.1016/j.devcel.2011.02.006
- Mizushima N, Yoshimori T. How to interpret LC3 immunoblotting. Autophagy 2007; 3:542-5; PMID:17611390; http://dx.doi.org/10.4161/auto.4600
- Tanida I, Waguri S. Measurement of autophagy in cells and tissues. Methods Mol Biol 2010; 648:193-214; PMID:20700714; http://dx.doi.org/10.1007/978-1-60761-756-3_13
- Itakura E, Kishi-Itakura C, Mizushima N. The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell 2012; 151:1256-69; PMID:23217709; http://dx.doi.org/10.1016/j.cell.2012.11.001
- Itakura E, Mizushima N. Syntaxin 17: the autophagosomal SNARE. Autophagy 2013; 9:917-9; PMID:23466629; http://dx.doi.org/10.4161/auto.24109
- Wang RC, Wei Y, An Z, Zou Z, Xiao G, Bhagat G, White M, Reichelt J, Levine B. Akt-mediated regulation of autophagy and tumorigenesis through Beclin 1 phosphorylation. Science 2012; 338:956-9; PMID:23112296; http://dx.doi.org/10.1126/science.1225967
- Glas J, Konrad A, Schmechel S, Dambacher J, Seiderer J, Schroff F, Wetzke M, Roeske D, Torok HP, Tonenchi L, et al. The ATG16L1 gene variants rs2241879 and rs2241880 (T300A) are strongly associated with susceptibility to Crohn's disease in the German population. Am J Gastroenterol 2008; 103:682-91; PMID:18162085; http://dx.doi.org/10.1111/j.1572-0241.2007.01694.x
- Van LJ, Russell RK, Nimmo ER, Drummond HE, Smith L, Anderson NH, Davies G, Gillett PM, McGrogan P, Weaver LT, et al. Autophagy gene ATG16L1 influences susceptibility and disease location but not childhood-onset in Crohn's disease in Northern Europe. Inflamm Bowel Dis 2008; 14:338-46; PMID:18088053; http://dx.doi.org/10.1002/ibd.20340
- Weersma RK, Zhernakova A, Nolte IM, Lefebvre C, Rioux JD, Mulder F, van Dullemen HM, Kleibeuker JH, Wijmenga C, Dijkstra G. ATG16L1 and IL23R are associated with inflammatory bowel diseases but not with celiac disease in the Netherlands. Am J Gastroenterol 2008; 103:621-7; PMID:18047540; http://dx.doi.org/10.1111/j.1572-0241.2007.01660.x
- Roberts RL, Gearry RB, Hollis-Moffatt JE, Miller AL, Reid J, Abkevich V, Timms KM, Gutin A, Lanchbury JS, Merriman TR, et al. IL23R R381Q and ATG16L1 T300A are strongly associated with Crohn's disease in a study of New Zealand Caucasians with inflammatory bowel disease. Am J Gastroenterol 2007; 102:2754-61; PMID:17894849; http://dx.doi.org/10.1111/j.1572-0241.2007.01525.x
- Baldassano RN, Bradfield JP, Monos DS, Kim CE, Glessner JT, Casalunovo T, Frackelton EC, Otieno FG, Kanterakis S, Shaner JL, et al. Association of the T300A non-synonymous variant of the ATG16L1 gene with susceptibility to paediatric Crohn's disease. Gut 2007; 56:1171-3; PMID:17625155; http://dx.doi.org/10.1136/gut.2007.122747
- Prescott NJ, Fisher SA, Franke A, Hampe J, Onnie CM, Soars D, Bagnall R, Mirza MM, Sanderson J, Forbes A, et al. A nonsynonymous SNP in ATG16L1 predisposes to ileal Crohn's disease and is independent of CARD15 and IBD5. Gastroenterology 2007; 132:1665-71; PMID:17484864; http://dx.doi.org/10.1053/j.gastro.2007.03.034
- Rioux JD, Xavier RJ, Taylor KD, Silverberg MS, Goyette P, Huett A, Green T, Kuballa P, Barmada MM, Datta LW, et al. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat Genet 2007; 39:596-604; PMID:17435756; http://dx.doi.org/10.1038/ng2032
- Lu XC, Tao Y, Wu C, Zhao PL, Li K, Zheng JY, Li LX. Association between variants of the autophagy related gene-IRGM and susceptibility to Crohn's disease and ulcerative colitis: a meta-analysis. PLoS One 2013; 8:e80602; PMID:24232856; http://dx.doi.org/10.1371/journal.pone.0080602
- Barrett JC, Hansoul S, Nicolae DL, Cho JH, Duerr RH, Rioux JD, Brant SR, Silverberg MS, Taylor KD, Barmada MM, et al. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn's disease. Nat Genet 2008; 40:955-62; PMID:18587394; http://dx.doi.org/10.1038/ng.175
- Kaser A, Lee AH, Franke A, Glickman JN, Zeissig S, Tilg H, Nieuwenhuis EE, Higgins DE, Schreiber S, Glimcher LH, et al. XBP1 links ER stress to intestinal inflammation and confers genetic risk for human inflammatory bowel disease. Cell 2008; 134:743-56; PMID:18775308; http://dx.doi.org/10.1016/j.cell.2008.07.021
- Franke A, McGovern DP, Barrett JC, Wang K, Radford-Smith GL, Ahmad T, Lees CW, Balschun T, Lee J, Roberts R, et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn's disease susceptibility loci. Nat Genet 2010; 42:1118-25; PMID:21102463; http://dx.doi.org/10.1038/ng.717
- Henckaerts L, Cleynen I, Brinar M, John JM, Van SK, Rutgeerts P, Vermeire S. Genetic variation in the autophagy gene ULK1 and risk of Crohn's disease. Inflamm Bowel Dis 2011; 17:1392-7; PMID:21560199; http://dx.doi.org/10.1002/ibd.21486
- Liu LY, Schaub MA, Sirota M, Butte AJ. Transmission distortion in Crohn's disease risk gene ATG16L1 leads to sex difference in disease association. Inflamm Bowel Dis 2012; 18:312-22; PMID:21618365; http://dx.doi.org/10.1002/ibd.21781
- Douroudis K, Kingo K, Traks T, Ratsep R, Silm H, Vasar E, Koks S. ATG16L1 gene polymorphisms are associated with palmoplantar pustulosis. Hum Immunol 2011; 72:613-5; PMID:21513755; http://dx.doi.org/10.1016/j.humimm.2011.03.009
- Ishibashi K, Fujita N, Kanno E, Omori H, Yoshimori T, Itoh T, Fukuda M. Atg16L2, a novel isoform of mammalian Atg16L that is not essential for canonical autophagy despite forming an Atg12-5-16L2 complex. Autophagy 2011; 7:1500-13; PMID:22082872; http://dx.doi.org/10.4161/auto.7.12.18025
- Yang SK, Hong M, Zhao W, Jung Y, Baek J, Tayebi N, Kim KM, Ye BD, Kim KJ, Park SH, et al. Genome-wide association study of Crohn's disease in Koreans revealed three new susceptibility loci and common attributes of genetic susceptibility across ethnic populations. Gut 2014; 63:80-7; PMID:23850713; http://dx.doi.org/10.1136/gutjnl-2013-305193
- Lacher M, Schroepf S, Ballauff A, Lohse P, von SD, Kappler R, Koletzko S. Autophagy 16-like 1 rs2241880 G allele is associated with Crohn's disease in German children. Acta Paediatr 2009; 98:1835-40; PMID:19659808; http://dx.doi.org/10.1111/j.1651-2227.2009.01438.x
- Yamazaki K, Onouchi Y, Takazoe M, Kubo M, Nakamura Y, Hata A. Association analysis of genetic variants in IL23R, ATG16L1 and 5p13.1 loci with Crohn's disease in Japanese patients. J Hum Genet 2007; 52:575-83; PMID:17534574; http://dx.doi.org/10.1007/s10038-007-0156-z
- Yang SK, Park M, Lim J, Park SH, Ye BD, Lee I, Song K. Contribution of IL23R but not ATG16L1 to Crohn's disease susceptibility in Koreans. Inflamm Bowel Dis 2009; 15:1385-90; PMID:19334001; http://dx.doi.org/10.1002/ibd.20921
- Zhi J, Zhi FC, Chen ZY, Yao GP, Guan J, Lin Y, Zhang YC. [Correlation of the autophagosome gene ATG16L1 polymorphism and inflammatory bowel disease]. Nan Fang Yi Ke Da Xue Xue Bao 2008; 28:649-51; PMID:18495612
- Murthy A, Li Y, Peng I, Reichelt M, Katakam AK, Noubade R, Roose-Girma M, DeVoss J, Diehl L, Graham RR, et al. A Crohn's disease variant in Atg16l1 enhances its degradation by caspase 3. Nature 2014; 506:456-62; PMID:24553140; http://dx.doi.org/10.1038/nature13044
- Lassen KG, Kuballa P, Conway KL, Patel KK, Becker CE, Peloquin JM, Villablanca EJ, Norman JM, Liu TC, Heath RJ, et al. Atg16L1 T300A variant decreases selective autophagy resulting in altered cytokine signaling and decreased antibacterial defense. Proc Natl Acad Sci U S A 2014; 111:7741-6; PMID:24821797; http://dx.doi.org/10.1073/pnas.1407001111
- Zhai Z, Wu F, Dong F, Chuang AY, Messer JS, Boone DL, Kwon JH. Human autophagy gene ATG16L1 is post-transcriptionally regulated by MIR142-3p. Autophagy 2014; 10:468-79; PMID:24401604; http://dx.doi.org/10.4161/auto.27553
- Nguyen HT, Dalmasso G, Muller S, Carriere J, Seibold F, Darfeuille-Michaud A. Crohn's disease-associated adherent invasive Escherichia coli modulate levels of microRNAs in intestinal epithelial cells to reduce autophagy. Gastroenterology 2014; 146:508-19; PMID:24148619; http://dx.doi.org/10.1053/j.gastro.2013.10.021
- Cadwell K, Liu JY, Brown SL, Miyoshi H, Loh J, Lennerz JK, Kishi C, Kc W, Carrero JA, Hunt S, et al. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature 2008; 456:259-63; PMID:18849966; http://dx.doi.org/10.1038/nature07416
- Saitoh T, Fujita N, Jang MH, Uematsu S, Yang BG, Satoh T, Omori H, Noda T, Yamamoto N, Komatsu M, et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature 2008; 456:264-8; PMID:18849965; http://dx.doi.org/10.1038/nature07383
- Barker N, van Es JH, Kuipers J, Kujala P, van den Born M, Cozijnsen M, Haegebarth A, Korving J, Begthel H, Peters PJ, et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature 2007; 449:1003-7; PMID:17934449; http://dx.doi.org/10.1038/nature06196
- Thachil E, Hugot JP, Arbeille B, Paris R, Grodet A, Peuchmaur M, Codogno P, Barreau F, Ogier-Denis E, Berrebi D, et al. Abnormal activation of autophagy-induced crinophagy in Paneth cells from patients with Crohn's disease. Gastroenterology 2012; 142:1097-9; PMID:22285936; http://dx.doi.org/10.1053/j.gastro.2012.01.031
- Vandussen KL, Liu TC, Li D, Towfic F, Modiano N, Winter R, Haritunians T, Taylor KD, Dhall D, Targan SR, et al. Genetic variants synthesize to produce paneth cell phenotypes that define subtypes of Crohn's disease. Gastroenterology 2014; 146:200-9; PMID:24076061; http://dx.doi.org/10.1053/j.gastro.2013.09.048
- Cadwell K, Patel KK, Komatsu M, Virgin HW, Stappenbeck TS. A common role for Atg16L1, Atg5 and Atg7 in small intestinal Paneth cells and Crohn disease. Autophagy 2009; 5:250-2; PMID:19139628; http://dx.doi.org/10.4161/auto.5.2.7560
- Harris J, Hartman M, Roche C, Zeng SG, O'Shea A, Sharp FA, Lambe EM, Creagh EM, Golenbock DT, Tschopp J, et al. Autophagy controls IL-1beta secretion by targeting pro-IL-1beta for degradation. J Biol Chem 2011; 286:9587-97; PMID:21228274; http://dx.doi.org/10.1074/jbc.M110.202911
- Crisan TO, Plantinga TS, van de Veerdonk FL, Farcas MF, Stoffels M, Kullberg BJ, van der Meer JW, Joosten LA, Netea MG. Inflammasome-independent modulation of cytokine response by autophagy in human cells. PLoS One 2011; 6:e18666; PMID:21490934; http://dx.doi.org/10.1371/journal.pone.0018666
- Lee J, Kim HR, Quinley C, Kim J, Gonzalez-Navajas J, Xavier R, Raz E. Autophagy suppresses interleukin-1beta (IL-1beta) signaling by activation of p62 degradation via lysosomal and proteasomal pathways. J Biol Chem 2012; 287:4033-40; PMID:22167182; http://dx.doi.org/10.1074/jbc.M111.280065
- Puleston DJ, Simon AK. Autophagy in the immune system. Immunology 2014; 141:1-8; PMID:23991647; http://dx.doi.org/10.1111/imm.12165
- Yano T, Mita S, Ohmori H, Oshima Y, Fujimoto Y, Ueda R, Takada H, Goldman WE, Fukase K, Silverman N, et al. Autophagic control of listeria through intracellular innate immune recognition in drosophila. Nat Immunol 2008; 9:908-16; PMID:18604211; http://dx.doi.org/10.1038/ni.1634
- Kim JJ, Lee HM, Shin DM, Kim W, Yuk JM, Jin HS, Lee SH, Cha GH, Kim JM, Lee ZW, et al. Host cell autophagy activated by antibiotics is required for their effective antimycobacterial drug action. Cell Host Microbe 2012; 11:457-68; PMID:22607799; http://dx.doi.org/10.1016/j.chom.2012.03.008
- Jia K, Thomas C, Akbar M, Sun Q, Adams-Huet B, Gilpin C, Levine B. Autophagy genes protect against Salmonella typhimurium infection and mediate insulin signaling-regulated pathogen resistance. Proc Natl Acad Sci U S A 2009; 106:14564-9; PMID:19667176; http://dx.doi.org/10.1073/pnas.0813319106
- Tung SM, Unal C, Ley A, Pena C, Tunggal B, Noegel AA, Krut O, Steinert M, Eichinger L. Loss of Dictyostelium ATG9 results in a pleiotropic phenotype affecting growth, development, phagocytosis and clearance and replication of Legionella pneumophila. Cell Microbiol 2010; 12:765-80; PMID:20070309; http://dx.doi.org/10.1111/j.1462-5822.2010.01432.x
- Marchiando AM, Ramanan D, Ding Y, Gomez LE, Hubbard-Lucey VM, Maurer K, Wang C, Ziel JW, van RN, Nunez G, et al. A deficiency in the autophagy gene Atg16L1 enhances resistance to enteric bacterial infection. Cell Host Microbe 2013; 14:216-24; PMID:23954160; http://dx.doi.org/10.1016/j.chom.2013.07.013
- Strisciuglio C, Miele E, Wildenberg ME, Giugliano FP, Andreozzi M, Vitale A, Capasso F, Camarca A, Barone MV, Staiano A, et al. T300A variant of autophagy ATG16L1 gene is associated with decreased antigen sampling and processing by dendritic cells in pediatric Crohn's disease. Inflamm Bowel Dis 2013; 19:2339-48; PMID:24022642; http://dx.doi.org/10.1097/MIB.0b013e3182a6a11c
- Kuballa P, Huett A, Rioux JD, Daly MJ, Xavier RJ. Impaired autophagy of an intracellular pathogen induced by a Crohn's disease associated ATG16L1 variant. PLoS One 2008; 3:e3391; PMID:18852889; http://dx.doi.org/10.1371/journal.pone.0003391
- Homer CR, Richmond AL, Rebert NA, Achkar JP, McDonald C. ATG16L1 and NOD2 interact in an autophagy-dependent antibacterial pathway implicated in Crohn's disease pathogenesis. Gastroenterology 2010; 139:1630-41, 1641; PMID:20637199; http://dx.doi.org/10.1053/j.gastro.2010.07.006
- Strisciuglio C, Duijvestein M, Verhaar AP, Vos AC, van den Brink GR, Hommes DW, Wildenberg ME. Impaired autophagy leads to abnormal dendritic cell-epithelial cell interactions. J Crohns Colitis 2013; 7:534-41; PMID:22981596; http://dx.doi.org/10.1016/j.crohns.2012.08.009
- Wildenberg ME, Vos AC, Wolfkamp SC, Duijvestein M, Verhaar AP, te Velde AA, van den Brink GR, Hommes DW. Autophagy attenuates the adaptive immune response by destabilizing the immunologic synapse. Gastroenterology 2012; 142:1493-503; PMID:22370477; http://dx.doi.org/10.1053/j.gastro.2012.02.034
- Lapaquette P, Bringer MA, Darfeuille-Michaud A. Defects in autophagy favour adherent-invasive Escherichia coli persistence within macrophages leading to increased pro-inflammatory response. Cell Microbiol 2012; 14:791-807; PMID:22309232; http://dx.doi.org/10.1111/j.1462-5822.2012.01768.x
- Wolfkamp SC, Verseyden C, Vogels EW, Meisner S, Boonstra K, Peters CP, Stokkers PC, te Velde AA. ATG16L1 and NOD2 polymorphisms enhance phagocytosis in monocytes of Crohn's disease patients. World J Gastroenterol 2014; 20:2664-72; PMID:24627602; http://dx.doi.org/10.3748/wjg.v20.i10.2664
- Raju D, Hussey S, Ang M, Terebiznik MR, Sibony M, Galindo-Mata E, Gupta V, Blanke SR, Delgado A, Romero-Gallo J, et al. Vacuolating cytotoxin and variants in Atg16L1 that disrupt autophagy promote Helicobacter pylori infection in humans. Gastroenterology 2012; 142:1160-71; PMID:22333951; http://dx.doi.org/10.1053/j.gastro.2012.01.043
- Deuring JJ, Fuhler GM, Konstantinov SR, Peppelenbosch MP, Kuipers EJ, de HC, van der Woude CJ. Genomic ATG16L1 risk allele-restricted Paneth cell ER stress in quiescent Crohn's disease. Gut 2013; [Epub ahead of print]:PMID:23964099; http://dx.doi.org/10.1136/gutjnl-2012-303527
- Frank DN, Robertson CE, Hamm CM, Kpadeh Z, Zhang T, Chen H, Zhu W, Sartor RB, Boedeker EC, Harpaz N, et al. Disease phenotype and genotype are associated with shifts in intestinal-associated microbiota in inflammatory bowel diseases. Inflamm Bowel Dis 2011; 17:179-84; PMID:20839241; http://dx.doi.org/10.1002/ibd.21339
- Gutierrez A, Scharl M, Sempere L, Holler E, Zapater P, Almenta I, Gonzalez-Navajas JM, Such J, Wiest R, Rogler G, et al. Genetic susceptibility to increased bacterial translocation influences the response to biological therapy in patients with Crohn's disease. Gut 2014; 63:272-80; PMID:23376290; http://dx.doi.org/10.1136/gutjnl-2012-303557
- Murdoch TB, Xu W, Stempak JM, Landers C, Targan SR, Rotter JI, Silverberg MS. Pattern recognition receptor and autophagy gene variants are associated with development of antimicrobial antibodies in Crohn's disease. Inflamm Bowel Dis 2012; 18:1743-8; PMID:22275320; http://dx.doi.org/10.1002/ibd.22884
- Sorbara MT, Ellison LK, Ramjeet M, Travassos LH, Jones NL, Girardin SE, Philpott DJ. The Protein ATG16L1 Suppresses Inflammatory Cytokines Induced by the Intracellular Sensors Nod1 and Nod2 in an Autophagy-Independent Manner. Immunity 2013; 14:858-73; PMID:24238340; http://dx.doi.org/10.1016/j.immuni.2013.10.013
- Lipinski S, Rosenstiel P. Debug Your Bugs - How NLRs Shape Intestinal Host-Microbe Interactions. Front Immunol 2013; 4:479; PMID:24409180; http://dx.doi.org/10.3389/fimmu.2013.00479
- Ramjeet M, Hussey S, Philpott DJ, Travassos LH. 'Nodophagy': New crossroads in Crohn disease pathogenesis. Gut Microbes 2010; 1:307-15; PMID:21327039; http://dx.doi.org/10.4161/gmic.1.5.13295
- Hollien J. Evolution of the unfolded protein response. Biochim Biophys Acta 2013; 1833:2458-63; PMID:23369734; http://dx.doi.org/10.1016/j.bbamcr.2013.01.016
- Heazlewood CK, Cook MC, Eri R, Price GR, Tauro SB, Taupin D, Thornton DJ, Png CW, Crockford TL, Cornall RJ, et al. Aberrant mucin assembly in mice causes endoplasmic reticulum stress and spontaneous inflammation resembling ulcerative colitis. PLoS Med 2008; 5:e54; PMID:18318598; http://dx.doi.org/10.1371/journal.pmed.0050054
- Adolph TE, Tomczak MF, Niederreiter L, Ko HJ, Bock J, Martinez-Naves E, Glickman JN, Tschurtschenthaler M, Hartwig J, Hosomi S, et al. Paneth cells as a site of origin for intestinal inflammation. Nature 2013; 503:272-6; PMID:24089213; http://dx.doi.org/10.1038/nature12599
- Lin YC, Kuo HC, Wang JS, Lin WW. Regulation of inflammatory response by 3-methyladenine involves the coordinative actions on Akt and glycogen synthase kinase 3beta rather than autophagy. J Immunol 2012; 189:4154-64; PMID:22972931; http://dx.doi.org/10.4049/jimmunol.1102739
- Buning C, Genschel J, Buhner S, Kruger S, Kling K, Dignass A, Baier P, Bochow B, Ockenga J, Schmidt HH, et al. Mutations in the NOD2/CARD15 gene in Crohn's disease are associated with ileocecal resection and are a risk factor for reoperation. Aliment Pharmacol Ther 2004; 19:1073-8; PMID:15142196; http://dx.doi.org/10.1111/j.1365-2036.2004.01967.x
- Gazouli M, Pachoula I, Panayotou I, Mantzaris G, Chrousos G, Anagnou NP, Roma-Giannikou E. NOD2/CARD15, ATG16L1 and IL23R gene polymorphisms and childhood-onset of Crohn's disease. World J Gastroenterol 2010; 16:1753-8; PMID:20380008; http://dx.doi.org/10.3748/wjg.v16.i14.1753
- Amre DK, Mack DR, Morgan K, Krupoves A, Costea I, Lambrette P, Grimard G, Dong J, Feguery H, Bucionis V, et al. Autophagy gene ATG16L1 but not IRGM is associated with Crohn's disease in Canadian children. Inflamm Bowel Dis 2009; 15:501-7; PMID:18985712; http://dx.doi.org/10.1002/ibd.20785
- Strisciuglio C, Auricchio R, Martinelli M, Staiano A, Giugliano FP, Andreozzi M, De RM, Giannetti E, Gianfrani C, Izzo P, et al. Autophagy genes variants and paediatric Crohn's disease phenotype: A single-centre experience. Dig Liver Dis 2014; [Epub ahead of print]:PMID:24656308; http://dx.doi.org/10.1016/j.dld.2014.02.016
- Massey DC, Bredin F, Parkes M. Use of sirolimus (rapamycin) to treat refractory Crohn's disease. Gut 2008; 57:1294-6; PMID:18719139; http://dx.doi.org/10.1136/gut.2008.157297
- Yin H, Li X, Zhang B, Liu T, Yuan B, Ni Q, Hu S, Gu H. Sirolimus ameliorates inflammatory responses by switching the regulatory T/T helper type 17 profile in murine colitis. Immunology 2013; 139:494-502; PMID:23480027; http://dx.doi.org/10.1111/imm.12096
- Matsuda C, Ito T, Song J, Mizushima T, Tamagawa H, Kai Y, Hamanaka Y, Inoue M, Nishida T, Matsuda H, et al. Therapeutic effect of a new immunosuppressive agent, everolimus, on interleukin-10 gene-deficient mice with colitis. Clin Exp Immunol 2007; 148:348-59; PMID:17437423; http://dx.doi.org/10.1111/j.1365-2249.2007.03345.x
- Reinisch W, Panes J, Lemann M, Schreiber S, Feagan B, Schmidt S, Sturniolo GC, Mikhailova T, Alexeeva O, Sanna L, et al. A multicenter, randomized, double-blind trial of everolimus versus azathioprine and placebo to maintain steroid-induced remission in patients with moderate-to-severe active Crohn's disease. Am J Gastroenterol 2008; 103:2284-92; PMID:18671816; http://dx.doi.org/10.1111/j.1572-0241.2008.02024.x